Polypropylene Glycol-Polyoxytetramethylene Glycol Multiblock Copolymers with High Molecular Weight: Synthesis, Characterization, and Silanization

Abstract
:1. Introduction
2. Results and Discussion
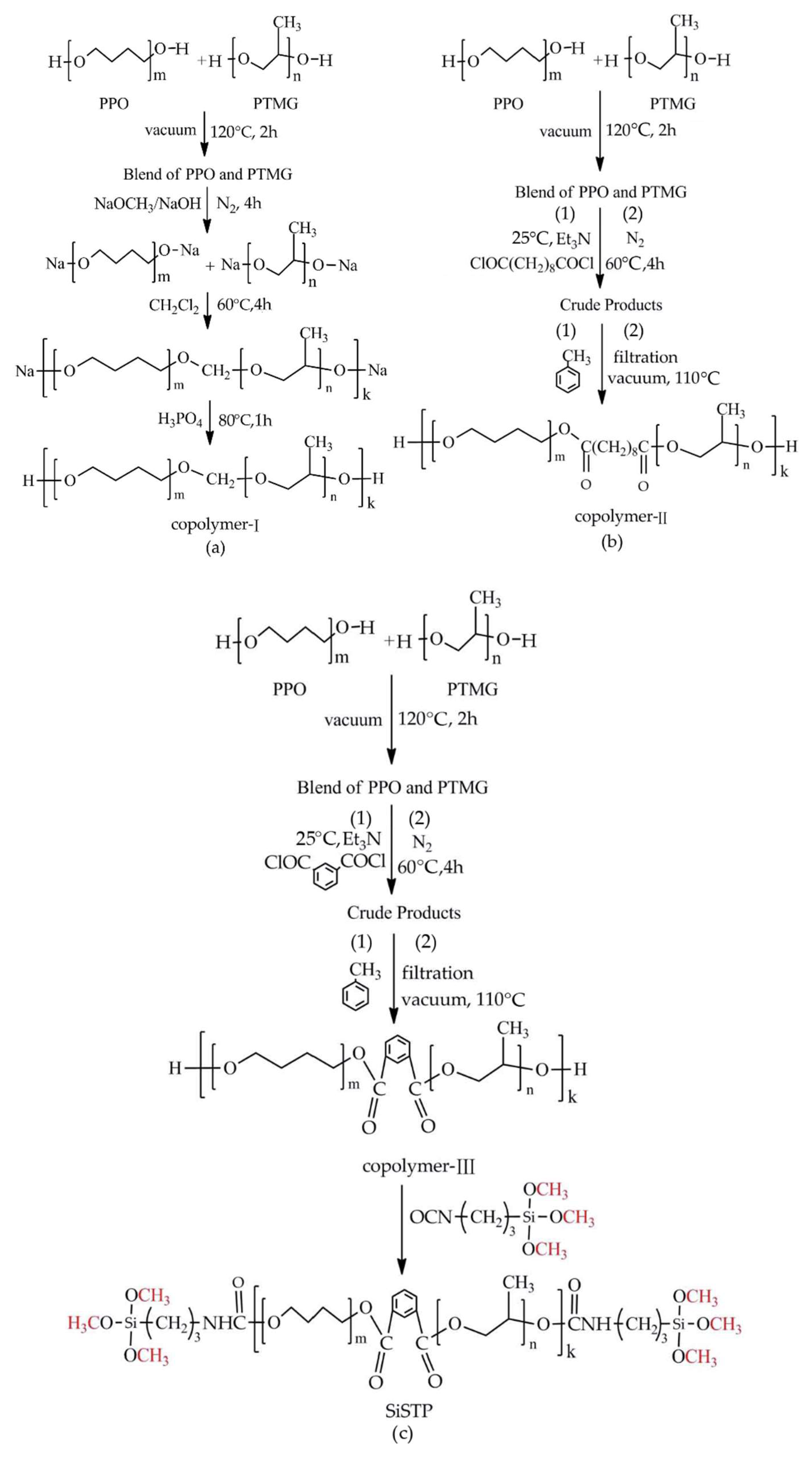
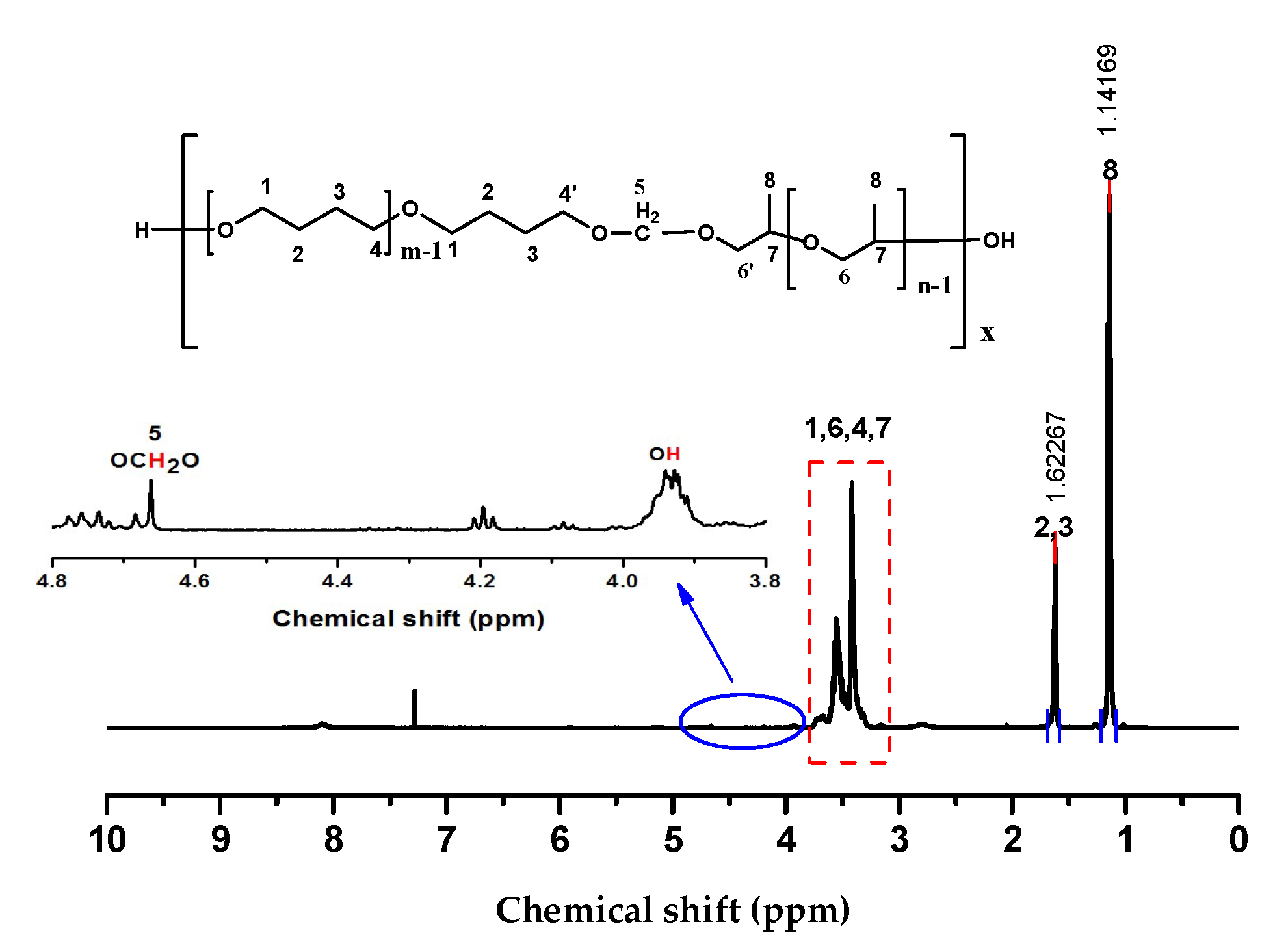
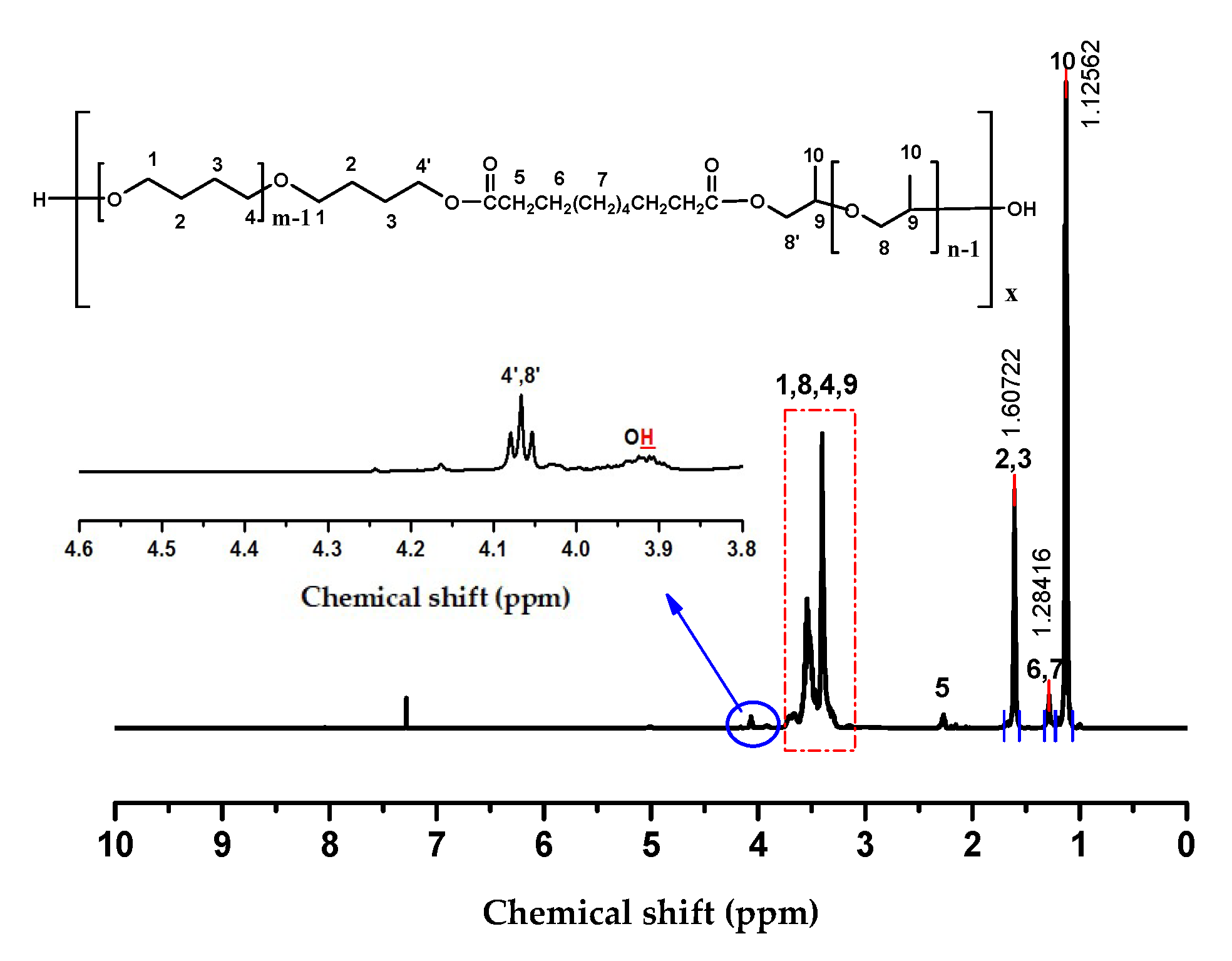
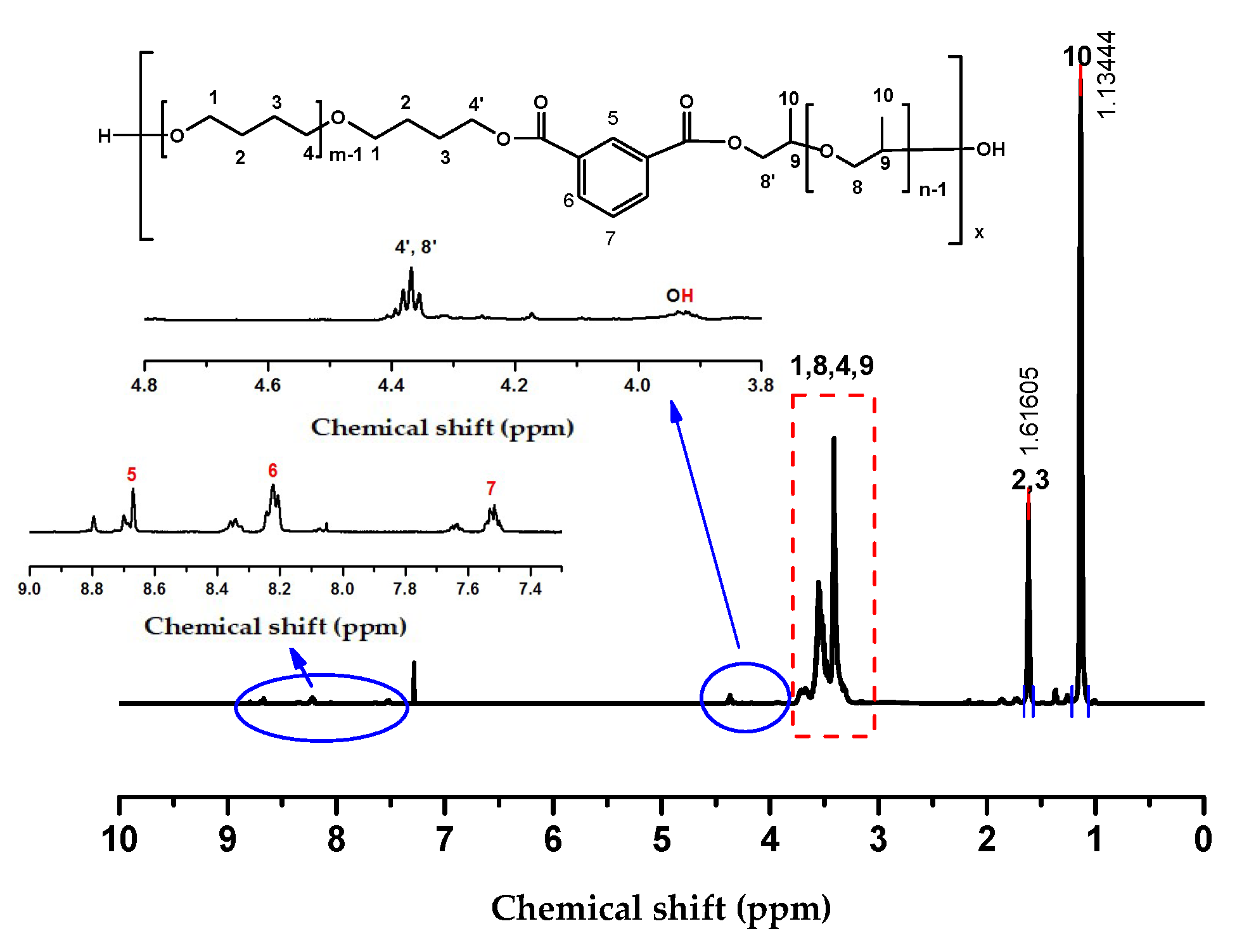
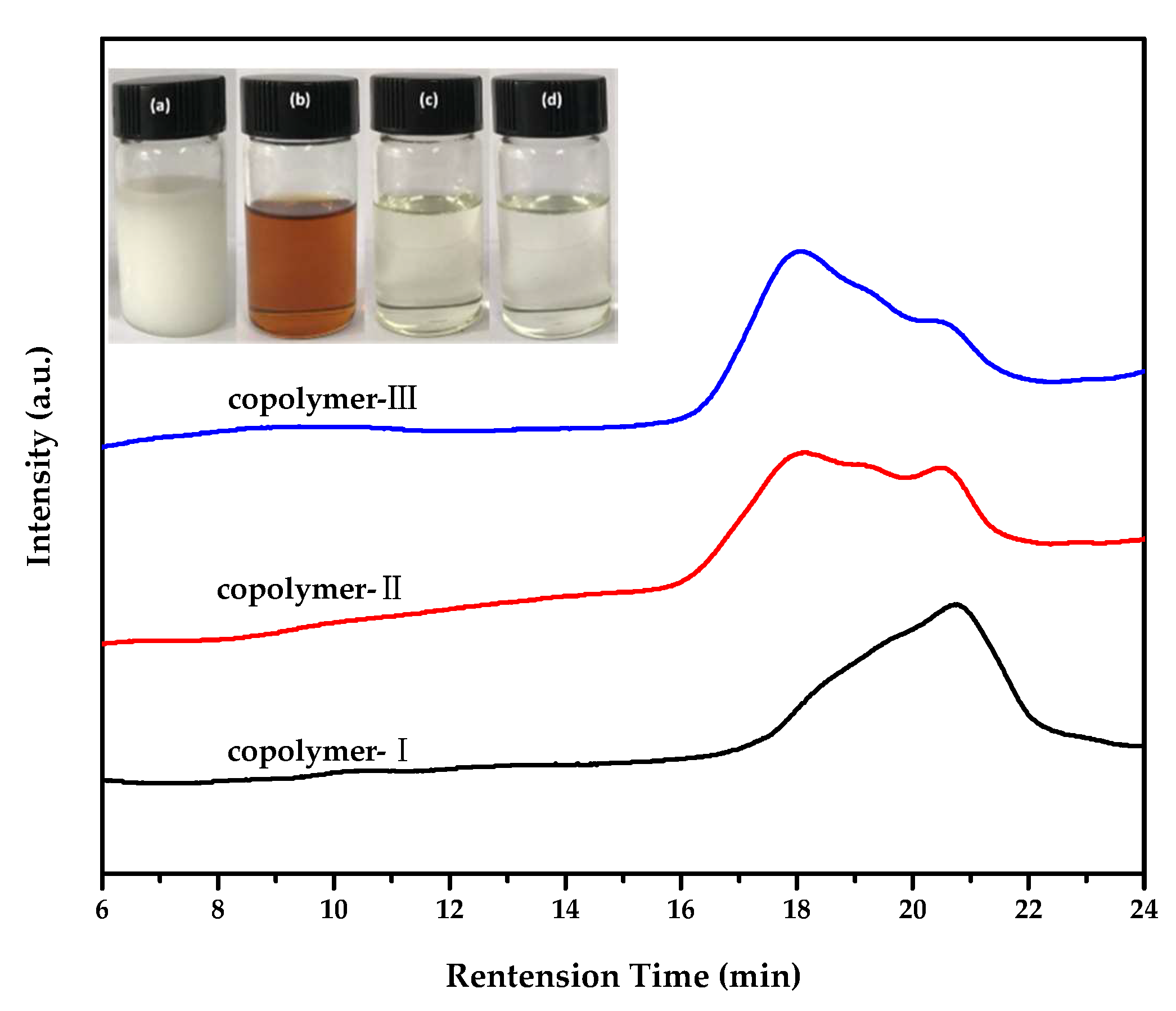
2.1. Choice of Chain Extenders
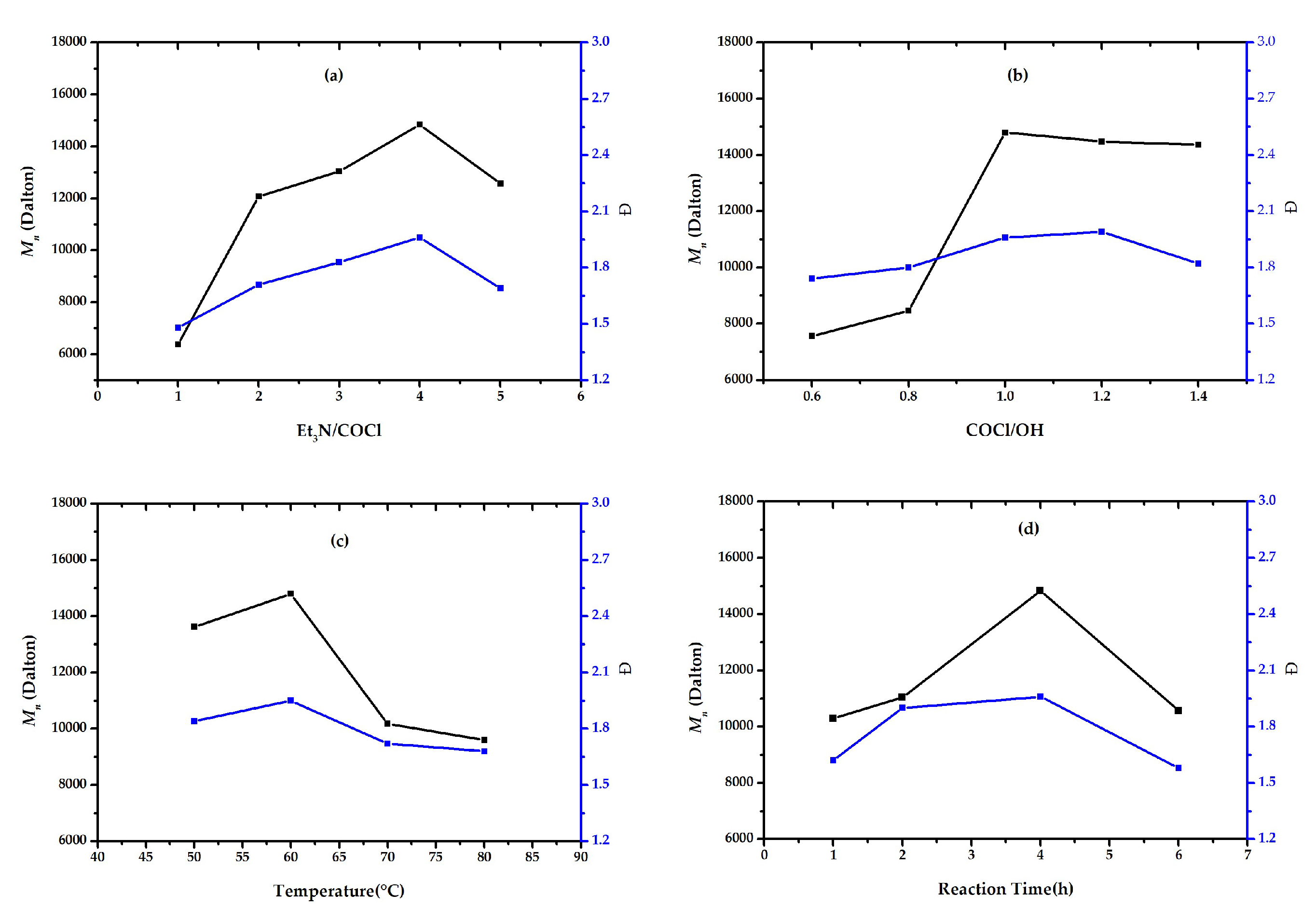
2.2. Optimizing Reaction Conditions
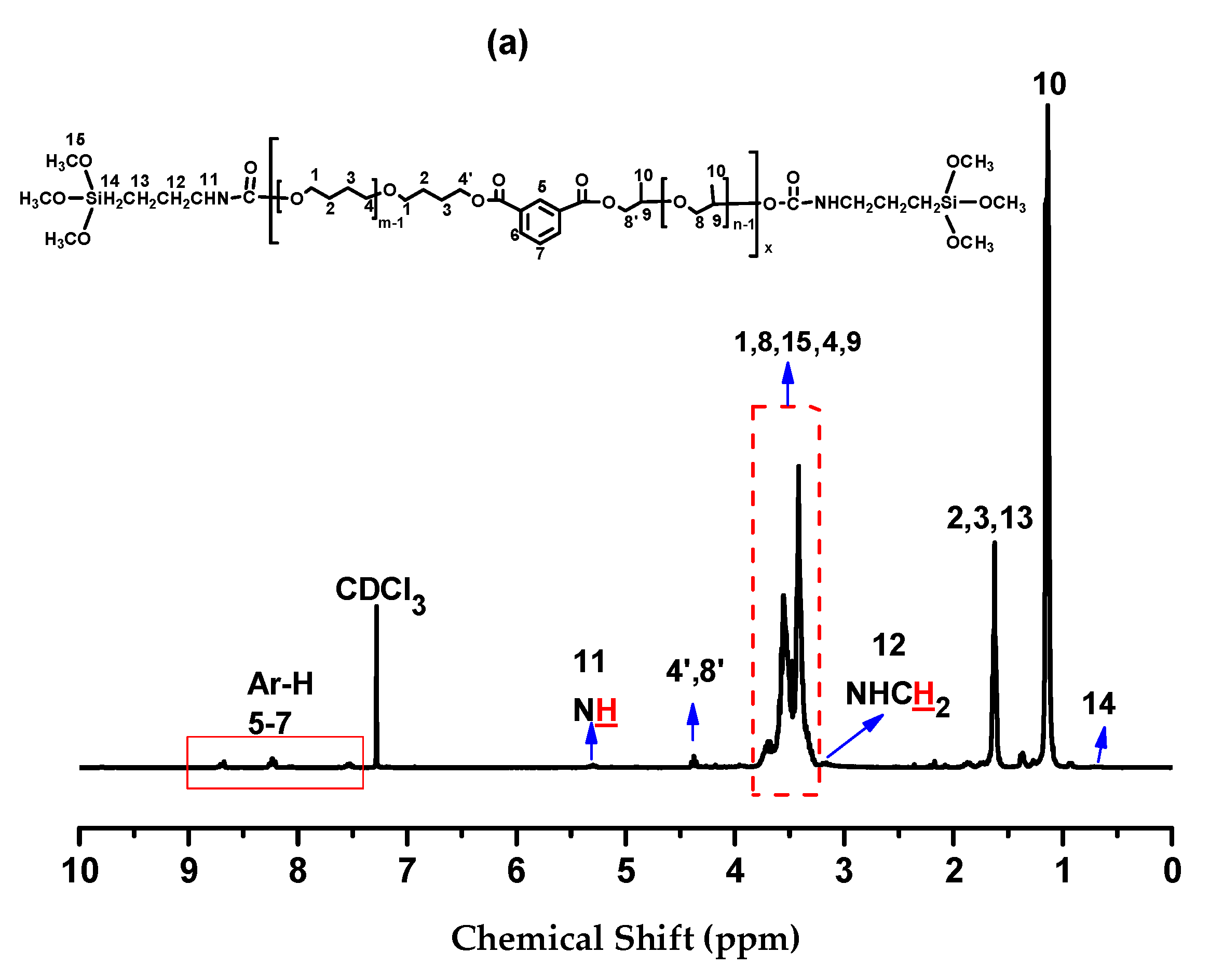
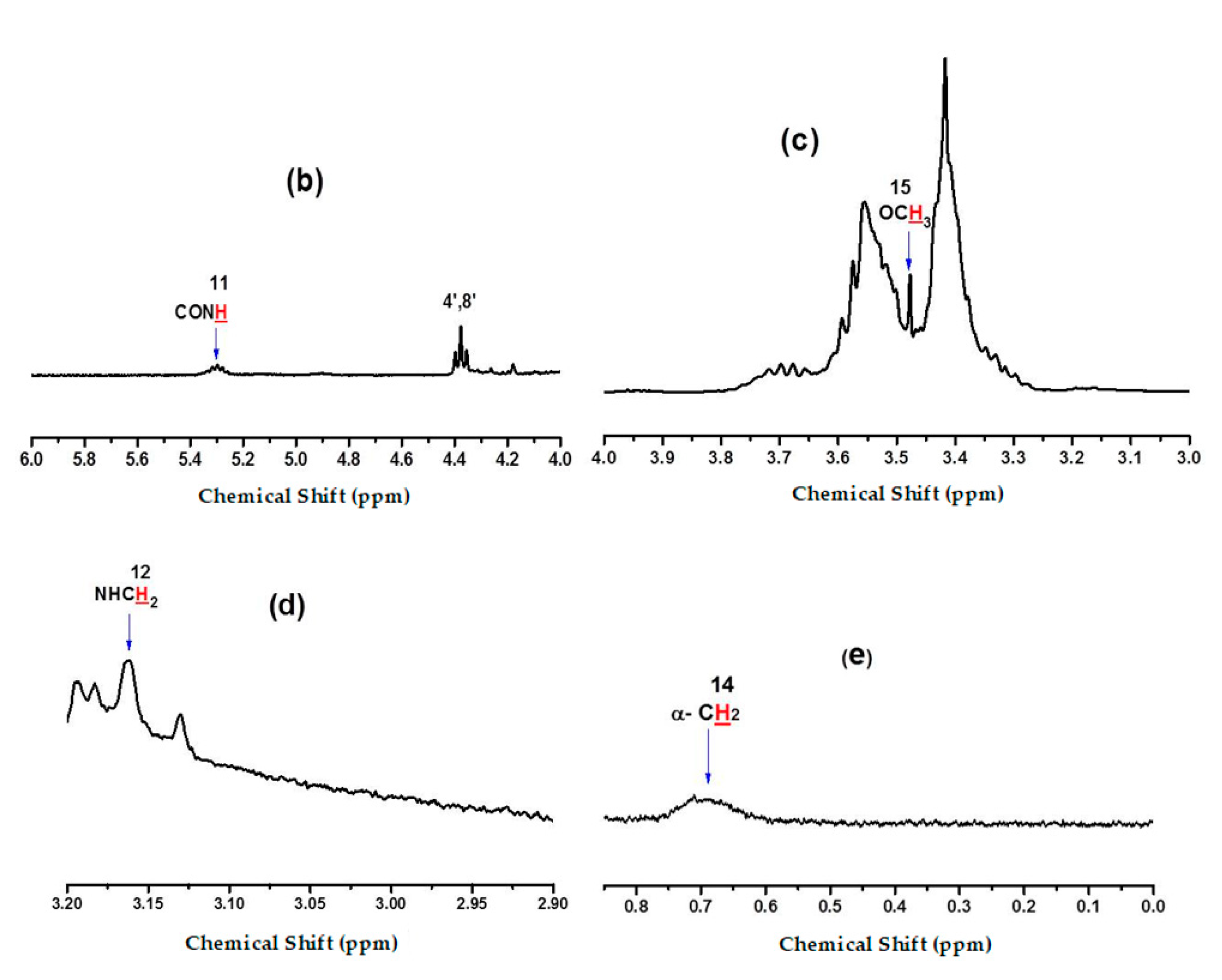
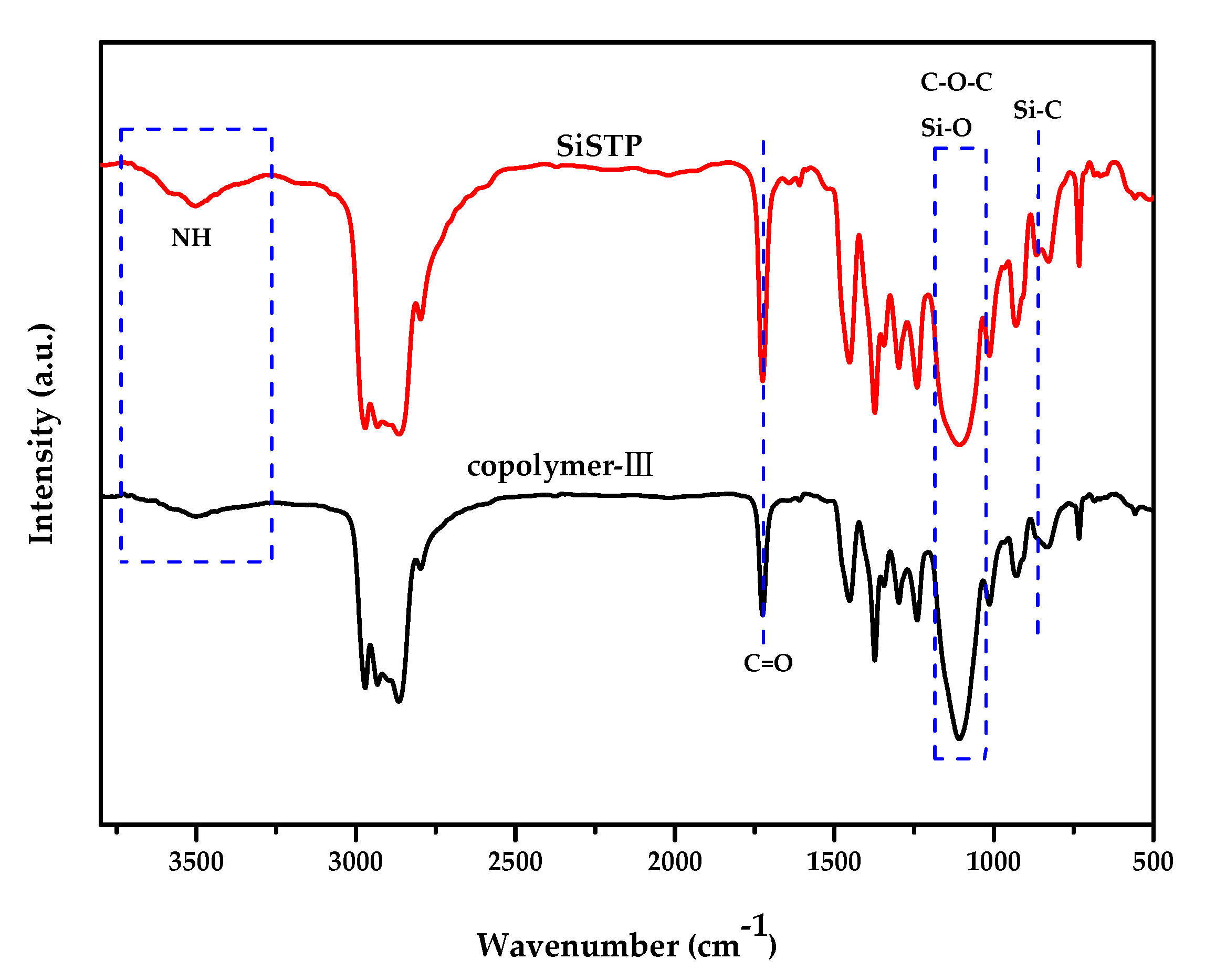
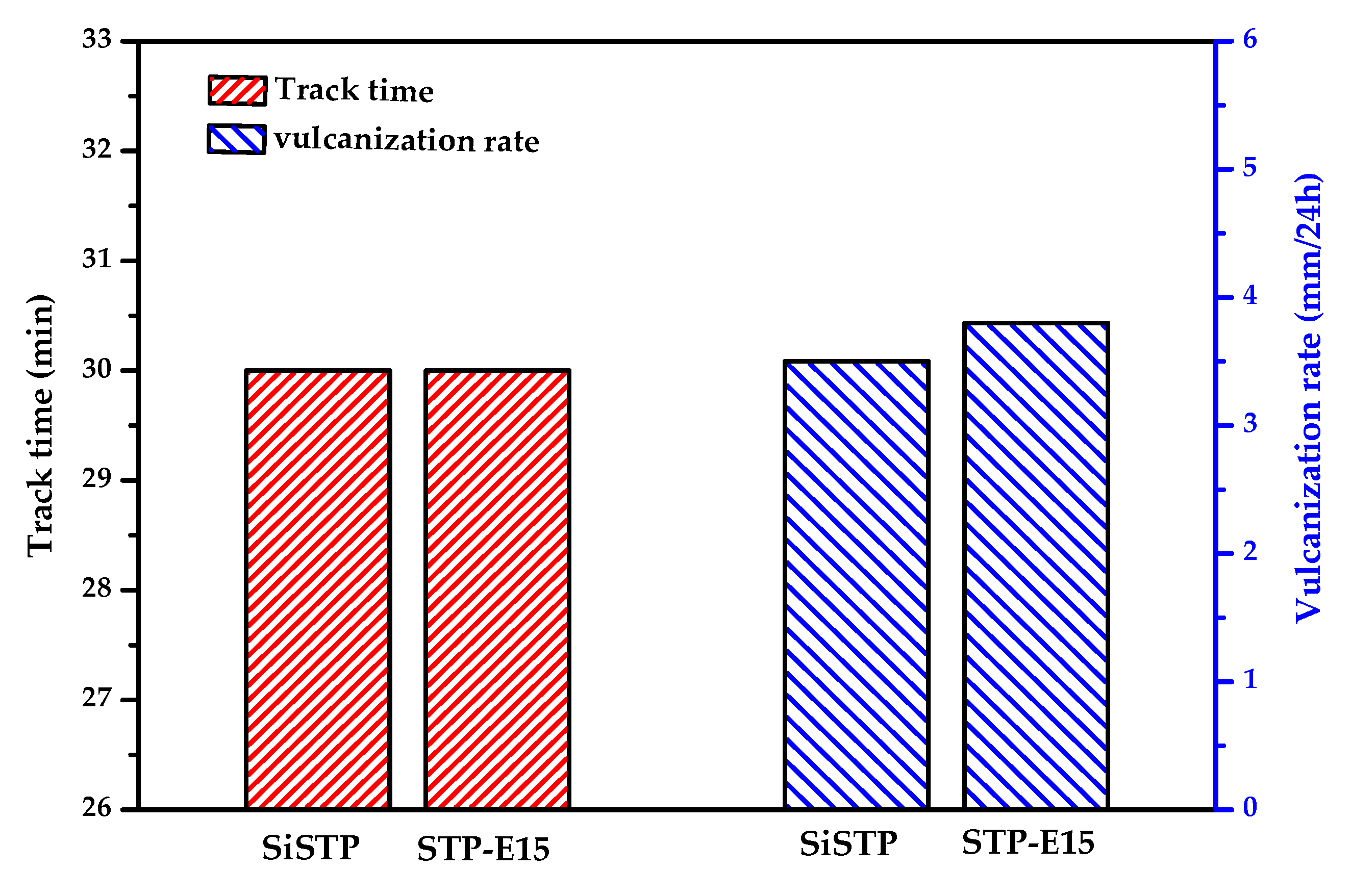
2.3. Silanization of Block Polyether
3. Experimental Section
3.1. Materials
3.2. Synthesis of PPO-PTMG Multiblock Copolyethers Using 1,2-dichloromethane
3.3. Synthesis of PPO-PTMG Multiblock Copolyethers Using Diacid Chlorides
3.4. Silanization of Block Copolyether
3.5. Measurement
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klein, R.; Wurm, F.R. Aliphatic Polyethers: Classical polymers for the 21st century. Macromo. Rapid Comm. 2015, 36, 1147. [Google Scholar] [CrossRef] [PubMed]
- Pruckmayr, G.; Palmer, C.F.; Lodoen, G.A. Copolymers of tetrahydrofuran, ethylene oxide and an additional cyclic ether. U.S. Patent 6989432, 2006. [Google Scholar]
- Oguro, K.; Kun, N.; Nishimura, H.; Mitaka, K.; Takao, D. Modified PTMG based thermoplastic polyurethane elastomers. J. Elastom. Plast. 1985, 17, 261–272. [Google Scholar] [CrossRef]
- James, R.; Wolfe, J.R. Elastomeric polyether-ester block copolymers: properties as a function of the structure and concentration of the ester group. Adv. Chem. Ser. 1979, 129–151. [Google Scholar] [CrossRef]
- Bednarek, M.; Przemysław, K.A.; Penczek, S. Coexistence of activated monomer and active chain end mechanisms in cationic copolymerization of tetrahydrofuran with ethylene oxide. Macromolecules 1999, 32, 5257–5263. [Google Scholar] [CrossRef]
- Bednarek, M.; Przemysław, K. Mechanism of cyclics formation in the cationic copolymerization of tetrahydrofuran with ethylene oxide in the presence of diols. Macromol. Chem. Phys. 1999, 200, 2443–2447. [Google Scholar] [CrossRef]
- Bednarek, M.; Przemysław, K. Cationic copolymerization of tetrahydrofuran with ethylene oxide in the presence of diols: Composition, microstructure, and properties of copolymers. J. Polym. Sci. Pol. Chem. 1999, 37, 3455–3463. [Google Scholar] [CrossRef]
- Blanchard, L.P.; Kondo, S.; Moinard, J.; Pierson, J.F.; Tahiani, F. Copolymerization of tetrahydrofuran with propylene oxide. III. 1,2,3-propanetriol as cocatalyst. J. Polym. Sci. Pol. Chem. 1972, 10, 399–412. [Google Scholar] [CrossRef]
- Alvarez, E.J.; Hornof, V.; Blanchard, L.P. Cationic copolymerization of tetrahydrofuran with propylene oxide. IV. Use of antimony pentachloride as catalyst. J. Polym. Sci. Pol. Chem. 1972, 10, 1895–1903. [Google Scholar] [CrossRef]
- Alvarez, E.J.; Hornof, V.; Blanchard, L.P. Cationic copolymerization of tetrahydrofuran with propylene oxide. V. Effect of diol cocatalyst structure and concentration with antimony pentachloride as catalyst. J. Polym. Sci. Pol. Chem. 1972, 10, 2237–2244. [Google Scholar] [CrossRef]
- Turovskaya, L.N.; Matveyeva, N.G.; Berlin, A.A. Copolymerization of tetrahydrofuran with propylene oxide in the presence of methacrylic anhydride. Polym. Sci. U.S.S.R. 1973, 15, 2074–2079. [Google Scholar] [CrossRef]
- Berlin, A.A.; Turovskaya, L.N.; Matveyeva, N.G. The mechanism of co-oligomerization of tetrahydrofuran with propylene oxide in the presence of methacrylic anhydride. Polym. Sci. U.S.S.R. 1977, 19, 495–501. [Google Scholar] [CrossRef]
- Kuzayev, A.I.; Komratov, G.N.; Korovina, G.V.; Entelis, S. Copolymerization of tetrahydrofuran and epichlorohydrin on boron trifluoride tetrahydrofuranate. Polym. Sci. U.S.S.R. 1970, 12, 1124–1131. [Google Scholar] [CrossRef]
- Misir, M.; Ozturk, T.; Emirik, M.; Yilmaz, S.S. Synthesis of novel tetrahydrofuran-epichlorohydrin [poly(THF-b-ECH)] macromonomeric peroxy initiators by cationic copolymerization and the quantum chemically investigation of initiation system effects. J. Polym. Sci. Pol. Chem. 2010, 48, 2896–2909. [Google Scholar] [CrossRef]
- Christ, E.; Herzberger, J.; Montigny, M.; Tremel, W.; Frey, H. Poly(THF-co-cyano ethylene oxide): cyano ethylene oxide (CEO) copolymerization with THF leading to multifunctional and water-soluble polyTHF polyelectrolytes. Macromolecules 2016, 49, 3681–3695. [Google Scholar] [CrossRef]
- Baijal, M.D.; Blanchard, L.P. Kinetic aspects of the copolymerization of tetrahydrofuran with propylene oxide. Part II. J. Polym. Sci. Pol. Symp. 1968, 23, 157–167. [Google Scholar] [CrossRef]
- Hammond, J.M.; Hooper, J.F.; Robertson, W.G.P. Cationic copolymerization of tetrahydrofuran with epoxides. I. Polymerization mechanism in the presence of a glycol. J. Polym. Sci. Pol. Chem. 1971, 9, 265–279. [Google Scholar] [CrossRef]
- Hammond, J.M.; Hooper, F.; Robertson, W.G.P. Cationic copolymerization of tetrahydrofuran with epoxides. II. Characterization and gel-permeation chromatography of by-products formed during polymerization. J. Polym. Sci. Pol. Chem. 1971, 9, 281–294. [Google Scholar] [CrossRef]
- Wang, C.D.; Luo, Y.; Xia, M. Synthesis and characterization of the hydroxyl-terminated block copolyether of PPO-PTHF-PPO by macroinitiator method. Polym. Mater. Sci. Eng. 2011, 27, 1–4. [Google Scholar]
- Zhang, X.; Fan, W.; Liao, X.; Fan, X. Synthesis process of poly(ethylene oxide)-poly(tetrahydrofuran)-poly(ethylene oxide) block copolymers with narrow molecular weight distribution. Polym. Mater. Sci. Eng. 2013, 29, 37–45. [Google Scholar]
- Wang, C.; Pan, H.; Su, L.; Zhang, L. Synthesis of hydroxyl-terminated block copolyether of polytetrahydrofuran-polypropylene via oxonium ions. Polym. Mater. Sci. Eng. 2013, 29, 15–18. [Google Scholar]
- Yang, H.; Xu, J.; Pispas, S.; Zhang, G. Hybrid copolymerization of ε-caprolactone and methyl methacrylate. Macromolecules 2012, 45, 3312–3317. [Google Scholar] [CrossRef]
- Schawe, J.E.K.; Höhne, G.W.H. Modulated temperature DSC measurements relating to the cold crystallization process of poly(ethylene terephtalate). J. Therm. Anal. 1996, 46, 893–903. [Google Scholar] [CrossRef]
- Xu, C.L.; Zeng, J.B.; Zhu, Q.Y.; Wang, Y.Z. Poly(ethylene succinate)-b-poly(butylene succinate) multiblock copolyesters: The effects of block length and composition on physical properties. Ind. Eng. Chem. Res. 2013, 52, 13669−13676. [Google Scholar] [CrossRef]
- Okunev, P.A.; Okuneva, A.G.; Tarakanov, O.G.; Vakhtina, I.A. Influence of copolymerization conditions on the molecular structure and molecular weight distribution in tertrahydrofuran: Propylene oxide copolymers. Polym. Sci. U.S.S.R. 1969, 11, 403–408. [Google Scholar] [CrossRef]
- Zhang, C.H.; Hu, M.Q. Synthesis of hydroxyl terminated polylactic acid chain extended by hexyl dichloride. Fine Chem. 2014, 31, 137–141. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Chain Extender | Mn | Đ | OH value (OHV)/mgKOH/g | cf | dTc (°C) | eTm (°C) | |
|---|---|---|---|---|---|---|---|---|
| a Ther. | b Titr. | |||||||
| I | CH2Cl2 | 7200 | 1.71 | 15.52 | 9.70 | 1.25 | −3.1 | 25.5 |
| II | sebacoyl chloride | 10,700 | 1.85 | 10.44 | 7.54 | 1.44 | −18.3 | 22.3 |
| III | m-phthaloyl chloride | 12,000 | 1.85 | 9.29 | 8.28 | 1.78 | - | 21.2 |
| Copolymers | Feed Ratio of Et3N/COCl/OH | Temp. (°C) | Time (h) | Mn | Đ | OHV (mgKOH/g) | f | |
|---|---|---|---|---|---|---|---|---|
| a Ther. | b Titr. | |||||||
| III | 2/1/1 | 60 | 4 | 12,000 | 1.71 | 9.29 | 8.28 | 1.78 |
| IIIa | 4/1/1 | 60 | 4 | 14,800 | 1.96 | 7.56 | 4.16 | 1.10 |
| IIIb | 4/1/1.2 | 60 | 4 | 14,400 | 1.99 | 7.74 | 4.36 | 1.13 |
| IIIc | 4/1/1 | 50 | 4 | 13,600 | 1.84 | 8.21 | 7.08 | 1.72 |
| IIId | 4/1/1 | 60 | 2 | 11,000 | 1.90 | 10.15 | 9.46 | 1.86 |
| Reaction Condition Order | Feed Ratios | Temperature (°C) | Time (h) | |
|---|---|---|---|---|
| Et3N/COCl a | COCl/OH b | |||
| 1 | 1/1 | 0.6/1 | 50 | 1 |
| 2 | 2/1 | 0.8/1 | 60 | 2 |
| 3 | 3/1 | 1/1 | 70 | 4 |
| 4 | 4/1 | 1.2/1 | 80 | 6 |
| 5 | 5/1 | 1.4/1 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Wang, L.; Wang, Q.; Luan, A.; Mai, Y.; Huang, L.; Chen, Y.; Yan, S.; Xiong, W. Polypropylene Glycol-Polyoxytetramethylene Glycol Multiblock Copolymers with High Molecular Weight: Synthesis, Characterization, and Silanization. Molecules 2019, 24, 4317. https://doi.org/10.3390/molecules24234317
Hu W, Wang L, Wang Q, Luan A, Mai Y, Huang L, Chen Y, Yan S, Xiong W. Polypropylene Glycol-Polyoxytetramethylene Glycol Multiblock Copolymers with High Molecular Weight: Synthesis, Characterization, and Silanization. Molecules. 2019; 24(23):4317. https://doi.org/10.3390/molecules24234317
Chicago/Turabian StyleHu, Wei, Lei Wang, Quanyong Wang, Anbo Luan, Yuliang Mai, Linjia Huang, Yongjun Chen, Shijing Yan, and Wenjie Xiong. 2019. "Polypropylene Glycol-Polyoxytetramethylene Glycol Multiblock Copolymers with High Molecular Weight: Synthesis, Characterization, and Silanization" Molecules 24, no. 23: 4317. https://doi.org/10.3390/molecules24234317