
Efficient Synthesis of UDP-Furanoses via 4,5-Dicyanoimidazole(DCI)-Promoted Coupling of Furanosyl-1-Phosphates with Uridine Phosphoropiperidate


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. A Phosphoramidite Approach for Furanosyl-1-Phosphate Synthesis
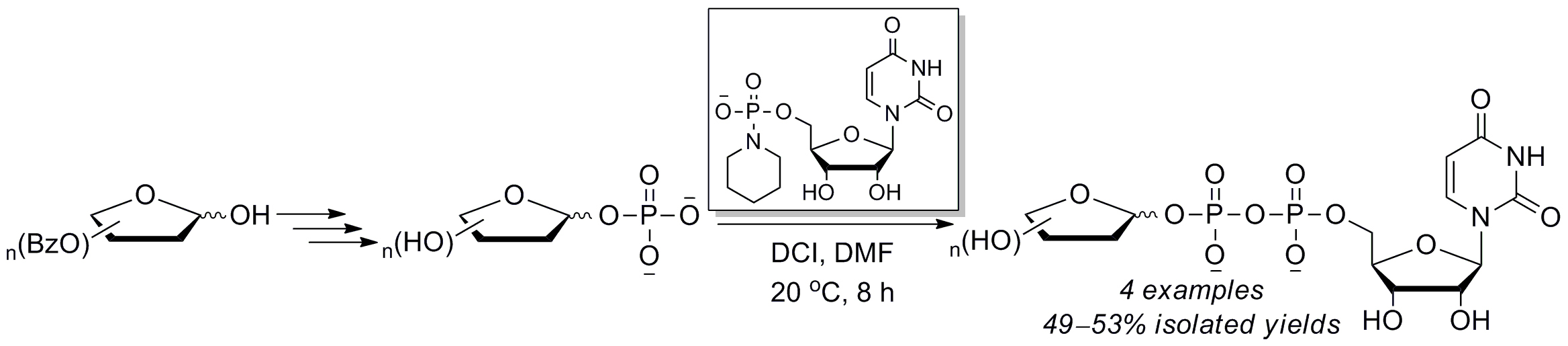
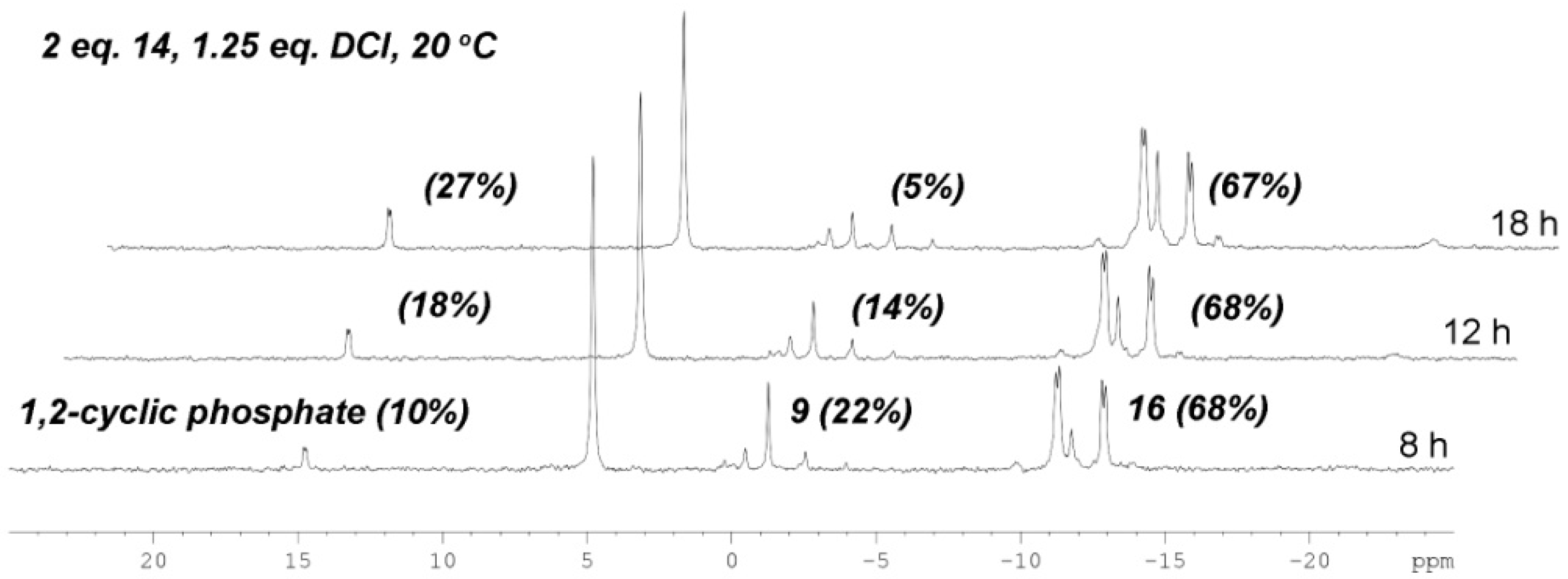
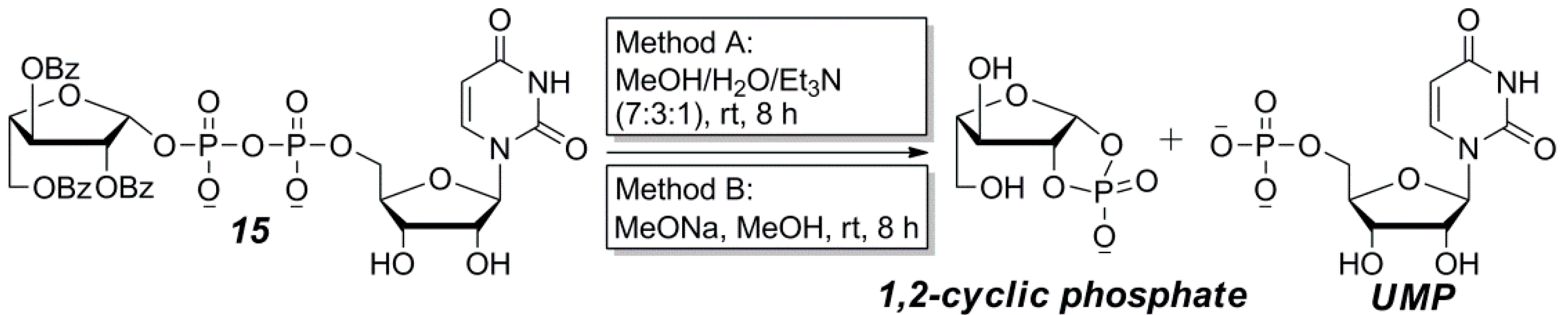
2.2. The Attempt to Synthesize UDP-l-Araf (16) from Benzoyl-Protected l-Arabinofuranosyl-1-Phosphate (13)
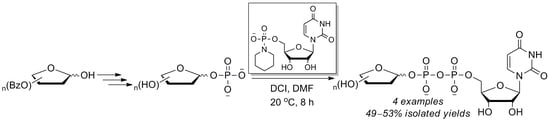
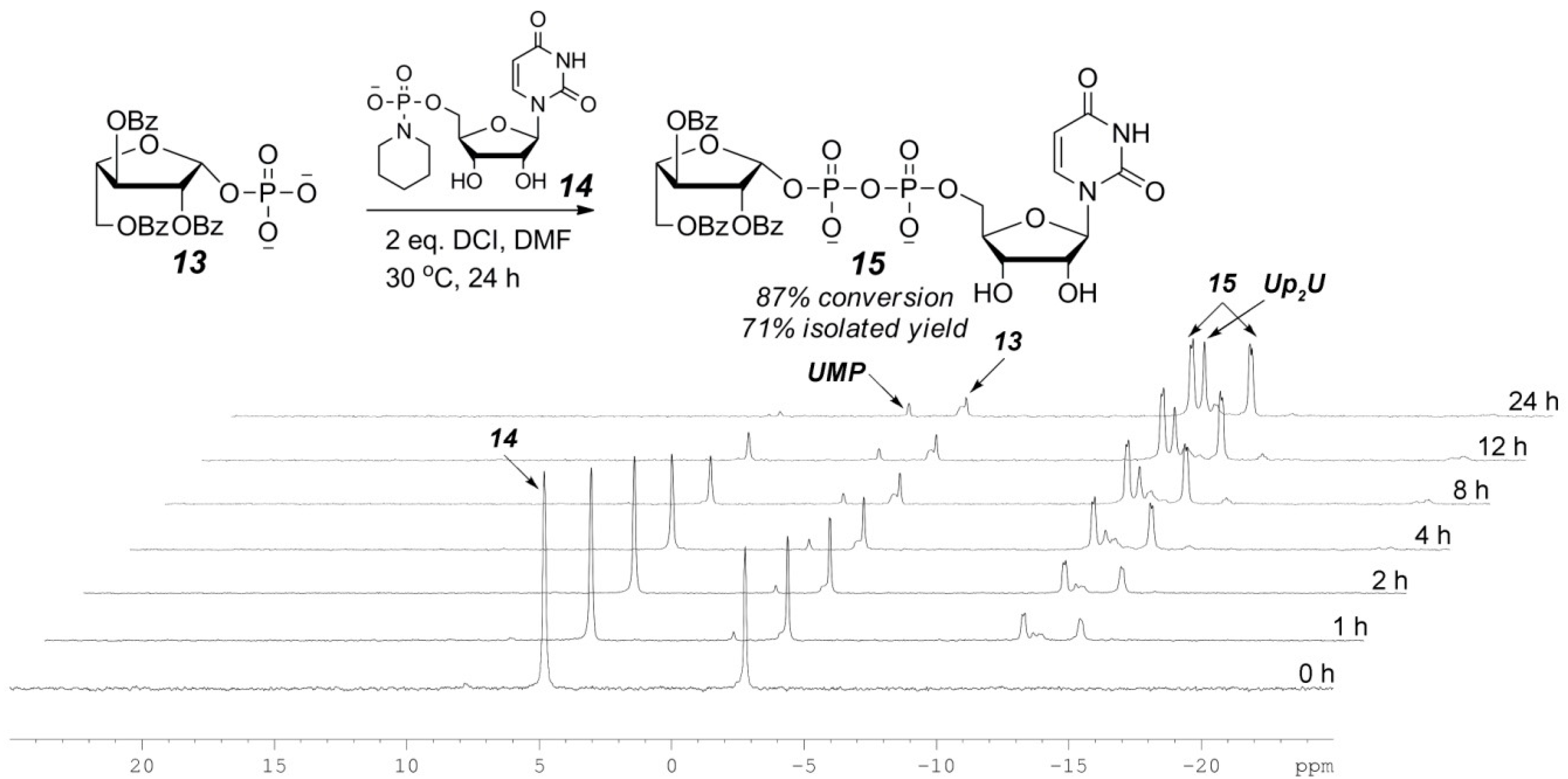
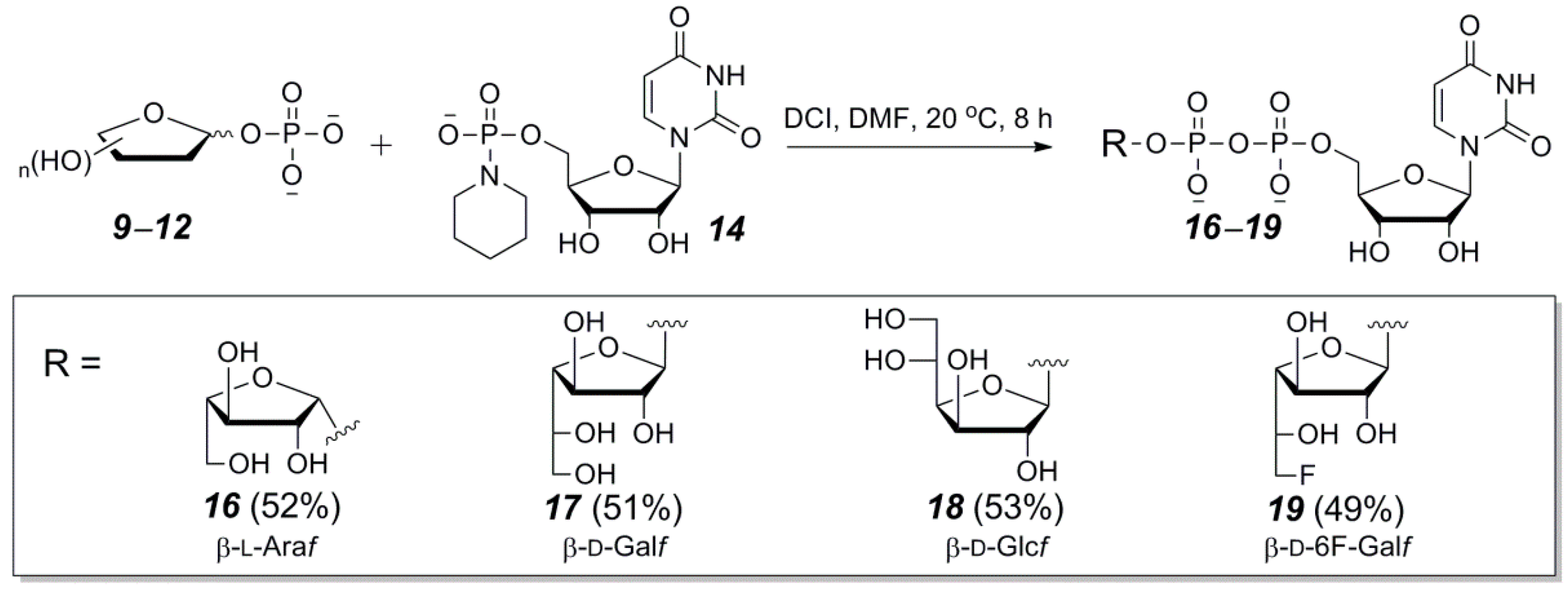
2.3. The P(V)-N Activation Method for the Synthesis UDP-Furanoses from Furanosyl-1-Phosphates
3. Materials and Methods
3.1. General Methods
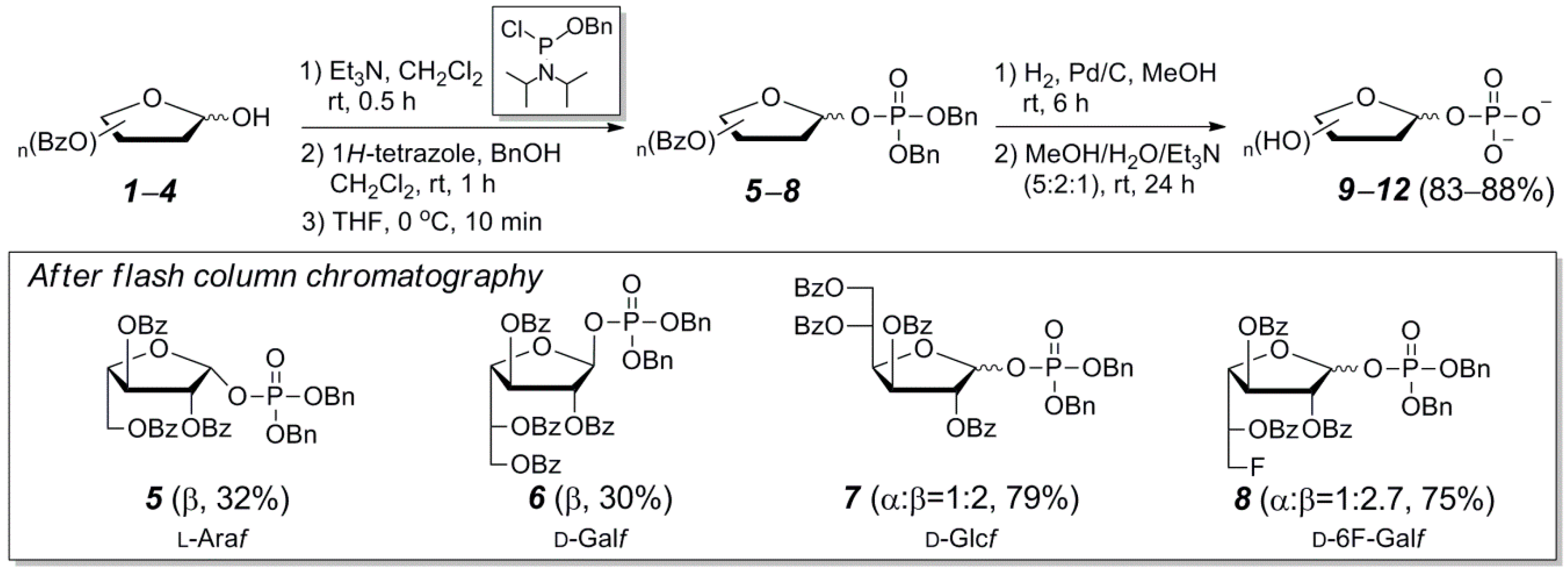
3.2. General Synthetic Procedure and Characterization of Protected Furanosyl-1-Phosphates 5–8
3.3. General Synthetic Procedure and Characterization of Furanosyl-1-Phosphates 9–13
3.4. General Synthetic Procedure and Characterization of UDP-Furanoses 15–19
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- de Lederkremer, R.M.; Colli, W. Galactofuranose-containing glycoconjugates in trypanosomatids. Glycobiology. 1995, 5, 547–552. [Google Scholar] [CrossRef]
- Bernnan, P.J.; Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Previato, J.O.; Mendonca-Previato, L.; Jones, C.; Wait, R. Structure of the carbohydrate moiety of the glycophosphosphingolipid of Endotrypanum schaudinni. Glycoconjugate. J. 1993, 10, 340. [Google Scholar] [CrossRef]
- Moriguchi, T.; Wada, T.; Sekine, M. New nucleoside-sugar conjugates: 6-N-Glycosyloxyphosphorylated adenosine derivatives as partial structures of agrocin 84. J. Org. Chem. 1996, 61, 9223–9228. [Google Scholar] [CrossRef]
- Sato, N.; Nakazawa, F.; Sato, M.; Hoshino, E.; Ito, T. Structural studies of the antigenic polysaccharide of Eubacterium saburreum, strain T19. Carbohydr. Res. 1993, 245, 105–111. [Google Scholar] [CrossRef]
- Micovic, V.M.; Hranisavljevic-Jakovljevic, M.; Miljkovic-Stojanovic, J. Structural study of polysaccharides from the oak lichen Evernia prunastri (L) Ach.: Part I. An alkali-soluble galatomannan. Carbohydr. Res. 1969, 10, 525–533. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A.; Teta, R. Terpioside from the marine sponge Terpios sp., the first glycosphingolipid having an l-fucofuranose unit. Eur. J. Org. Chem. 2008, 8, 2130–2134. [Google Scholar] [CrossRef]
- Fincher, G.B.; Stone, B.A.; Clarke, A.E. Arabinogalactan-proteins: Structure, biosynthesis, and function. Annu. Rev. Plant Physiol. 1983, 34, 47–70. [Google Scholar] [CrossRef]
- Peltier, P.; Euzen, R.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Recent knowledge and innovations related to hexofuranosides: Structure, synthesis and applications. Carbohydr. Res. 2008, 343, 1897–1923. [Google Scholar] [CrossRef]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Chlubnova, I.; Legentil, L.; Dureau, R.; Pennec, A.; Almendros, M.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Specific and non-specific enzymes for furanosyl-containing conjugates: Biosynthesis, metabolism, and chemo-enzymatic synthesis. Carbohydr. Res. 2012, 356, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Jackson, M.; Ma, Y.; McNeil, M. Cell wall core galactofuran synthesis is essential for growth of mycobacteria. J. Bacteriol. 2001, 183, 3991–3998. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.R.; Lowary, T.L. Chemistry and biology of galactofuranose-containing polysaccharides. ChemBioChem. 2009, 10, 1920–1938. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.L.; Tourco, S.J. Galactofuranose metabolism: A potential target for antimicrobial chemotherapy. Cell. Mol. Life. Sci. 2003, 60, 259–266. [Google Scholar] [PubMed]
- Nassau, P.M.; Martin, S.L.; Brown, R.E.; Weston, A.; Monsey, D.; McNeil, M.R.; Duncan, K. Galactofuranose biosynthesis in Escherichia coli K-12: Identification and cloning of UDP-galactopyranose mutase. J. Bacteriol. 1996, 178, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.; Kleczka, B.; Gerardy-Schahn, R.; Routier, F.H. Identification and partial characterization of two eukaryotic UDP-galactopyranose mutases. Biol. Chem. 2005, 386, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Poulin, M.B.; Nothaft, H.; Hug, I.; Feldman, M.F.; Szymanski, C.M.; Lowary, T.L. Characterization of a bifunctional pyranose-furanose mutase from Campylobacter jejuni 11168. J. Bio. Chem. 2010, 285, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Belanova, M.; Dianiskova, P.; Brennan, P.J.; Completo, G.C.; Rose, N.L.; Lowary, T.L.; Mikusova, K. Galactosyl transferases in mycobacterial cell wall synthesis. J. Bacteriol. 2008, 190, 1141–1145. [Google Scholar] [CrossRef]
- Lee, R.; Monsey, D.; Weston, A.; Duncan, J.K.; Rithner, C.; McNeil, M. Enzymatic synthesis of UDP-galactofuranose and an assay for UDP-galactopyranose mutase based on high-performance liquid chromatography. Anal. Biochem. 1996, 242, 1–7. [Google Scholar] [CrossRef]
- de Lederkremer, R.M.; Nahmad, V.B.; Varela, O. Synthesis of α-d-galactofuranosyl phosphate. J. Org. Chem. 1994, 59, 690–692. [Google Scholar] [CrossRef]
- Kovensky, J.; McNeil, M.; Sinay, P. d-Galactofuranosylphosphonates. First synthesis of UDP-C-d-galactofuranose. J. Org. Chem. 1999, 64, 6202–6205. [Google Scholar] [CrossRef]
- Tsvetkov, Y.E.; Nikolaev, A.V. The first chemical synthsis of UDP-α- d-galactofuranose. J. Chem. Soc. Perkin Trans. 2000, 1, 889–891. [Google Scholar] [CrossRef]
- Mariño, K.; Marino, C.; Lima, C.; Baldoni, L.; de Lederkremer, R.M. The first chemical synthesis of UDP[6 -3H]-α- d-galactofuranose. Eur. J. Org. Chem. 2005, 14, 2958–2964. [Google Scholar]
- Snitynsky, R.B.; Lowary, T.L. Synthesis of nitrogen-containing furanose sugar nucleotides for use as enzymatic probes. Org. Lett. 2014, 16, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.B.; Liu, H.-W. Studies of UDP-galactopyranose mutase from Escherichia coli: An unusual role of reduced FAD in its catalysis. J. Am. Chem. Soc. 2000, 122, 9065–9070. [Google Scholar] [CrossRef]
- Zhang, Q.B.; Liu, H.-W. Chemical synthesis of UDP-β-l-arabinofuranose and its turnover to UDP-β-l-arabinopyranose by UDP-galactopyranose mutase. Bioorg. Med. Chem. Lett. 2001, 11, 145–149. [Google Scholar] [CrossRef]
- Marlow, A.L.; Kiessling, L.L. Improved chemical synthesis of UDP-galactofuranose. Org. Lett. 2001, 3, 2517–2519. [Google Scholar] [CrossRef]
- Brown, C.D.; Rusek, M.S.; Kiessling, L.L. Fluorosugar chain termination agents as probes of the sequence specificity of a carbohydrate polymerase. J. Am. Chem. Soc. 2012, 134, 6552–6555. [Google Scholar] [CrossRef]
- Peltier, P.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Versatile synthesis of rare nucleotide furanoses. Org. Lett. 2007, 9, 5227–5230. [Google Scholar] [CrossRef]
- Peltier, P.; Guegan, J.-P.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Stereoselective chemoenzymatic synthesis of UDP-1,2-cis-furanoses from α,β-furanosyl 1-phosphates. Eur. J. Org. Chem. 2008, 5988–5994. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, S.-S.; Sun, J.; Liu, S.; Xiao, Q.; Pu, S.-Z. A P(V)–N activation strategy for synthesis of nucleoside polyphosphates. J. Org. Chem. 2013, 78, 8417–8426. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Gong, S.-S.; Sun, J.; Wang, C.-J.; Liu, S.; Liu, G.-D.; Ma, C. Efficient synthesis of nucleoside 5′-triphosphates and their β,γ-bridging oxygen-modified analogs from nucleoside 5′-phosphates. Tetrahedron Lett. 2014, 55, 2114–2118. [Google Scholar] [CrossRef]
- Sun, Q.; Sun, J.; Gong, S.-S.; Wang, C.-J.; Pu, S.-Z.; Feng, F.-D. Efficient synthesis of 5-hydroxymethyl-, 5-formyl-, and 5-carboxyl-2′-deoxycytidine and their triphosphates. RSC Adv. 2014, 4, 36036–36039. [Google Scholar] [CrossRef]
- Sun, Q.; Li, X.-J.; Sun, J.; Gong, S.-S.; Liu, G.; Liu, G.-D. An improved P(V)-N activation strategy for the synthesis of nucleoside diphosphate 6-deoxy-l-sugars. Tetrahedron 2014, 70, 294–300. [Google Scholar] [CrossRef]
- Ferrières, V.; Blanchard, S.; Fischer, D.; Plusquellec, D. A novel synthesis of d-galactofuranosyl, d-glucofuranosyl and d-mannofuranosyl 1-phosphates based on remote activation of new and free hexofuranosyl donors. Bioorg. Med. Chem. Lett. 2012, 12, 3515–3518. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, Q.-K.; Gong, S.-S.; Fu, Q.-L.; Xiao, Q. Synthesis and enzymatic evaluation of phosphoramidon and its β anomer: Anomerization of α-l-rhamnose triacetate upon phosphitylation. Bioorg. Med. Chem. 2013, 21, 6778–6787. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-S.; Wang, R.; Chen, W.-J.; Chen, J.-Z.; Gong, S.-S.; Sun, Q. The first chemical synthesis of pyrazofurin 5′-triphosphate. Tetrahedron Lett. 2018, 59, 3423–3427. [Google Scholar] [CrossRef]
- Zamyatina, A.; Gronow, S.; Oertelt, C.; Puchberger, M.; Brade, H.; Kosma, P. Efficient chemical synthesis of the two anomers of ADP-l-glycero- and d-glycero-d-manno-heptopyranose allows the determination of the substrate specificities of bacterial heptosyltransferases. Angew. Chem. Int. Ed. 2000, 39, 4150–4153. [Google Scholar] [CrossRef]
- Dinev, Z.; Wardak, A.Z.; Brownleed, R.T.C.; Williams, S.J. A convenient gram-scale synthesis of uridine diphospho(13C6)glucose. Carbohydr. Res. 2006, 341, 1743–1747. [Google Scholar] [CrossRef]
- Lubineau, A.; Fischer, J.-C. High-yielding one-step conversion of d-glucose and d-galactose to the corresponding α and β methyl- d-glucofuranosides and galactofuranosides. Synth. Commun. 1991, 21, 815–818. [Google Scholar] [CrossRef]
- Euzen, R.; Lopez, G.; Nugier-Chauvin, C.; Ferrières, V.; Plusquellec, D.; Remond, C.; O’Donohue, M. A chemoenzymatic approach for the synthesis of unnatural disaccharides containing d-galacto- or d-fucofuranosides. Eur. J. Org. Chem. 2005, 22, 4860–4869. [Google Scholar] [CrossRef]
- Wei, G.; Zhang, L.; Cai, C.; Cheng, S.; Du, Y. Selective cleavage of sugar anomeric O-acyl groups using FeCl3·6H2O. Tetrahedron Lett. 2008, 49, 5488–5491. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-J.; Han, S.-B.; Xie, Z.-B.; Huang, H.-S.; Jiang, D.-H.; Gong, S.-S.; Sun, Q. Efficient Synthesis of UDP-Furanoses via 4,5-Dicyanoimidazole(DCI)-Promoted Coupling of Furanosyl-1-Phosphates with Uridine Phosphoropiperidate. Molecules 2019, 24, 655. https://doi.org/10.3390/molecules24040655
Chen W-J, Han S-B, Xie Z-B, Huang H-S, Jiang D-H, Gong S-S, Sun Q. Efficient Synthesis of UDP-Furanoses via 4,5-Dicyanoimidazole(DCI)-Promoted Coupling of Furanosyl-1-Phosphates with Uridine Phosphoropiperidate. Molecules. 2019; 24(4):655. https://doi.org/10.3390/molecules24040655
Chicago/Turabian StyleChen, Wei-Jie, Shuai-Bo Han, Zhen-Biao Xie, Hua-Shan Huang, Duo-Hua Jiang, Shan-Shan Gong, and Qi Sun. 2019. "Efficient Synthesis of UDP-Furanoses via 4,5-Dicyanoimidazole(DCI)-Promoted Coupling of Furanosyl-1-Phosphates with Uridine Phosphoropiperidate" Molecules 24, no. 4: 655. https://doi.org/10.3390/molecules24040655
APA StyleChen, W.-J., Han, S.-B., Xie, Z.-B., Huang, H.-S., Jiang, D.-H., Gong, S.-S., & Sun, Q. (2019). Efficient Synthesis of UDP-Furanoses via 4,5-Dicyanoimidazole(DCI)-Promoted Coupling of Furanosyl-1-Phosphates with Uridine Phosphoropiperidate. Molecules, 24(4), 655. https://doi.org/10.3390/molecules24040655