
Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
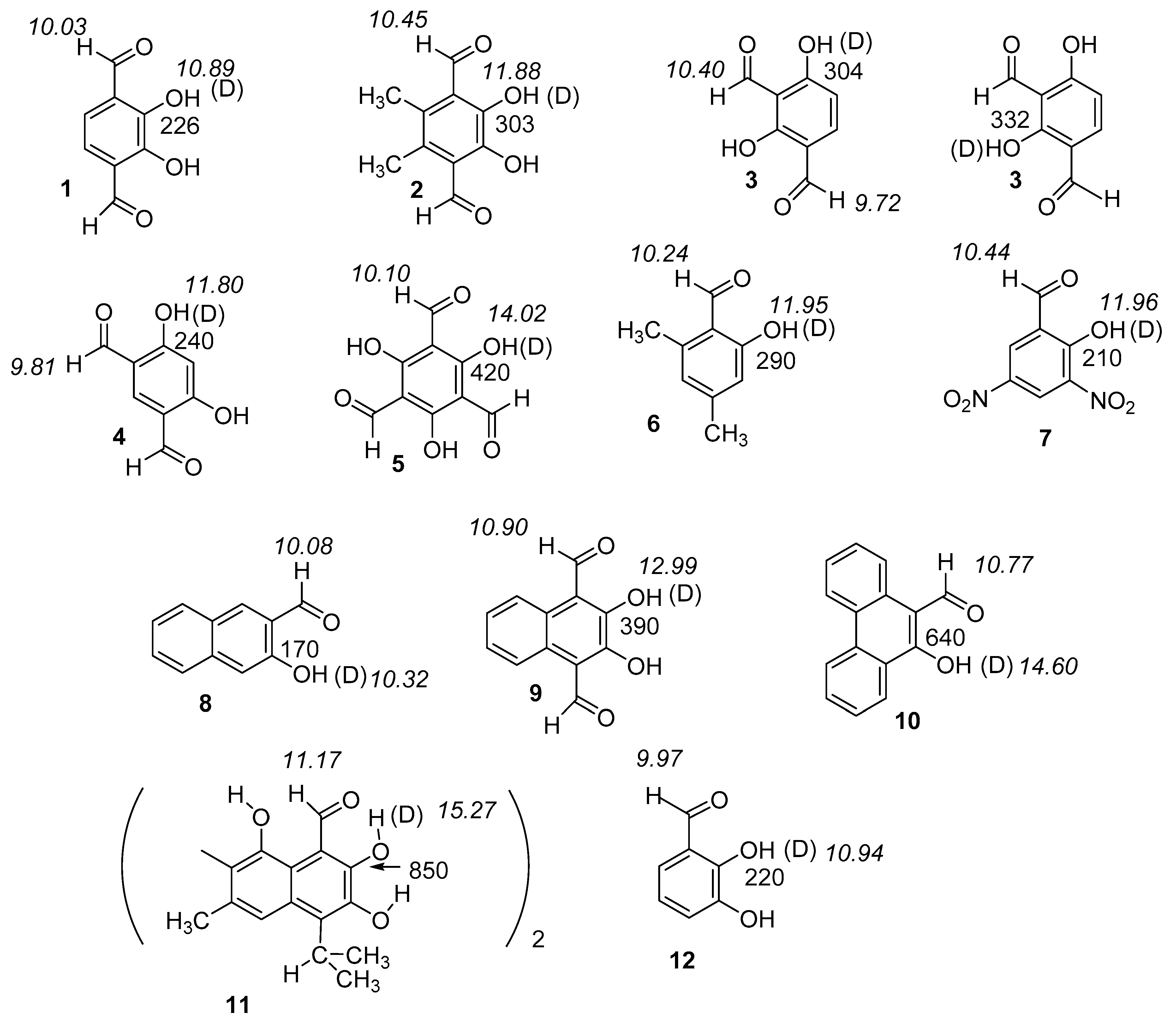
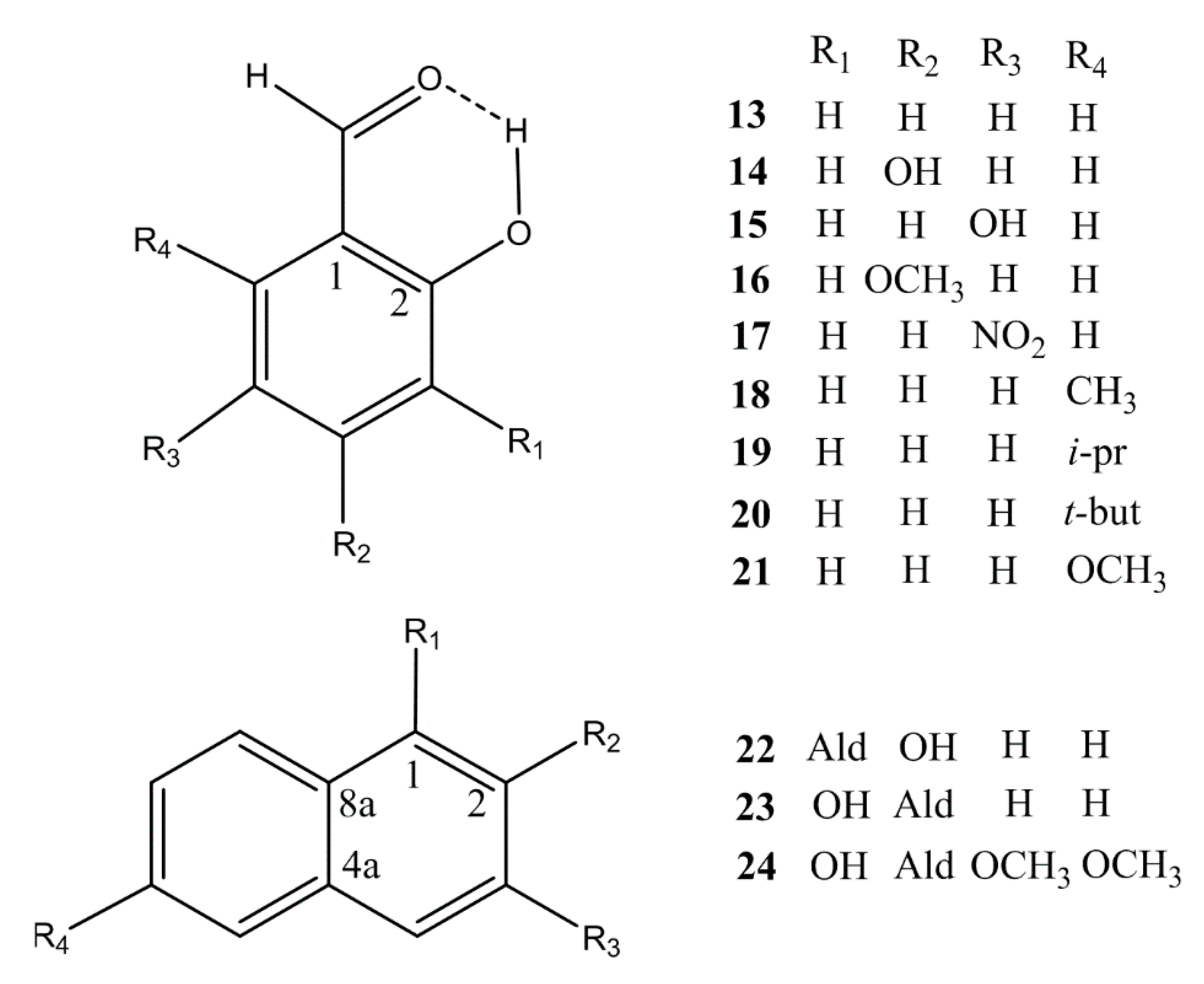
2. Results
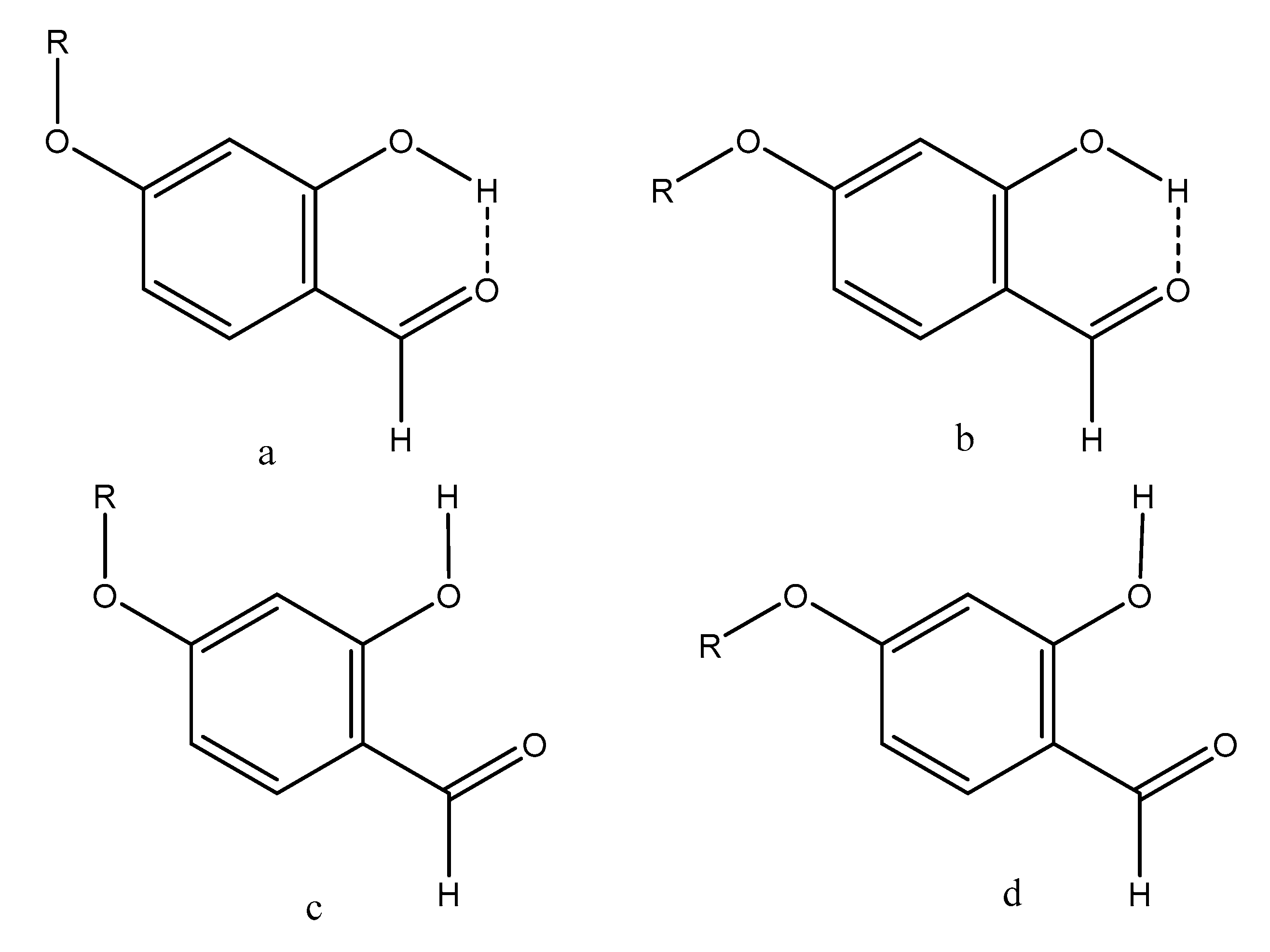
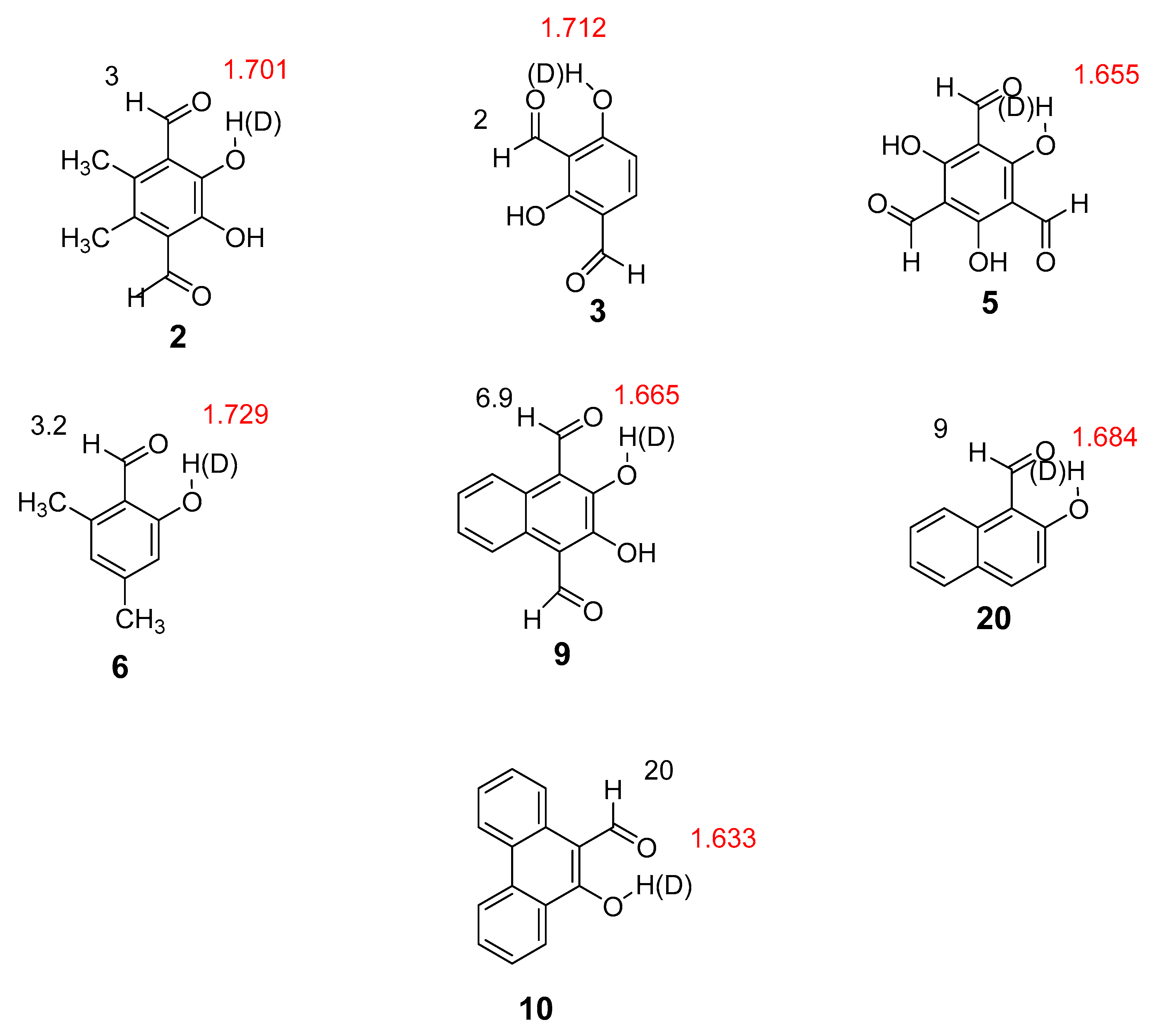
2.1. Structures and Energies
2.2. Deuterium Isotope Effects on Chemical Shifts
2.2.1. Deuterium Isotope Effects on 13C Chemical Shifts
2.2.2. Deuterium Isotope Effects on 1H Chemical Shifts
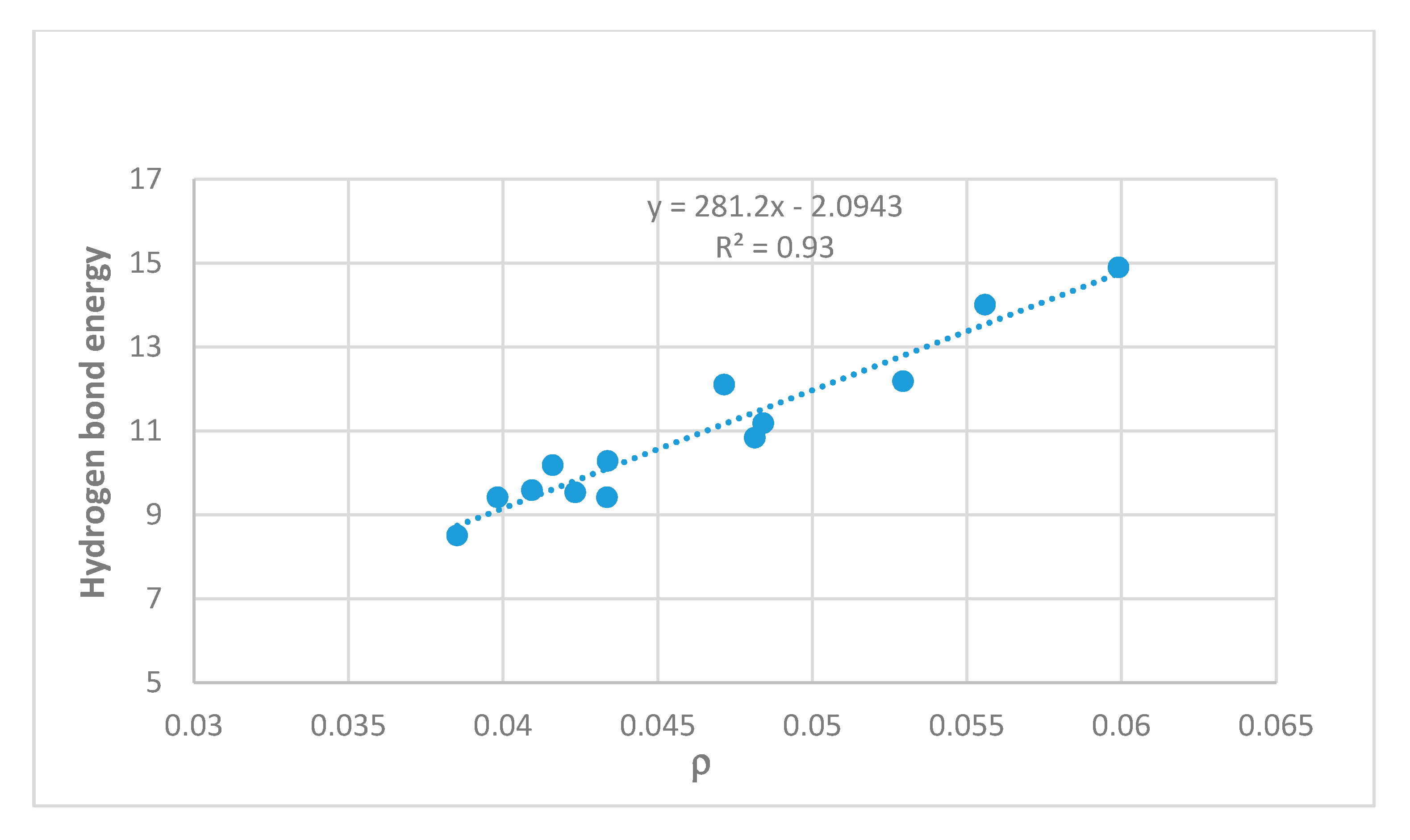
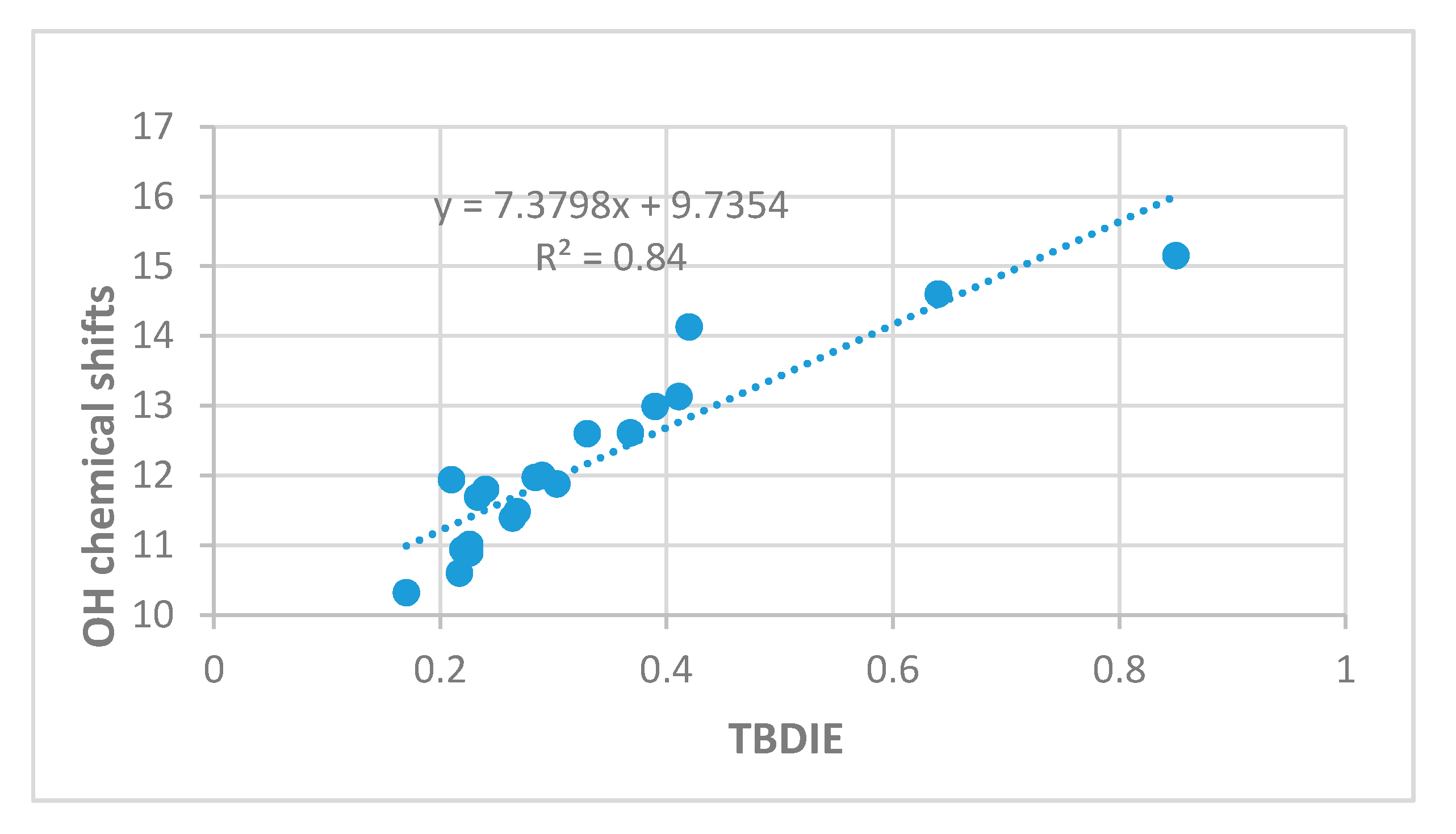
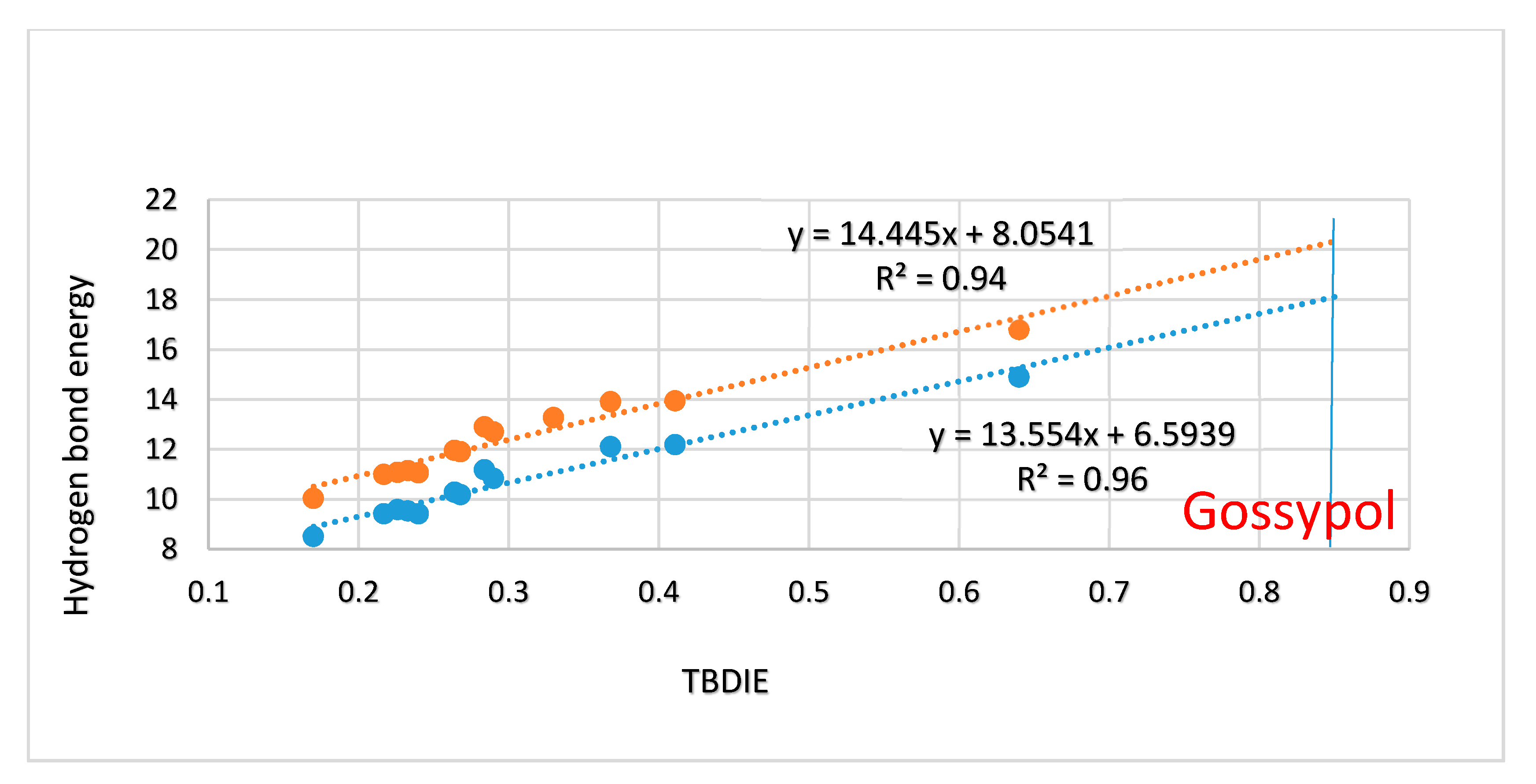
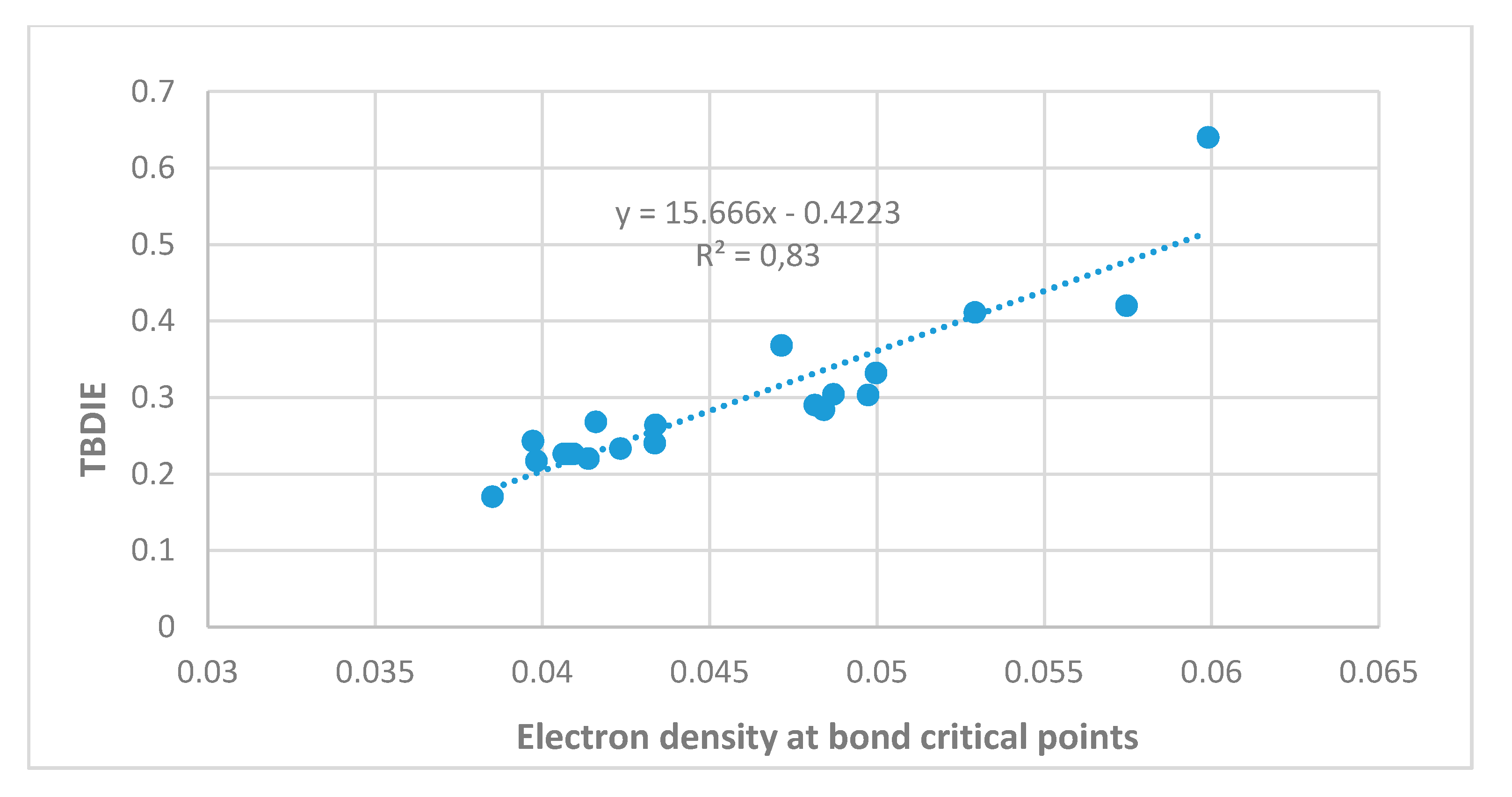
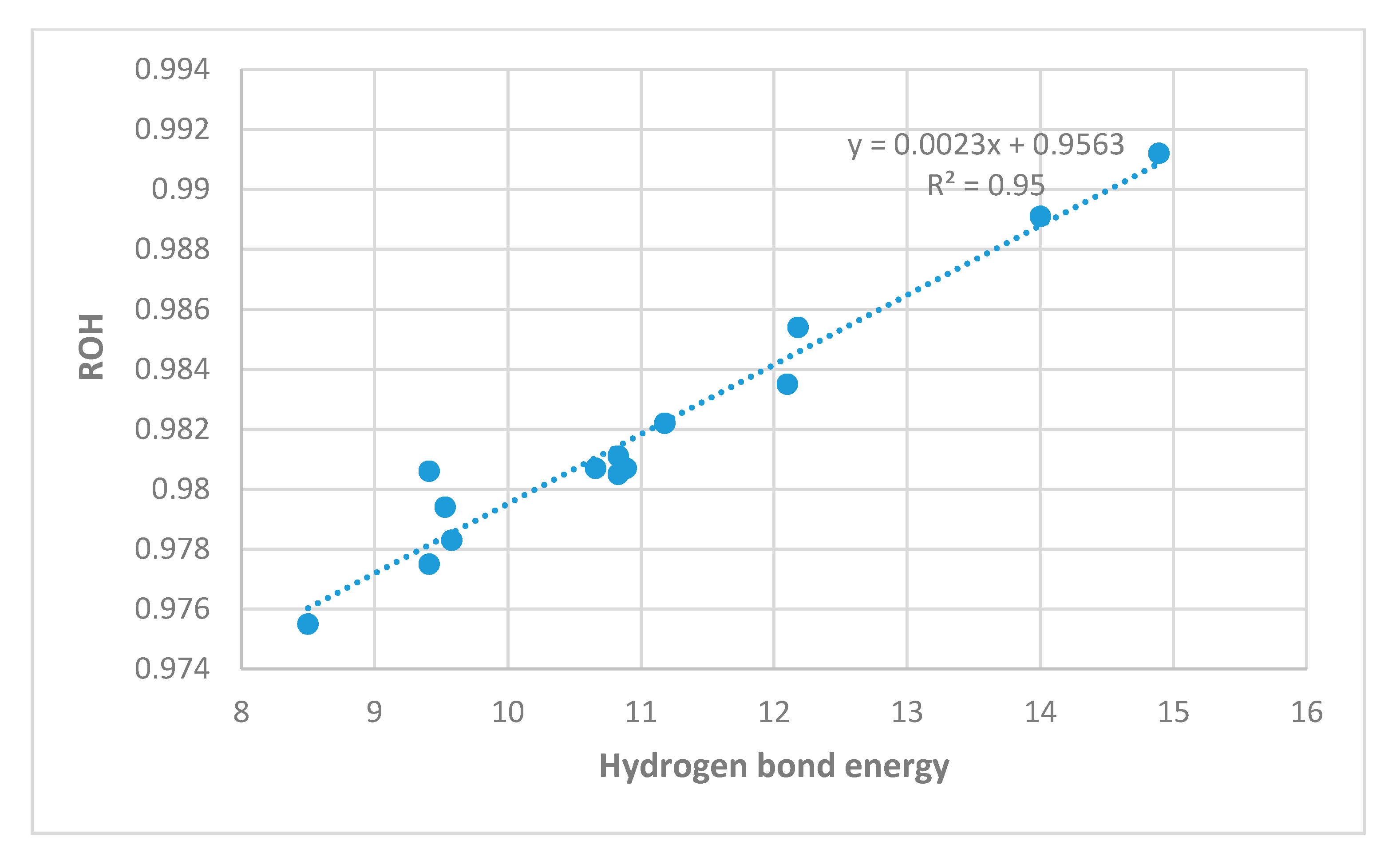
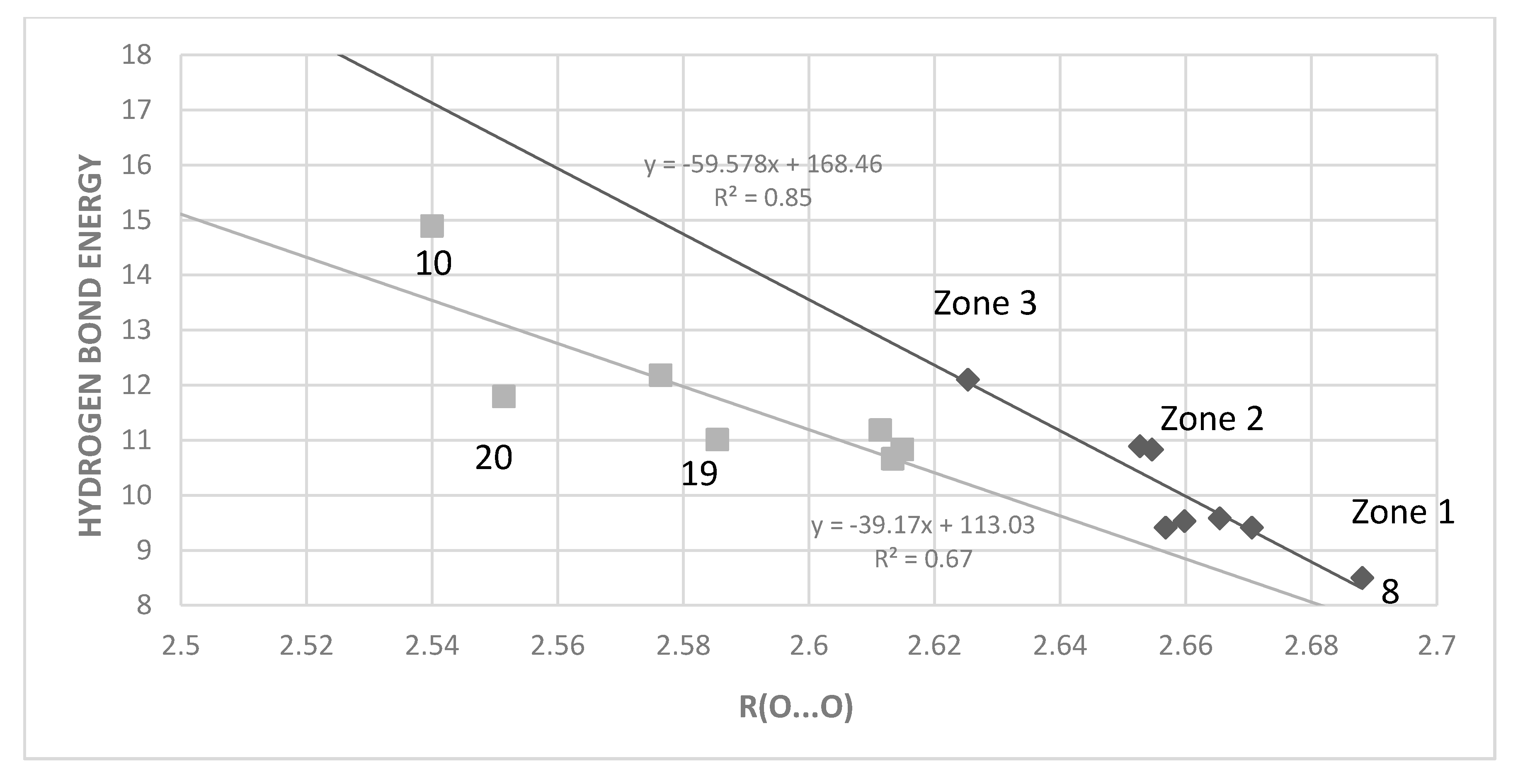
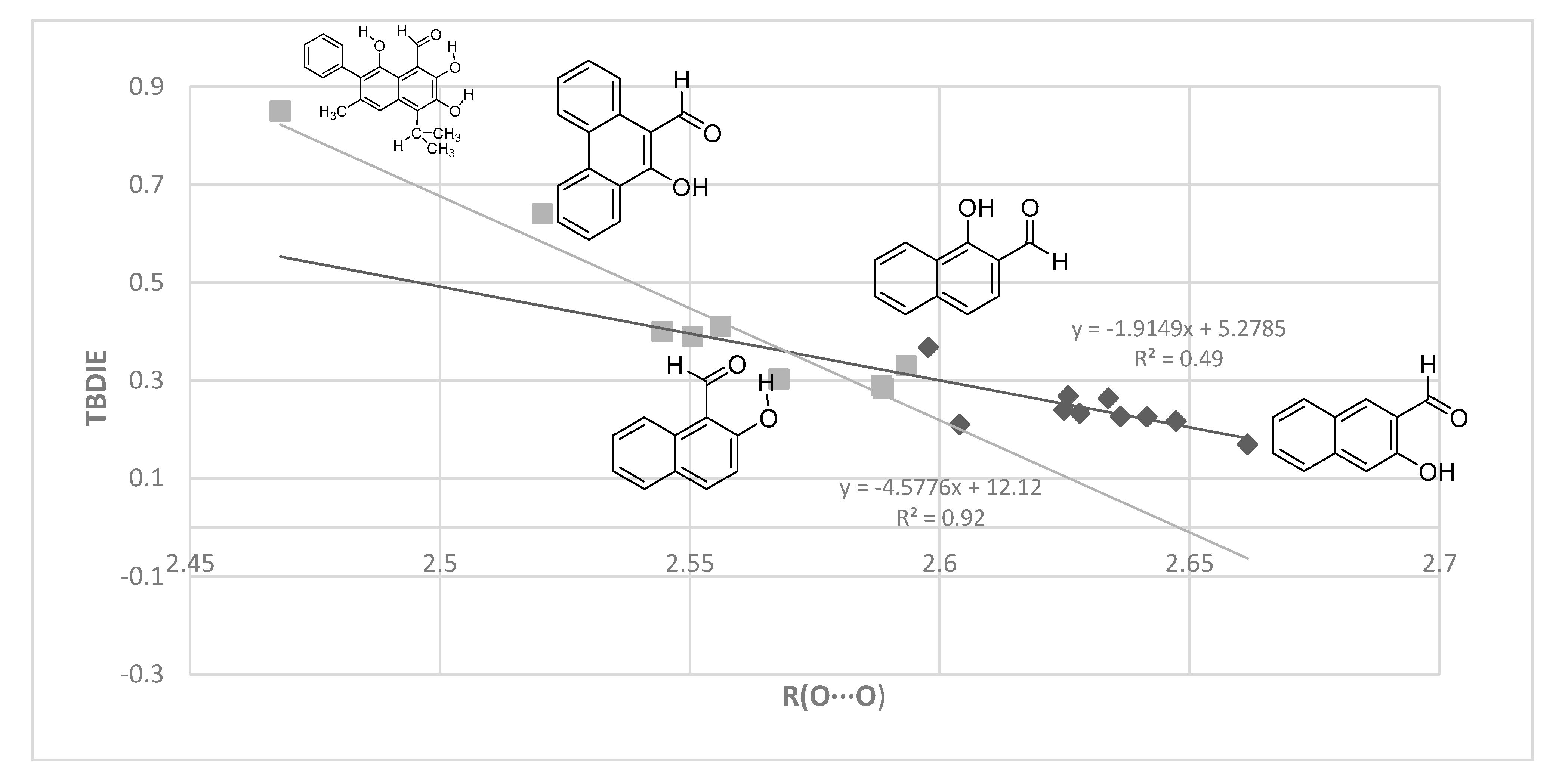
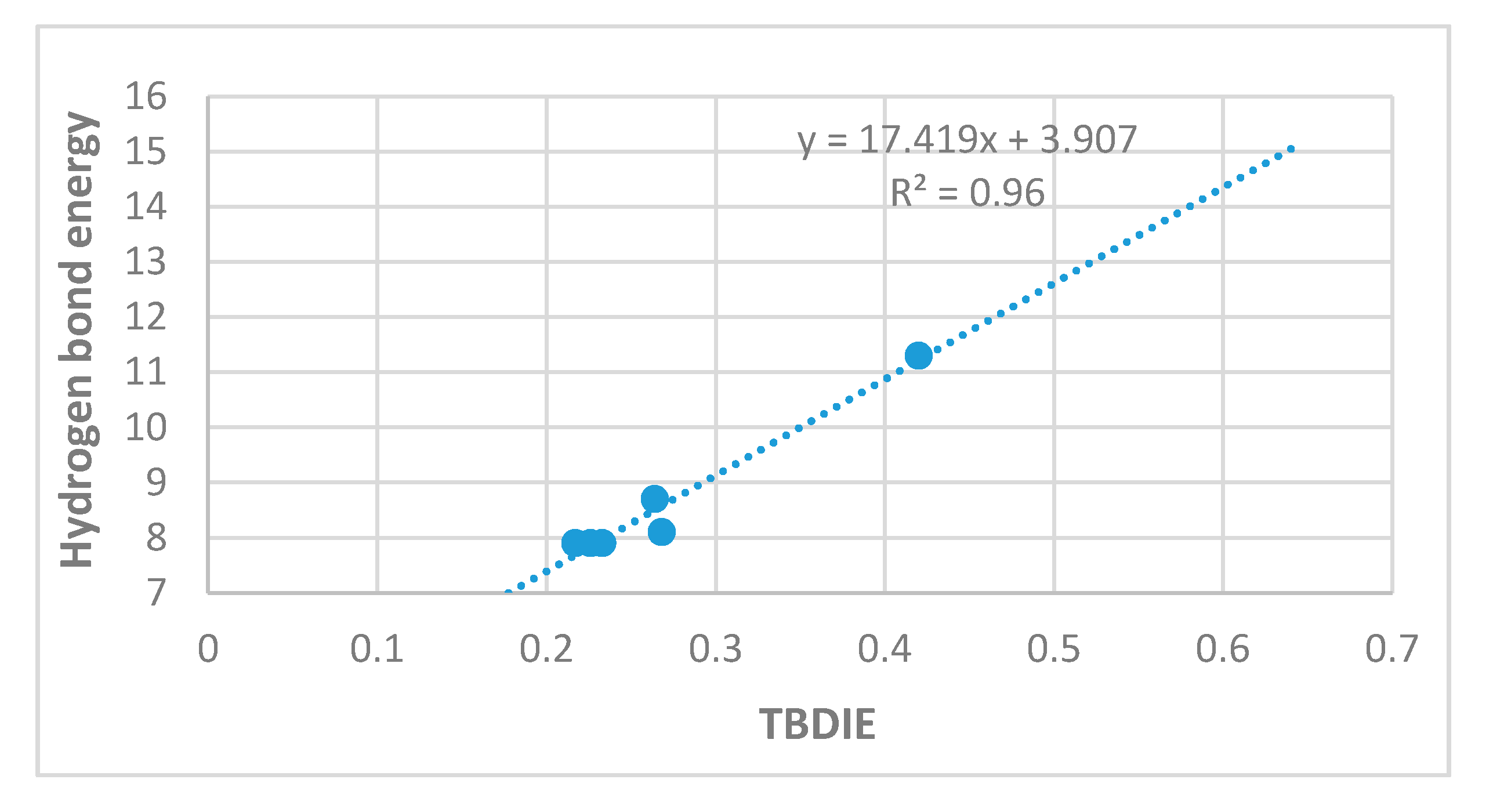
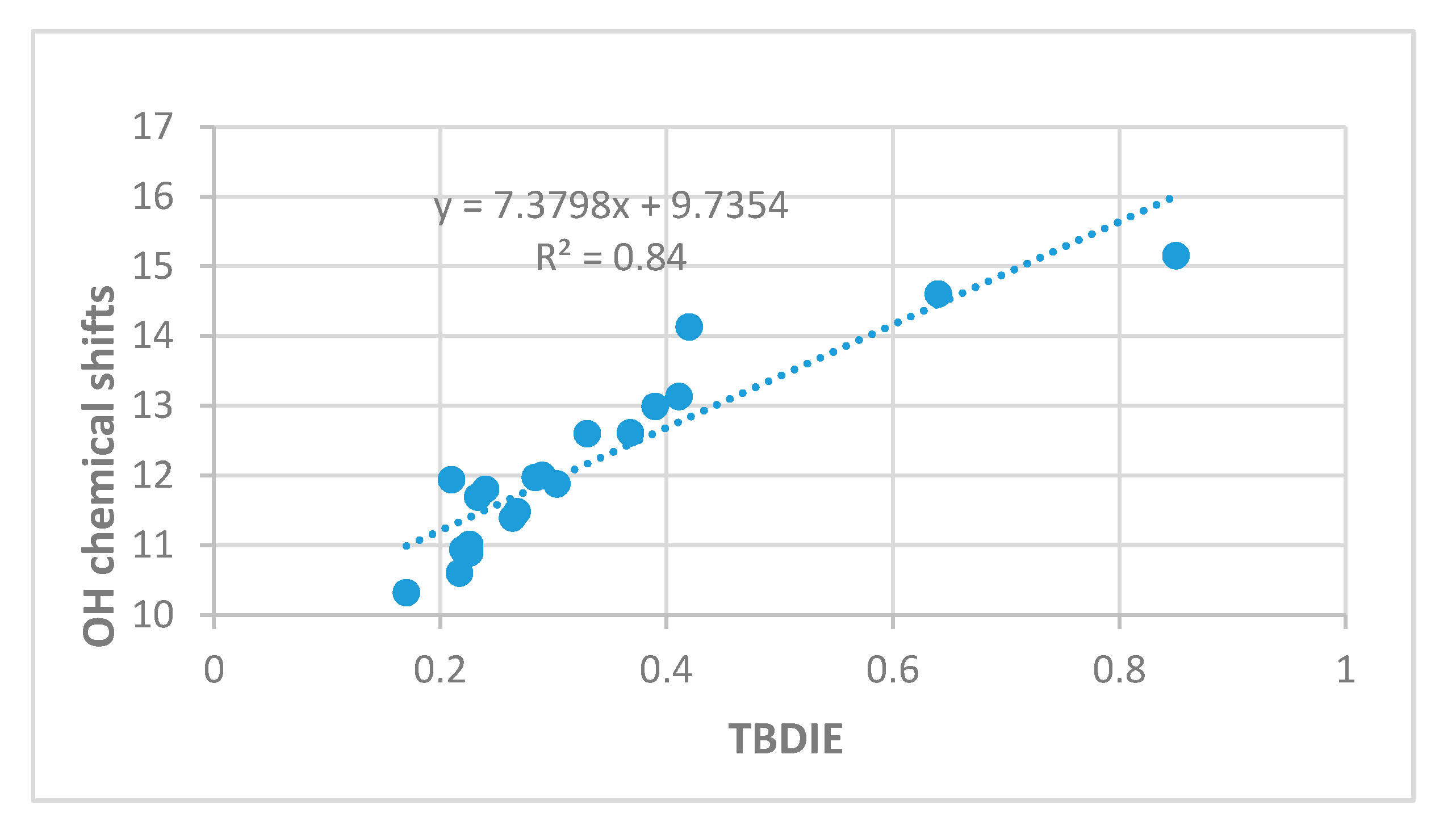
2.3. Correlations
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis
4.3. Deuteration
4.4. Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perrin, C.L.; Nielson, J.B. “Strong” Hydrogen Bonds in Chemistry and Biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L. Are Short, Low-Barrier Hydrogen Bonds unusually Strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR investigations of strong intramolecular hydrogen bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiner, S. Special issue: Intramolecular Hydrogen Bonding 2017. Molecules 2017, 22, 1521. [Google Scholar] [CrossRef] [Green Version]
- Afonin, A.V.; Vashenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H-NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting Hydrogen-Bond Strength from Acid-Base Molecular properties. The pKa slide rule: Toward the Solution of a Long-Lasting problem. Acc. Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef]
- Grabowski, S.J. An estimation of strength of intramolecular hydrogen bonds-ab initio and AIM studies. J. Mol. Struct. 2001, 562, 137–143. [Google Scholar] [CrossRef]
- Guillaumes, L.; Simon, S.; Fonseca Guerra, C. The Role of Aromaticity, Hybridization, Electrostatics, and Covalency in Resonance-Assisted Hydrogen Bonds of Adenine-Thymine (AT) Base Pairs and Their Mimics. ChemistryOpen 2015, 4, 318–327. [Google Scholar] [CrossRef]
- Sharif, S.; Huot, M.C.; Tolstoy, P.M.; Toney, M.D.; Jonsson, K.H.M.; Limbach, H.-H. 15N Nuclear magnetic Resonance Studies of Acid-Base properties of Pyridoxal-5′-phospate Aldimines in Aqeous solution. J. Phys. Chem. B 2007, 111, 3869–3876. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendation 2011). Pure Appl. Chem. 2011, 83, 1537–1641. [Google Scholar] [CrossRef]
- Hansen, P.E. NMR Studies of Compounds with Intramolecular Hydrogen bonds. In Isotope Effects in Chemistry and Biology; Kohen, A., Limbach, H.-H., CRC, Eds.; Taylor and Francis: Milton Park, UK, 2006; pp. 253–280. [Google Scholar]
- Cuma, M.; Scheiner, S.; Kar, T. Competition between rotamerization and proton transfer in o-hydroxybenzaldehyde. J. Am. Chem. Soc. 1998, 120, 10497–10503. [Google Scholar] [CrossRef]
- Grabowski, J.J. Theoretical studies of strong hydrogen bonds. Annu. Rep. Prog. Chem. Sect. C 2006, 102, 131–165. [Google Scholar] [CrossRef]
- Gadre, S.R.; Shirsat, R.N.; Limaye, A.C. Molecular Tailoring approach for Simulation of electrostatic Properties. J. Phys. Chem. 1994, 98, 9165–9169. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D. Energy of Intramolecular Hydrogen Bonding in ortho-Hydroxybenzaldehydes, Phenones and Quinones. Transfer of Aromaticity from ipso-Benzene ring to the Enol System(s). Molecules 2017, 22, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Reuben, J. Intramolecular Hydrogen Bonding as Reflected in the Deuterium Isotope effects on Carbon-13 Chemical shifts. Correlation with Hydrogen bond Energies. J. Am. Chem. Soc. 1986, 108, 1735–1738. [Google Scholar] [CrossRef]
- Schaefer, T. A Relationship between Hydroxyl proton Chemical shifts and torsional Frequencies in some Ortho-Substituted phenol Derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Hansen, P.E.; Tüchsen, E. Deuterium Isotope Effects on Carbonyl Chemical Shifts of BPTI. Hydrogen-bonding and Structure Determination in Proteins. Acta Chem. Scand. 1989, 43, 710–712. [Google Scholar] [CrossRef]
- Tüchsen, E.; Hansen, P.E. Hydrogen Bonding Monitored by Deuterium Isotope Effects on Carbonyl 13C Chemical Shifts in Basic Pancreatic Trypsin Inhibitor. Intra Residue Hydrogen Bonds in Antiparallel ß-sheet. Int. J. Biol. Macromol. 1991, 13, 2–8. [Google Scholar]
- Hansen, P.E. Isotope Effects on Nuclear Shielding. Intra-molecular Hydrogen- bonded Ketones, Aldehydes and Esters. Magn. Reson. Chem. 1993, 31, 23–37. [Google Scholar] [CrossRef]
- Hansen, P.E.; Bolvig, S.; Wozniak, K. Steric compression and twist in o-hydroxy acyl aromatics with intramolecular hydrogen bonding. J. Mol. Struct. 2005, 749, 155–168. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yanez, M.; Elguero, J. Resonance-assisted hydrogen bonds: A critical examination. Structure and stability of the enols of beta-diketones and beta-enaminones. J. Phys. Chem. A 2007, 11, 3585–3591. [Google Scholar] [CrossRef]
- That, Q.T.; Phung, K.P.; Hansen, P.E. Schiff Bases of Gossypol. A NMR and DFT Study. Magn. Reson. Chem. 2005, 43, 302–308. [Google Scholar] [CrossRef]
- Gdaniec, M.; Ibragimov, B.T.; Talipov, S.A. Lattice inclusion-compounds of Gossypol-Structure of the 2,3-Gossypol-Benzaldehyde Coordinatoclathrate. Acta Crystallogr. 1991, C47, 573–577. [Google Scholar]
- Pareras, G.; Palusiak, M.; Duran, M.; Solà, M.; Simon, S. Tuning of strength of the Resonance-assisted Hydrogen Bond in o-Hydroxybenzaldehyde by Substitution in the Aromatic Ring. J. Phys. Chem. A 2018, 122, 2279–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant, A.J.; Yun, M.; Sauer, M.; Yeung, C.S.; Maclachlan, M. Tautomerization in Naphthalenediimines: A Keto-Enamine Schiff Base Macromolecule. Org. Lett. 2005, 7, 4827–4830. [Google Scholar] [CrossRef]
- Shopsowitz, K.E.; Edwards, D.; Gallant, A.J.; MacLachlan, M.J. Highly substituted Schiff base microcycles via hexasubstituted benzene: A convenient double Duff formylation of catechol derivatives. Tetrahedron 2009, 65, 8113–8119. [Google Scholar] [CrossRef]
- Sauer, M.; Yeung, C.; Chong, J.H.; Patrick, B.O.; Maclachlan, M.J. N-Salidylidenes: Tautomers for Formation of Hydrogen-Bonded Capsules, clefts and Chains. J. Org. Chem. 2005, 71, 775–788. [Google Scholar] [CrossRef]
- Knight, P.D.; Clarkson, G.; Hammond, M.L.; Kimberley, B.S.; Scott, P. Radical and migratory insertion reaction mechanisms in Schiff base zirconium alkyls. J. Organomet. Chem. 2005, 690, 2125–5144. [Google Scholar] [CrossRef]
- Legouin, B.; Gayral, M.; Uriac, P.; Cupifia, J.-F.; Levoin, N.; Toupet, L.; van de Weghe, P. Molecular Tweezers: Synthesis and Formation of Host-Guest complexes. Eur. J. Org. Chem. 2010, 28, 5503–5508. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent Molecular-orbital methods. 9. Extended Gaussian-type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis-sets for anion calculations. 3. The 3-21+G basis set for 1st-row elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent Molecular-orbital methods. 25. Supplementary functions for Gaussian-Basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- AIMAll (17.11.14), Todd, A. Keneth, TK Gristmill Software. Available online: aim.tkgristmill.com (accessed on 4 December 2019).
Sample Availability: Samples of the compounds may be available from the authors. Please inquire. |












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, P.E.; Kamounah, F.S.; Saeed, B.A.; MacLachlan, M.J.; Spanget-Larsen, J. Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength. Molecules 2019, 24, 4533. https://doi.org/10.3390/molecules24244533
Hansen PE, Kamounah FS, Saeed BA, MacLachlan MJ, Spanget-Larsen J. Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength. Molecules. 2019; 24(24):4533. https://doi.org/10.3390/molecules24244533
Chicago/Turabian StyleHansen, Poul Erik, Fadhil S. Kamounah, Bahjat A. Saeed, Mark J. MacLachlan, and Jens Spanget-Larsen. 2019. "Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength" Molecules 24, no. 24: 4533. https://doi.org/10.3390/molecules24244533
APA StyleHansen, P. E., Kamounah, F. S., Saeed, B. A., MacLachlan, M. J., & Spanget-Larsen, J. (2019). Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength. Molecules, 24(24), 4533. https://doi.org/10.3390/molecules24244533