
Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy

 ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Interaction Analysis of CDK2-Ligand Crystal Structures
2.2. Screening of CDK2 Inhibitor-Like Compounds
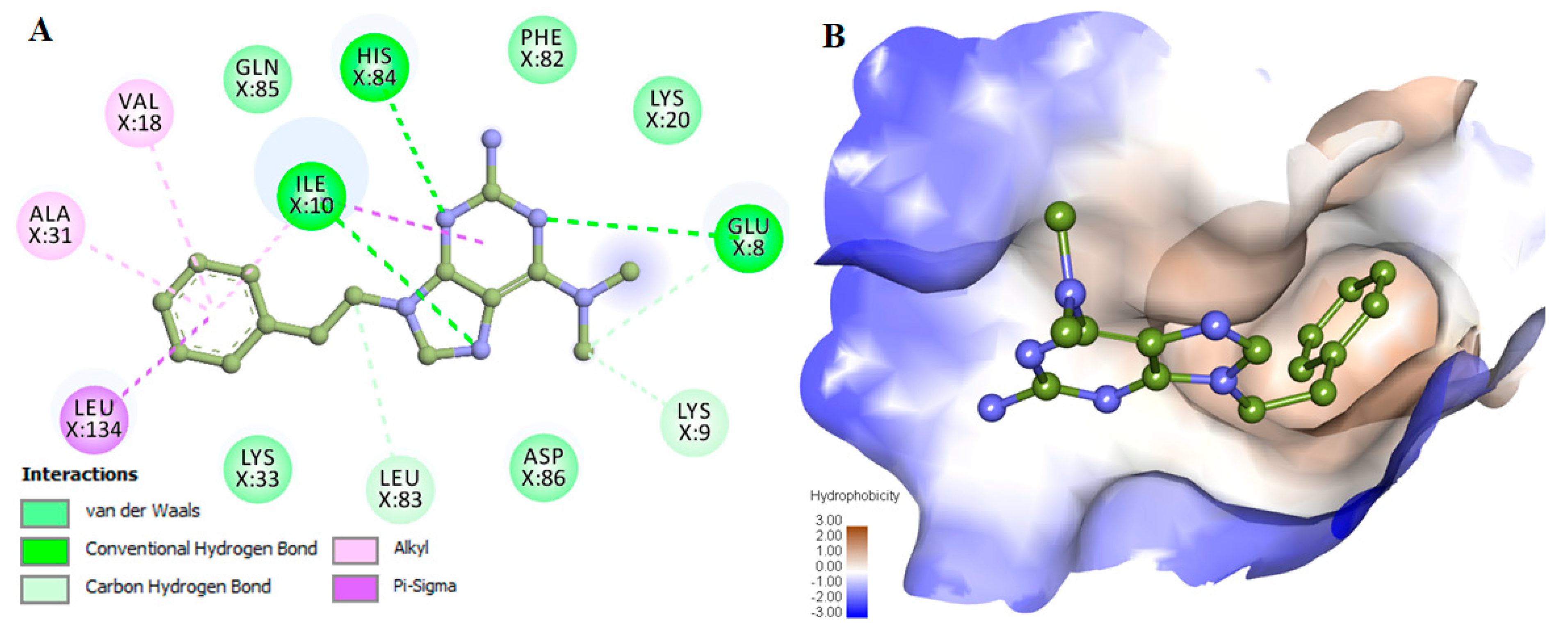
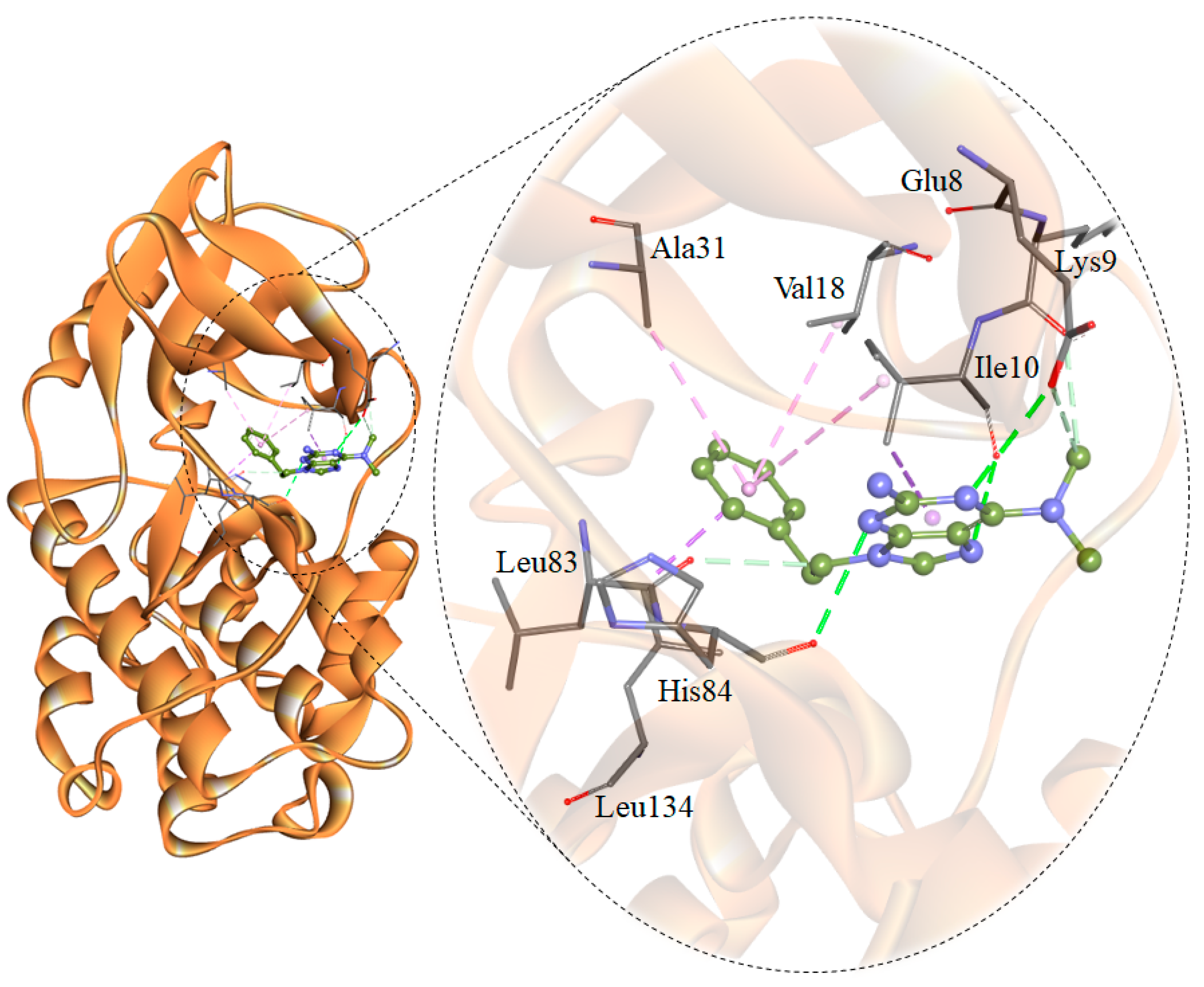
2.3. Assessment and Interaction Analysis
2.4. The Potential Energy of the Systems
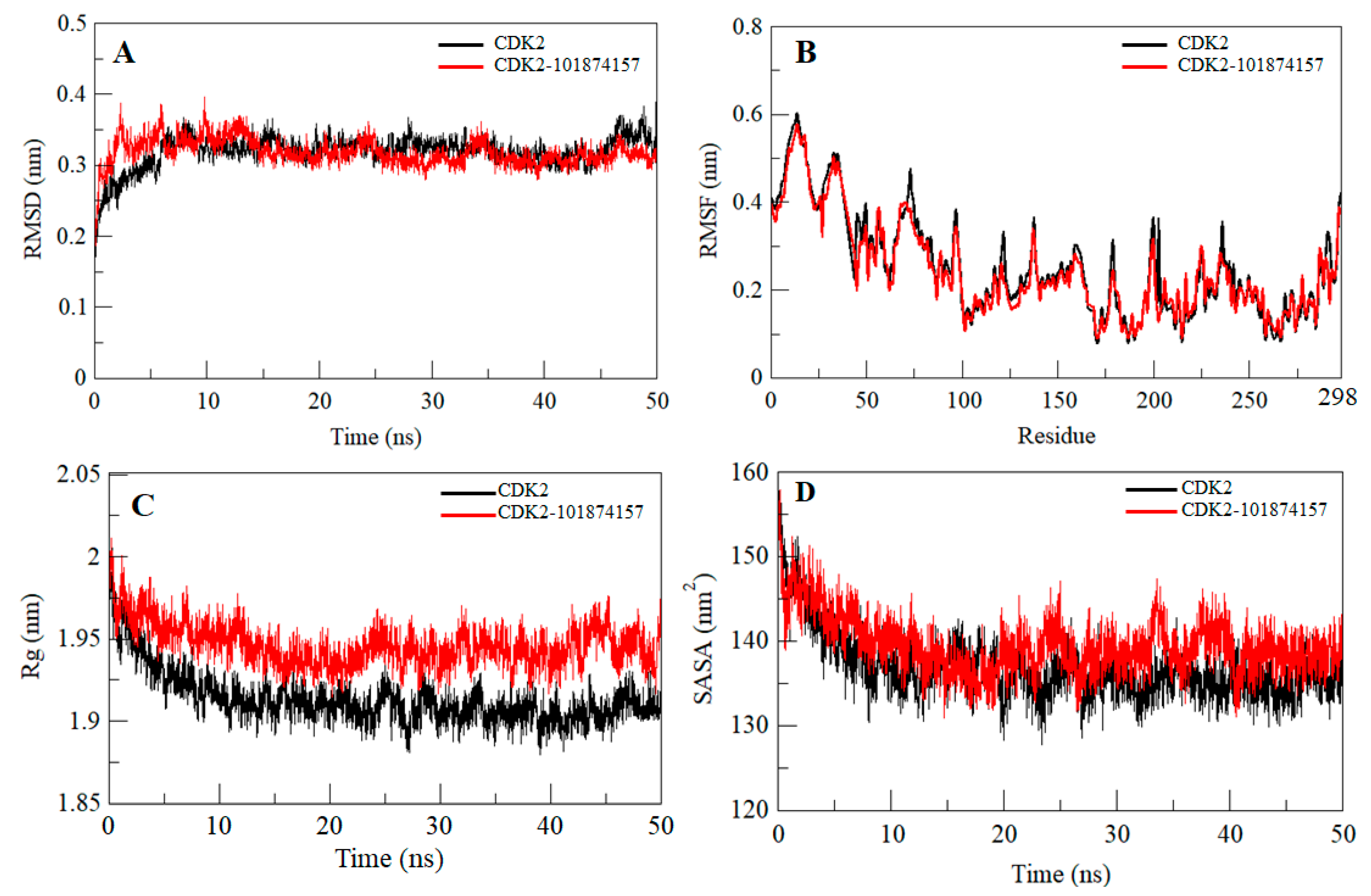
2.5. Structural Deviation and Compactness
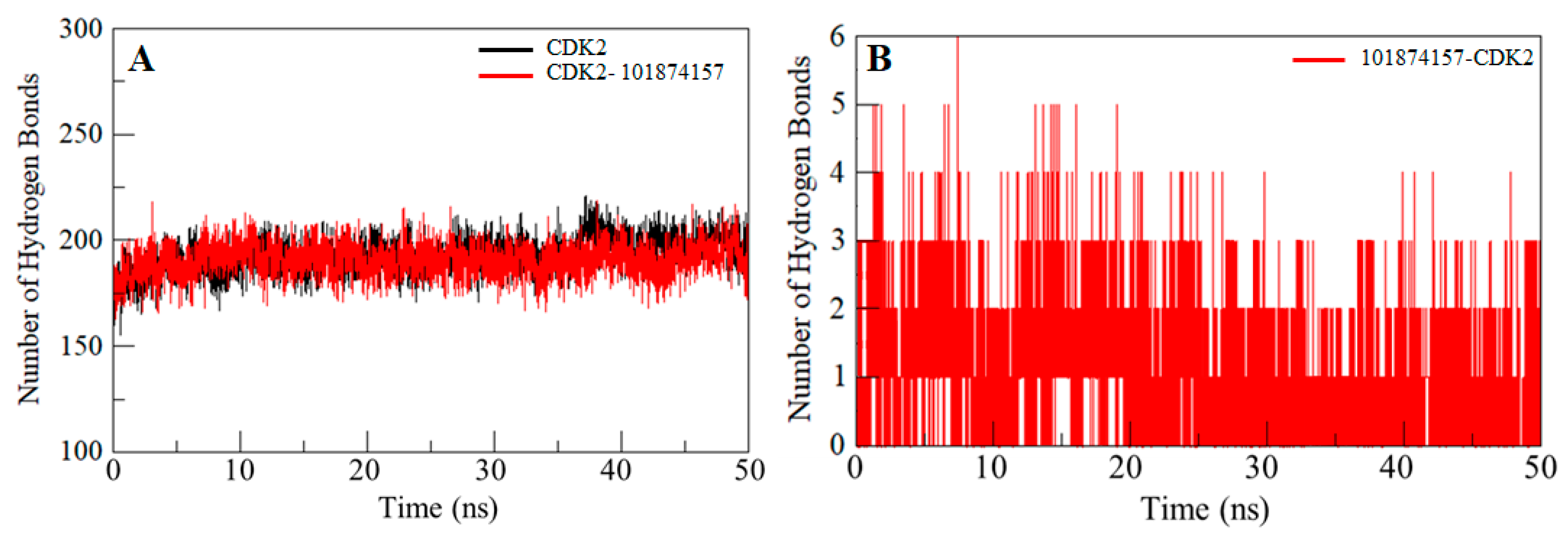
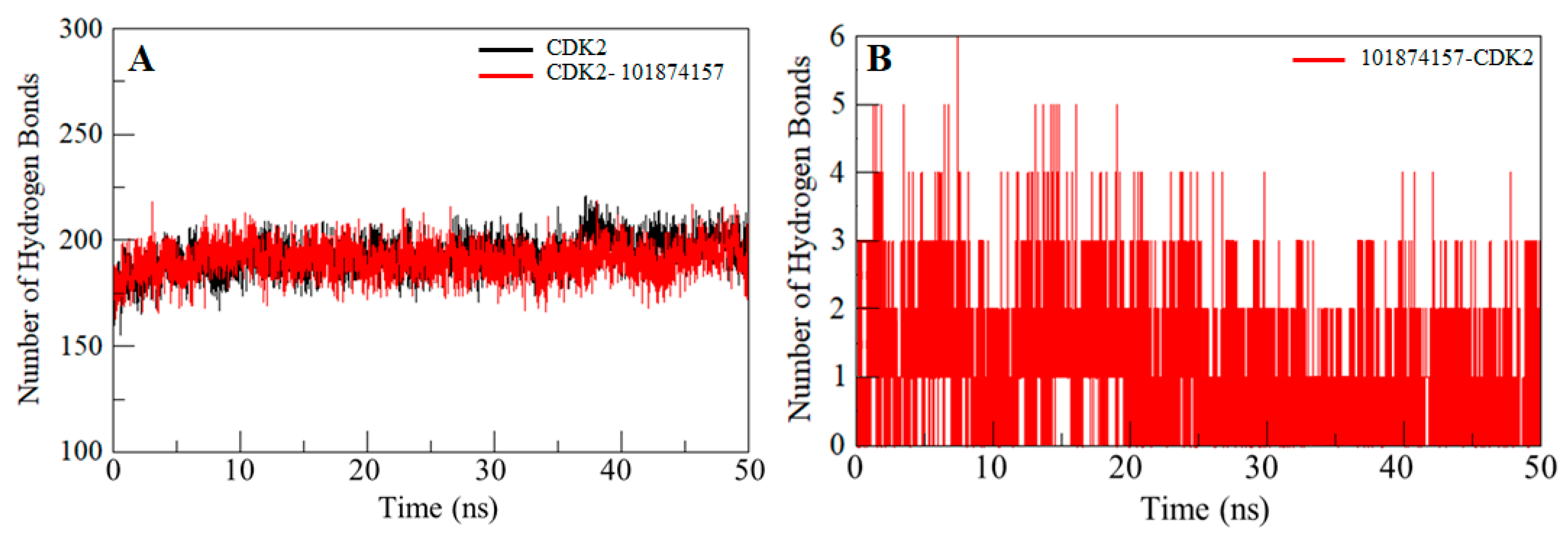
2.6. Hydrogen Bonds Analysis
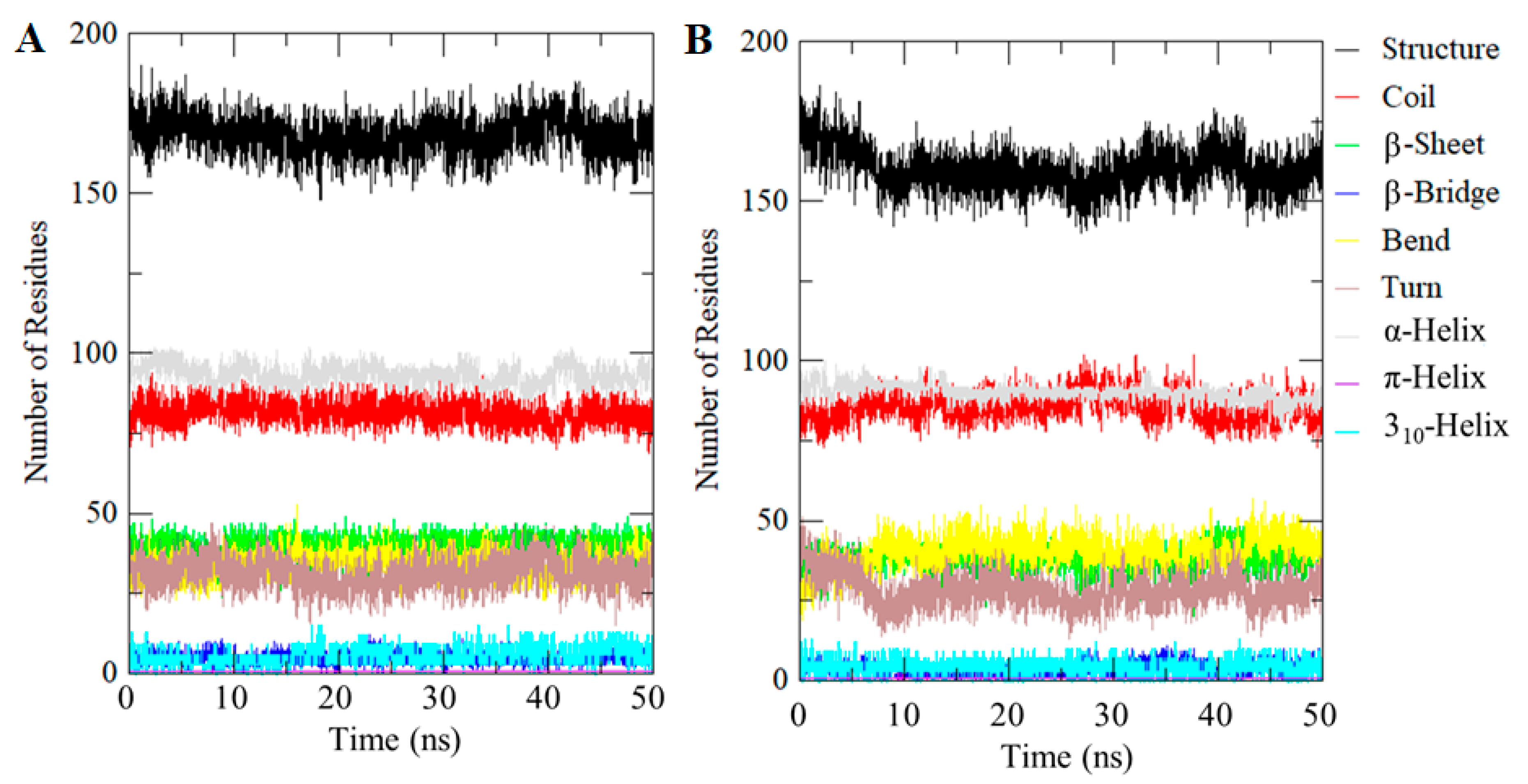
2.7. Evaluation of Secondary Structures
3. Materials and Methods
3.1. Materials
3.2. Interaction Analysis of Co-Crystallized Ligands
3.3. Retrieval of CDK2 Inhibitor-Like Compounds
3.4. Virtual Screening Using Molecular Docking Approach
3.5. Hit-Selection and Drug-Ability Assessment
3.6. MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gu, Y.; Rosenblatt, J.; Morgan, D.O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992, 11, 3995–4005. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.R.; Lowe, E.D.; Petri, E.; Skamnaki, V.; Antrobus, R.; Johnson, L. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle 2007, 6, 1350–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettencourt-Dias, M.; Hildebrandt, F.; Pellman, D.; Woods, G.; Godinho, S.A. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chohan, T.A.; Qian, H.; Pan, Y.; Chen, J.-Z. Cyclin-dependent kinase-2 as a target for cancer therapy: Progress in the development of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2015, 22, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Gladden, A.B.; Diehl, J.A. Cell cycle progression without cyclin E/CDK2: Breaking down the walls of dogma. Cancer Cell 2003, 4, 160–162. [Google Scholar] [CrossRef] [Green Version]
- Lane, M.E.; Yu, B.; Rice, A.; Lipson, K.E.; Liang, C.; Sun, L.; Tang, C.; McMahon, G.; Pestell, R.G.; Wadler, S. A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res. 2001, 61, 6170–6177. [Google Scholar]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561. [Google Scholar] [CrossRef]
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, J. Progression through the G1-phase of the on-going cell cycle. J. Cell. Biochem. 2003, 90, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.; Reinert, T.; Cristofanilli, M. Emerging innovative therapeutic approaches leveraging cyclin-dependent kinase inhibitors to treat advanced breast cancer. Clin. Pharmacol. Ther. 2018, 103, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgant, D.O.; Kim, S.-H. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595. [Google Scholar] [CrossRef] [PubMed]
- Connell-Crowley, L.; Solomon, M.; Wei, N.; Harper, J.W. Phosphorylation independent activation of human cyclin-dependent kinase 2 by cyclin A in vitro. Mol. Biol. Cell 1993, 4, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Ann. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: An update. J. Med. Chem. 2018, 62, 4233–4251. [Google Scholar] [CrossRef]
- Beale, G.; Haagensen, E.J.; Thomas, H.D.; Wang, L.-Z.; Revill, C.H.; Payne, S.L.; Golding, B.T.; Hardcastle, I.R.; Newell, D.R.; Griffin, R.J. Combined PI3K and CDK2 inhibition induces cell death and enhances in vivo antitumour activity in colorectal cancer. Br. J. Cancer 2016, 115, 682. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Welburn, J.P.; Tucker, J.A.; Johnson, T.; Lindert, L.; Morgan, M.; Willis, A.; Noble, M.E.; Endicott, J.A. How tyrosine 15 phosphorylation inhibits the activity of cyclin-dependent kinase 2-cyclin A. J. Biol. Chem. 2007, 282, 3173–3181. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.Q.; Jacobsen, D.M.; Young, M.A. Briefly bound to activate: Transient binding of a second catalytic magnesium activates the structure and dynamics of CDK2 kinase for catalysis. Structure 2011, 19, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Schulze-Gahmen, U.; Brandsen, J.; Jones, H.D.; Morgan, D.O.; Meijer, L.; Vesely, J.; Kim, S.H. Multiple modes of ligand recognition: Crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopentenyladenine. Proteins Struct. Funct. Bioinform. 1995, 22, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Thunnissen, A.-M.; White, A.; Garnier, M.; Nikolic, M.; Tsai, L.; Walter, J.; Cleverley, K.; Salinas, P.; Wu, Y. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Yu, B.; Lane, M.E.; Wadler, S. SU9516, a cyclin-dependent kinase 2 inhibitor, promotes accumulation of high molecular weight E2F complexes in human colon carcinoma cells. Biochemical pharmacology 2002, 64, 1091–1100. [Google Scholar] [CrossRef]
- Nguyen, T.; Hawkins, E.; Kolluri, A.; Kmieciak, M.; Park, H.; Lin, H.; Grant, S. Synergism between bosutinib (SKI-606) and the Chk1 inhibitor (PF-00477736) in highly imatinib-resistant BCR/ABL+ leukemia cells. Leuk. Res. 2015, 39, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischmann, T.O.; Hruza, A.; Duca, J.S.; Ramanathan, L.; Mayhood, T.; Windsor, W.T.; Le, H.V.; Guzi, T.J.; Dwyer, M.P.; Paruch, K. Structure-guided discovery of cyclin-dependent kinase inhibitors. Biopolym. Orig. Res. Biomol. 2008, 89, 372–379. [Google Scholar] [CrossRef]
- Beg, A.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. High throughput screening, docking, and molecular dynamics studies to identify potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. J. Biomol. Struct. Dyn. 2019, 37, 2179–2192. [Google Scholar] [CrossRef]
- Dahiya, R.; Mohammad, T.; Roy, S.; Anwar, S.; Gupta, P.; Haque, A.; Khan, P.; Kazim, S.N.; Islam, A.; Ahmad, F.; et al. Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy. Int. J. Biol. Macromol. 2019, 136, 1076–1085. [Google Scholar] [CrossRef]
- Fatima, S.; Mohammad, T.; Jairajpuri, D.S.; Rehman, M.T.; Hussain, A.; Samim, M.; Ahmad, F.J.; Alajmi, M.F.; Hassan, M.I. Identification and evaluation of glutathione conjugate gamma-l-glutamyl-l-cysteine for improved drug delivery to the brain. J. Biomol. Struct. Dyn. 2019, 1–11. [Google Scholar] [CrossRef]
- Gulzar, M.; Ali, S.; Khan, F.I.; Khan, P.; Taneja, P.; Hassan, M.I. Binding mechanism of caffeic acid and simvastatin to the integrin linked kinase for therapeutic implications: A comparative docking and MD simulation studies. J. Biomol. Struct. Dyn. 2019, 37, 4327–4337. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Naz, F.; Shahbaaz, M.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. Designing New Kinase Inhibitor Derivatives as Therapeutics Against Common Complex Diseases: Structural Basis of Microtubule Affinity-Regulating Kinase 4 (MARK4) Inhibition. OMICS 2015, 19, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Naz, F.; Shahbaaz, M.; Khan, S.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. PKR-inhibitor binds efficiently with human microtubule affinity-regulating kinase 4. J. Mol. Graph. Model. 2015, 62, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Naz, H.; Shahbaaz, M.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. Effect of pH on the structure, function, and stability of human calcium/calmodulin-dependent protein kinase IV: Combined spectroscopic and MD simulation studies. Biochem. Cell Biol. 2016, 94, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, H.; Shahbaaz, M.; Haque, M.A.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. Urea-induced denaturation of human calcium/calmodulin-dependent protein kinase IV: A combined spectroscopic and MD simulation studies. J. Biomol. Struct. Dyn. 2017, 35, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.B.; Shahbaaz, M.; Khan, S.H.; Srivastava, S.; Islam, A.; Ahmad, F.; Hassan, M.I. Estimation of pH effect on the structure and stability of kinase domain of human integrin-linked kinase. J. Biomol. Struct. Dyn. 2019, 37, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.I.; Wang, Y. Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies. Int. J. Mol. Sci. 2019, 20, 884. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.I.; Shahbaaz, M.; Bisetty, K.; Waheed, A.; Sly, W.S.; Ahmad, F.; Hassan, M.I. Large scale analysis of the mutational landscape in beta-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene 2016, 576, 36–44. [Google Scholar] [CrossRef]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menendez, C.; Hernandez, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic beta-fructosidase enzymes. J. Mol. Model. 2015, 21, 2772. [Google Scholar] [CrossRef]
- Ali, S.A.; Hassan, M.I.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In eLS; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2001. [Google Scholar]
- Hoda, N.; Naz, H.; Jameel, E.; Shandilya, A.; Dey, S.; Hassan, M.I.; Ahmad, F.; Jayaram, B. Curcumin specifically binds to the human calcium-calmodulin-dependent protein kinase IV: Fluorescence and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2016, 34, 572–584. [Google Scholar] [CrossRef]
- Idrees, D.; Shahbaaz, M.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. Effect of pH on structure, function, and stability of mitochondrial carbonic anhydrase VA. J. Biomol. Struct. Dyn. 2017, 35, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821. [Google Scholar] [CrossRef] [PubMed]
- Veselý, J.; Havliček, L.; Strnad, M.; Blow, J.J.; Donella-Deana, A.; Pinna, L.; Letham, D.S.; Kato, J.Y.; Detivaud, L.; Leclerc, S. Inhibition of cyclin-dependent kinases by purine analogues. Eur. J. Biochem. 1994, 224, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, S.M. ChemBioOffice Ultra 2010 Suite; ACS Publications: Washington, DC, USA, 2010. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, B.; Roshan, M.; Shaahul Hameed, B.; Sumathi, K.; Senthilkumar, R.; Udayakumar, A.; Venkatesh Babu, K.; Kalaivani, M.; Sowmiya, G.; Sivasankari, P. PSAP: Protein structure analysis package. J. Appl. Crystallogr. 2007, 40, 773–777. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Zhang, L.; Ai, H.; Chen, W.; Yin, Z.; Hu, H.; Zhu, J.; Zhao, J.; Zhao, Q.; Liu, H. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef]
- Merkel, R.; Nassoy, P.; Leung, A.; Ritchie, K.; Evans, E. Energy landscapes of receptor–ligand bonds explored with dynamic force spectroscopy. Nature 1999, 397, 50. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification and evaluation of bioactive natural products as potential inhibitors of human microtubule affinity-regulating kinase 4 (MARK4). J. Biomol. Struct. Dyn. 2019, 37, 1813–1829. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Mohammad, T.; Dahiya, R.; Roy, S.; Noman, O.M.A.; Alajmi, M.F.; Hussain, A.; Hassan, M.I. Evaluation of binding and inhibition mechanism of dietary phytochemicals with sphingosine kinase 1: Towards targeted anticancer therapy. Sci. Rep. 2019, 10, 18727. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorption | Distribution | Metabolism | Excretion | Carcinogenicity | |||
|---|---|---|---|---|---|---|---|
| WS * | HIA (%) | BBB Permeability | CNS Permeability | CYP Subs./Inh. | Clearance | Renal OCT2 Substrate | |
| Soluble | 95.93 | −0.83 | −2.63 | No | 0.89 | Yes | No |
| System | RMSD (nm) | RMSF (nm) | Rg (nm) | SASA (nm2) | Kinetic Energy | Enthalpy | Volume (nm3) | Density (g/L) |
|---|---|---|---|---|---|---|---|---|
| CDK2 | 0.32 | 0.25 | 1.91 | 136.81 | 140,218 | −723,310 | 571.86 | 1024.83 |
| CDK2-101874157 | 0.31 | 0.23 | 1.94 | 139.49 | 140,064 | −714,423 | 583.33 | 1004.62 |
| System | Percentage of Protein Secondary Structure | |||||||
|---|---|---|---|---|---|---|---|---|
| Structure * | Coil | β-sheet | β-bridge | Bend | Turn | α-Helix | Other # | |
| CDK2 | 58 | 28 | 14 | 1 | 12 | 11 | 32 | 2 |
| CDK2-101874157 | 55 | 30 | 13 | 1 | 14 | 10 | 31 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, T.; Batra, S.; Dahiya, R.; Baig, M.H.; Rather, I.A.; Dong, J.-J.; Hassan, I. Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy. Molecules 2019, 24, 4589. https://doi.org/10.3390/molecules24244589
Mohammad T, Batra S, Dahiya R, Baig MH, Rather IA, Dong J-J, Hassan I. Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy. Molecules. 2019; 24(24):4589. https://doi.org/10.3390/molecules24244589
Chicago/Turabian StyleMohammad, Taj, Sagar Batra, Rashmi Dahiya, Mohammad Hassan Baig, Irfan Ahmad Rather, Jae-June Dong, and Imtaiyaz Hassan. 2019. "Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy" Molecules 24, no. 24: 4589. https://doi.org/10.3390/molecules24244589
APA StyleMohammad, T., Batra, S., Dahiya, R., Baig, M. H., Rather, I. A., Dong, J. -J., & Hassan, I. (2019). Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy. Molecules, 24(24), 4589. https://doi.org/10.3390/molecules24244589