
Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis



{kind=link}
{kind=link}
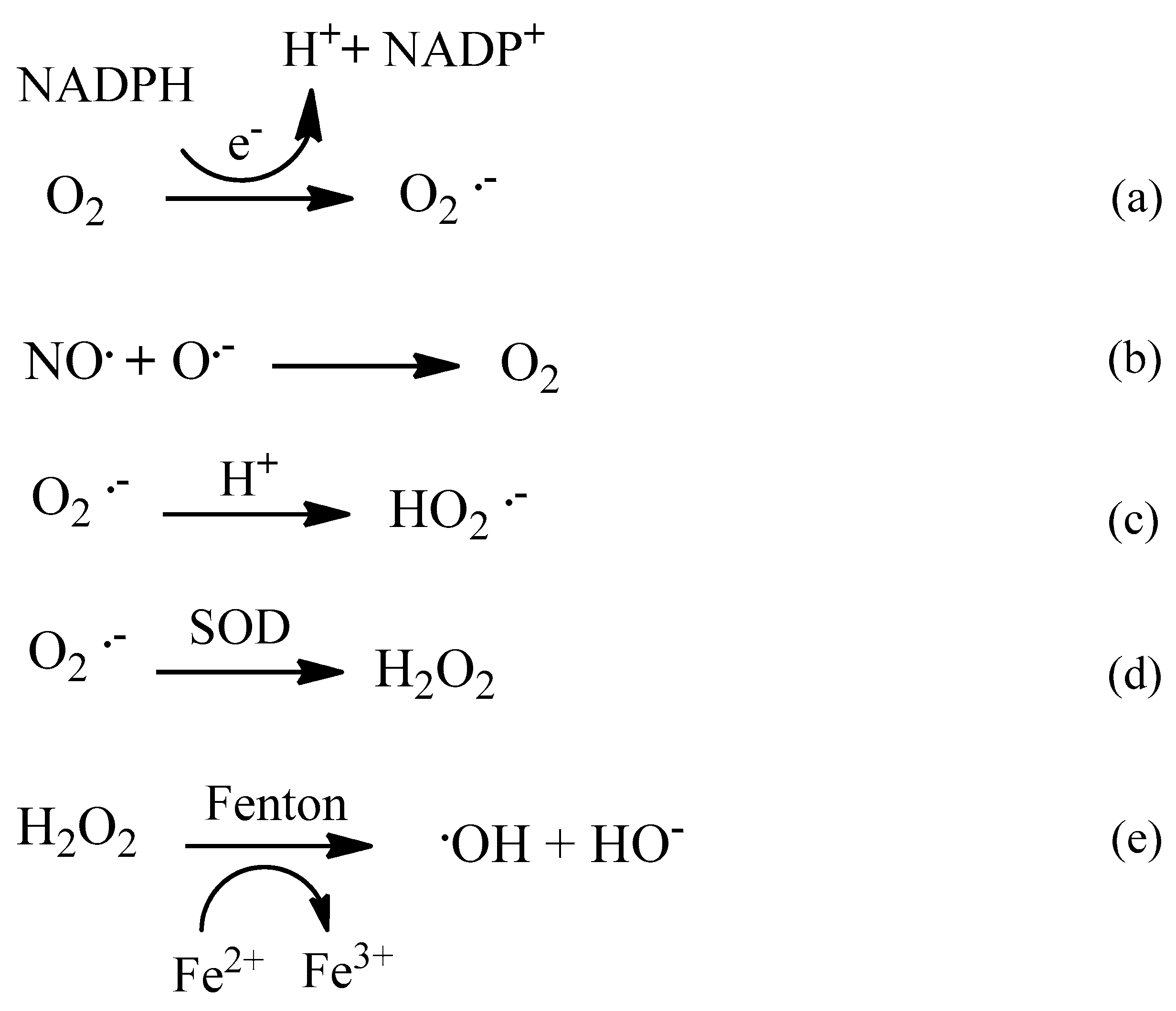{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
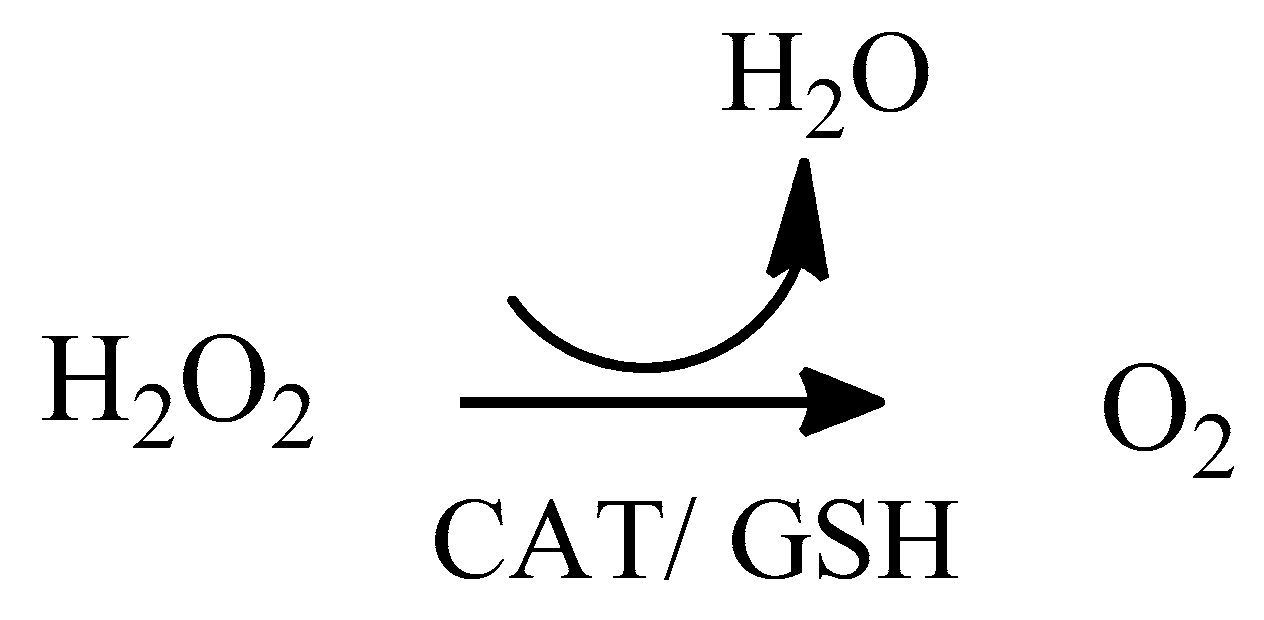
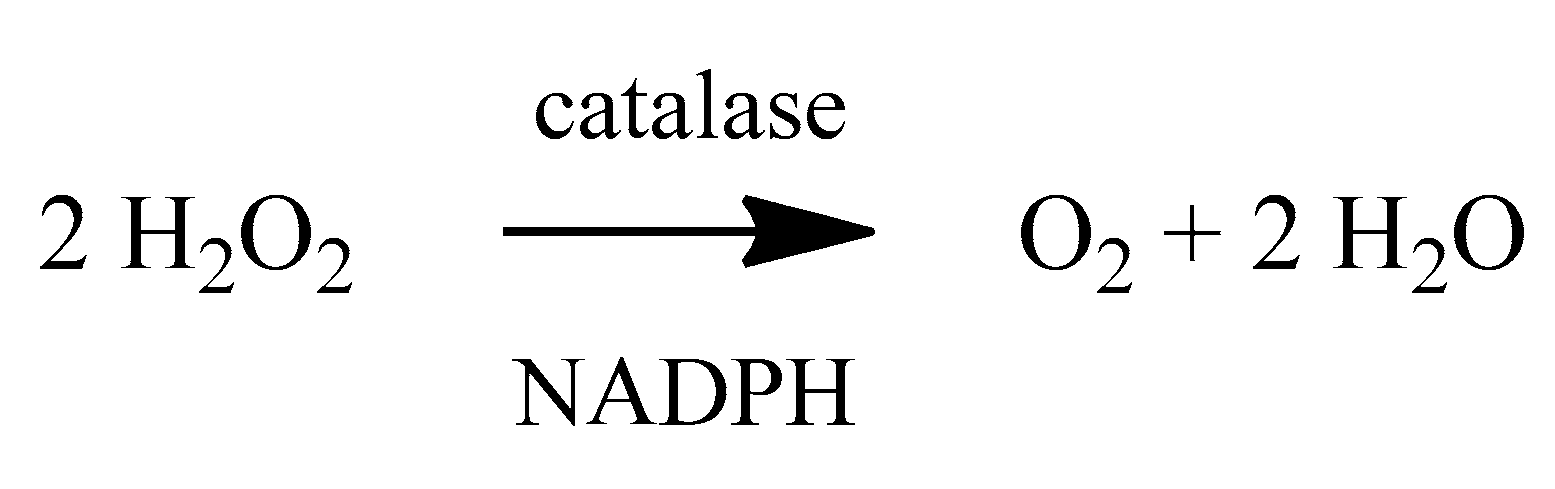
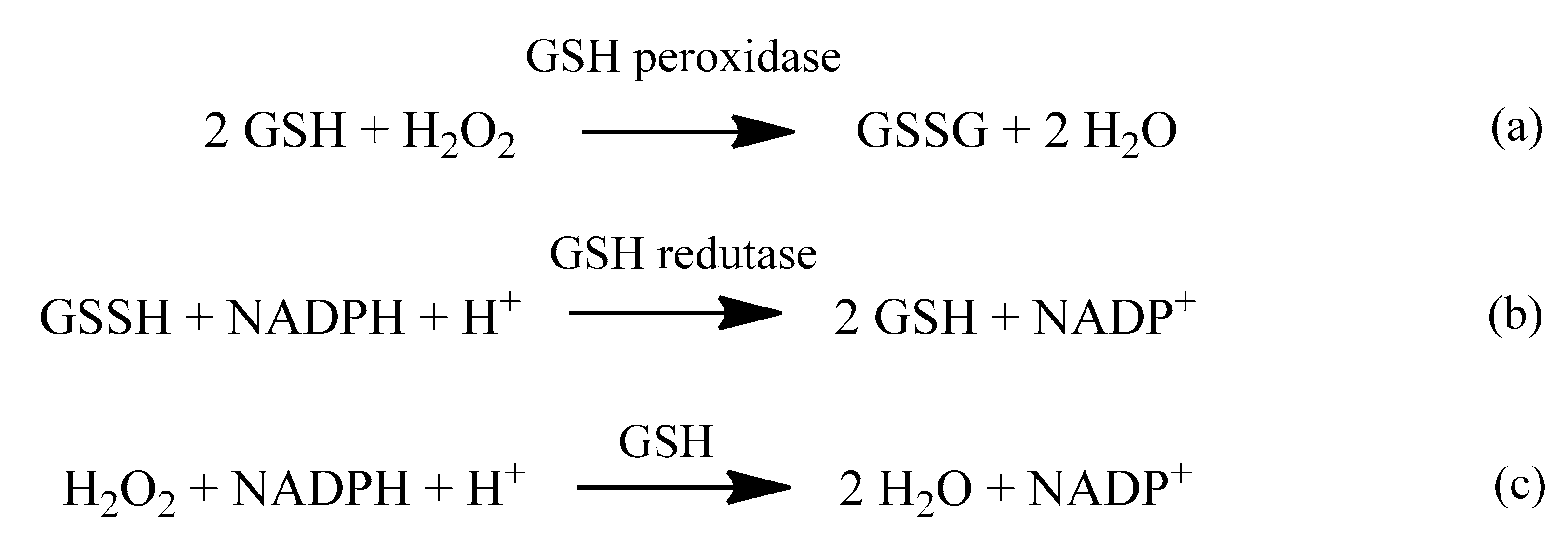
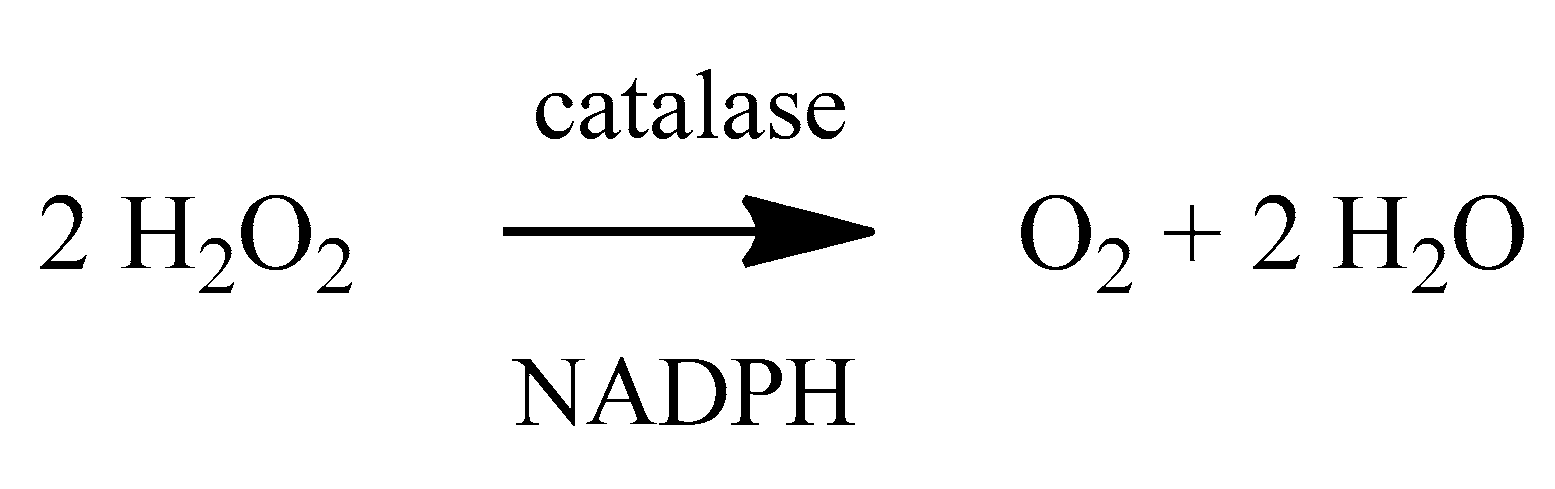
2. Oxidative Stress
2.1. Reactive Oxygen Species (ROS)
2.2. The Mitochondria and ROS Generation
3. AD-Related Hypotheses
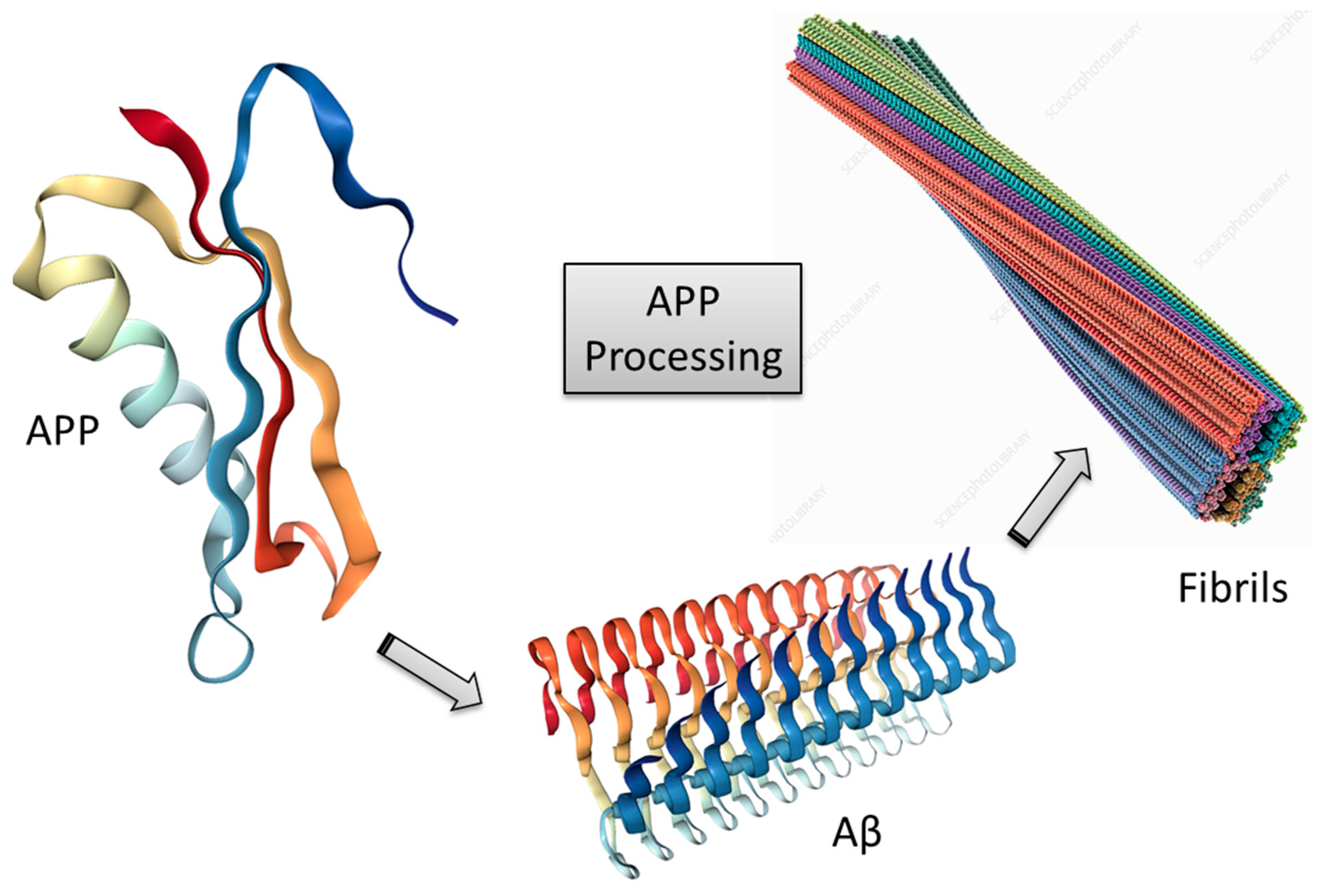
3.1. Amyloid-β Cascade Hypothesis
3.2. Tau Protein Hypothesis
3.3. Cholinergic Hypothesis
4. Antioxidants
5. Other Relevant Therapeutic Approaches in AD Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de Castro, A.A.; Soares, F.V.; Pereira, A.F.; Polisel, D.A.; Caetano, M.S.; Leal, D.H.S.; da Cunha, E.F.F.; Nepovimova, E.; Kuca, K.; Ramalho, T.C. Non-conventional compounds with potential therapeutic effects against Alzheimer’s disease. Expert Rev. Neurother. 2019, 19, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.; Green, C.; Jones, R.W.; Förstl, H.; Simsek, D.; de Reydet de Vulpillieres, F.; Luthman, S.; Adlard, N.; Bhattacharyya, S.; Wimo, A. Current issues and future research priorities for health economic modelling across the full continuum of Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Patience, A.A.; Sharma, N.; Khurana, N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur. J. Pharmacol. 2017, 815, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef]
- De Castro, A.A.; da Cunha, E.F.F.; Pereira, A.F.; Soares, F.V.; Leal, D.H.S.; Kuca, K.; Ramalho, T.C. Insights into the Drug Repositioning Applied to the Alzheimer’s Disease Treatment and Future Perspectives. Curr. Alzheimer Res. 2018, 15, 1161–1178. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [Green Version]
- Kishida, K.T.; Klann, E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 2007, 9, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Rego, A.C.; Oliveira, C.R. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 2003, 28, 1563–1574. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 531–549. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont- Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and mitochondrial dysfunction in alzheimer disease. Biochim. et Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s disease. J. Alzheimer Diases. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative Stress as a biomarker for alzheimer disease. Biomarker Med. 2018, 12, 201–203. [Google Scholar] [CrossRef]
- Wojtunik- Kulesza, K.; Oniszczuck, A.; Oniszczuck, T.; Waksmundzka-Hajnos, M. The influence of common free radicals and antoxidants on development of alzheimer’s disease. Biomed. Pharmacoth. 2016, 78, 39–49. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yon, S. The role of oxidative stress in neurodegenerative disease. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Hung, C.H.L.; Cheng, S.S.Y.; Cheung, Y.T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.S.; Lee, S.M.Y.; Chang, R.C.C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative Stress: A major pathogenises and potential therapeutics target of antioxidative agents in Parkinson’s disease and Alzheimer disease. Prog. Nurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of alzheimer’s disease: Understanding the therapeutics strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Dutordoir, R.M.; Bates, D.A.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. et. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.L.; Hanney, M.; Francis, P.T.; Ballard, C.G. Amyloid beta concentrations in older people with Down syndrome and dementia. Neurosci. Lett. 2009, 451, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Broadstock, M.; Ballard, C.; Corbett, A. Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert Opin. Pharmacother. 2014, 15, 1797–1810. [Google Scholar] [CrossRef] [PubMed]
- Saez-Orellana, F.; Godoy, P.A.; Bastidas, C.Y.; Silva-Grecchi, T.; Guzman, L.; Aguayo, L.G.; Fuentealba, J. ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology 2016, 100, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Godyn, J.; Jonczyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Golde, T.E. The pathogenesis of Alzheimer’s disease and the role of Abeta42. CNS Spectr. 2007, 12, 4–6. [Google Scholar] [CrossRef]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Abeta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Sales, T.A.; Prandi, I.G.; de Castro, A.A.; Leal, D.H.S.; da Cunha, E.F.F.; Kuca, K.; Ramalho, T.C. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci. 2019, 20, 1829. [Google Scholar] [CrossRef] [Green Version]
- Grill, J.D.; Cummings, J.L. Current therapeutic targets for the treatment of Alzheimer’s disease. Expert Rev. Neurother. 2010, 10, 711–728. [Google Scholar] [CrossRef] [Green Version]
- Bachurin, S.O.; Bovina, E.V.; Ustyugov, A.A. Drugs in Clinical Trials for Alzheimer’s Disease: The Major Trends. Med. Res. Rev. 2017, 37, 1186–1225. [Google Scholar] [CrossRef]
- Levey, A.I.; Kitt, C.A.; Simonds, W.F.; Price, D.L.; Brann, M.R. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 1991, 11, 3218–3226. [Google Scholar] [CrossRef] [Green Version]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.M.; Polanco, J.-C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta. Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.-Y.; Trojanowski, J.Q. The disordered neuronal cytoskeleton in Alzheimer’s disease. Curr. Opin. Neurobiol. 1992, 2, 653–656. [Google Scholar] [CrossRef]
- Clark, C.M.; Xie, S.; Chittams, J.; Ewbank, D.; Peskind, E.; Galasko, D.; Morris, J.C.; McKeel, D.W.J.; Farlow, M.; Weitlauf, S.L.; et al. Cerebrospinal fluid tau and beta-amyloid: How well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch. Neurol. 2003, 60, 1696–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell. 2007, 18, 5060–5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Mandelkow, E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics 2008, 5, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illenberger, S.; Zheng-Fischhofer, Q.; Preuss, U.; Stamer, K.; Baumann, K.; Trinczek, B.; Biernat, J.; Godemann, R.; Mandelkow, E.M.; Mandelkow, E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol. Biol. Cell. 1998, 9, 1495–1512. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. 2005, 112, 813–838. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Wu, X.-L.; Pina-Crespo, J.; Zhang, Y.-W.; Chen, X.-C.; Xu, H.-X. Tau-mediated Neurodegeneration and Potential Implications in Diagnosis and Treatment of Alzheimer’s Disease. Chin. Med. J. (Engl.) 2017, 130, 2978–2990. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet (London, England) 1976, 2, 1403. [Google Scholar] [CrossRef]
- Kar, S.; Issa, A.M.; Seto, D.; Auld, D.S.; Collier, B.; Quirion, R. Amyloid beta-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. J. Neurochem. 1998, 70, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Kar, S.; Quirion, R. Beta-amyloid peptides as direct cholinergic neuromodulators: A missing link? Trends Neurosci. 1998, 21, 43–49. [Google Scholar] [CrossRef]
- Nordberg, A.; Alafuzoff, I.; Winblad, B. Nicotinic and muscarinic subtypes in the human brain: Changes with aging and dementia. J. Neurosci. Res. 1992, 31, 103–111. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
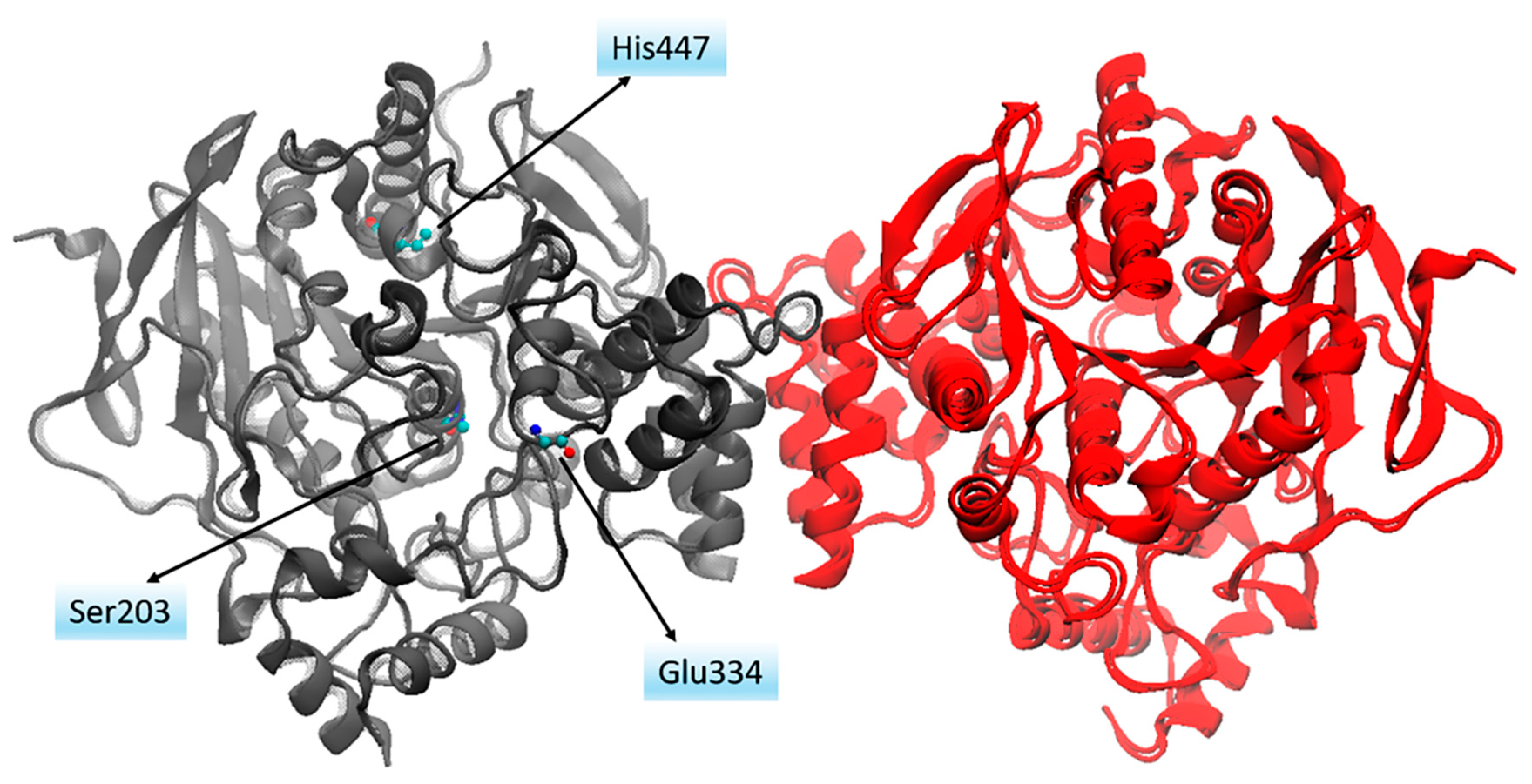
- Franklin, M.C.; Rudolph, M.J.; Ginter, C.; Cassidy, M.S.; Cheung, J. Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins 2016, 84, 1246–1256. [Google Scholar] [CrossRef]
- Lleo, A.; Greenberg, S.M.; Growdon, J.H. Current pharmacotherapy for Alzheimer’s disease. Ann. Rev. Med. 2006, 57, 513–533. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.L.; Farlow, M.R.; Doody, R.S.; Mohs, R.; Friedhoff, L.T. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Donepezil Study Group. Neurology 1998, 50, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey-Bloom, J.; Anand, R.; Veach, J. A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int. J. Geriatr. Psyopharmacol. 1998, 1, 55–65. [Google Scholar]
- Tariot, P.N.; Solomon, P.R.; Morris, J.C.; Kershaw, P.; Lilienfeld, S.; Ding, C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. The Galantamine USA-10 Study Group. Neurology 2000, 54, 2269–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L. Cholinesterase inhibitors: A new class of psychotropic compounds. Am. J. Psychiatry 2000, 157, 4–15. [Google Scholar] [CrossRef]
- Ryan, J.; Scali, J.; Carriere, I.; Ritchie, K.; Ancelin, M.-L. Hormonal treatment, mild cognitive impairment and Alzheimer’s disease. Int. Psychogeriatr. 2008, 20, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Giacobini, E. Cholinesterase inhibitors stabilize Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 920, 321–327. [Google Scholar] [CrossRef]
- Machado, L.P.; Kohaygawa, A.; Saito, M.E.; da Silveira, V.F.; Yonezawa, L.A. Lesão Oxidativa eritrocitária e mecanismos antioxidantes. Rev. Ciências Agroveterinárias 2009, 8, 84–94. [Google Scholar]
- Barreiros, A.L.B.S.; David, J.M.; David, J.P. Estresse oxidativo: relaçÃ\poundso entre geraçÃ\poundso de espÃ\copyrightcies reativas e defesa do organismo. QuÃ\-mica Nov. 2006, 29, 113–123. [Google Scholar]
- de Oliveira, M.C.; Schoffen, J.P.F. Oxidative stress action in cellular aging. Braz. Arch. Biol. Technol. 2010, 53, 1333–1342. [Google Scholar] [CrossRef] [Green Version]
- Celi, P. The role of oxidative stress in small ruminants’ health and production. Rev. Bras. Zootec. 2010, 39, 348–363. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.L.A.; Matsubara, L.S. Radicais livres: Conceitos, doenÃ\Sas relacionadas, sistema de defesa e estresse oxidativo. Rev. da Assoc. MÃ\copyrightdica Bras. 1997, 43, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Clement, M.V.; Long, L.H. Hydrogen peroxide in the human body. FEBS Lett. 2000, 486, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M. Superoxide: A two-edged sword. Braz. J. Med. Biol. Res. 1997, 30, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Vina, J.; Lloret, A.; Orti, R.; Alonso, D. Molecular bases of the treatment of Alzheimer’s disease with antioxidants: Prevention of oxidative stress. Mol. Aspects Med. 2004, 25, 117–123. [Google Scholar] [CrossRef]
- Aliev, G.; Obrenovich, M.E.; Tabrez, S.; Jabir, N.R.; Reddy, V.P.; Li, Y.; Burnstock, G.; Cacabelos, R.; Kamal, M.A. Link between cancer and Alzheimer disease via oxidative stress induced by nitric oxide-dependent mitochondrial DNA overproliferation and deletion. Oxid. Med. Cell. Longev. 2013, 2013, 962984. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet (London, England) 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Hauptmann, S.; Keil, U.; Scherping, I.; Bonert, A.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 2006, 41, 668–673. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Argellati, F.; Pronzato, M.A.; Domenicotti, C. Vitamin E and neurodegenerative diseases. Mol. Aspects Med. 2007, 28, 591–606. [Google Scholar] [CrossRef]
- Bianchi, M.d.L.P.; Antunes, L.M.G. Radicais livres e os principais antioxidantes da dieta. Rev. Nutr. 1999, 12, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, A.B.; Usman, M.; Ali, F.; Satti, S.A. Vitamin supplementation as an ajuvant treatment for Alzheimer disease. J. Clin. Diagn. Res. 2016, 10, 7–11. [Google Scholar]
- Ono, K.; Yamada, M. Vitamin A and Alzheimer’s disease. Geriatrics GerontolIntl. 2012, 12, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with vitamin B12-Underestimated neurological significance. Nutrients 2013, 5, 5031–5045. [Google Scholar]
- Presse, N.; Belleville, S.; Gaudreau, P.; Greenwood, C.E.; Kergoat, M.J.; Morais, J.A. Vitamin K status and cognitive function in healthy older adults. Neurobiol. Aging 2013, 34, 2777–2783. [Google Scholar] [CrossRef]
- Barbosa, K.B.F.; Costa, N.M.B.; Alfenas, R.d.C.G.; De Paula, S.O.; Minim, V.P.R.; Bressan, J. Estresse oxidativo: Conceito, implicaçÃ\mues e fatores modulatÃ\textthreesuperiorrios. Rev. Nutr. 2010, 23, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Jordao, A.; Chiarello, P.G.; Meirelles Bernardes, M.S.; Vannucchi, H.; Paulo, S. Peroxidação lipídica e etanol: Papel da glutationa reduzida e da vitamina E. Medicina (Ribeirao Preto. Online) 1998, 31. [Google Scholar]
- Scarmeas, N.; Luchsinger, J.A.; Schupf, N.; Brickman, A.M.; Cosentino, S.; Tang, M.X.; Stern, Y. Physical activity, diet, and risk of Alzheimer disease. JAMA 2009, 302, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S. Mediterranean diet and late-life cognitive impairment: A taste of benefit. JAMA 2009, 302, 686–687. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, B.R.; Cozzolino, S.M.F. Oxidative stress in Alzheimer’s disease: The role of vitamins C and E. Nutr.—Rev. da Soc. Bras. Aliment. e Nutr. 2009, 34, 249–259. [Google Scholar]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akabar, M.; Akabar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.; Lucariello, A.; Contieri, M.; Esposito, T.; Luca, A.; Guerra, G.; Perna, A. Therapeutic effects of turmeric in several diseases: An overview. Chem.-Biol. Interact. 2019, 310. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.M.; Lopes, T.; Oleastro, M.; Gato, I.V.; Floch, P.; Benejat, L.; Chaves, P.; Pereira, T.; Seixas, E.; Machado, J.; et al. Curcumin inhibits gastric inflammation induced by Helicobacter pylori infection in a mouse model. Nutrients 2015, 7, 306–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Jeynes, B.; Provias, J. Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis. Curr. Alzheimer Res. 2008, 5, 432–437. [Google Scholar] [CrossRef]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2006, 13, 195–208. [Google Scholar] [CrossRef]
- Peters, F.; Salihoglu, H.; Rodrigues, E.; Herzog, E.; Blume, T.; Filser, S.; Dorostkar, M.; Shimshek, D.R.; Brose, N.; Neumann, U.; et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 2018, 135, 695–710. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Schade, N.; Opitz, A.; Hilbrich, I.; Stieler, J.; Vogel, T.; Neukel, V.; Oberstadt, M.; Totzke, F.; Schächtele, C.; et al. Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay. Molecules 2018, 23, 2335. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, C.; Koo, C.-Y.; Yankova, N.; Tavares, I.A.; Wray, S.; Noble, W.; Hanger, D.P.; Morris, J.D.H. A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol. Commun. 2018, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadikar, H.; Torres, I.; Aiello, G.; Kurup, M.; Yang, Z.; Lin, F.; Kobeissy, F.; Yost, R.; Wang, K.K. Screening of Tau Protein Kinase Inhibitors in a Tauopathy-relevant cell-based model of Tau Hyperphosphorylation and Oligomerization. bioRxiv 2019, 821389. [Google Scholar]
- Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Jomova, K.; Kollar, V.; Rusko, M.; Valko, M. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch. Toxicol. 2019, 93, 2491–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacoppo, J.O.S.; França, T.C.C.; Kuča, K.; Da Cunha, E.F.F.; Abagyan, R.; Mancini, D.T.; Ramalho, T.C. Molecular modeling and in vitro reactivation study between the oxime BI-6 and acetylcholinesterase inhibited by different nerve agents. J. Biomol. Struct. Dyn. 2015, 33, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.F.; de Castro, A.A.; Soares, F.V.; Soares Leal, D.H.; da Cunha, E.F.F.; Mancini, D.T.; Ramalho, T.C. Development of technologies applied to the biodegradation of warfare nerve agents: Theoretical evidence for asymmetric homogeneous catalysis. Chem. Biol. Interact. 2019, 308, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Polisel, D.A.; de Castro, A.A.; Mancini, D.T.; da Cunha, E.F.F.; França, T.C.C.; Ramalho, T.C.; Kuca, K. Slight difference in the isomeric oximes K206 and K203 makes huge difference for the reactivation of organophosphorus-inhibited AChE: Theoretical and experimental aspects. Chem. Biol. Interact. 2019, 309, 108671. [Google Scholar] [CrossRef]
- Prandi, I.G.; Ramalho, T.C.; França, T.C.C. Esterase 2 as a fluorescent biosensor for the detection of organophosphorus compounds: Docking and electronic insights from molecular dynamics. Mol. Simul. 2019, 45, 1432–1436. [Google Scholar] [CrossRef]
- Da Silva, J.A.V.; Nepovimova, E.; Ramalho, T.C.; Kuca, K.; Celmar Costa França, T. Molecular modeling studies on the interactions of 7-methoxytacrine-4-pyridinealdoxime, 4-PA, 2-PAM, and obidoxime with VX-inhibited human acetylcholinesterase: A near attack conformation approach. J. Enzyme Inhib. Med. Chem. 2019, 34, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Soares, F.V.; de Castro, A.A.; Pereira, A.F.; Leal, D.H.S.; Mancini, D.T.; Krejcar, O.; Ramalho, T.C.; da Cunha, E.F.F.; Kuca, K. Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication. Int. J. Mol. Sci. 2018, 19, 1257. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.F.; Ramalho, T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules 2019, 24, 4410. https://doi.org/10.3390/molecules24234410
Teixeira JP, de Castro AA, Soares FV, da Cunha EFF, Ramalho TC. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules. 2019; 24(23):4410. https://doi.org/10.3390/molecules24234410
Chicago/Turabian StyleTeixeira, Jéssika P., Alexandre A. de Castro, Flávia V. Soares, Elaine F. F. da Cunha, and Teodorico C. Ramalho. 2019. "Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis" Molecules 24, no. 23: 4410. https://doi.org/10.3390/molecules24234410
APA StyleTeixeira, J. P., de Castro, A. A., Soares, F. V., da Cunha, E. F. F., & Ramalho, T. C. (2019). Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules, 24(23), 4410. https://doi.org/10.3390/molecules24234410