
Concepts and Core Principles of Fragment-Based Drug Design


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Library Construction, Preselection, and General Considerations
2.1. General Principles for Library Design
2.2. The Rule of Three
- The molecular weight is ≤300 Da;
- The number of hydrogen-bond donors ≤3;
- Number of hydrogen-bond acceptors ≤3;
- The logP is ≤3.
2.3. Application of Electrophilic Fragments
3. Fragment Hit Identification
3.1. Virtual Screening and Pan-Assay Interference Compounds Filters
3.2. Biophysical Detection Methods for Fragment Screening
3.2.1. Surface Plasmon Resonance
3.2.2. Thermal Shift Assay
3.2.3. Microscale Thermophoresis
3.2.4. X-ray Methods
3.2.5. Nuclear Magnetic Resonance Methods
Saturation-Transfer Difference NMR
Chemical-Shift Perturbation NMR Spectroscopy
19 Fluorine NMR Spectroscopy
3.2.6. Isothermal Titration Calorimetry
3.2.7. Bio-Layer Interferometry
4. Fragment-Hit Qualification
4.1. General Considerations
4.2. Enzyme Targeting
4.3. Functional Enzyme Assays
5. Fragment-To-Lead Optimization
5.1. Growing
5.2. Merging
5.3. Linking
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Erlanson, D.A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, X.; Wang, A.; Zheng, Y.; Gao, Y.; Zhou, J. Evolutions in fragment-based drug design: The deconstruction–reconstruction approach. Drug Discov. Today 2014, 20, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Jahnke, W. Fragment-based Drug Discovery. Lessons and Outlook; Wiley-VCH: Weinheim, Germany, 2016; ISBN 9783527337750. [Google Scholar]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187. [Google Scholar] [CrossRef] [PubMed]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The rule of three for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644. [Google Scholar] [CrossRef]
- Wermuth, C.G. The Practice of Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780124172050. [Google Scholar]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105. [Google Scholar] [CrossRef]
- Schuffenhauer, A.; Ruedisser, S.; Marzinzik, A.; Jahnke, W.; Selzer, P.; Jacoby, E. Library Design for Fragment Based Screening. CTMC 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef]
- Shi, Y.; Itzstein, M.V. How Size Matters: Diversity for Fragment Library Design. Molecules 2019, 24, 2838. [Google Scholar] [CrossRef]
- Heidrich, J.; Sperl, L.E.; Boeckler, F.M. Embracing the Diversity of Halogen Bonding Motifs in Fragment-Based Drug Discovery-Construction of a Diversity-Optimized Halogen-Enriched Fragment Library. Front. Chem. 2019, 7, 9. [Google Scholar] [CrossRef]
- Liu, M.; Quinn, R.J. Fragment-based screening with natural products for novel anti-parasitic disease drug discovery. Expert Opin. Drug Discov. 2019, 14, 1283–1295. [Google Scholar] [CrossRef]
- Kutchukian, P.S.; So, S.-S.; Fischer, C.; Waller, C.L. Fragment library design: Using cheminformatics and expert chemists to fill gaps in existing fragment libraries. Methods Mol. Biol. 2015, 1289, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Sandomenico, A.; Caporale, A.; Doti, N.; Cross, S.; Cruciani, G.; Chambery, A.; de Falco, S.; Ruvo, M. Synthetic Peptide Libraries. From random mixtures to in Vivo testing. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Guillon, R.; Rahimova, R.; Preeti; Egron, D.; Rouanet, S.; Dumontet, C.; Aghajari, N.; Jordheim, L.P.; Chaloin, L.; Peyrottes, S. Lead optimization and biological evaluation of fragment-based cN-II inhibitors. Eur. J. Med. Chem. 2019, 168, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Ferenczy, G.G.; Keserű, G.M. On the enthalpic preference of fragment binding. Med. Chem. Commun. 2016, 7, 332–337. [Google Scholar] [CrossRef]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660. [Google Scholar] [CrossRef] [PubMed]
- Brown, N. In Silico Medicinal Chemistry; Royal Society of Chemistry: Cambridge, UK, 2015; ISBN 978-1-78262-163-8. [Google Scholar]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef]
- Prothiwa, M.; Englmaier, F.; Böttcher, T. Competitive Live-Cell Profiling Strategy for Discovering Inhibitors of the Quinolone Biosynthesis of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2018, 140, 14019–14023. [Google Scholar] [CrossRef]
- Tanaka, D. Fragment-based Drug Discovery: Concept and Aim. Yakugaku Zasshi 2010, 130, 315–323. [Google Scholar] [CrossRef]
- Wasley, J.W. Book Review of Fragment-Based Drug Discovery. A Practical Approach. J. Med. Chem. 2009, 52, 6168. [Google Scholar] [CrossRef]
- Schultes, S.; de Graaf, C.; Haaksma, E.E.; de Esch, I.J.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, e147–e202. [Google Scholar] [CrossRef]
- Orita, M.; Ohno, K.; Warizaya, M.; Amano, Y.; Niimi, T. Lead generation and examples opinion regarding how to follow up hits. Meth. Enzymol. 2011, 493, 383–419. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Abell, C. Fragment-based Drug Discovery; The Royal Society of Chemistry: Camebridge, UK, 2015; ISBN 9781849739085. [Google Scholar]
- Singh, M.; Tam, B.; Akabayov, B. NMR-Fragment Based Virtual Screening: A Brief Overview. Molecules 2018, 23, 233. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Bielska, E.; Lucas, X.; Czerwoniec, A.; Kasprzak, J.M.; Kaminska, K.H.; Bujnicki, J.M. Virtual screening strategies in drug design-methods and applications. BioTechnologia 2011, 92, 249–264. [Google Scholar] [CrossRef]
- Yamaotsu, N.; Hirono, S. In silico fragment-mapping method: A new tool for fragment-based/structure-based drug discovery. J. Comput. Aided Mol. Des. 2018, 32, 1229–1245. [Google Scholar] [CrossRef]
- Wang, T.; Wu, M.-B.; Chen, Z.-J.; Chen, H.; Lin, J.-P.; Yang, L.-R. Fragment-Based Drug Discovery and Molecular Docking in Drug Design. Curr. Pharm. Biotechnol. 2015, 16, 11–25. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.-C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef]
- Melville, J.L.; Burke, E.K.; Hirst, J.D. Machine learning in virtual screening. Comb. Chem. High Throughput Screen. 2009, 12, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment Screening by Surface Plasmon Resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.-P.; Neumann, T.; van Hijfte, L. Fragment-Based Drug Discovery. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W., Hamley, P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 221–249. ISBN 9781118771723. [Google Scholar]
- Grädler, U.; Schwarz, D.; Blaesse, M.; Leuthner, B.; Johnson, T.L.; Bernard, F.; Jiang, X.; Marx, A.; Gilardone, M.; Lemoine, H.; et al. Discovery of novel Cyclophilin D inhibitors starting from three dimensional fragments with millimolar potencies. Bioorg. Med. Chem. Lett. 2019, 126717. [Google Scholar] [CrossRef]
- Cooper, M.A.; Mayr, L. Label-Free Technologies for Drug Discovery; Wiley-Blackwell: Oxford, UK, 2011; ISBN 0470746831. [Google Scholar]
- Schneider, C.S.; Bhargav, A.G.; Perez, J.G.; Wadajkar, A.S.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Surface plasmon resonance as a high throughput method to evaluate specific and non-specific binding of nanotherapeutics. J. Control. Release 2015, 219, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Bleiweis, A.S. Identification of cariogenic bacteria by fluorescent antibody and other techniques: An international symposium. New York City, April 3-4, 1975. Preface. J. Dent. Res. 1976, 55, A4. [Google Scholar] [CrossRef]
- Kroeck, K.G.; Sacco, M.D.; Smith, E.W.; Zhang, X.; Shoun, D.; Akhtar, A.; Darch, S.E.; Cohen, F.; Andrews, L.D.; Knox, J.E.; et al. Discovery of dual-activity small-molecule ligands of Pseudomonas aeruginosa LpxA and LpxD using SPR and X-ray crystallography. Sci. Rep. 2019, 9, 15450. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.B.; Roughley, S.D.; Matassova, N.; Brough, P.A. Off-rate screening (ORS) by surface plasmon resonance. An efficient method to kinetically sample hit to lead chemical space from unpurified reaction products. J. Med. Chem. 2014, 57, 2845–2850. [Google Scholar] [CrossRef]
- Wang, Y.; Edalji, R.P.; Panchal, S.C.; Sun, C.; Djuric, S.W.; Vasudevan, A. Are We There Yet? Applying Thermodynamic and Kinetic Profiling on Embryonic Ectoderm Development (EED) Hit-to-Lead Program. J. Med. Chem. 2017, 60, 8321–8335. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery; Wiley: London, UK, 2013; ISBN 9781118488133. [Google Scholar]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, T.; Junker, H.-D.; Schmidt, K.; Sekul, R. SPR-based fragment screening: Advantages and applications. Curr. Top. Med. Chem. 2007, 7, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Chavanieu, A.; Pugnière, M. Developments in SPR Fragment Screening. Expert Opin. Drug Discov. 2016, 11, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Senisterra, G.; Chau, I.; Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev. Technol. 2012, 10, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Grøftehauge, M.K.; Hajizadeh, N.R.; Swann, M.J.; Pohl, E. Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI). Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 36–44. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212. [Google Scholar] [CrossRef]
- Zhang, R.; Monsma, F. Fluorescence-based thermal shift assays. Curr. Opin. Drug Discov. Devel. 2010, 13, 389–402. [Google Scholar]
- Silvestre, H.L.; Blundell, T.L.; Abell, C.; Ciulli, A. Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc. Natl. Acad. Sci. USA 2013, 110, 12984–12989. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, L.; Gulati, S.; Sears, A.; Stewart, P.L.; Palczewski, K. An effective thiol-reactive probe for differential scanning fluorimetry with a standard real-time polymerase chain reaction device. Anal. Biochem. 2016, 499, 63–65. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, S.; Lapidus, L.J. Fluorescent Probe DCVJ Shows High Sensitivity for Characterization of Amyloid β-Peptide Early in the Lag Phase. Chembiochem 2017, 18, 2205–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, D.; Cardew, E.; Freitag-Pohl, S.; Pohl, E. How to Stabilize Protein: Stability Screens for Thermal Shift Assays and Nano Differential Scanning Fluorimetry in the Virus-X Project. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, A.O.; Szekrenyi, A.; Joosten, H.-J.; Finnigan, J.; Charnock, S.; Fessner, W.-D. nanoDSF as screening tool for enzyme libraries and biotechnology development. FEBS J. 2019, 286, 184–204. [Google Scholar] [CrossRef] [Green Version]
- Mork-Jansson, A.E.; Eichacker, L.A. A strategy to characterize chlorophyll protein interaction in LIL3. Plant Methods 2019, 15, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowska, J.; Żamojć, K.; Wyrzykowski, D.; Wiczk, W.; Chmurzyński, L. Copper(II) complexation by fragment of central part of FBP28 protein from Mus musculus. Biophys. Chem. 2018, 241, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Mashalidis, E.H.; Śledź, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.M.; Breitsprecher, D.; Duhr, S.; Baaske, P.; Schubert, T.; Längst, G. MicroScale Thermophoresis: A Rapid and Precise Method to Quantify Protein-Nucleic Acid Interactions in Solution. Methods Mol. Biol. 2017, 1654, 151–164. [Google Scholar] [CrossRef]
- Schubert, T.; Längst, G. Studying epigenetic interactions using MicroScale Thermophoresis (MST). AIMS Biophys. 2015, 2, 370–380. [Google Scholar] [CrossRef]
- Asmari, M.; Ratih, R.; Alhazmi, H.A.; El Deeb, S. Thermophoresis for characterizing biomolecular interaction. Methods 2018, 146, 107–119. [Google Scholar] [CrossRef]
- Rainard, J.M.; Pandarakalam, G.C.; McElroy, S.P. Using Microscale Thermophoresis to Characterize Hits from High-Throughput Screening: A European Lead Factory Perspective. SLAS Discov. 2018, 23, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, P.; Amaning, K.; Maschberger, M.; Vallee, F.; Steier, V.; Baaske, P.; Duhr, S.; Breitsprecher, D.; Rak, A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screen. 2016, 21, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls. Int. J. Mol. Sci. 2012, 13, 12857–12879. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Hyvönen, M.; Arnold, E. Fragment-based Drug Discovery and X-ray Crystallography; Springer: Heidelberg, Germany, 2012; ISBN 9783642275401. [Google Scholar]
- Müller, I. Guidelines for the successful generation of protein–ligand complex crystals. Acta Crystallogr. D Struct. Biol. 2017, 73, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tounge, B.A.; Parker, M.H. Designing a diverse high-quality library for crystallography-based FBDD screening. Meth. Enzymol. 2011, 493, 3–20. [Google Scholar] [CrossRef]
- Source, D.L. Fragment Screening-XChem-MX-Diamond Light Source. Available online: https://diamond.ac.uk/Instruments/Mx/Fragment-Screening.html (accessed on 18 June 2019).
- Hartshorn, M.J.; Murray, C.W.; Cleasby, A.; Frederickson, M.; Tickle, I.J.; Jhoti, H. Fragment-based lead discovery using X-ray crystallography. J. Med. Chem. 2005, 48, 403–413. [Google Scholar] [CrossRef]
- Schiebel, J.; Krimmer, S.G.; Röwer, K.; Knörlein, A.; Wang, X.; Park, A.Y.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Radeva, N.; et al. High-Throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure 2016, 24, 1398–1409. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J Appl Crystallogr. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; Bradley, A.R.; Collins, P.; Nowak, R.P.; Talon, R.; Marsden, B.D.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123. [Google Scholar] [CrossRef] [PubMed]
- FragMAX-BioMAX Fragment Screening Platform. Available online: https://maxiv.lu.se/accelerators-beamlines/beamlines/biomax/user-access/fragmax-biomax-fragment-screening-platform/ (accessed on 11 November 2019).
- Dias, D.M.; Ciulli, A. NMR approaches in structure-based lead discovery: Recent developments and new frontiers for targeting multi-protein complexes. Prog. Biophys. Mol. Biol. 2014, 116, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, C.; ten Brink, T.; Guichou, J.-F.; Cala, O.; Krimm, I. Comparing binding modes of analogous fragments using NMR in fragment-based drug design: Application to PRDX5. PLoS ONE 2014, 9, e102300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, D.M.; Harris, R.K. Encyclopedia of Magnetic Resonance EMR; Wiley InterScience: New York, NY, USA, 2007; ISBN 9780470034590. [Google Scholar]
- Cala, O.; Krimm, I. Ligand-Orientation Based Fragment Selection in STD NMR Screening. J. Med. Chem. 2015, 58, 8739–8742. [Google Scholar] [CrossRef] [PubMed]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Bhunia, A.; Bhattacharjya, S.; Chatterjee, S. Applications of saturation transfer difference NMR in biological systems. Drug Discov. Today 2012, 17, 505–513. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Pellecchia, M.; Sem, D.S.; Wüthrich, K. Nmr in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211. [Google Scholar] [CrossRef]
- Arntson, K.E.; Pomerantz, W.C.K. Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem. 2016, 59, 5158–5171. [Google Scholar] [CrossRef]
- Okaru, A.O.; Brunner, T.S.; Ackermann, S.M.; Kuballa, T.; Walch, S.G.; Kohl-Himmelseher, M.; Lachenmeier, D.W. Application of 19F NMR Spectroscopy for Content Determination of Fluorinated Pharmaceuticals. J. Anal. Methods Chem. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Tengel, T.; Fex, T.; Emtenas, H.; Almqvist, F.; Sethson, I.; Kihlberg, J. Use of 19F NMR spectroscopy to screen chemical libraries for ligands that bind to proteins. Org. Biomol. Chem. 2004, 2, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Martino, R.; Gilard, V.; Desmoulin, F.; Malet-Martino, M. Interest of fluorine-19 nuclear magnetic resonance spectroscopy in the detection, identification and quantification of metabolites of anticancer and antifungal fluoropyrimidine drugs in human biofluids. Chemotherapy 2006, 52, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Dalvit, C. Design and generation of highly diverse fluorinated fragment libraries and their efficient screening with improved (19) F NMR methodology. ChemMedChem 2013, 8, 2057–2069. [Google Scholar] [CrossRef]
- Leavitt, S.; Freire, E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560–566. [Google Scholar] [CrossRef]
- Damian, L. Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol. Biol. 2013, 1008, 103–118. [Google Scholar] [CrossRef]
- Ward, W.H.J.; Holdgate, G.A. 7 Isothermal Titration Calorimetry in Drug Discovery. In Progress in Medicinal Chemistry; King, F.D., Oxford, A.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 309–376. ISBN 9780444506368. [Google Scholar]
- Wiseman, T.; Williston, S.; Brandts, J.F.; Lin, L.N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [Google Scholar] [CrossRef]
- Schönauer, E.; Kany, A.M.; Haupenthal, J.; Hüsecken, K.; Hoppe, I.J.; Voos, K.; Yahiaoui, S.; Elsässer, B.; Ducho, C.; Brandstetter, H.; et al. Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. [Google Scholar] [CrossRef] [Green Version]
- Ferenczy, G.G.; Keserű, G.M. Thermodynamics of fragment binding. J. Chem. Inf. Model. 2012, 52, 1039–1045. [Google Scholar] [CrossRef]
- Williams, G.; Ferenczy, G.G.; Ulander, J.; Keserű, G.M. Binding thermodynamics discriminates fragments from druglike compounds: A thermodynamic description of fragment-based drug discovery. Drug Discov. Today 2017, 22, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Rees, D. Fragment-based Drug Discovery: Lessons and Outlook. Edited by Daniel, A. Erlanson and Wolfgang Jahnke; Series Editors: Raimund Mannhold, Hugo Kubinyi, and Gerd Folkers. ChemMedChem 2016, 11, 1667. [Google Scholar] [CrossRef]
- Torres, F.E.; Recht, M.I.; Coyle, J.E.; Bruce, R.H.; Williams, G. Higher throughput calorimetry: Opportunities, approaches and challenges. Curr. Opin. Struct. Biol. 2010, 20, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.W.; Verdonk, M.L. Entropic consequences of linking ligands. In Fragment-Based Approaches in Drug Discovery; WILEY-VCG: Weinheim, Germany, 2006; ISBN 9783527312917. [Google Scholar]
- Davis, B.J.; Erlanson, D.A. Learning from our mistakes: The ‘unknown knowns’ in fragment screening. Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdiche, Y.; Malashock, D.; Pinkerton, A.; Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 2008, 377, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kamat, V.; Rafique, A. Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal. Biochem. 2017, 536, 16–31. [Google Scholar] [CrossRef]
- Yu, Y.; Mitchell, S.; Lynaugh, H.; Brown, M.; Nobrega, R.P.; Zhi, X.; Sun, T.; Caffry, I.; Cao, Y.; Yang, R.; et al. Understanding ForteBio’s Sensors for High-Throughput Kinetic and Epitope Screening for Purified Antibodies and Yeast Culture Supernatant. J. Biomol. Screen. 2016, 21, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Shultz, M.D. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [Google Scholar] [CrossRef]
- Mortenson, P.N.; Murray, C.W. Assessing the lipophilicity of fragments and early hits. J. Comput. Aided Mol. Des. 2011, 25, 663–667. [Google Scholar] [CrossRef]
- Marion, D. An introduction to biological NMR spectroscopy. Mol. Cell. Proteomics 2013, 12, 3006–3025. [Google Scholar] [CrossRef] [Green Version]
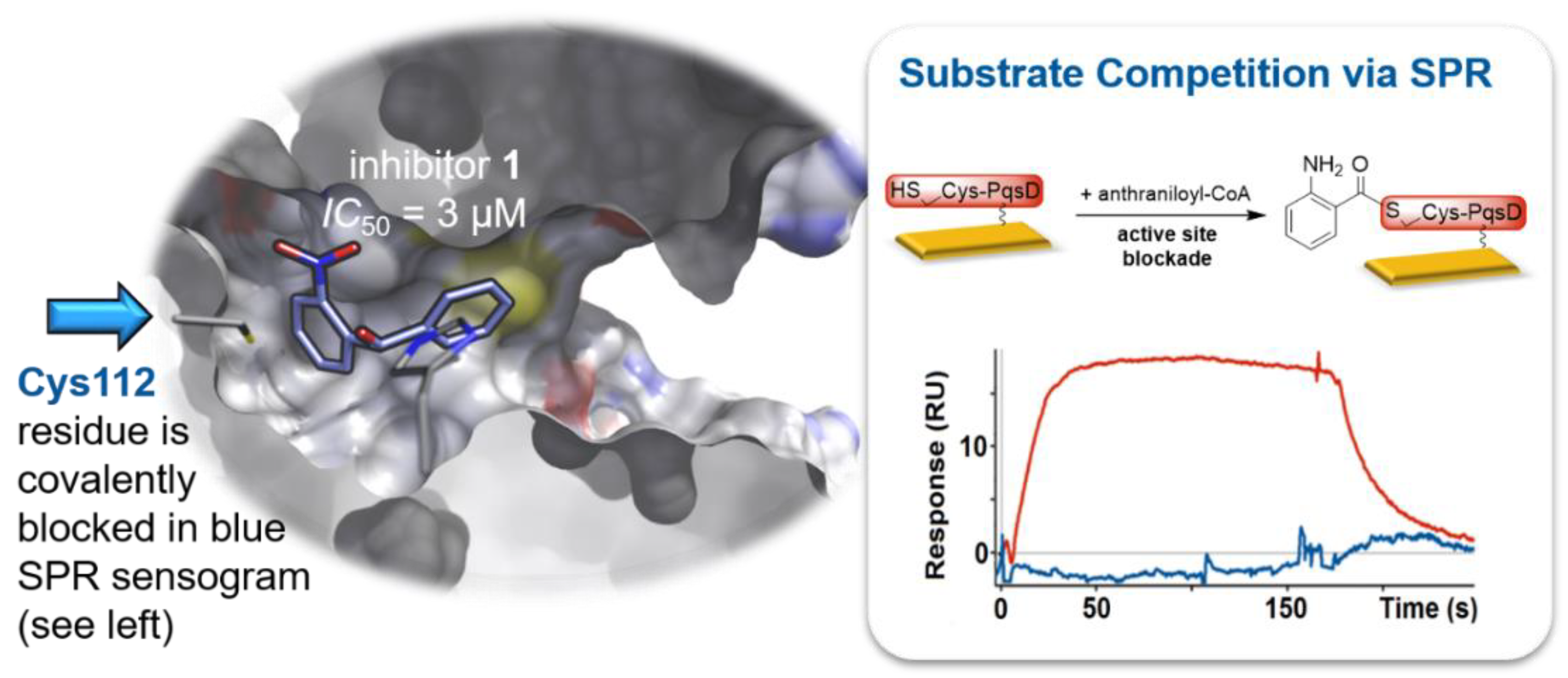
- Storz, M.P.; Brengel, C.; Weidel, E.; Hoffmann, M.; Hollemeyer, K.; Steinbach, A.; Müller, R.; Empting, M.; Hartmann, R.W. Biochemical and biophysical analysis of a chiral PqsD inhibitor revealing tight-binding behavior and enantiomers with contrary thermodynamic signatures. ACS Chem. Biol. 2013, 8, 2794–2801. [Google Scholar] [CrossRef] [PubMed]
- Storz, M.P.; Maurer, C.K.; Zimmer, C.; Wagner, N.; Brengel, C.; de Jong, J.C.; Lucas, S.; Müsken, M.; Häussler, S.; Steinbach, A.; et al. Validation of PqsD as an anti-biofilm target in Pseudomonas aeruginosa by development of small-molecule inhibitors. J. Am. Chem. Soc. 2012, 134, 16143–16146. [Google Scholar] [CrossRef] [PubMed]
- Allegretta, G.; Weidel, E.; Empting, M.; Hartmann, R.W. Catechol-based substrates of chalcone synthase as a scaffold for novel inhibitors of PqsD. Eur. J. Med. Chem. 2015, 90, 351–359. [Google Scholar] [CrossRef]
- Weidel, E.; Negri, M.; Empting, M.; Hinsberger, S.; Hartmann, R.W. Composing compound libraries for hit discovery--rationality-driven preselection or random choice by structural diversity? Future Med. Chem. 2014, 6, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M. A Bioinorganic Approach to Fragment-Based Drug Discovery Targeting Metalloenzymes. Acc. Chem. Res. 2017, 50, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Ciulli, A.; Abell, C. Fragment-based approaches to enzyme inhibition. Curr. Opin. Biotechnol. 2007, 18, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Ciulli, A.; Williams, G.; Smith, A.G.; Blundell, T.L.; Abell, C. Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J. Med. Chem. 2006, 49, 4992–5000. [Google Scholar] [CrossRef]
- Landon, M.R.; Lancia, D.R.; Yu, J.; Thiel, S.C.; Vajda, S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J. Med. Chem. 2007, 50, 1231–1240. [Google Scholar] [CrossRef]
- Deng, G.; Gu, R.-F.; Marmor, S.; Fisher, S.L.; Jahic, H.; Sanyal, G. Development of an LC-MS based enzyme activity assay for MurC: Application to evaluation of inhibitors and kinetic analysis. J. Pharm. Biomed. Anal. 2004, 35, 817–828. [Google Scholar] [CrossRef]
- Sundberg, S. High-throughput and ultra-high-throughput screening: Solution- and cell-based approaches. Curr. Opin. Biotechnol. 2000, 11, 47–53. [Google Scholar] [CrossRef]
- Moreno, A.; Kuzmiak-Glancy, S.; Jaimes, R.; Kay, M.W. Enzyme-dependent fluorescence recovery of NADH after photobleaching to assess dehydrogenase activity of isolated perfused hearts. Sci. Rep. 2017, 7, 45744. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.A.; Balaban, R.S. Direct Imaging of Dehydrogenase Activity within Living Cells Using Enzyme-Dependent Fluorescence Recovery after Photobleaching (ED-FRAP). Biophys. J. 2001, 80, 2018–2028. [Google Scholar] [CrossRef] [Green Version]
- Fahs, S.; Lujan, P.; Köhn, M. Approaches to Study Phosphatases. ACS Chem. Biol. 2016, 11, 2944–2961. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Jakob, V.; Oberhausen, K.; Stein, S.C.; Cucarro, I.; Schulz, T.F.; Empting, M. Fragment-Based Discovery of a Qualified Hit Targeting the Latency-Associated Nuclear Antigen of the Oncogenic Kaposi’s Sarcoma-Associated Herpesvirus/Human Herpesvirus 8. J. Med. Chem. 2019, 62, 3924–3939. [Google Scholar] [CrossRef] [PubMed]
- Sahner, J.H.; Brengel, C.; Storz, M.P.; Groh, M.; Plaza, A.; Müller, R.; Hartmann, R.W. Combining in silico and biophysical methods for the development of Pseudomonas aeruginosa quorum sensing inhibitors: An alternative approach for structure-based drug design. J. Med. Chem. 2013, 56, 8656–8664. [Google Scholar] [CrossRef] [PubMed]
- Sahner, J.H.; Empting, M.; Kamal, A.; Weidel, E.; Groh, M.; Börger, C.; Hartmann, R.W. Exploring the chemical space of ureidothiophene-2-carboxylic acids as inhibitors of the quorum sensing enzyme PqsD from Pseudomonas aeruginosa. Eur. J. Med. Chem. 2015, 96, 14–21. [Google Scholar] [CrossRef]
- Murray, C.W.; Verdonk, M.L. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J. Comput. Aided Mol. Des. 2002, 16, 741–753. [Google Scholar] [CrossRef]
- Mondal, M.; Radeva, N.; Fanlo-Virgós, H.; Otto, S.; Klebe, G.; Hirsch, A.K.H. Fragment Linking and Optimization of Inhibitors of the Aspartic Protease Endothiapepsin: Fragment-Based Drug Design Facilitated by Dynamic Combinatorial Chemistry. Angew. Chem. Int. Ed. Engl. 2016, 55, 9422–9426. [Google Scholar] [CrossRef] [Green Version]
- Mondal, M.; Hirsch, A.K.H. Dynamic combinatorial chemistry: A tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 2015, 44, 2455–2488. [Google Scholar] [CrossRef] [Green Version]
- Frei, P.; Hevey, R.; Ernst, B. Dynamic Combinatorial Chemistry: A New Methodology Comes of Age. Chemistry 2019, 25, 60–73. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. https://doi.org/10.3390/molecules24234309
Kirsch P, Hartman AM, Hirsch AKH, Empting M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules. 2019; 24(23):4309. https://doi.org/10.3390/molecules24234309
Chicago/Turabian StyleKirsch, Philine, Alwin M. Hartman, Anna K. H. Hirsch, and Martin Empting. 2019. "Concepts and Core Principles of Fragment-Based Drug Design" Molecules 24, no. 23: 4309. https://doi.org/10.3390/molecules24234309
APA StyleKirsch, P., Hartman, A. M., Hirsch, A. K. H., & Empting, M. (2019). Concepts and Core Principles of Fragment-Based Drug Design. Molecules, 24(23), 4309. https://doi.org/10.3390/molecules24234309