
Deficiency of Urokinase Plasminogen Activator May Impair β Cells Regeneration and Insulin Secretion in Type 2 Diabetes Mellitus

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. uPA-/- Mice Were Prone to HyperGlycemia and Development of T2DM
2.2. uPA-/- Mice Failed to Regain Normal Insulin Secretion and Regeneration
2.3. After Treatment with uPA Plasmid, uPA-/- Mice Were Less Likely to Develop T2DM
2.4. The uPA Secretion Rate Decelerated and uPA Expression on β Cells Declined in High-Glucose Condition
2.5. Insulin Secretion Rate Decelerated and Replication of β Cell Declined in High Glucose after Silencing uPA
2.6. Treatment with uPA Enhanced β Cell Proliferation in Normal Glucose, but Insulin Secretion Rate Decelerated in High Glucose after Treatment with Anti-uPA Antibody
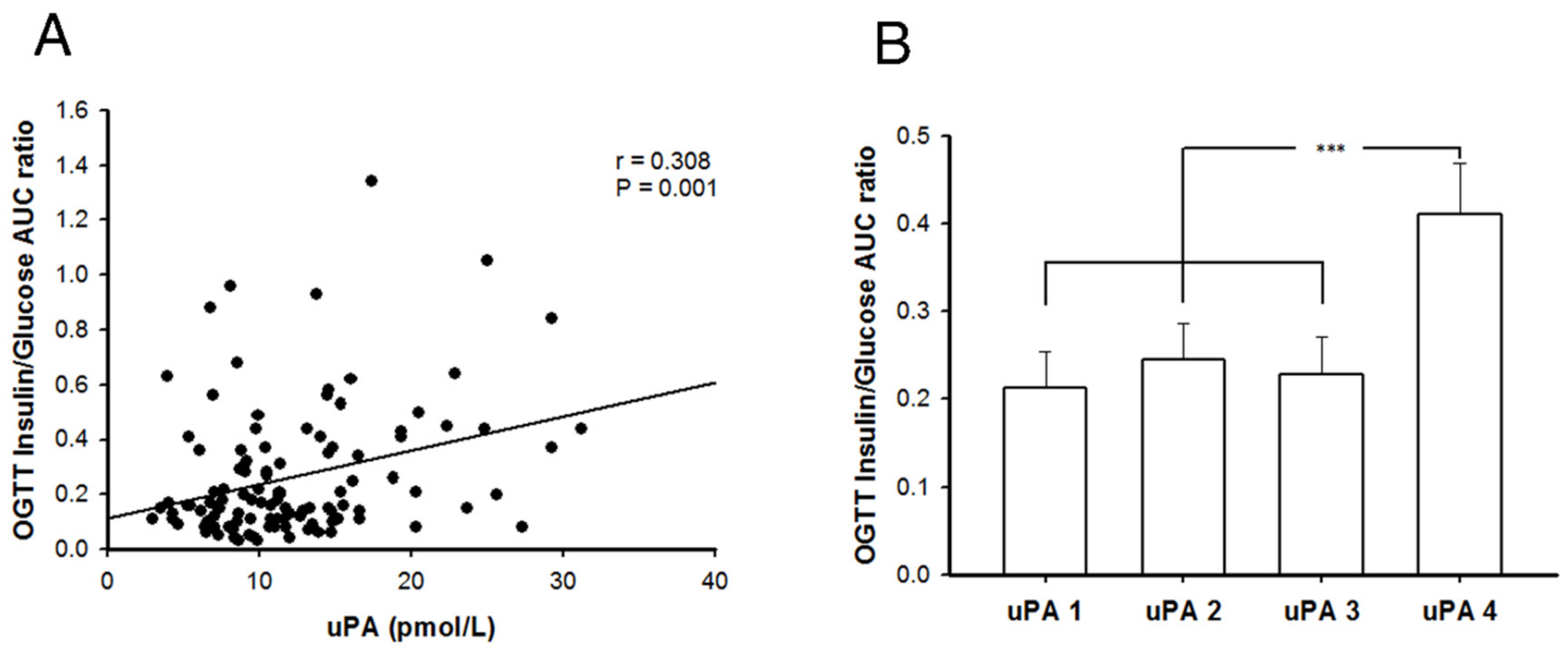
2.7. Better Insulin Secretion Capability after Oral Glucose Challenge in Patients with T2DM and Higher uPA Levels
2.8. Discussion
3. Materials and Methods
3.1. In Vivo Mice Study
3.1.1. Developmental of uPA-/- BALB/c Mice
Backcross Breeding
DNA from Tail Biopsy and Genotyping Identification
3.1.2. Induction of T2DM Mouse Model
3.1.3. uPA-/- Mice Treated with uPA Plasmid
3.1.4. Measurement of Murine uPA, Insulin, Glucagon, Insulin Resistance, and Secretion
3.1.5. Hematoxylin and Eosin (H&E) and IHC Stains of Islet for Insulin, Glucagon, uPA, and PCNA
3.2. In Vitro Cell Study
3.2.1. Assessing uPA Expression and Secretion of Murine β Cells in Normal or High Glucose Medium
3.2.2. Measurement of Insulin Secretion of β Cells with and without uPA Silencing
3.2.3. Measurement of β Cell Proliferation with and without uPA Silencing in High Glucose
3.2.4. Assessment of Insulin Secretion Rate and β Cell Proliferation after Treating with uPA
3.3. Clinical Investigation
3.3.1. Subjects
3.3.2. OGTT
3.3.3. Laboratory Measurement
3.4. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Consent for Publication
Availability of Data and Material
Abbreviations
References
- Levy, J.; Atkinson, A.B.; Bell, P.M.; McCance, D.R.; Hadden, D.R. Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: The 10-year follow-up of the belfast diet study. Diabet. Med. 1998, 15, 290–296. [Google Scholar] [CrossRef]
- U.K. Prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: A progressive disease. U.K. Prospective diabetes study group. Diabetes 1995, 44, 1249–1258. [CrossRef]
- Ahrén, B. Type 2 diabetes, insulin secretion and beta-cell mass. Curr. Mol. Med. 2005, 5, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Grant, P.J. Diabetes mellitus as a prothrombotic condition. J. Intern. Med. 2007, 262, 157–172. [Google Scholar] [CrossRef]
- Mondino, A.; Blasi, F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004, 25, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Bryer, S.C.; Fantuzzi, G.; Van Rooijen, N.; Koh, T.J. Urokinase-type plasminogen activator plays essential roles in macrophage chemotaxis and skeletal muscle regeneration. J. Immunol. 2008, 180, 1179–1188. [Google Scholar] [CrossRef]
- Lee, S.R.; Guo, S.Z.; Scannevin, R.H.; Magliaro, B.C.; Rhodes, K.J.; Wang, X.; Lo, E.H. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci. Lett. 2007, 417, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, N. The urokinase receptor (uPAR) and the upar-associated protein (uPARAP/Endo180): Membrane proteins engaged in matrix turnover during tissue remodeling. Biol. Chem. 2004, 385, 103–136. [Google Scholar] [CrossRef]
- Fuhrman, B. The urokinase system in the pathogenesis of atherosclerosis. Atherosclerosis 2012, 222, 8–14. [Google Scholar] [CrossRef]
- Fisher, E.J.; McLennan, S.V.; Yue, D.K.; Turtle, J.R. High glucose reduces generation of plasmin activity by mesangial cells. Microvasc. Res. 1997, 53, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.R.; Maquoi, E.; Demeulemeester, D.; Van Hoef, B.; Collen, D. Modulation of fibrinolytic and gelatinolytic activity during adipose tissue development in a mouse model of nutritionally induced obesity. Thromb Haemost. 2002, 88, 345–353. [Google Scholar] [PubMed]
- Jimenez, P.A.; Teliska, M.; Liu, B.; Antonaccio, M.J. Urokinase-type plasminogen activator stimulates wound healing in the diabetic mouse. Inflamm. Res. 1997, 46, S169–S170. [Google Scholar] [CrossRef] [PubMed]
- Teramura, Y.; Iwata, H. Islets surface modification prevents blood-mediated inflammatory responses. Bioconjug Chem. 2008, 19, 1389–1395. [Google Scholar] [CrossRef]
- Hayashi, K.; Kojima, R.; Ito, M. Strain differences in the diabetogenic activity of streptozotocin in mice. Biol. Pharm. Bull. 2006, 29, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Fearnside, J.F.; Dumas, M.E.; Rothwell, A.R.; Wilder, S.P.; Cloarec, O.; Toye, A.; Blancher, C.; Holmes, E.; Tatoud, R.; Barton, R.H. Phylometabonomic patterns of adaptation to high fat diet feeding in inbred mice. PLoS ONE 2008, 3, e1668. [Google Scholar] [CrossRef]
- Christow, S.P.; Bychkov, R.; Schroeder, C.; Dietz, R.; Haller, H.; Dumler, I.; Gulba, D.C. Urokinase activates calcium-dependent potassium channels in u937 cells via calcium release from intracellular stores. Eur. J. Biochem. 1999, 265, 264–272. [Google Scholar] [CrossRef]
- Bonner-Weir, S.; Trent, D.F.; Honey, R.N.; Weir, G.C. Responses of neonatal rat islets to streptozotocin: Limited B-cell regeneration and hyperglycemia. Diabetes 1981, 30, 64–69. [Google Scholar] [CrossRef]
- De Koning, E.J.; Bonner-Weir, S.; Rabelink, T.J. Preservation of beta-cell function by targeting beta-cell mass. Trends Pharm. Sci. 2008, 29, 218–227. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Bonner-Weir, S. Pancreatic β Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab. 2017, 27, 30492–30498. [Google Scholar] [CrossRef]
- Shimizu, M.; Hara, A.; Okuno, M.; Matsuno, H.; Okada, K.; Ueshima, S.; Matsuo, O.; Niwa, M.; Akita, K.; Yamada, Y.; et al. Mechanism of retarded liver regeneration in plasminogen activator-deficient mice: Impaired activation of hepatocyte growth factor after fas-mediated massive hepatic apoptosis. Hepatology 2001, 33, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Lluís, F.; Roma, J.; Suelves, M.; Parra, M.; Aniorte, G.; Gallardo, E.; Illa, I.; Rodríguez, L.; Hughes, S.M.; Carmeliet, P.; et al. Urokinase-dependent plasminogen activation is required for efficient skeletal muscle regeneration in vivo. Blood 2001, 97, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Nguyen, M.H.; Yu, B.; Novak, M.L.; Simon, R.H.; Koh, T.J. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood 2009, 114, 5052–5061. [Google Scholar] [CrossRef] [PubMed]
- Mekkawy, A.H.; Pourgholami, M.H.; Morris, D.L. Involvement of urokinase-type plasminogen activator system in cancer: An overview. Med. Res. Rev. 2014, 34, 918–956. [Google Scholar] [CrossRef] [PubMed]
- Cantero, D.; Friess, H.; Deflorin, J.; Zimmermann, A.; Bründler, M.A.; Riesle, E.; Korc, M.; Büchler, M.W. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer. 1997, 75, 388–395. [Google Scholar] [CrossRef]
- Wu, C.Z.; Chang, L.C.; Lin, Y.F.; Hung, Y.J.; Pei, D.; Chu, N.F.; Chen, J.S. Urokinase plasminogen activator receptor and its soluble form in common biopsy-proven kidney diseases and in staging of diabetic nephropathy. Clin. Biochem. 2015, 48, 1324–1329. [Google Scholar] [CrossRef]
- Sherif, E.M.; El Maksood, A.A.A.; Youssef, O.I.; Salah El-Din, N.Y.; Khater, O.K.M. Soluble urokinase plasminogen activator receptor in type 1 diabetic children, relation to vascular complications. J. Diabetes Complications 2019, 33, 628–633. [Google Scholar] [CrossRef]
- Samman Tahhan, A.; Hayek, S.S.; Sandesara, P.; Hajjari, J.; Hammadah, M.; O’Neal, W.T.; Kelli, H.M.; Alkhoder, A.; Ghasemzadeh, N.; Ko, Y.A.; et al. Circulating soluble urokinase plasminogen activator receptor levels and peripheral arterial disease outcomes. Atherosclerosis 2017, 264, 108–114. [Google Scholar] [CrossRef]
- Rotbain Curovic, V.; Theilade, S.; Winther, S.A.; Tofte, N.; Eugen-Olsen, J.; Persson, F.; Hansen, T.W.; Jeppesen, J.; Rossing, P. Soluble Urokinase Plasminogen Activator Receptor Predicts Cardiovascular Events, Kidney Function Decline, and Mortality in Patients With Type 1 Diabetes. Diabetes Care. 2019, 42, 1112–1119. [Google Scholar] [CrossRef]
- Wei, X.; Zhu, D.; Feng, C.; Chen, G.; Mao, X.; Wang, Q.; Wang, J.; Liu, C. Inhibition of peptidyl-prolyl cis-trans isomerase B mediates cyclosporin A-induced apoptosis of islet β cells. Exp. Med. 2018, 16, 3959–3964. [Google Scholar] [CrossRef]
- Tian, H.; Wang, Z.Y. Zinc Chelator Inhibits Zinc-Induced Islet Amyloid Polypeptide Deposition and Apoptosis in INS-1 Cells. Biol. Trace Elem. Res. 2019, 189, 201–208. [Google Scholar] [CrossRef]
- Masiello, P.; Broca, C.; Gross, R.; Roye, M.; Manteghetti, M.; Hillaire-Buys, D.; Novelli, M.; Ribes, G. Experimental NIDDM: Development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 1998, 47, 224–229. [Google Scholar] [CrossRef]
- Nakamura, T.; Terajima, T.; Ogata, T.; Ueno, K.; Hashimoto, N.; Ono, K.; Yano, S. Establishment and pathophysiological characterization of type 2 diabetic mouse model produced by streptozotocin and nicotinamide. Biol. Pharm. Bull. 2006, 29, 1167–1174. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
Sample Availability: Samples are available from the first or corresponding author. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| uPA1 | uPA2 | uPA3 | uPA4 | All | |
|---|---|---|---|---|---|
| n | 28 | 28 | 28 | 28 | 112 |
| uPA (pmol/L) | 6.1 ± 1.42,3,4 | 9.5 ± 0.81,3,4 | 12.9 ± 1.31,2,4 | 20.5 ± 4.91,2,3 | 12.2 ± 6.0 |
| Sex (M/F) | 16/12 | 15/13 | 18/10 | 14/14 | 63/49 |
| Age (year) | 52.6 ± 10.5 | 53.2 ± 9.9 | 52.6 ± 9.2 | 49.4 ± 11.6 | 51.9 ± 10.4 |
| BMI (kg/m2) | 26.1 ± 4.3 | 26.7 ± 4.2 | 26.9 ± 3.8 | 27.2 ± 3.3 | 26.7 ± 3.9 |
| HgbA1c (%) | 8.1 ± 1.22,4 | 7.2 ± 1.11,3 | 8.2 ± 1.82,4 | 6.9 ± 1.21,3 | 7.6 ± 1.4 |
| FPG (mmol/L) | 8.6 ± 2.1 | 8.2 ± 2.4 | 9.6 ± 3.54 | 7.2 ± 3.23 | 8.4 ± 3.0 |
| SBP (mmHg) | 122.8 ± 13.8 | 127.4 ± 13.8 | 125.7 ± 9.5 | 124.6 ± 1.6 | 125.1 ± 14.3 |
| DBP (mmHg) | 74.4 ± 10.2 | 77.6 ± 12.0 | 78.4 ± 8.5 | 78.5 ± 10.5 | 77.2 ± 10.4 |
| TG (mmol/L) | 1.60 ± 0.89 | 1.58 ± 0.91 | 1.95 ± 0.90 | 1.77 ± 1.12 | 1.72 ± 0.96 |
| HDL (mmol/L) | 1.07 ± 0.33 | 1.16 ± 0.35 | 1.16 ± 0.62 | 1.05 ± 0.37 | 1.11 ± 0.42 |
| LDL (mmol/L) | 2.59 ± 0.934 | 2.64 ± 0.654 | 2.79 ± 0.86 | 3.14 ± 0.891,2 | 2.79 ± 0.86 |
| HOMA-β | 32.8 ± 43.8 | 56.6 ± 61.5 | 36.6 ± 42.7 | 108.3 ± 79.81,2,3 | 58.6 ± 65.5 |
| HOMA-IR | 4.76 ± 3.23 | 6.56 ± 5.74 | 7.59 ± 5.73 | 6.30 ± 6.37 | 6.25 ± 5.42 |
| FFA (μmol/L) | 83.4 ± 29.3 | 84.1 ± 29.1 | 89.6 ± 29.4 | 84.6 ± 24.5 | 85.4 ± 27.8 |
| C-peptide (pmol/L) | 676.5 ± 337.2 | 643.6 ± 330.5 | 652.1 ± 322.8 | 826.8 ± 378.0 | 700.4 ± 346.5 |
| Adiponectin | 7.70 ± 4.14 | 9.19 ± 7.78 | 8.28 ± 5.91 | 6.87 ± 3.63 | 8.01 ± 5.61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-Z.; Ou, S.-H.; Chang, L.-C.; Lin, Y.-F.; Pei, D.; Chen, J.-S. Deficiency of Urokinase Plasminogen Activator May Impair β Cells Regeneration and Insulin Secretion in Type 2 Diabetes Mellitus. Molecules 2019, 24, 4208. https://doi.org/10.3390/molecules24234208
Wu C-Z, Ou S-H, Chang L-C, Lin Y-F, Pei D, Chen J-S. Deficiency of Urokinase Plasminogen Activator May Impair β Cells Regeneration and Insulin Secretion in Type 2 Diabetes Mellitus. Molecules. 2019; 24(23):4208. https://doi.org/10.3390/molecules24234208
Chicago/Turabian StyleWu, Chung-Ze, Shih-Hsiang Ou, Li-Chien Chang, Yuh-Feng Lin, Dee Pei, and Jin-Shuen Chen. 2019. "Deficiency of Urokinase Plasminogen Activator May Impair β Cells Regeneration and Insulin Secretion in Type 2 Diabetes Mellitus" Molecules 24, no. 23: 4208. https://doi.org/10.3390/molecules24234208
APA StyleWu, C.-Z., Ou, S.-H., Chang, L.-C., Lin, Y.-F., Pei, D., & Chen, J.-S. (2019). Deficiency of Urokinase Plasminogen Activator May Impair β Cells Regeneration and Insulin Secretion in Type 2 Diabetes Mellitus. Molecules, 24(23), 4208. https://doi.org/10.3390/molecules24234208