
Position-Selective Synthesis and Biological Evaluation of Four Isomeric A-Ring Amino Derivatives of the Alkaloid Luotonin A

 ,
, 

Abstract
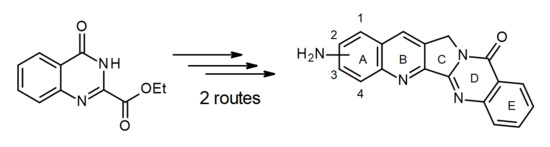
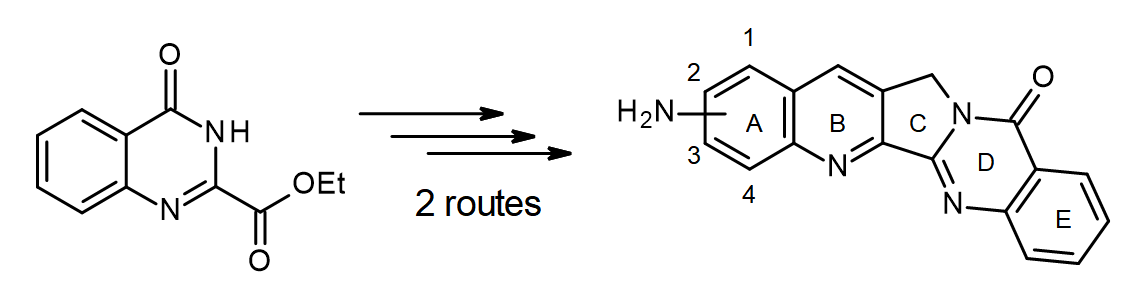
1. Introduction
2. Results and Discussion
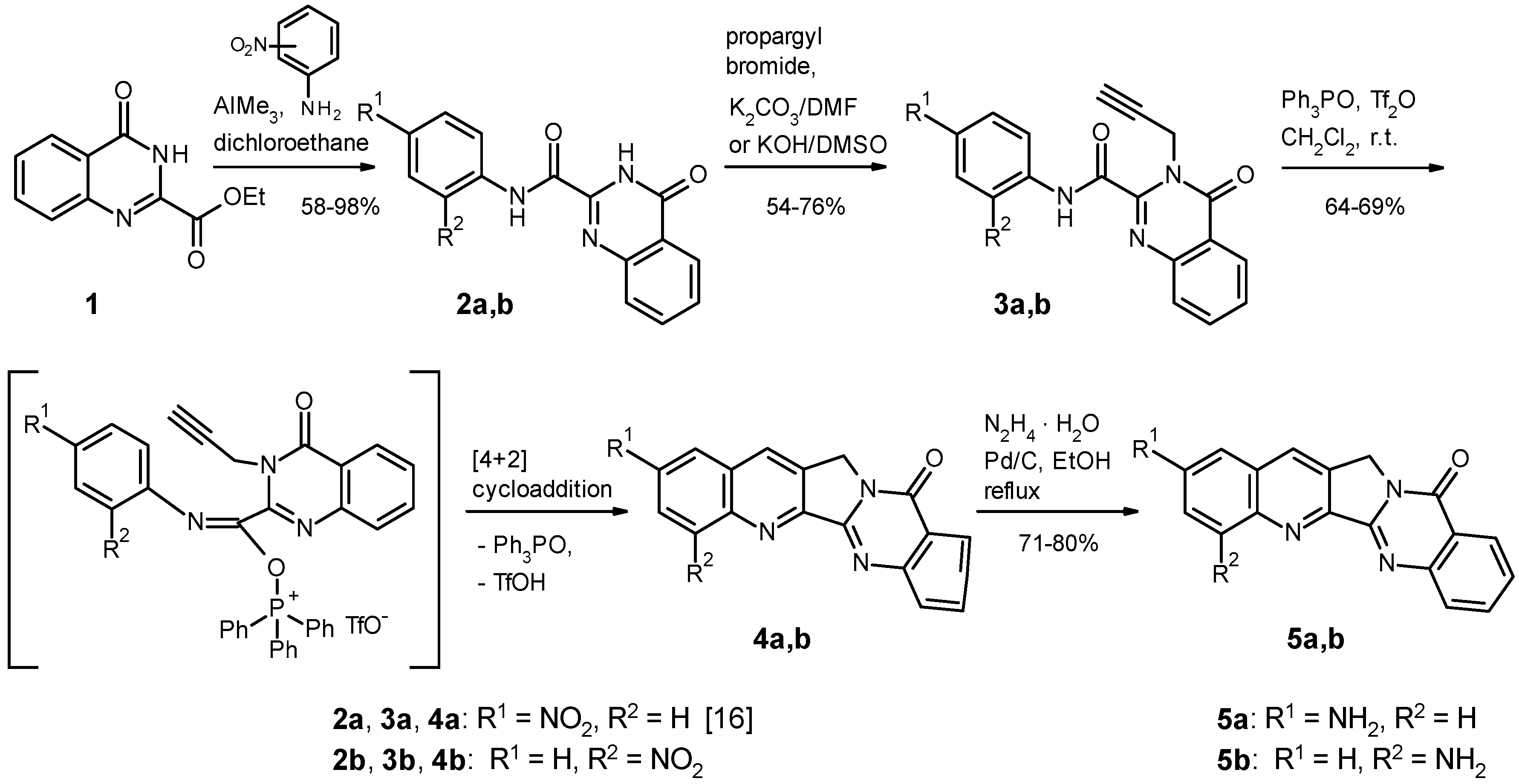
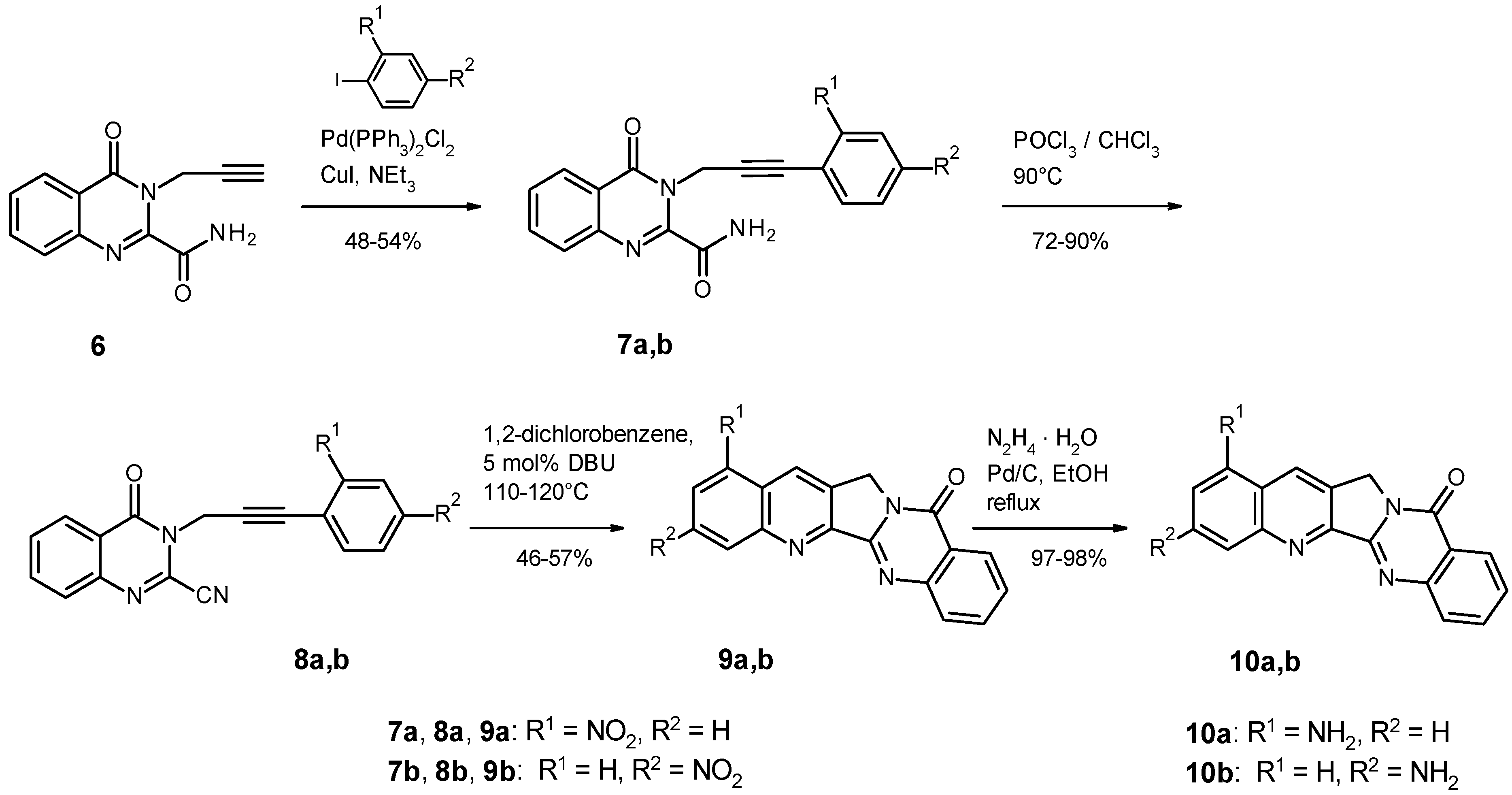
2.1. Chemistry
2.2. Biological Evaluation
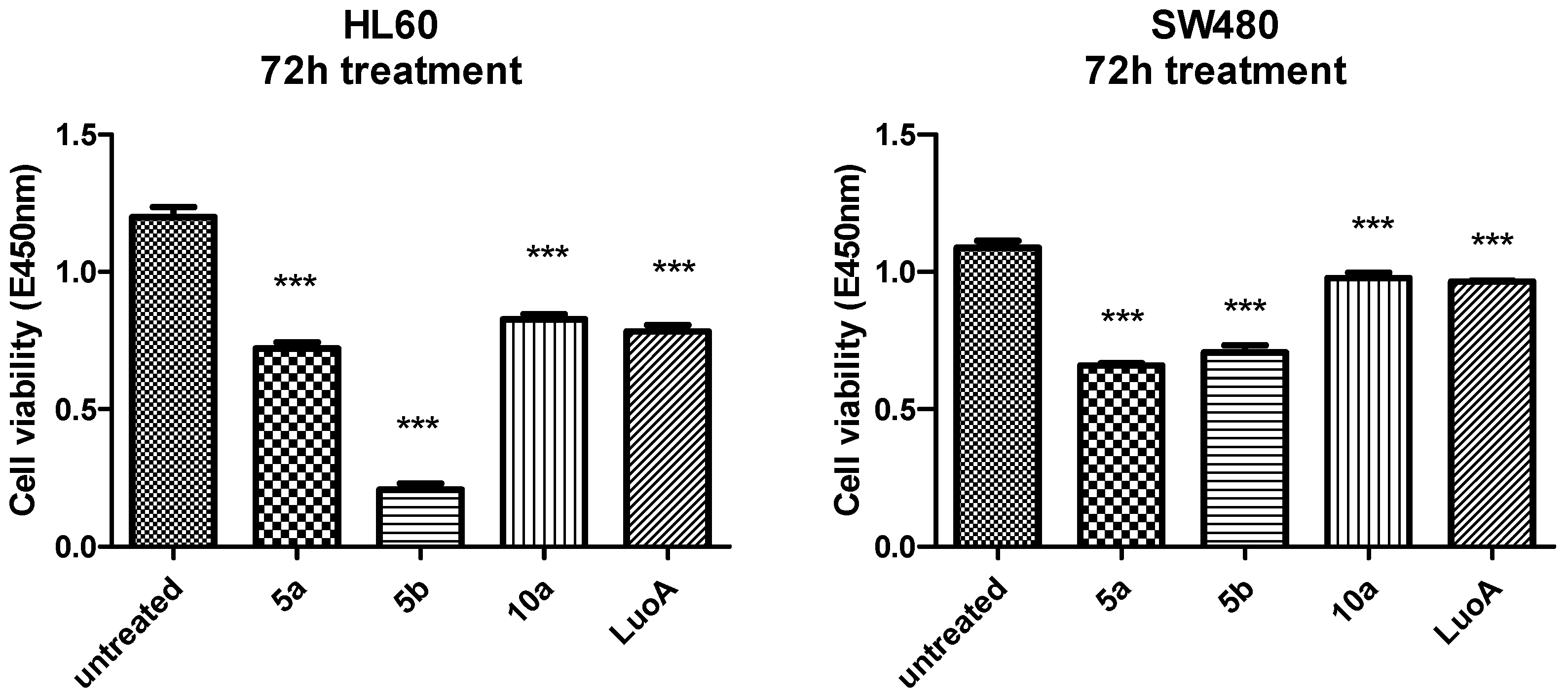
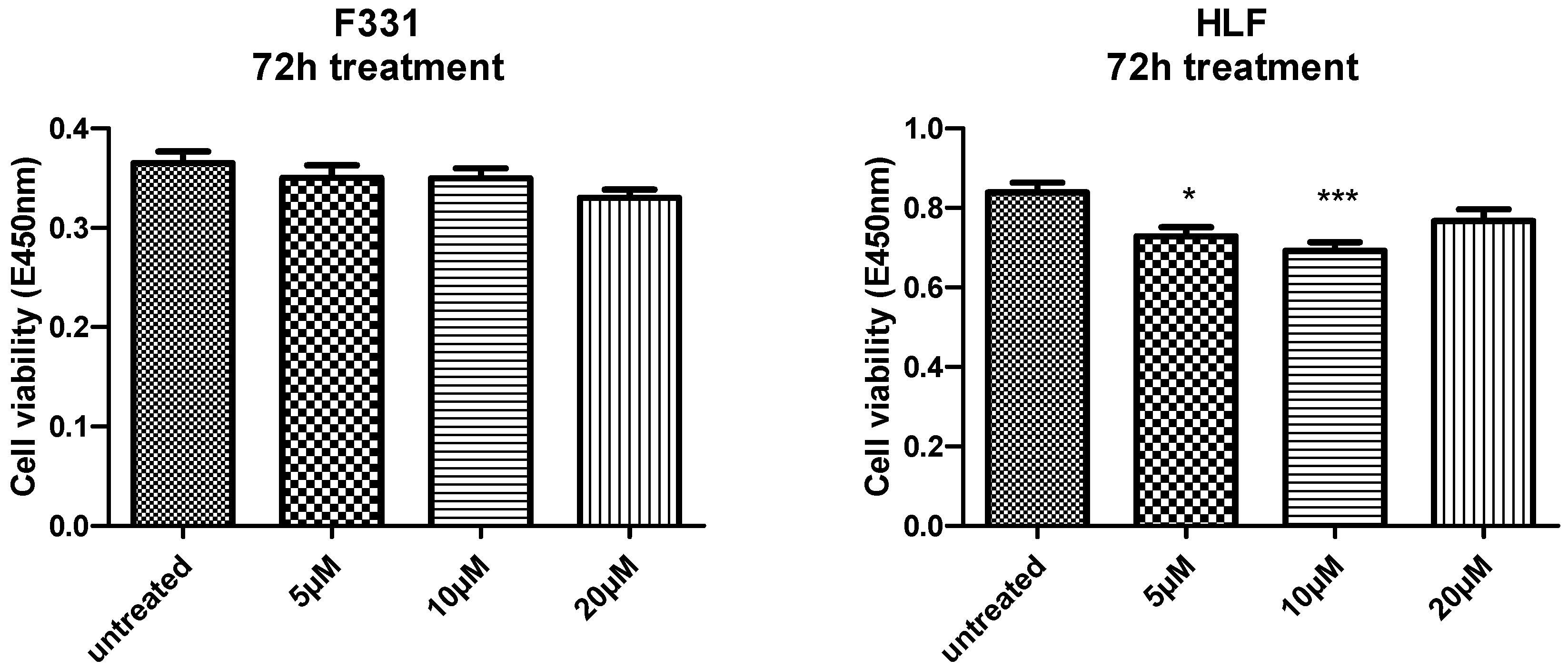
2.2.1. Evaluation of Cell Viability and Toxicity
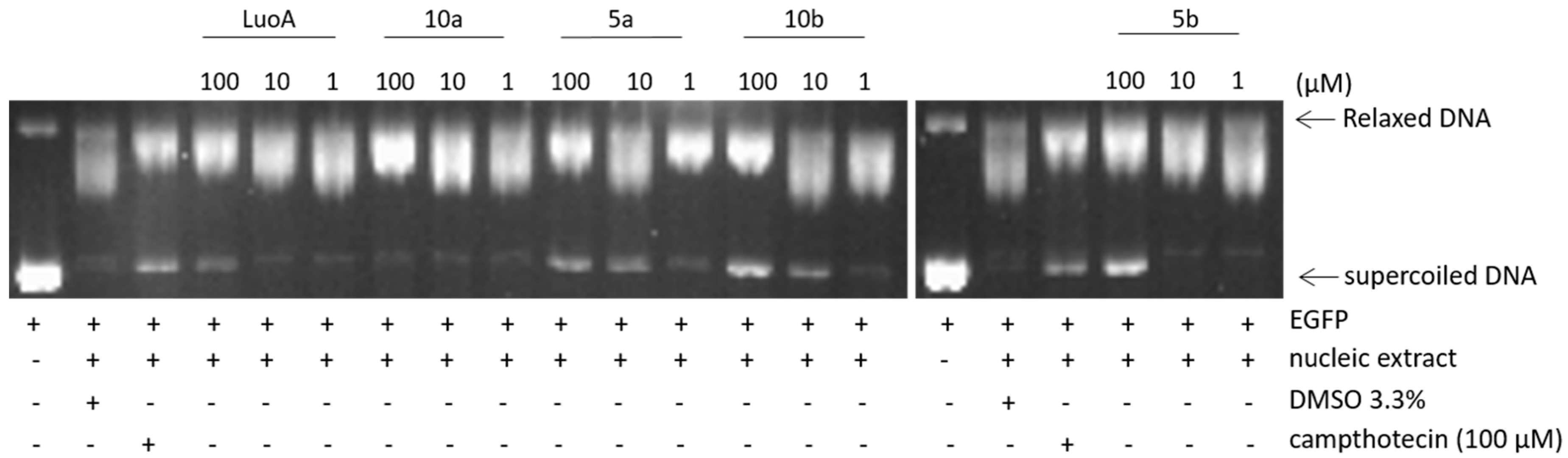
2.2.2. Top1 Inhibition
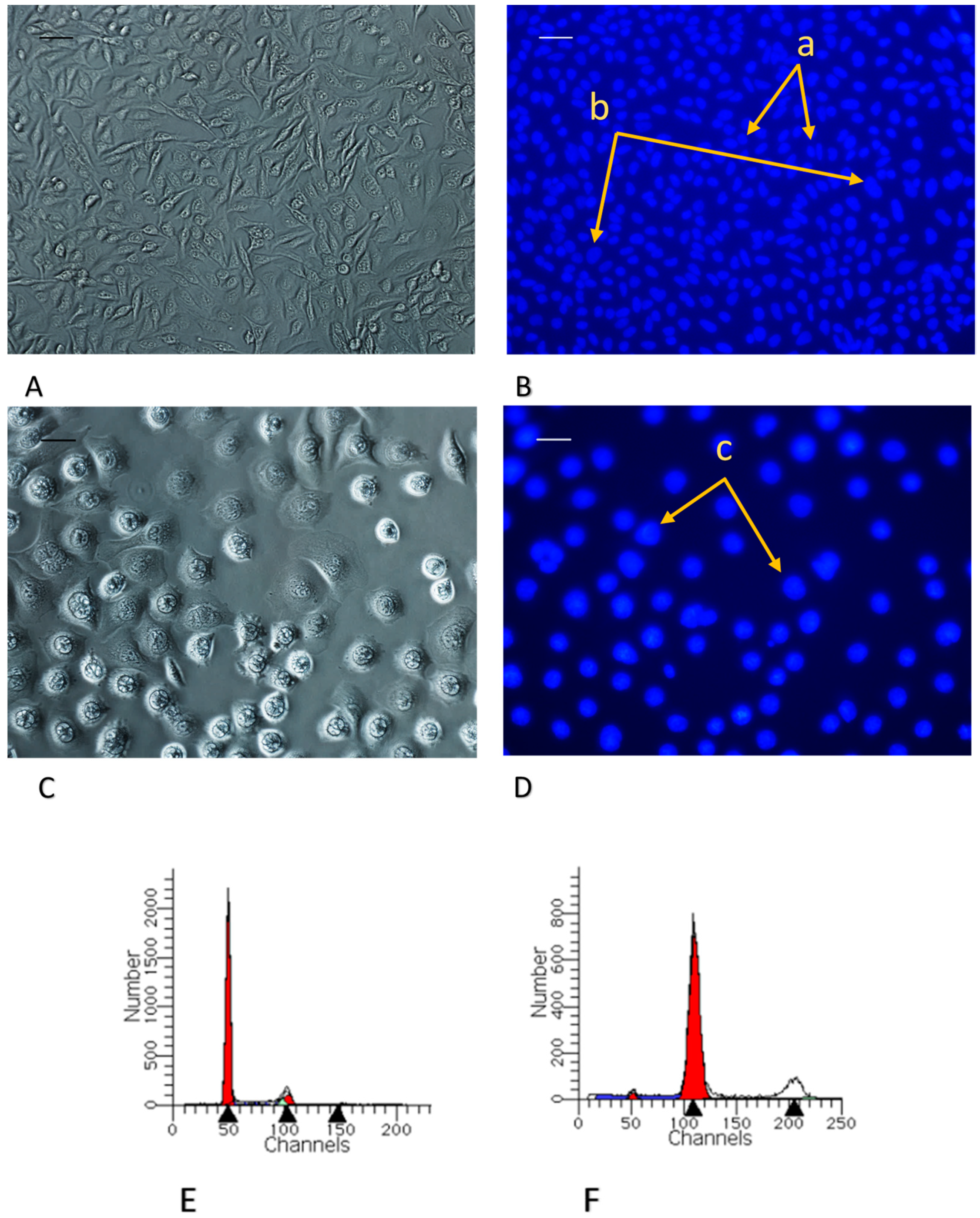
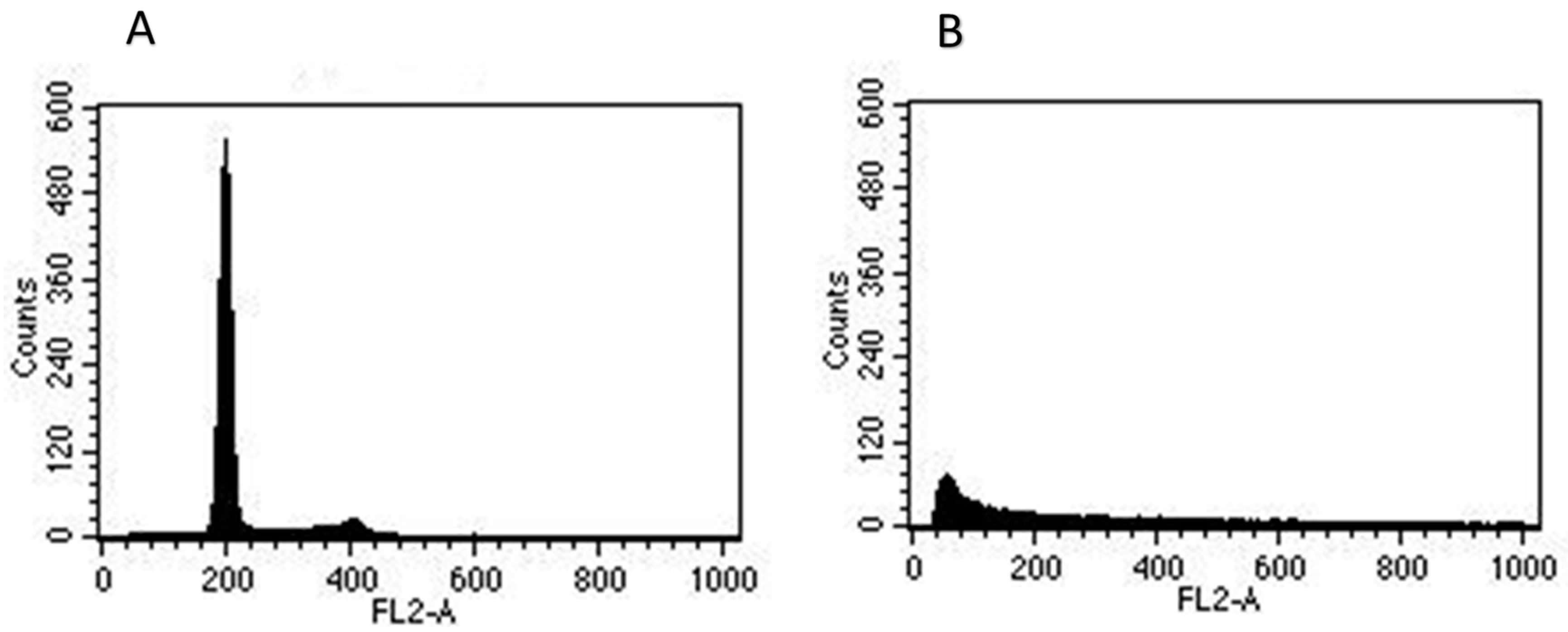
2.2.3. Morphological Alterations and Cell Cycle Analysis: Induction of G2/M Arrest
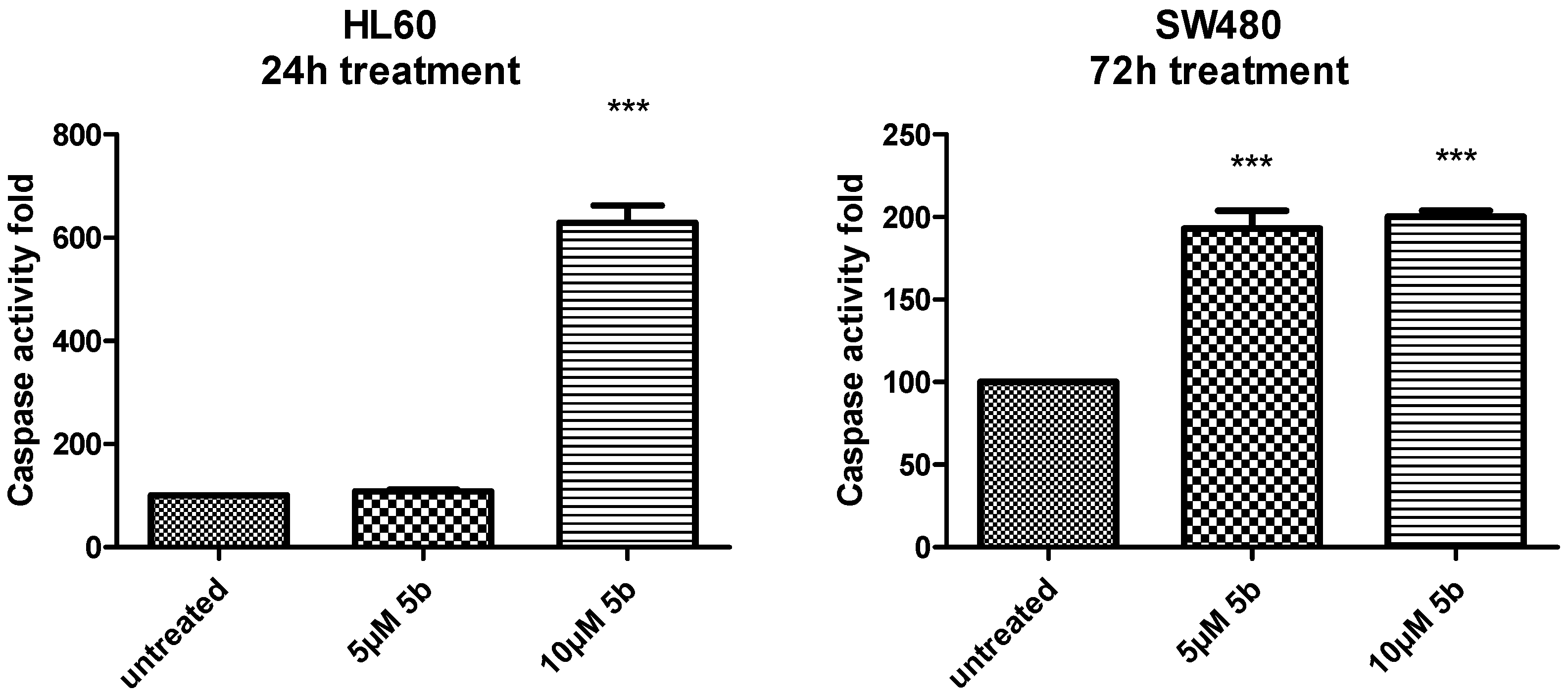
2.2.4. Stimulation of Caspase Activity by Compound 5b
3. Conclusions
4. Experimental Section
4.1. Chemistry
4.1.1. General
4.1.2. Syntheses
4.2. Biology
4.2.1. Cell Culture
4.2.2. Cell Viability Assay
4.2.3. DNA Relaxation Assay
4.2.4. Cell Cycle Distribution Assay (FACS)
4.2.5. Caspase Assay
4.2.6. Nuclear Staining
4.2.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pommier, Y. DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Mross, K.; Jakob, A.; Kanz, L.; Bokemeyer, C. Topotecan—A novel topoisomerase I inhibitor: Pharmacology and clinical experience. Oncology 1999, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Ahlawat, P.; Srinivas, N.R. Irinotecan and its active metabolite, SN-38: Review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed. Chromatogr. 2010, 24, 104–123. [Google Scholar] [CrossRef]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Pilon, A.A.; Smith, M.; Hickey, R.; Malkas, L.; Pommier, Y. Conversion of topoisomerase 1 cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000, 20, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.L.; Cha, H.C.; Jahng, Y. Recent advances in the studies on luotonins. Molecules 2011, 16, 4861–4883. [Google Scholar] [CrossRef]
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M. Luotonin A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef]
- Ma, Z.Z.; Hano, Y.S.; Nomura, T.; Chen, Y.J. Novel quinazoline-quinoline alkaloids with cytotoxic and DNA topoisomerase II inhibitory activities. Bioorg. Med. Chem. Lett. 2004, 14, 1193–1196. [Google Scholar] [CrossRef]
- Nacro, K.; Zha, C.C.; Guzzo, P.R.; Jason Herr, R.; Peace, D.; Friedrich, T.D. Synthesis and topoisomerase poisoning activity of A-ring and E-ring substituted luotonin A derivatives. Bioorg. Med. Chem. 2007, 15, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Bao, J.L.; Chen, X.P.; Huang, M.; Wang, Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid.-Based Complement. Alternat. Med. 2012, 2012, 485042. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Pan, S.L.; Guh, J.H.; Chang, Y.L.; Pai, H.C.; Lin, C.H.; Teng, C.M. Antitumor mechanism of evodiamine, a constituent from Chinese herb Evodiae fructus, in human multiple-drug resistant breast cancer NCI/ADR-RES cells in vitro and in vivo. Carcinogenesis 2005, 26, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.F.; Huang, W.J.; Lin, L.C.; Wang, P.S. Inhibitory effects of evodiamine on the growth of human prostate cancer cell line LNCaP. Int. J. Cancer 2004, 110, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Guh, J.H.; Teng, C.M. Induction of mitotic arrest and apoptosis by evodiamine in human leukemic T-lymphocytes. Life Sci. 2004, 75, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Nuss, S. Weinreb amidation as the cornerstone of an improved synthetic route to A-ring-modified derivatives of luotonin A. Molecules 2012, 17, 11363–11378. [Google Scholar] [CrossRef]
- Atia, M.; Bogdan, D.; Brugger, M.; Haider, N.; Matyus, P. Remarkable regioselectivities in the course of the synthesis of two new Luotonin A derivatives. Tetrahedron 2017, 73, 3231–3239. [Google Scholar] [CrossRef]
- Ibric, A.; Dutter, K.; Marian, B.; Haider, N. A Facile Oxidative Opening of the C-Ring in Luotonin A and Derivatives. Molecules 2017, 22, 1540. [Google Scholar] [CrossRef]
- Haider, N.; Meng, G.; Roger, S.; Wank, S. An efficient and selective access to 1-substituted and 3-substituted derivatives of luotonin A. Tetrahedron 2013, 69, 7066–7072. [Google Scholar] [CrossRef]
- Cagir, A.; Eisenhauer, B.M.; Gao, R.; Thomas, S.J.; Hecht, S.M. Synthesis and topoisomerase I inhibitory properties of luotonin A analogues. Bioorg. Med. Chem. 2004, 12, 6287–6299. [Google Scholar] [CrossRef]
- Zhou, H.B.; Liu, G.S.; Yao, Z.J. Short and efficient total synthesis of luotonin a and 22-hydroxyacuminatine using a common cascade strategy. J. Org. Chem. 2007, 72, 6270–6272. [Google Scholar] [CrossRef] [PubMed]
- Basha, A.; Lipton, M.; Weinreb, S.M. Mild, General Method for Conversion of Esters to Amides. Tetrahedron Lett. 1977, 4171–4174. [Google Scholar] [CrossRef]
- Baker, B.R.; Almaula, P.I. Nonclassical Antimetabolites .10. Facile Synthesis of 4-Quinazolone-2-Carboxylic Acid and Structure of Bogerts Ammonium Salt. J. Org. Chem. 1962, 27, 4672–4674. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Singer, M.; Hussoin, M.S. Direct Borohydride Reduction of Alcohols to Alkanes with Phosphonium Anhydride Activation. J. Org. Chem. 1993, 58, 6913–6914. [Google Scholar] [CrossRef]
- Dallavalle, S.; Merlini, L.; Beretta, G.L.; Tinelli, S.; Zunino, F. Synthesis and cytotoxic activity of substituted luotonin A derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5757–5761. [Google Scholar] [CrossRef] [PubMed]
- Deady, L.W.; Sette, R.M.D. Lithiation of pivaloylamino derivatives of dibenzofuran and 9-methylcarbazole. Aust. J. Chem. 2001, 54, 177–180. [Google Scholar] [CrossRef]
- Haider, N.; Tropper, K. Dimethyl 6-Amino-1-methyl-9H-carbazole-2,3-dicarboxylate. Molbank 2015, M849. [Google Scholar] [CrossRef]
- Varadi, A.; Horvath, P.; Kurtan, T.; Mandi, A.; Toth, G.; Gergely, A.; Kokosi, J. Synthesis and configurational assignment of 1,2-dihydroimidazo[5,1-b] quinazoline-3,9-diones: Novel NMDA receptor antagonists. Tetrahedron 2012, 68, 10365–10371. [Google Scholar] [CrossRef]
- Usifoh, C.O.; Scriba, G.K.E. Synthesis and anticonvulsant activity of acetylenic quinazolinone derivatives. Arch Pharm 2000, 333, 261–266. [Google Scholar] [CrossRef]
- Khanna, I.K.; Yu, Y.; Huff, R.M.; Weier, R.M.; Xu, X.D.; Koszyk, F.J.; Collins, P.W.; Cogburn, J.N.; Isakson, P.C.; Koboldt, C.M.; et al. Selective cyclooxygenase-2 inhibitors: Heteroaryl modified 1,2-diarylimidazoles are potent, orally active antiinflammatory agents. J. Med. Chem. 2000, 43, 3168–3185. [Google Scholar] [CrossRef]
- Kuo, C.W.; Zhu, J.L.; Wu, J.D.; Chu, C.M.; Yao, C.F.; Shia, K.S. A convenient new procedure for converting primary amides into nitriles. Chem. Commun. 2007, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.X.; Petersen, J.L.; Wang, K.K. Synthesis of the parent and substituted tetracyclic ABCD ring cores of camptothecins via 1-(3-aryl-2-propynyl)-1,6-dihydro-6-oxo-2-pyridinecarbonitriles. Org. Lett. 2006, 8, 4665–4667. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harbor Protocols 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Kaufmann, S.H. Cytotoxic synergy between flavopiridol (NSC 649890, L86-8275) and various antineoplastic agents: The importance of sequence of administration. Cancer Res. 1997, 57, 3375–3380. [Google Scholar] [PubMed]
- Tyagi, A.K.; Singh, R.P.; Agarwal, C.; Chan, D.C.F.; Agarwal, R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin. Cancer Res. 2002, 8, 3512–3519. [Google Scholar] [PubMed]
- Ling, Y.H.; ElNaggar, A.K.; Priebe, W.; PerezSoler, R. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34(cdc2)/cyclin B-1 activity induced by doxorubicin in synchronized P388 cells. Mol. Pharmacol. 1996, 49, 832–841. [Google Scholar]
- Potter, A.J.; Gollahon, K.A.; Palanca, B.J.A.; Harbert, M.J.; Choi, Y.M.; Moskovitz, A.H.; Potter, J.D.; Rabinovitch, P.S. Flow cytometric analysis of the cell cycle phase specificity of DNA damage induced by radiation, hydrogen peroxide and doxorubicin. Carcinogenesis 2002, 23, 389–401. [Google Scholar] [CrossRef]
- DiPaola, R.S. To arrest or not to G(2)-M cell-cycle arrest. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Brit. J. Cancer 2008, 98, 523–528. [Google Scholar] [CrossRef]
- Kainz, K.P.; Krenn, L.; Erdem, Z.; Kaehlig, H.; Zehl, M.; Bursch, W.; Berger, W.; Marian, B. 2-Deprenyl-Rheediaxanthone B Isolated from Metaxya rostrata Induces Active Cell Death in Colorectal Tumor Cells. PLoS ONE 2013, 8, e65745. [Google Scholar] [CrossRef]
- Gu, J.J.; Kaufman, G.P.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Mitotic catastrophe and cell cycle arrest are alternative cell death pathways executed by bortezomib in rituximab resistant B-cell lymphoma cells. Oncotarget 2017, 8, 12741–12753. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Sacher, J.; Hohenegger, M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Brit. J. Pharmacol. 2004, 143, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Tamm, C.; Heidari, N.; Orrenius, S.; Zhivotovsky, B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ. 2008, 15, 555–566. [Google Scholar] [CrossRef]
- Skwarska, A.; Augustin, E.; Konopa, J. Sequential induction of mitotic catastrophe followed by apoptosis in human leukemia MOLT4 cells by imidazoacridinone C-1311. Apoptosis 2007, 12, 2245–2257. [Google Scholar] [CrossRef]
- Cliby, W.A.; Lewis, K.A.; Lilly, K.K.; Kaufmann, S.H. S phase and G(2) arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 2002, 277, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Goldwasser, F.; Shimizu, T.; Jackman, J.; Hoki, Y.; OConnor, P.M.; Kohn, K.W.; Pommier, Y. Correlations between S and G(2) arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996, 56, 4430–4437. [Google Scholar]
- Wu, N.; Wu, X.W.; Agama, K.; Pommier, Y.; Du, J.; Li, D.; Gu, L.Q.; Huang, Z.S.; An, L.K. A Novel DNA Topoisomerase I Inhibitor with Different Mechanism from Camptothecin Induces G2/M Phase Cell Cycle Arrest to K562 Cells. Biochemistry 2010, 49, 10131–10136. [Google Scholar] [CrossRef]
- Kalabis, J.; Patterson, M.J.; Enders, G.H.; Marian, B.; Iozzo, R.V.; Rogler, G.; Gimotty, P.A.; Herlyn, M. Stimulation of human colonic epithelial cells by leukemia inhibitory factor is dependent on collagen-embedded fibroblasts in organotypic culture. Faseb J. 2003, 17, 1115–1117. [Google Scholar] [CrossRef]
- Dornetshuber, R.; Heffeter, P.; Lemmens-Gruber, R.; Elbling, L.; Marko, D.; Micksche, M.; Berger, W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009, 53, 1112–1122. [Google Scholar] [CrossRef]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Signal | 10a (1-amino) | 5a (2-amino) | 10b (3-amino) | 5b (4-amino) |
|---|---|---|---|---|
| 1-H | – | 6.91 (d, J = 2.3) | 7.81 (d, J = 8.0) | 7.22 (dd, J = 8.2, 1.0) |
| 1-C | 149.7 | 104.1 | 128.9 | 113.8 |
| 2-H | 6.86 (dd, J = 7.6) | – | 7.16 (dd, J = 8.8, 2.0) | 7.45 (t, J = 7.8) |
| 2-C | 108.8 | 149.0 | 120-9 | 129.7 |
| 3-H | 7.57 (t, J = 8.1) | 7.31 (dd, J = 9.1, 2.4) | – | 6.98 (dd, J = 7.6, 1.1) |
| 3-C | 131.3 | 123.0 | 151.1 a | 109.1 |
| 4-H | 7.43 (d, J = 8.2) | 7.93 (d, J = 9.3) | 7.12 (d, unresolved) | – |
| 4-C | 116.7 | 130.7 | 105.9 | 146.2 |
| 4a-C | 145.7 | 142.8 | 151.0 a | 138.1 |
| 5a-C | 150.7 | 145.5 | 150.6 | 147.9 |
| 5b-C | 153.4 | 153.6 | 153.7 | 153.2 |
| 6a-C | 149.1 | 149.3 | 149.2 | 149.1 |
| 7-H | 7.97–7.90 (m) | 7.90–7.86 (m) | 7.95–7.91 (m) | 7.96–7.90 (m) |
| 7-C | 128.0 | 127.7 | 127.9 | 127.8 |
| 8-H | 7.97–7.90 (m) | 7.90–7.86 (m) | 7.95–7.91 (m) | 7.96–7.90 (m) |
| 8-C | 134.5 | 134.3 | 134.4 | 134.5 |
| 9-H | 7.66–7.61 (m) | 7.58 (ddd, J = 8.0, 6.1, 2.2) | 7.63–7.59 (m) | 7.62 (ddd, J = 7.7, 5.7, 2.5) |
| 9-C | 127.1 | 126.5 | 126.9 | 127.0 |
| 10-H | 8.30 (dd, J = 7.8, 1.0) | 8.26 (d, J = 7.1) | 8.28 (d, J = 8.0) | 8.30 (d, J = 7.7) |
| 10-C | 125.9 | 125.8 | 125.9 | 125.9 |
| 10a-C | 121.0 | 120.7 | 121.0 | 121.0 |
| 11-C | 159.7 | 159.7 | 159.8 | 159.7 |
| 13-H | 5.30 (s) | 5.20 (s) | 5.19 (s) | 5.28 (s) |
| 13-C | 47.5 | 47.3 | 47.3 | 47.4 |
| 13a-C | 128.4 | 131.2 b | 126.4 | 131.1 |
| 14-H | 8.92 (s) | 8.25 (s) | 8.41 (s) | 8.55 (s) |
| 14-C | 126.8 | 127.3 | 131.0 | 131.3 |
| 14a-C | 118.2 | 131.1 b | 121.6 | 129.3 |
| NH2 | 6.17 (br s) | 6.07 (br s) | 6.06 (br s) | 6.18 (br s) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibric, A.; Eckerstorfer, S.; Eder, M.; Louko, I.; Tunjic, L.; Heffeter, P.; Schueffl, H.H.; Marian, B.; Haider, N. Position-Selective Synthesis and Biological Evaluation of Four Isomeric A-Ring Amino Derivatives of the Alkaloid Luotonin A. Molecules 2019, 24, 716. https://doi.org/10.3390/molecules24040716
Ibric A, Eckerstorfer S, Eder M, Louko I, Tunjic L, Heffeter P, Schueffl HH, Marian B, Haider N. Position-Selective Synthesis and Biological Evaluation of Four Isomeric A-Ring Amino Derivatives of the Alkaloid Luotonin A. Molecules. 2019; 24(4):716. https://doi.org/10.3390/molecules24040716
Chicago/Turabian StyleIbric, Amra, Stefan Eckerstorfer, Martin Eder, Ivan Louko, Leopold Tunjic, Petra Heffeter, Hemma Henrike Schueffl, Brigitte Marian, and Norbert Haider. 2019. "Position-Selective Synthesis and Biological Evaluation of Four Isomeric A-Ring Amino Derivatives of the Alkaloid Luotonin A" Molecules 24, no. 4: 716. https://doi.org/10.3390/molecules24040716
APA StyleIbric, A., Eckerstorfer, S., Eder, M., Louko, I., Tunjic, L., Heffeter, P., Schueffl, H. H., Marian, B., & Haider, N. (2019). Position-Selective Synthesis and Biological Evaluation of Four Isomeric A-Ring Amino Derivatives of the Alkaloid Luotonin A. Molecules, 24(4), 716. https://doi.org/10.3390/molecules24040716