Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis

 ,
, 
 , ,
, , 
Abstract
:1. Introduction
- to provide the readers with a uniform assessment (ranking) of the affinity of fentanyl derivatives for the receptor,
- to translate the observed experimental trends in structure–activity relationships into structural terms by the means of molecular modeling, and
- using our experimental values, to see whether the popular scoring methods would be able to reproduce them with reasonable accuracy.
2. Results and Discussion
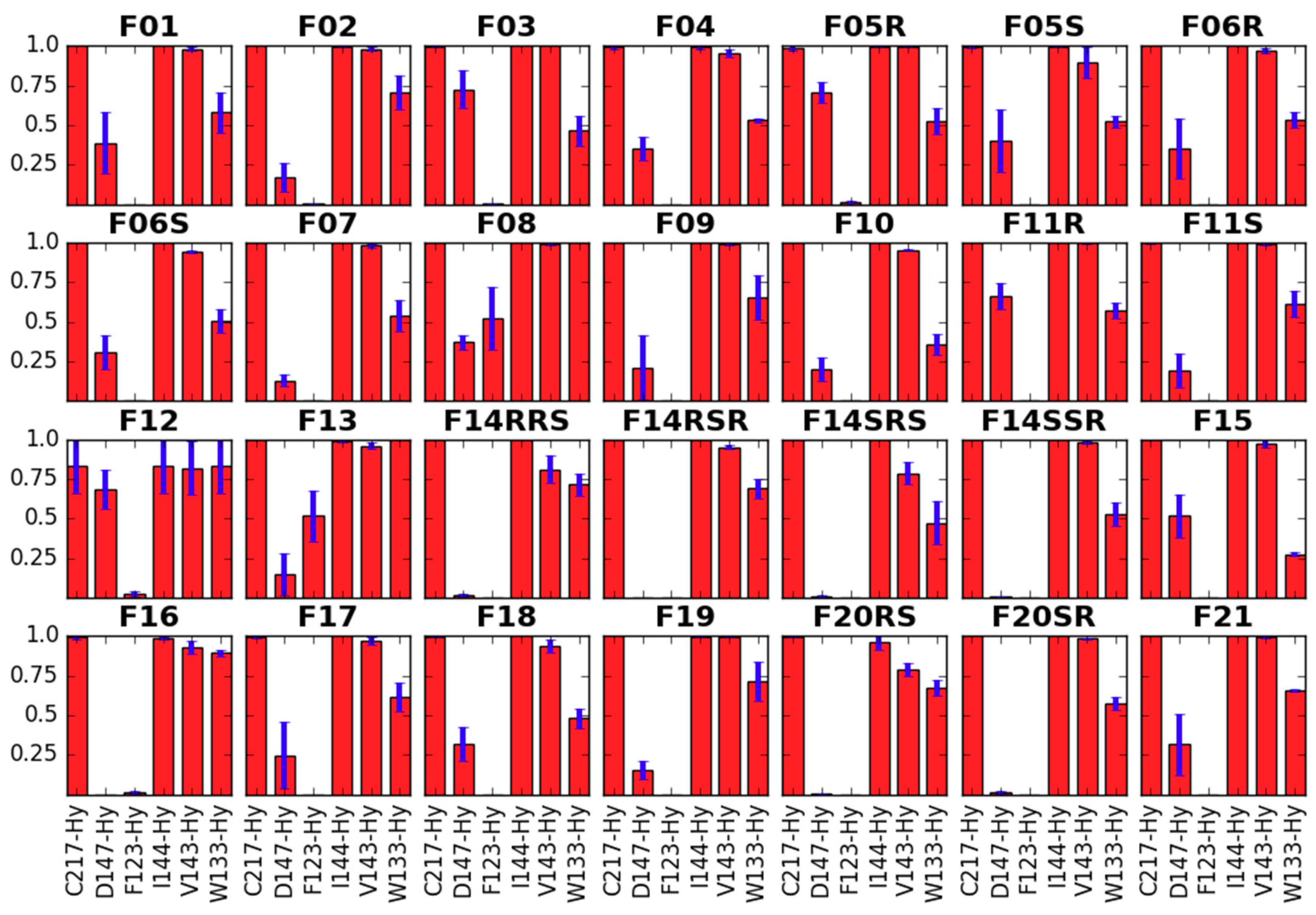
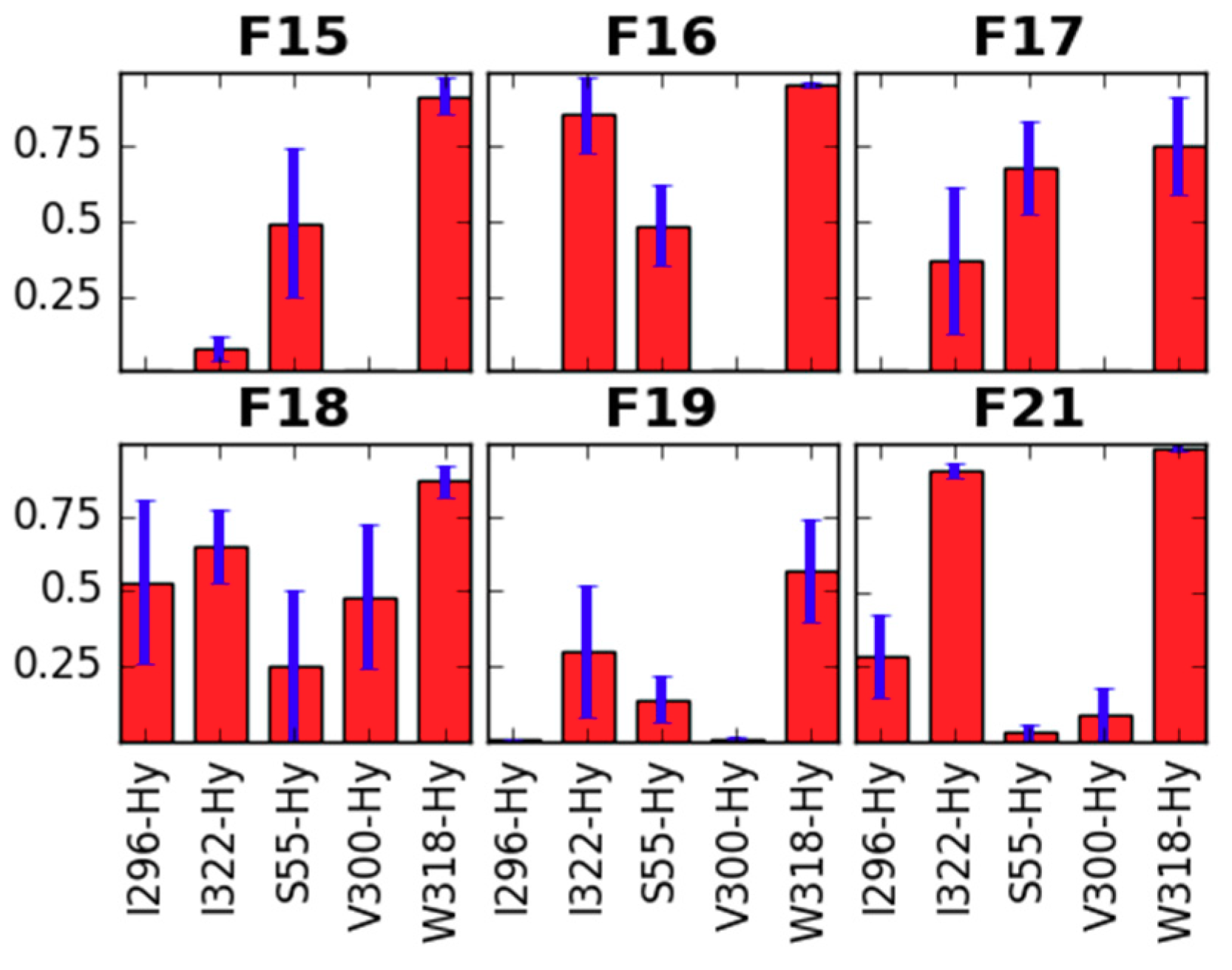
2.1. Receptor Binding Determination
2.2. Modeling of Fentanyl Derivatives Bound to the μ-Opioid Receptor
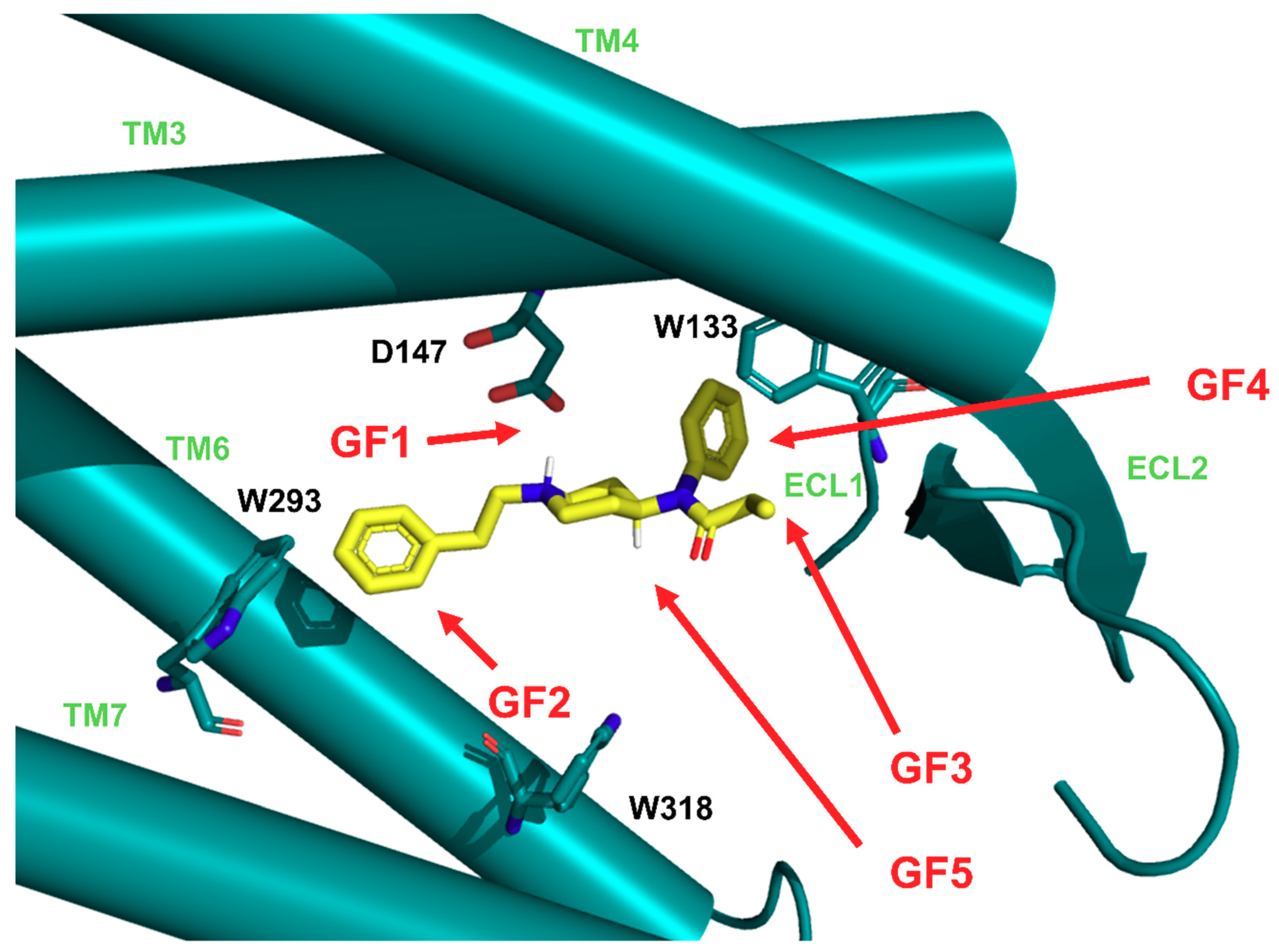
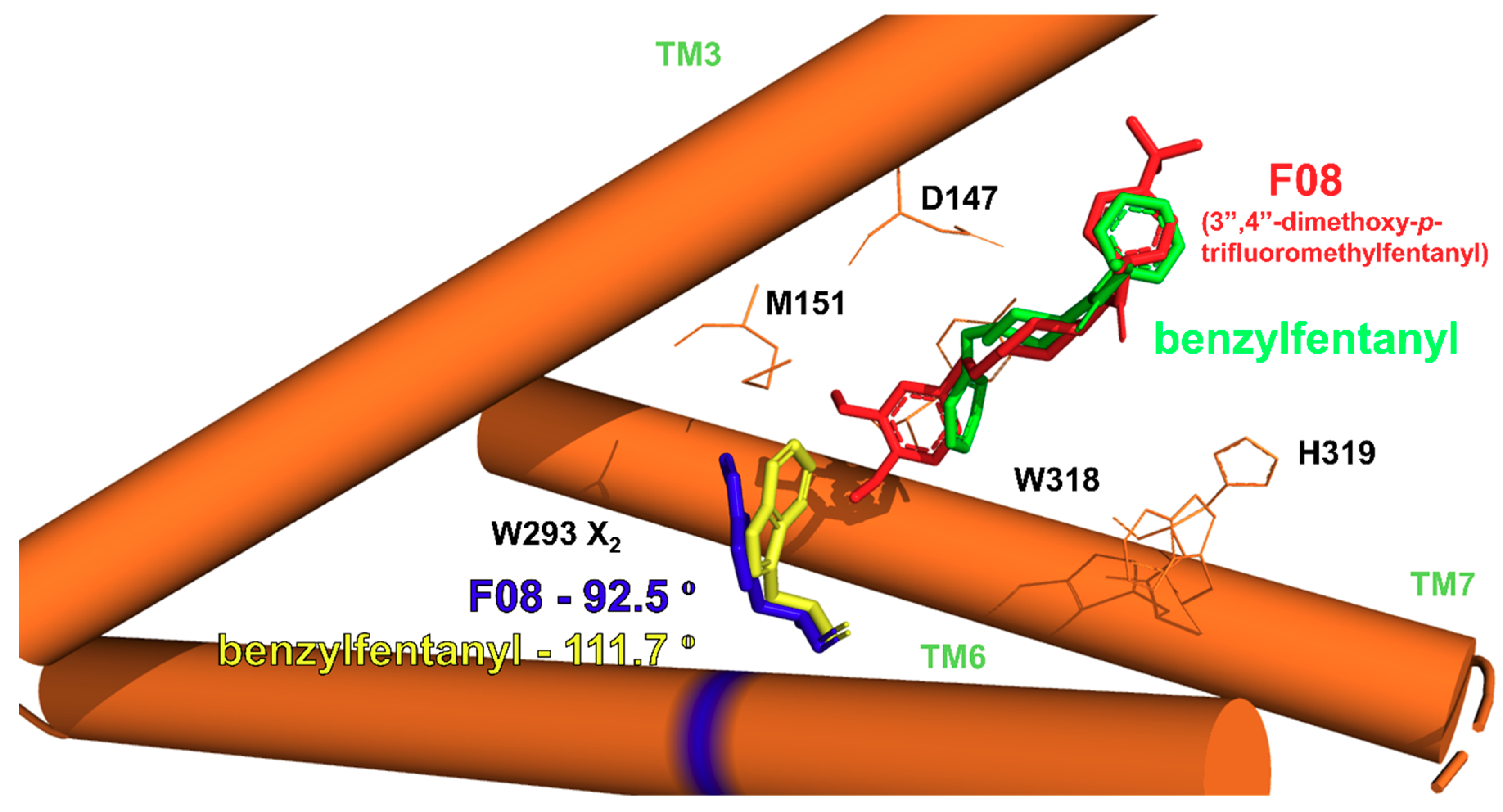
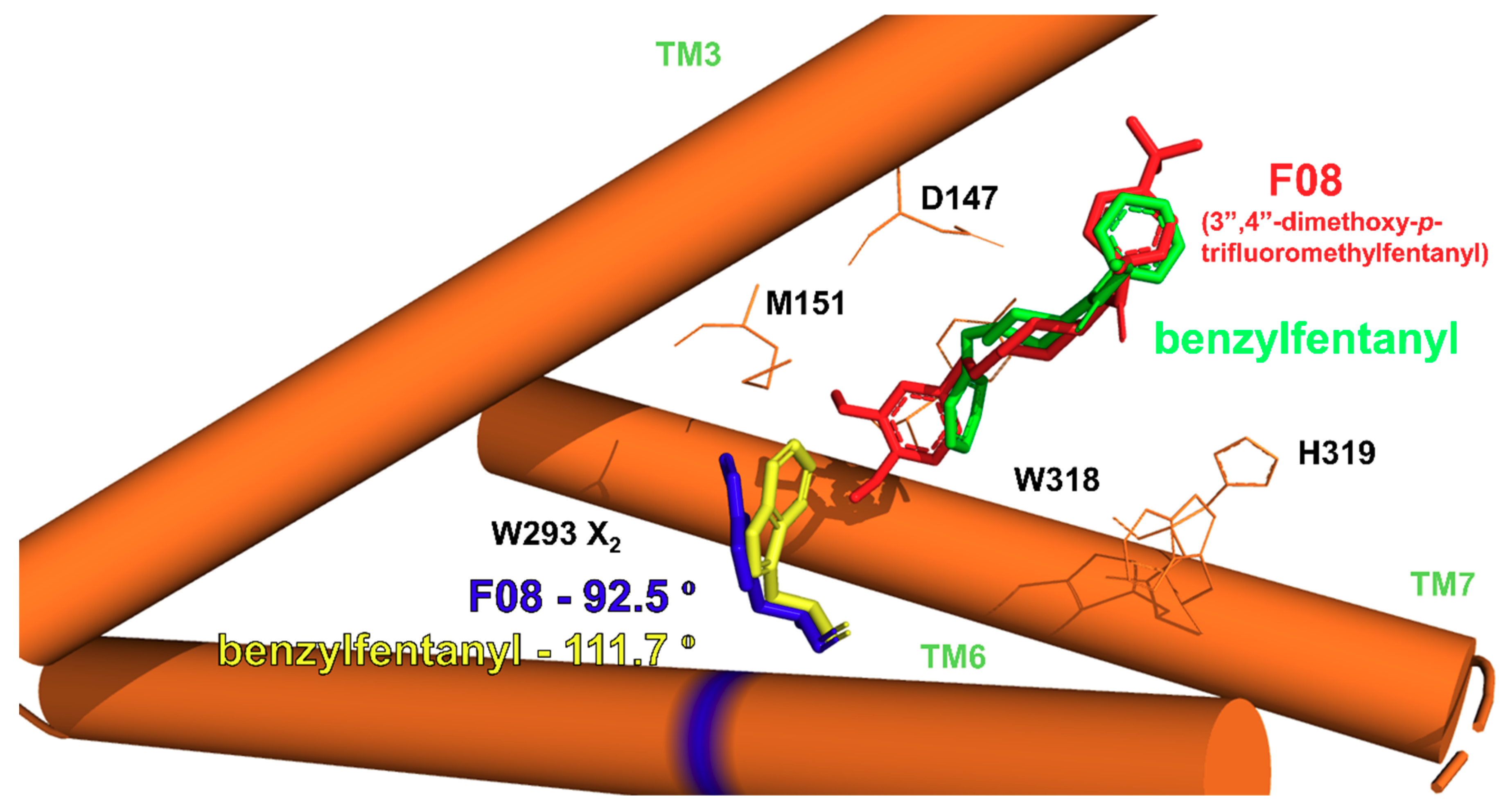
2.2.1. Binding Modes
- GF1. ionic interaction between D147 and the protonable amine of the ligands’ piperidine,
- GF2. orientation of the N-chain towards the interior of the receptor,
- GF3. positioning of the other ligands’ pole towards the receptor outlet,
- GF4. locating the aromatic ring of the anilide in the subpocket formed by transmembrane helices (TM) TM3, TM4 and extracellular loops (ECL): ECL1 and ECL2,
- GF5. 4-axial substituent (if present) directed towards W318.
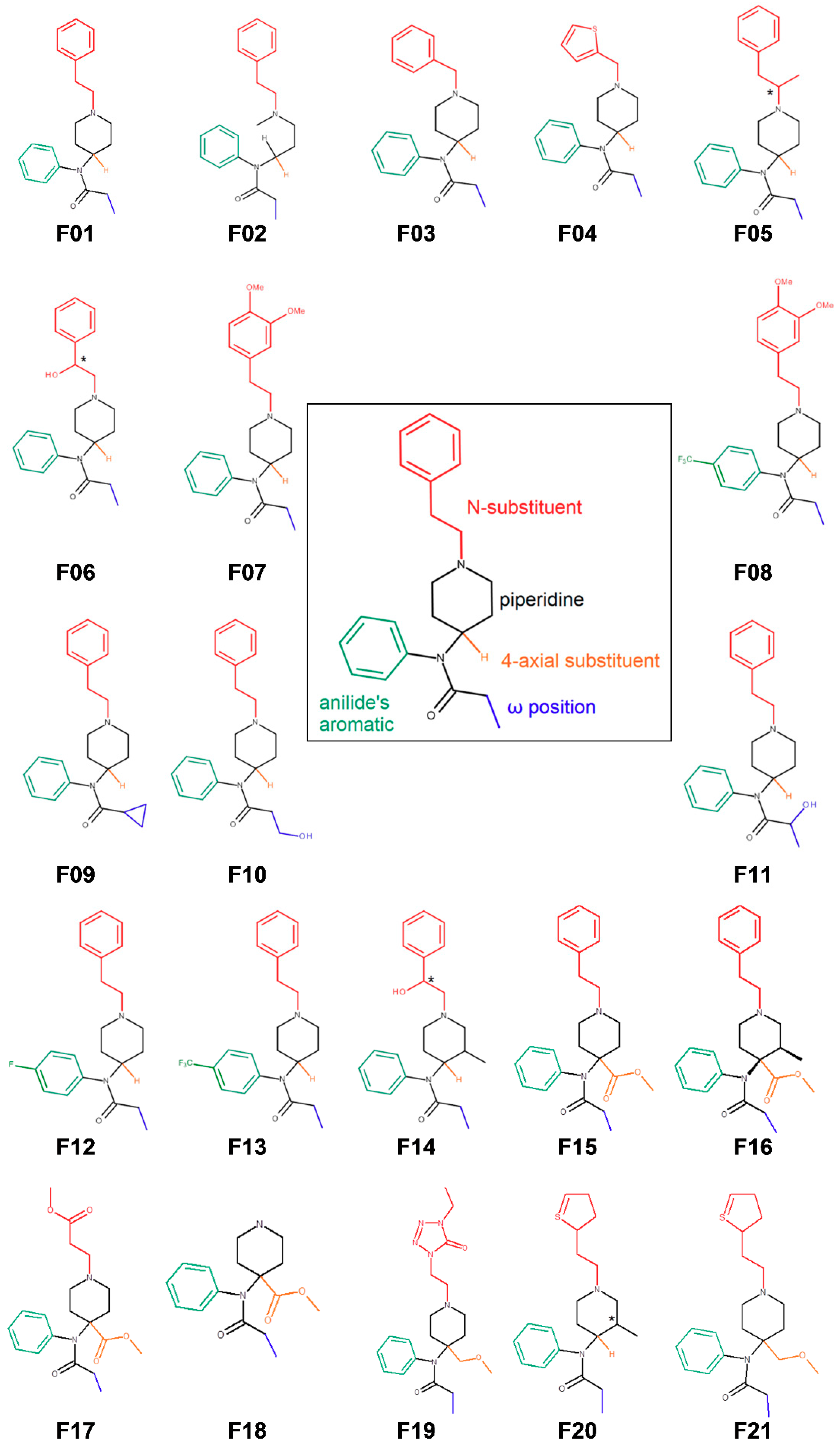
2.2.2. Binding Modes Compared to SAR Data
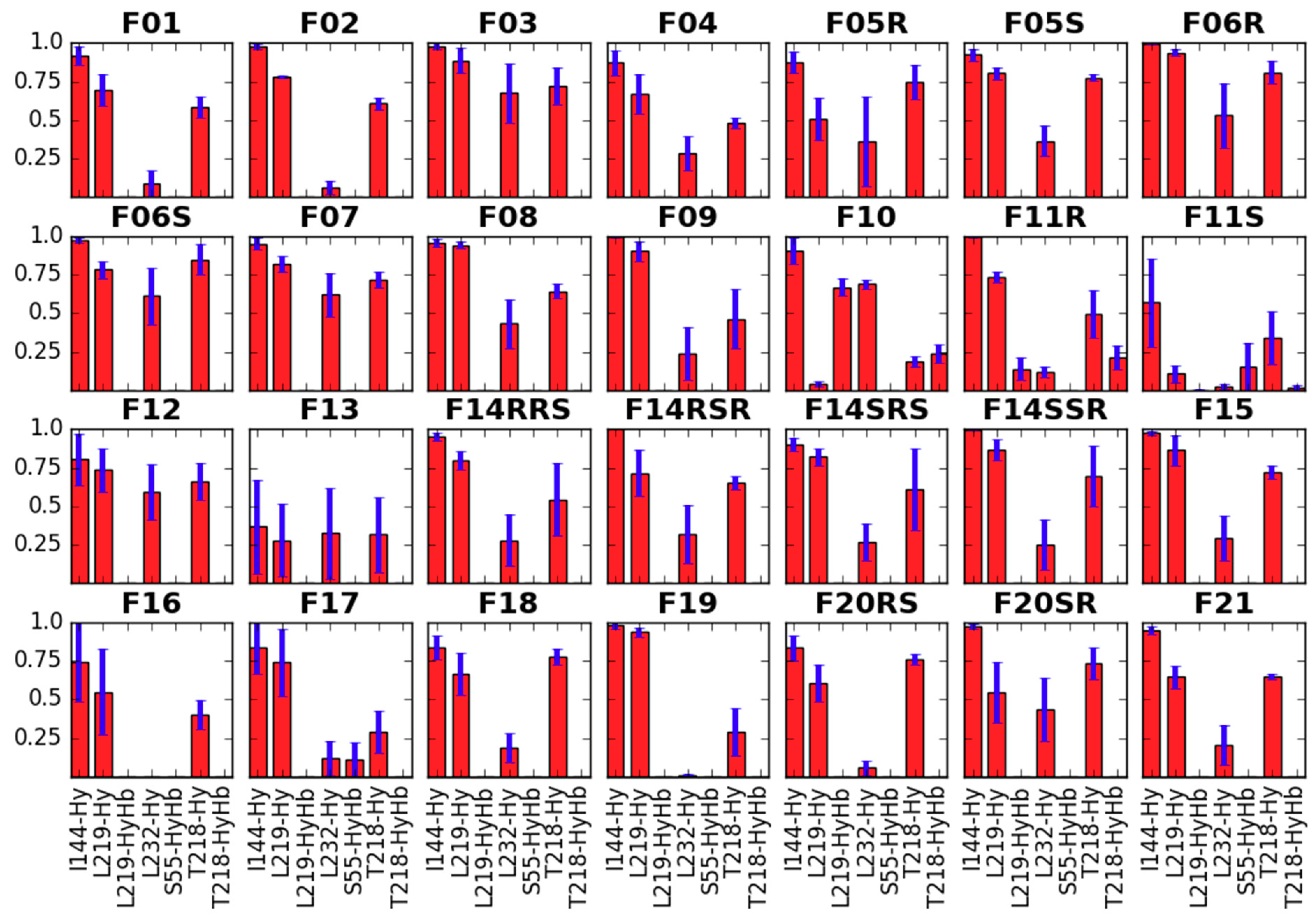
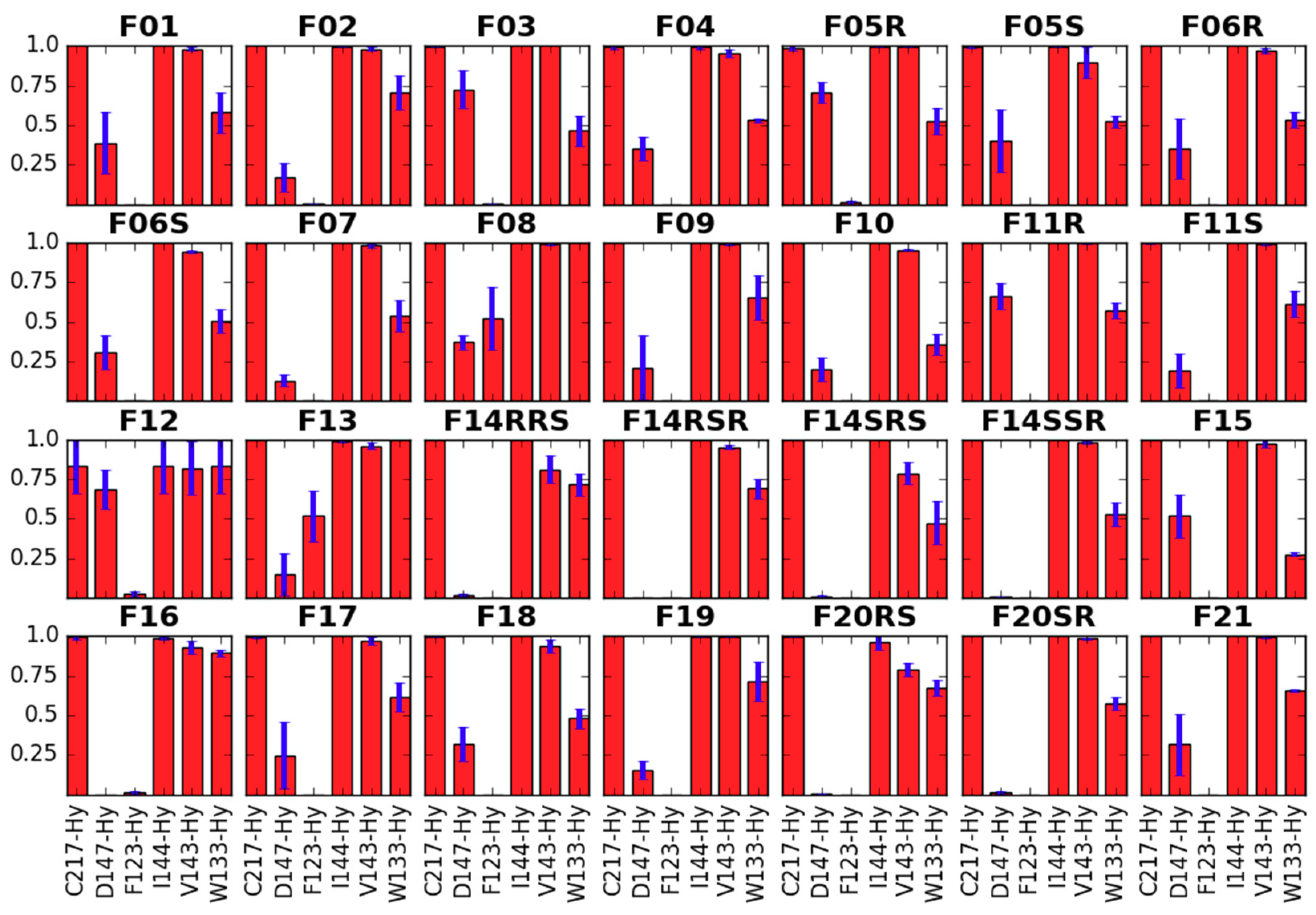
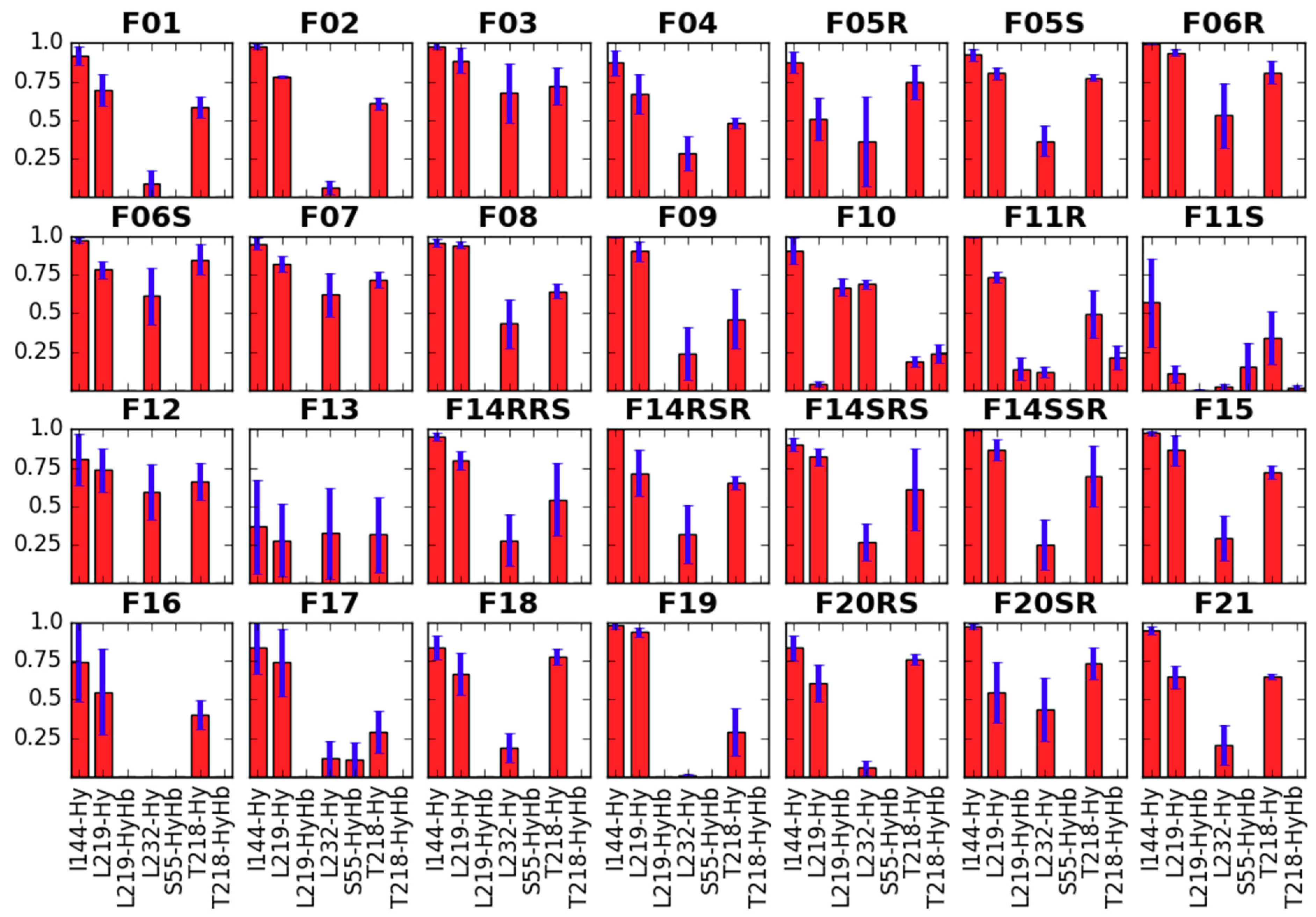
2.2.3. Receptor Characteristics from MD Simulations
- the RMSF of protein helices,
- side chain dihedral angles of key residues,
- the length of the so-called ‘3-7 lock’ and
- the hydration of D114 and Y336.
RMSF of Protein Helices
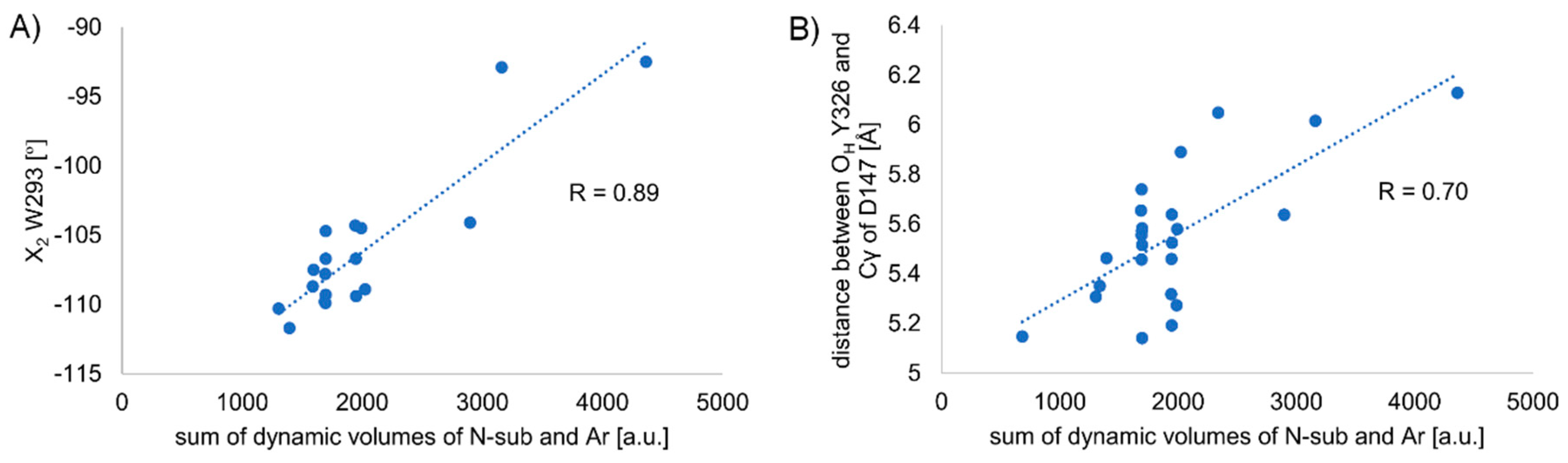
Side Chain Dihedral Angles of Key Residues
- interacting with the fentanyl piperidine (or close to it: D147, Y148),
- close to 4-axial substituent (W318, H319),
- close to the N-substituent (M151, W293, H297, Y326),
- close to anilide’s aromatic (Y133) or even
- outside the binding pocket (Y336).
Length of the so-Called ‘3-7 lock’
Hydration of D114 and Y336
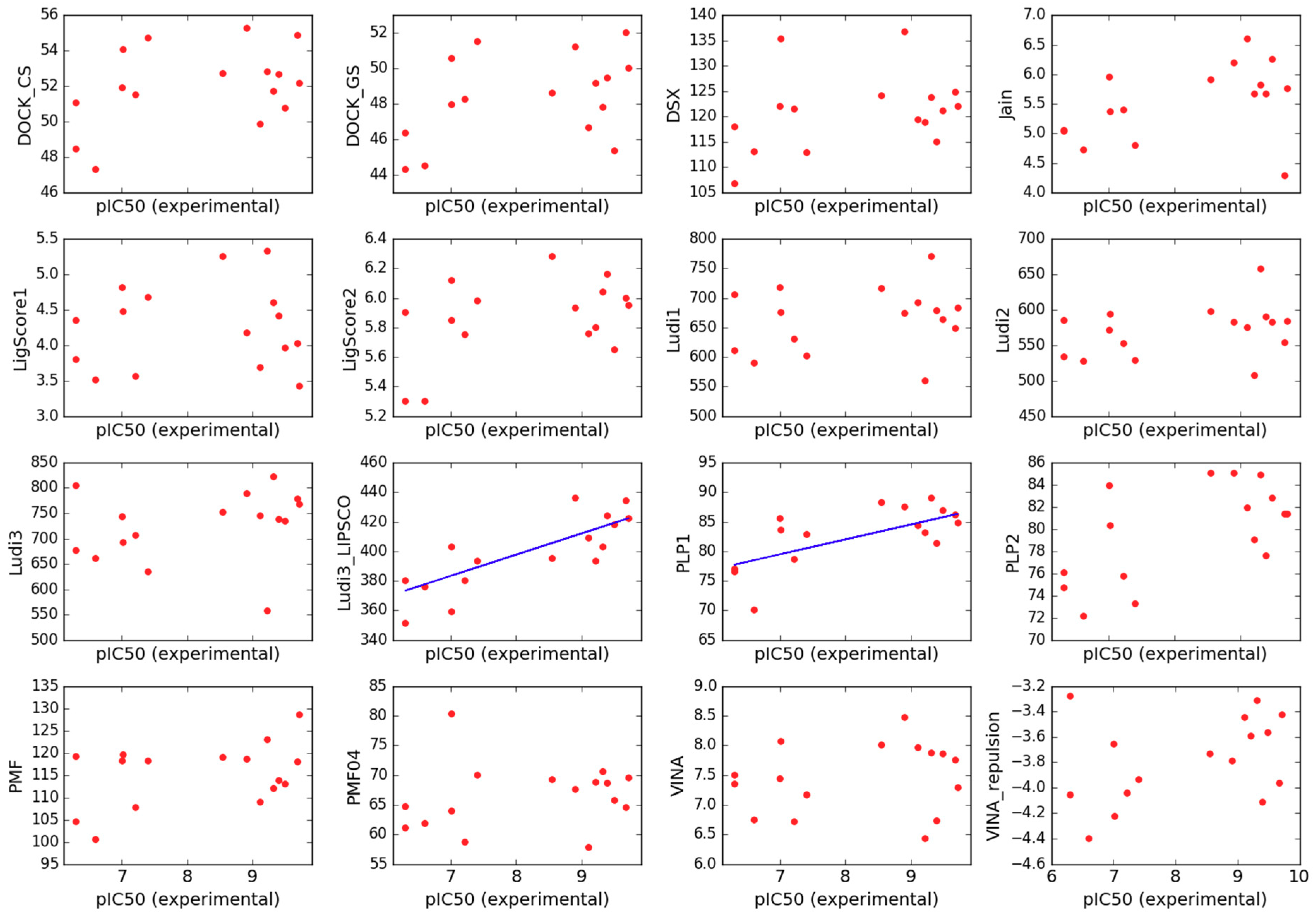
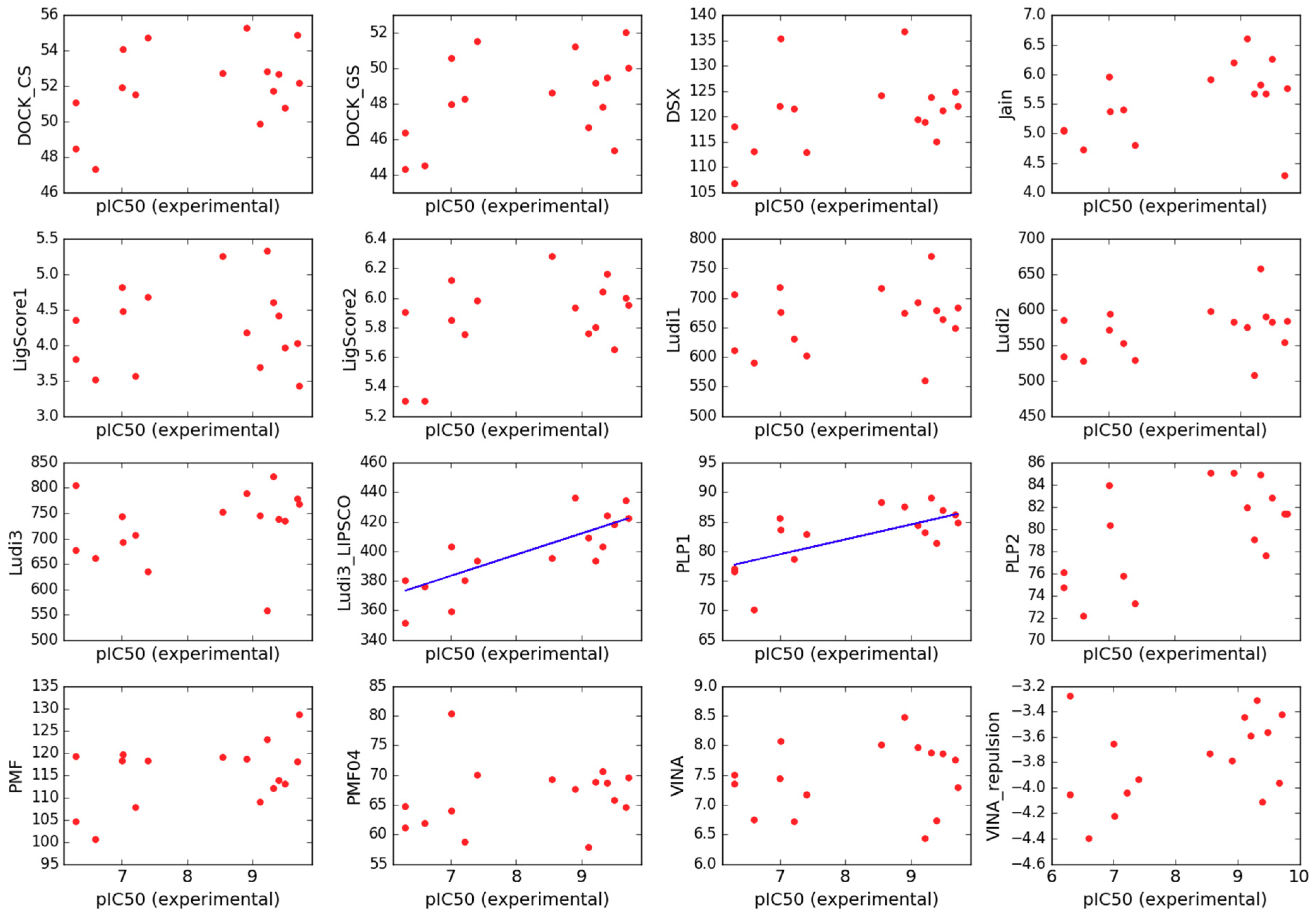
2.3. Scoring
3. Materials and Methods
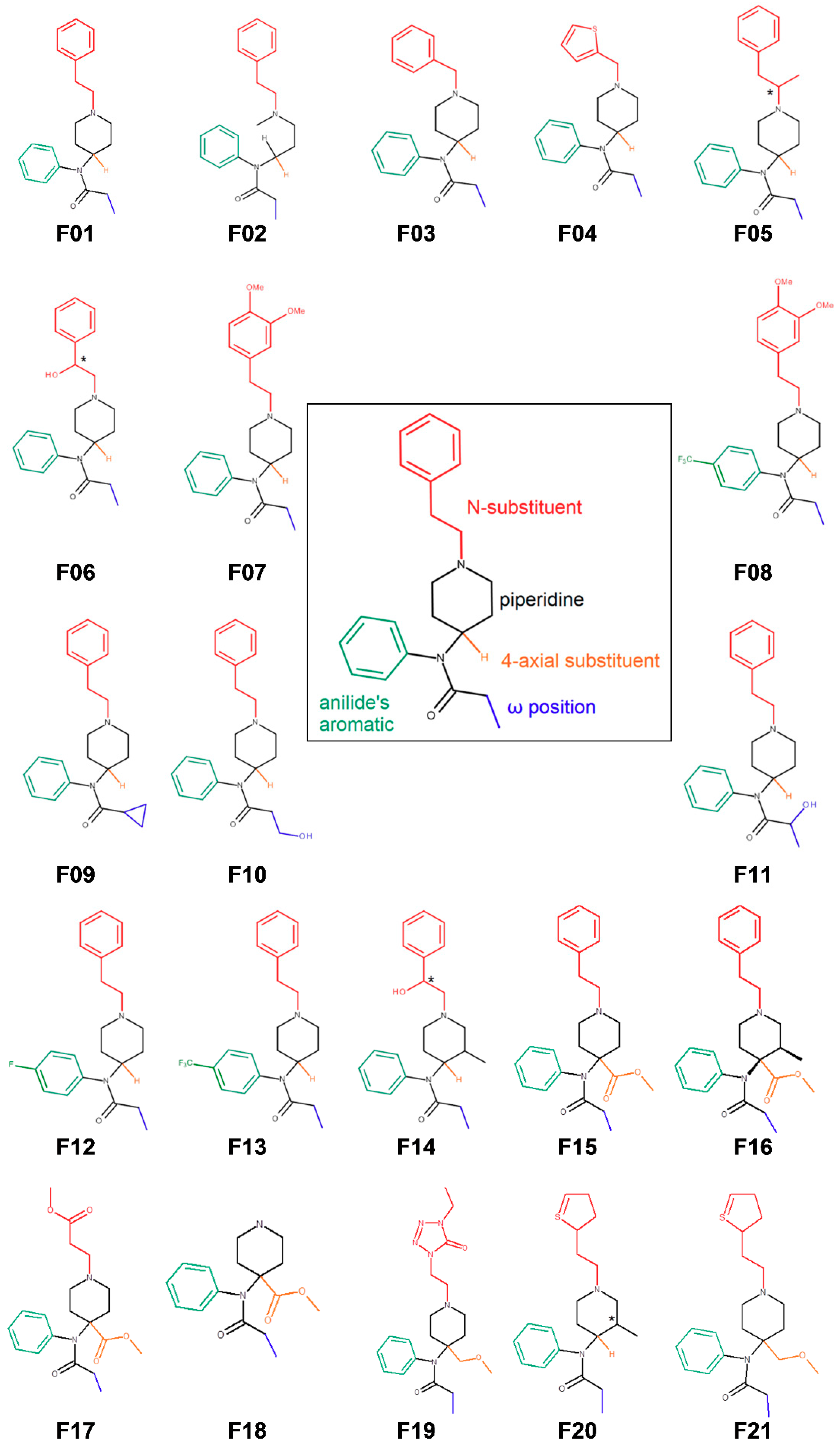
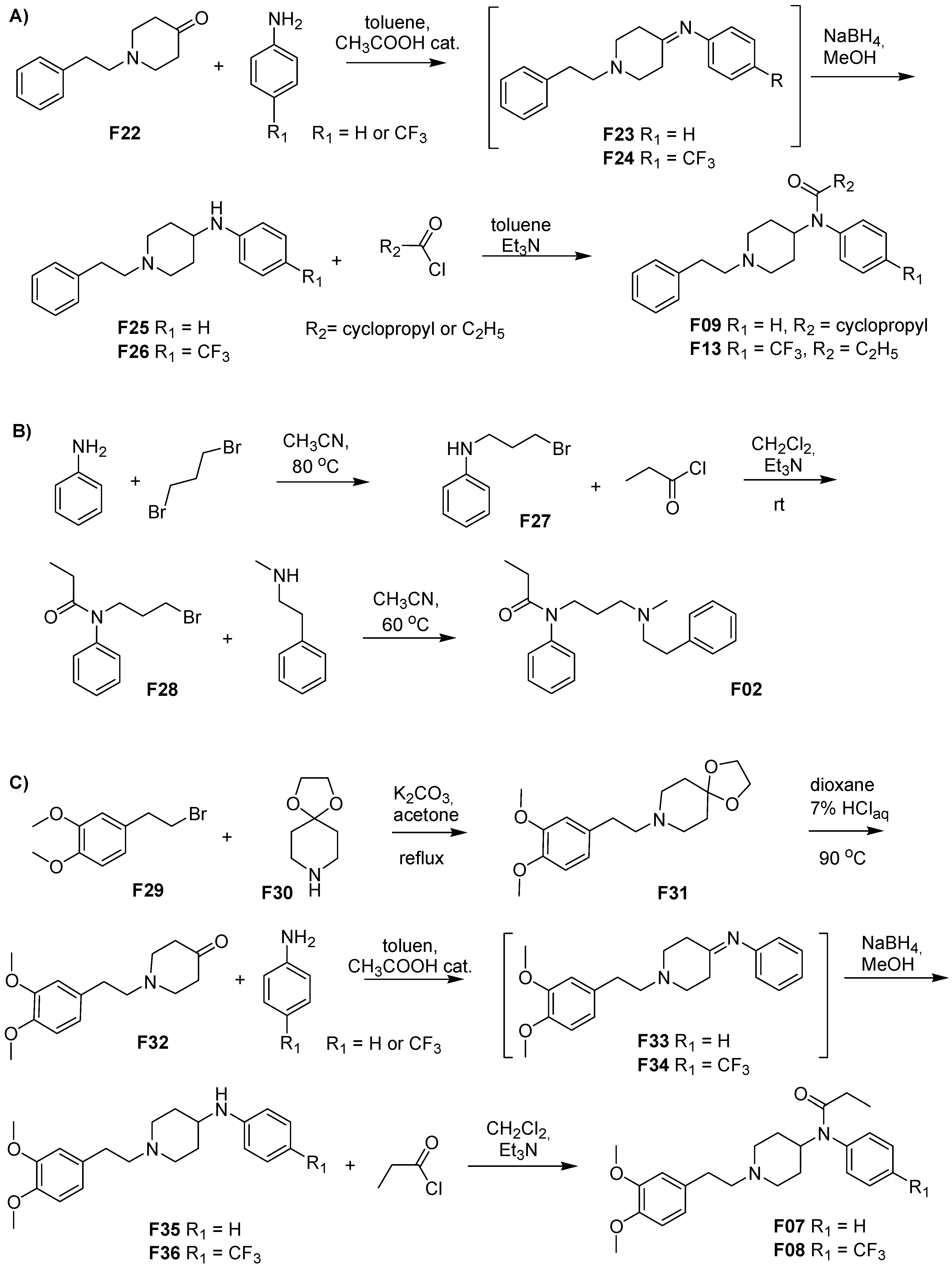
3.1. Fentanyl Derivatives
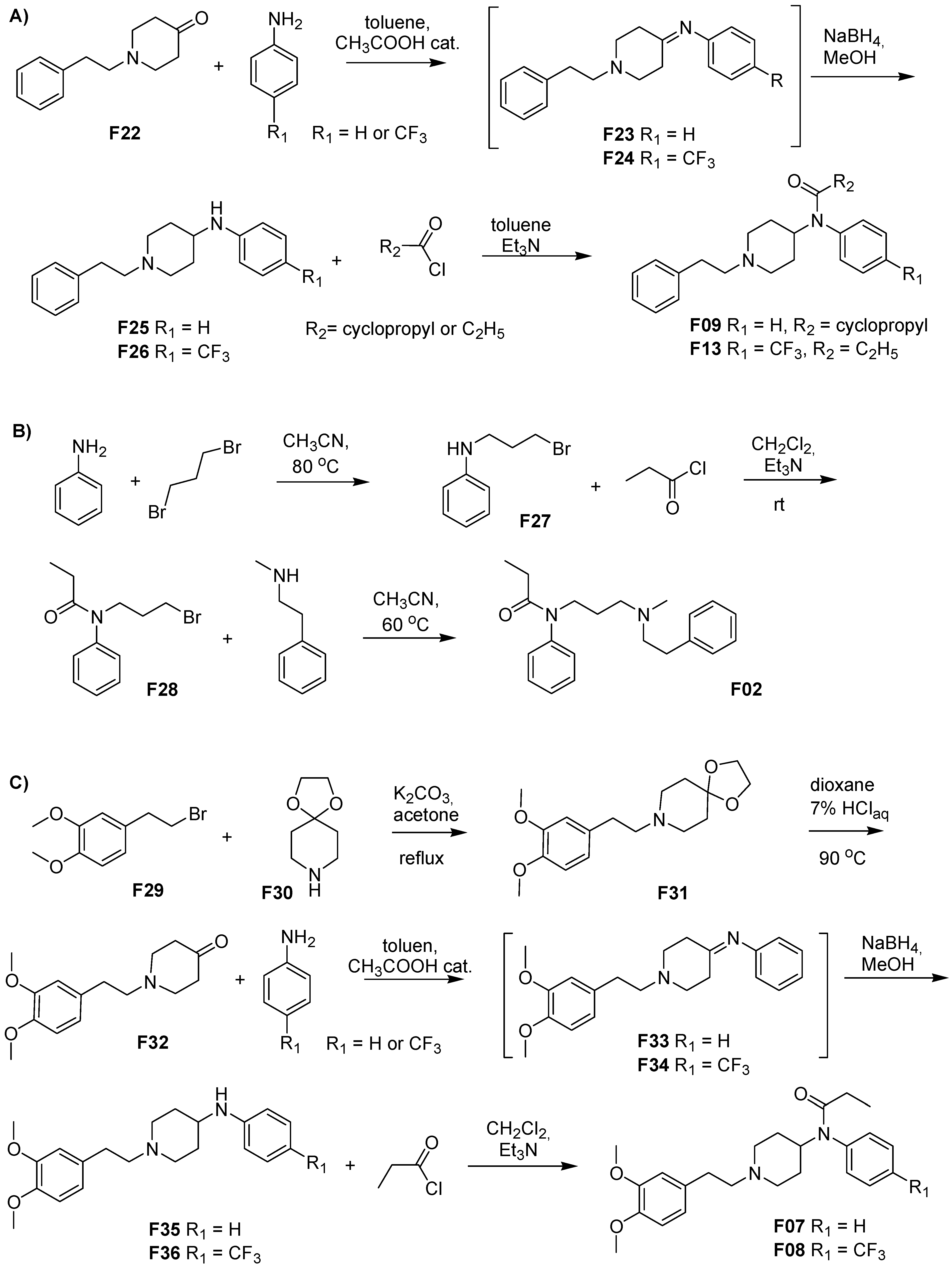
3.2. Chemistry
3.3. Synthesis
3.3.1. 8-[2-(3,4-Dimethoxyphenyl)ethyl]-1,4-dioxa-8-azaspiro[4.5]decane (F31)
3.3.2. 1-(3,4-Dimethoxyphenethyl)-piperidin-4-one (F32)
3.3.3. N-(1-Phenethyl-4-piperidinyl)-(3,4-dimethoxyphenyl)amine (F35)
3.3.4. 3″,4″-Dimethoxyfentanyl, N-(1-Phenethyl-4-piperidinyl)-N-(3,4-dimethoxyphenyl)-propionamide (F07)
3.3.5. N-(1-Phenethyl-4-piperidinyl)-(4-trifluoromethylphenyl)amine (F36)
3.3.6. 3″,4″-Dimethoxy-para-trifluoromethylfentanyl, N-(1-Phenethyl-4-piperidinyl)-N-(4-trifluoromethylphenyl)-propionamide (F08)
3.4. Binding Assays
3.5. Molecular Modeling
3.5.1. Receptor and Ligand Preparation
3.5.2. Molecular Dynamics
3.5.3. Analysis of Data from Molecular Dynamics
- root-mean-square-deviation (RMSD) of protein backbone atoms with a 0.0 ns structure taken as reference; RMSDs were calculated for separate helices, or for all them together; the residues considered for each helix are: 72-92 (H1), 104-128 (H2), 139-169 (H3), 185-204 (H4), 227-260 (H5), 275-304 (H6), 314-338 (H7);
- distance between ligands’ protonable nitrogen and Cγ atom of D147;
- X1 and X2 dihedral angles of: W133, D147, Y148, W293, H297, W318, H319, Y326, W336;
- distance between Cγ atom of D147 and OH atom of Y326;
- hydration of D114 and Y336 residues, understood as a number of water molecules found within 5.0 Å of any atom of a given residue.
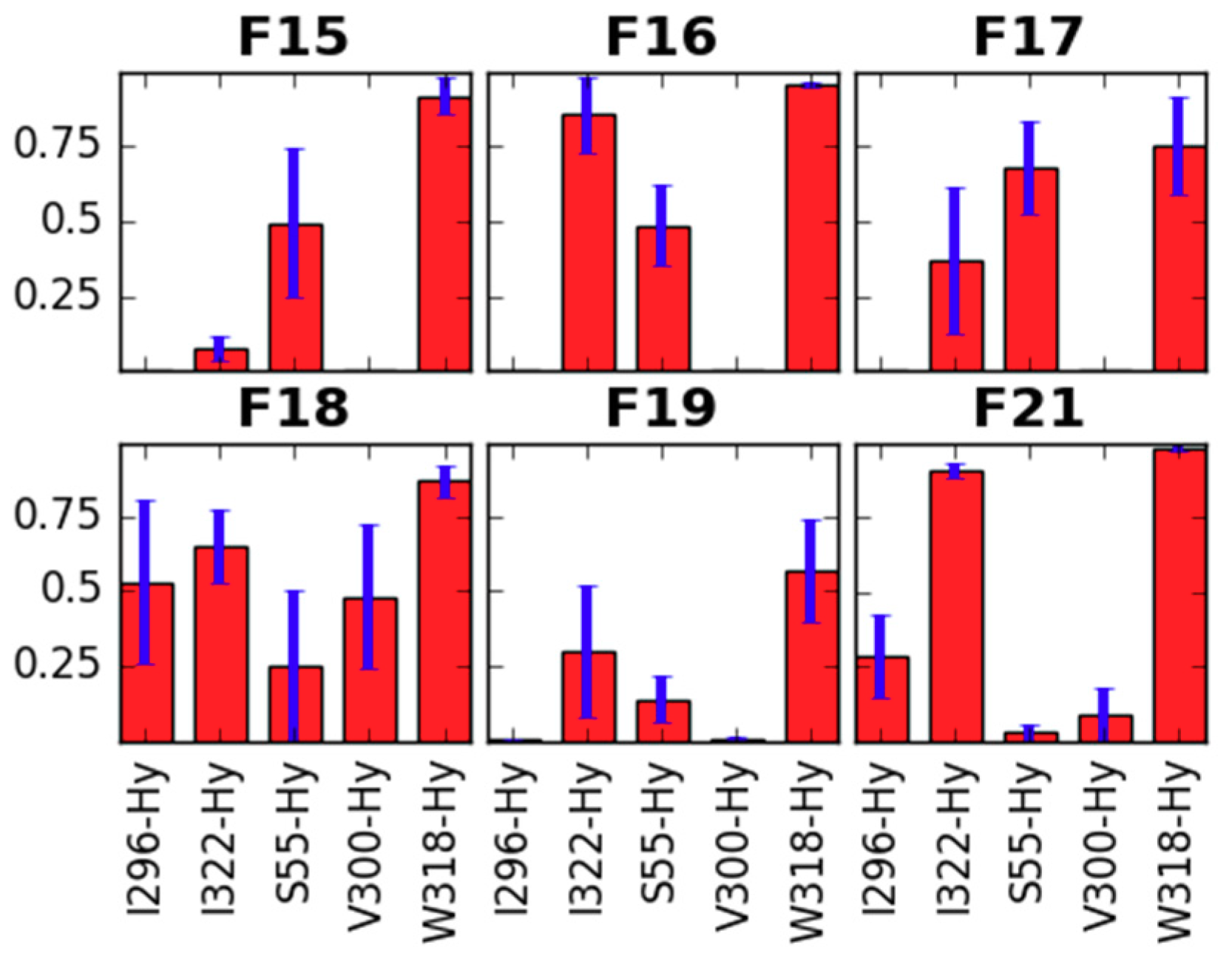
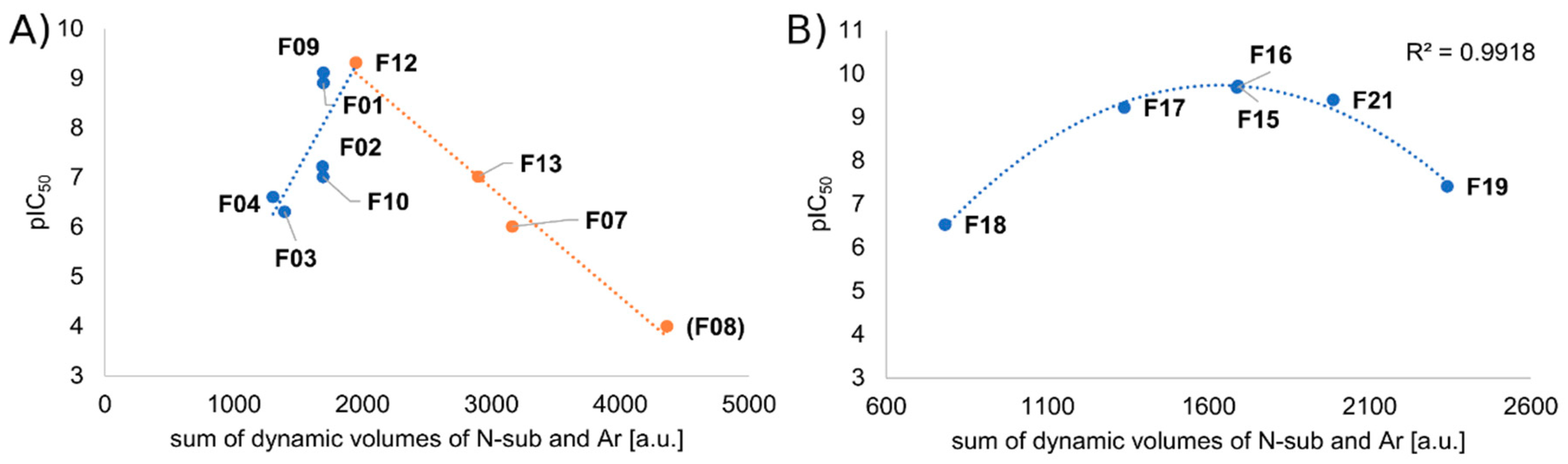
3.5.4. Volume Calculations
3.5.5. Scoring
- Jain scoring function (Jain) [85],
- Piecewise Linear Potential (PLP1 and PLP2) [86],
- LigScore scoring function (LigScore1 and LigScore2) [89],
- Ludi scoring function (Ludi1, Ludi2 and Ludi3) [90],
- VINA scoring function (VINA) [91],
- DSX scoring function (DSX) [92],
- DOCK continuous and grid scores (DOCK_CS and DOCK_GS) [93].
3.5.6. Regression Analysis and Molecular Graphics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cortazzo, M.H.; Copenhaver, D.; Fishman, S.M. Major Opioids and Chronic Opioid Therapy. In Practical Management of Pain; Elsevier: Amsterdam, The Netherlands, 2014; pp. 495.e3–507.e3. ISBN 9780323083409. [Google Scholar]
- Stanley, T.H. The Fentanyl Story. J. Pain 2014, 15, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.S.; Hruby, V.J. Fentanyl-related compounds and derivatives: Current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014, 6, 385–412. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.M.; Cunningham, C.W.; Mercer, S.L. DARK Classics in Chemical Neuroscience: Fentanyl. ACS Chem. Neurosci. 2018, 9, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.A.; Leif, R.N.; Mayer, B.P. An Efficient, Optimized Synthesis of Fentanyl and Related Analogs. PLoS ONE 2014, 9, e108250. [Google Scholar] [CrossRef] [PubMed]
- Jevtić, I.; Došen-Mićović, L.; Ivanović, E.; Todorović, N.; Ivanović, M. Synthesis of Orthogonally Protected (±)-3-Amino-4-anilidopiperidines and (±)-3-N-Carbomethoxyfentanyl. Synthesis (Stuttg.) 2017, 49, 3126–3136. [Google Scholar]
- Ogawa, N.; Furuishi, T.; Nagase, H.; Endo, T.; Takahashi, C.; Yamamoto, H.; Kawashima, Y.; Loftsson, T.; Kobayashi, M.; Ueda, H. Interaction of fentanyl with various cyclodextrins in aqueous solutions. J. Pharm. Pharmacol. 2016, 68, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Barbas, R.; Prohens, R.; Bauzá, A.; Franconetti, A.; Frontera, A. H-Bonded anion–anion complexes in fentanyl citrate polymorphs and solvates. Chem. Commun. 2019, 55, 115–118. [Google Scholar] [CrossRef]
- Bick, M.J.; Greisen, P.J.; Morey, K.J.; Antunes, M.S.; La, D.; Sankaran, B.; Reymond, L.; Johnsson, K.; Medford, J.I.; Baker, D. Computational design of environmental sensors for the potent opioid fentanyl. Elife 2017, 6, e28909. [Google Scholar] [CrossRef]
- Nami, M.; Salehi, P.; Dabiri, M.; Bararjanian, M.; Gharaghani, S.; Khoramjouy, M.; Al-Harrasi, A.; Faizi, M. Synthesis of novel norsufentanil analogs via a four-component Ugi reaction and in vivo, docking, and QSAR studies of their analgesic activity. Chem. Biol. Drug Des. 2018, 91, 902–914. [Google Scholar] [CrossRef]
- Váradi, A.; Palmer, T.C.; Haselton, N.; Afonin, D.; Subrath, J.J.; Le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Marrone, G.F.; Borics, A.; et al. Synthesis of Carfentanil Amide Opioids Using the Ugi Multicomponent Reaction. ACS Chem. Neurosci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef]
- Li, S.; Cohen-Karni, D.; Kovaliov, M.; Tomycz, N.; Cheng, B.; Whiting, D.; Averick, S. Synthesis and biological evaluation of fentanyl acrylic derivatives. RSC Adv. 2017, 7, 20015–20019. [Google Scholar] [CrossRef] [Green Version]
- Petrov, R.R.; Lee, Y.S.; Vardanyan, R.S.; Liu, L.; Ma, S.; Davis, P.; Lai, J.; Porreca, F.; Vanderah, T.W.; Hruby, V.J. Effect of anchoring 4-anilidopiperidines to opioid peptides. Bioorg. Med. Chem. Lett. 2013, 23, 3434–3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deekonda, S.; Wugalter, L.; Rankin, D.; Largent-Milnes, T.M.; Davis, P.; Wang, Y.; Bassirirad, N.M.; Lai, J.; Kulkarni, V.; Vanderah, T.W.; et al. Design and synthesis of novel bivalent ligands (MOR and DOR) by conjugation of enkephalin analogues with 4-anilidopiperidine derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 4683–4688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deekonda, S.; Rankin, D.; Davis, P.; Lai, J.; Vanderah, T.W.; Porecca, F.; Hruby, V.J. Design synthesis and structure–activity relationship of 5-substituted (tetrahydronaphthalen-2yl)methyl with N-phenyl-N-(piperidin-2-yl)propionamide derivatives as opioid ligands. Bioorg. Med. Chem. 2016, 24, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, L.; Stefanucci, A.; Pieretti, S.; Marzoli, F.; Fidanza, L.; Mollica, A.; Mirzaie, S.; Carradori, S.; De Petrocellis, L.; Schiano Moriello, A.; et al. Evaluation of the analgesic effect of 4-anilidopiperidine scaffold containing ureas and carbamates. J. Enzyme Inhib. Med. Chem. 2016, 31, 1638–1647. [Google Scholar] [CrossRef]
- Jevtic, I.; Penjisevic, J.; Ivanovic, M.; Kostic-Rajacic, S. Synthetic route towards potential bivalent ligands possessing opioid and D2/D3 pharmacophores. J. Serbian Chem. Soc. 2018, 105. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165.e13–1175.e13. [Google Scholar] [CrossRef]
- Kennedy, N.M.; Schmid, C.L.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Chen, Y.T.; Cameron, M.D.; Bohn, L.M.; Bannister, T.D. Optimization of a Series of Mu Opioid Receptor (MOR) Agonists with High G Protein Signaling Bias. J. Med. Chem. 2018, 61, 8895–8907. [Google Scholar] [CrossRef]
- Rodriguez-Gaztelumendi, A.; Spahn, V.; Labuz, D.; Machelska, H.; Stein, C. Analgesic effects of a novel pH-dependent μ-opioid receptor agonist in models of neuropathic and abdominal pain. Pain 2018, 159, 2277–2284. [Google Scholar] [CrossRef]
- Spahn, V.; Del Vecchio, G.; Rodriguez-Gaztelumendi, A.; Temp, J.; Labuz, D.; Kloner, M.; Reidelbach, M.; Machelska, H.; Weber, M.; Stein, C. Opioid receptor signaling, analgesic and side effects induced by a computationally designed pH-dependent agonist. Sci. Rep. 2018, 8, 8965. [Google Scholar] [CrossRef]
- Spahn, V.; Del Vecchio, G.; Labuz, D.; Rodriguez-Gaztelumendi, A.; Massaly, N.; Temp, J.; Durmaz, V.; Sabri, P.; Reidelbach, M.; Machelska, H.; et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355, 966–969. [Google Scholar] [CrossRef]
- Fisher, D.M.; Chang, P.; Wada, D.R.; Dahan, A.; Palmer, P.P. Pharmacokinetic Properties of a Sufentanil Sublingual Tablet Intended to Treat Acute Pain. Anesthesiology 2018, 128, 943–952. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Drug Abuse Overdose Death Rates. Available online: https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates (accessed on 4 January 2019).
- Mounteney, J.; Giraudon, I.; Denissov, G.; Griffiths, P. Fentanyls: Are we missing the signs? Highly potent and on the rise in Europe. Int. J. Drug Policy 2015, 26, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.; Poklis, J.; Poklis, A.; Wolf, C.; Mainland, M.; Hair, L.; Devers, K.; Chrostowski, L.; Arbefeville, E.; Merves, M. Postmortem Toxicology Findings of Acetyl Fentanyl, Fentanyl, and Morphine in Heroin Fatalities in Tampa, Florida. Acad. Forensic Pathol. 2015, 5, 676–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poklis, J.; Poklis, A.; Wolf, C.; Mainland, M.; Hair, L.; Devers, K.; Chrostowski, L.; Arbefeville, E.; Merves, M.; Pearson, J. Postmortem tissue distribution of acetyl fentanyl, fentanyl and their respective nor-metabolites analyzed by ultrahigh performance liquid chromatography with tandem mass spectrometry. Forensic Sci. Int. 2015, 257, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, B.P.; DeHope, A.J.; Mew, D.A.; Spackman, P.E.; Williams, A.M. Chemical Attribution of Fentanyl Using Multivariate Statistical Analysis of Orthogonal Mass Spectral Data. Anal. Chem. 2016, 88, 4303–4310. [Google Scholar] [CrossRef] [PubMed]
- Shaner, R.L.; Schulze, N.D.; Seymour, C.; Hamelin, E.I.; Thomas, J.D.; Johnson, R.C. Quantitation of fentanyl analogs in dried blood spots by flow-through desorption coupled to online solid phase extraction tandem mass spectrometry. Anal. Methods 2017, 9, 3876–3883. [Google Scholar] [CrossRef] [PubMed]
- Hikin, L.; Smith, P.R.; Ringland, E.; Hudson, S.; Morley, S.R. Multiple fatalities in the North of England associated with synthetic fentanyl analogue exposure: Detection and quantitation a case series from early 2017. Forensic Sci. Int. 2018, 282, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Elliott, S.P.; George, S. The analytical challenges of cyclopropylfentanyl and crotonylfentanyl: An approach for toxicological analysis. Drug Test. Anal. 2018, 10, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Vandergrift, G.W.; Hessels, A.J.; Palaty, J.; Krogh, E.T.; Gill, C.G. Paper spray mass spectrometry for the direct, semi-quantitative measurement of fentanyl and norfentanyl in complex matrices. Clin. Biochem. 2018, 54, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Skov-Skov Bergh, M.; Bogen, I.L.; Wilson, S.R.; Øiestad, Å.M.L. Addressing the Fentanyl Analog Epidemic by Multiplex UHPLC-MS/MS Analysis of Whole Blood. Ther. Drug Monit. 2018, 40, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.; Shaner, R.L.; Feyereisen, M.C.; Wharton, R.E.; Kaplan, P.; Hamelin, E.I.; Johnson, R.C. Determination of Fentanyl Analog Exposure Using Dried Blood Spots with LC–MS-MS. J. Anal. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Ciancetta, A.; Moro, S. Bridging Molecular Docking to Membrane Molecular Dynamics To Investigate GPCR–Ligand Recognition: The Human A 2A Adenosine Receptor as a Key Study. J. Chem. Inf. Model. 2014, 54, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.R.; Kruhlak, N.L.; Kim, M.T.; Hawkins, E.G.; Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE 2018, 13, e0197734. [Google Scholar] [CrossRef]
- Dyniewicz, J.; Lipiński, P.F.J.; Kosson, P.; Leśniak, A.; Bochyńska-Czyż, M.; Muchowska, A.; Tourwé, D.; Ballet, S.; Misicka, A.; Lipkowski, A.W. Hydrazone Linker as a Useful Tool for Preparing Chimeric Peptide/Nonpeptide Bifunctional Compounds. ACS Med. Chem. Lett. 2017, 8, 73–77. [Google Scholar] [CrossRef]
- Wright, W.B.; Brabander, H.J.; Hardy, R.A. Synthetic Analgesics. III. Basic Anilides and Carbanilates Containing the Phenalkyl Moiety 1. J. Org. Chem. 1961, 26, 485–490. [Google Scholar] [CrossRef]
- Ivanovic, M.; Micovic, I.V.; Vuckovic, S.; Prostran, M.; Todorovic, Z.; Ivanovic, E.; Kiricojevic, V.; Djordjevic, J.B.; Dosen-Micovic, L. The synthesis and pharmacological evaluation of (±)-2,3-seco-fentanyl analogues. J. Serb. Chem. Soc. 2004, 69, 955–968. [Google Scholar] [CrossRef]
- N.V. Research Laboratorium Dr P. Janssen N-(1-alkyl-4-piperidyl)-N-arylalkanoamides 1961. Available online: https://worldwide.espacenet.com/publicationDetails/originalDocument?CC=FR&NR=1517671A&KC=A&FT=D&ND=3&date=19680322&DB=EPODOC&locale=en_EP# (accessed on 18 January 2019).
- U.S. Drug Enforcement Administration. Three Factor Analysis for Temporary Scheduling of Cyclopropyl Fentanyl; U.S. Drug Enforcement Administration: Springfield, VA, USA, 2017.
- European Monitoring Centre for Drugs and Drug Addiction. EMCDDA–Europol Joint Report on a New Psychoactive Substance: N-phenyl-N-[1-(2-phenylethyl)piperidin-4-yl]cyclopropanecarboxamide (cyclopropylfentanyl); European Monitoring Centre for Drugs and Drug Addiction: Lisbon, Portugal, 2018. [Google Scholar]
- Volpe, D.A.; McMahon Tobin, G.A.; Mellon, R.D.; Katki, A.G.; Parker, R.J.; Colatsky, T.; Kropp, T.J.; Verbois, S.L. Uniform assessment and ranking of opioid μ receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol. 2011, 59, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.J.; Jarończyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular dynamics of fentanyl bound to μ-opioid receptor. J. Mol. Model. 2019. under review. [Google Scholar]
- Li, J.G.; Chen, C.; Yin, J.; Rice, K.; Zhang, Y.; Matecka, D.; de Riel, J.K.; DesJarlais, R.L.; Liu-Chen, L.Y. ASP147 in the third transmembrane helix of the rat mu opioid receptor forms ion-pairing with morphine and naltrexone. Life Sci. 1999, 65, 175–185. [Google Scholar] [CrossRef]
- Surratt, C.K.; Johnson, P.S.; Moriwaki, A.; Seidleck, B.K.; Blaschak, C.J.; Wang, J.B.; Uhl, G.R. mu opiate receptor: Charged transmembrane domain amino acids are critical for agonist recognition and intrinsic activity. J. Biol. Chem. 1994, 269, 20548–20553. [Google Scholar]
- Weltrowska, G.; Lemieux, C.; Chung, N.N.; Guo, J.J.; Wilkes, B.C.; Schiller, P.W. ‘Carba’-carfentanil (trans isomer): A μ opioid receptor (MOR) partial agonist with a distinct binding mode. Bioorg. Med. Chem. 2014, 22, 4581–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weltrowska, G.; Chung, N.N.; Lemieux, C.; Guo, J.; Lu, Y.; Wilkes, B.C.; Schiller, P.W. “Carba”-Analogues of Fentanyl are Opioid Receptor Agonists. J. Med. Chem. 2010, 53, 2875–2881. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the µ-opioid receptor–Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Zhu, Y.-C.; Jin, W.-Q.; Chen, X.-J.; Chen, J.; Ji, R.-Y.; Chi, Z.-Q. Stereoisomers of N-[1-(2-Hydroxy-2-phenylethyl)-3-methyl-4-piperidyl]-N-phenylpropanamide: Synthesis, Stereochemistry, Analgesic Activity, and Opioid Receptor Binding Characteristics. J. Med. Chem. 1995, 38, 3652–3659. [Google Scholar] [CrossRef]
- Jin, W.Q.; Wang, Z.X.; Chen, J.; Chen, X.J.; Chi, Z.Q. Analgesic activity and selectivity for opioid receptors of enantiomers of ohmefentanyl. Zhongguo Yao Li Xue Bao 1996, 17, 421–424. [Google Scholar]
- Mecozzi, S.; Rebek, J., Jr. The 55% Solution: A Formula for Molecular Recognition in the Liquid State. Chem. A Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Zürcher, M.; Diederich, F. Structure-Based Drug Design: Exploring the Proper Filling of Apolar Pockets at Enzyme Active Sites. J. Org. Chem. 2008, 73, 4345–4361. [Google Scholar] [CrossRef] [PubMed]
- Zürcher, M.; Gottschalk, T.; Meyer, S.; Bur, D.; Diederich, F. Exploring the Flap Pocket of the Antimalarial Target Plasmepsin II: The “55% Rule” Applied to Enzymes. ChemMedChem 2008, 3, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Casy, A.F.; Huckstep, M.R. Structure-Activity Studies of Fentanyl. J. Pharm. Pharmacol. 1988, 40, 605–608. [Google Scholar] [CrossRef]
- Kudzma, L.V.; Severnak, S.A.; Benvenga, M.J.; Ezell, E.F.; Ossipov, M.H.; Knight, V.V.; Rudo, F.G.; Spencer, H.K.; Spaulding, T.C. 4-Phenyl- and 4-heteroaryl-4-anilidopiperidines. A novel class of analgesic and anesthetic agents. J. Med. Chem. 1989, 32, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.R.; Wynn, R.L.; Rudo, F.G.; Doorley, B.M.; Spencer, H.K.; Spaulding, T. New 4-(heteroanilido)piperidines, structurally related to the pure opioidagonist fentanyl, with agonist and/or antagonist properties. J. Med. Chem. 1989, 32, 663–671. [Google Scholar] [CrossRef]
- Lalinde, N.; Moliterni, J.; Wright, D.; Spencer, H.K.; Ossipov, M.H.; Spaulding, T.C.; Rudo, F.G. Synthesis and pharmacological evaluation of a series of new 1,4-disubstituted 3-methyl-piperidine analgesics. J. Med. Chem. 1990, 33, 2876–2882. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Filipek, S.; Yuan, S. The Principles of Ligand Specificity on beta-2-adrenergic receptor. Sci. Rep. 2016, 6, 34736. [Google Scholar] [CrossRef] [Green Version]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of Molecular Switches in GPCRs—Theoretical and Experimental Studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef]
- Kolinski, M.; Filipek, S. Molecular Dynamics of μ Opioid Receptor Complexes with Agonists and Antagonists. Open Struct. Biol. J. 2008, 2, 8–20. [Google Scholar] [CrossRef]
- Hulme, E.C. GPCR activation: A mutagenic spotlight on crystal structures. Trends Pharmacol. Sci. 2013, 34, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Kellam, B.; Briddon, S.J.; Hill, S.J. Effect of a toggle switch mutation in TM6 of the human adenosine A 3 receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br. J. Pharmacol. 2014, 171, 3827–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, A.; Hempel, C.; Wanka, L.; Schubert, M.; Hamm, H.E.; Beck-Sickinger, A.G. G Protein Preassembly Rescues Efficacy of W 6.48 Toggle Mutations in Neuropeptide Y 2 Receptor. Mol. Pharmacol. 2018, 93, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Laroche, G.; Wang, X.; Ågren, H.; Bowman, G.R.; Giguère, P.M.; Tu, Y. Propagation of the Allosteric Modulation Induced by Sodium in the δ-Opioid Receptor. Chem. A Eur. J. 2017, 23, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, M.; Filipek, S. Study of a structurally similar kappa opioid receptor agonist and antagonist pair by molecular dynamics simulations. J. Mol. Model. 2010, 16, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W2466.48 opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. 2015, 54, 556–559. [Google Scholar]
- Yuan, S.; Palczewski, K.; Peng, Q.; Kolinski, M.; Vogel, H.; Filipek, S. The mechanism of ligand-induced activation or inhibition of μ- And κ-opioid receptors. Angew. Chem. Int. Ed. 2015, 54, 7560–7563. [Google Scholar] [CrossRef]
- Yuan, S.; Vogel, H.; Filipek, S. The role of water and sodium ions in the activation of the??-opioid receptor. Angew. Chem. Int. Ed. 2013, 52, 10112–10115. [Google Scholar] [CrossRef]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef]
- Enyedy, I.J.; Egan, W.J. Can we use docking and scoring for hit-to-lead optimization? J. Comput. Aided Mol. Des. 2008, 22, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardanyan, R.; Vijay, G.; Nichol, G.S.; Liu, L.; Kumarasinghe, I.; Davis, P.; Vanderah, T.; Porreca, F.; Lai, J.; Hruby, V.J. Synthesis and investigations of double-pharmacophore ligands for treatment of chronic and neuropathic pain. Bioorg. Med. Chem. 2009, 17, 5044–5053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thansandote, P.; Raemy, M.; Rudolph, A.; Lautens, M. Synthesis of Benzannulated N-Heterocycles by a Palladium-Catalyzed C−C/C−N Coupling of Bromoalkylamines. Org. Lett. 2007, 9, 5255–5258. [Google Scholar] [CrossRef] [PubMed]
- Ganellin, C.R.; Spickett, R.G.W. Compounds Affecting the Central Nervous System. I. 4-Piperidones and Related Compounds. J. Med. Chem. 1965, 8, 619–625. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01 2013; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Jain, A.N. Scoring noncovalent protein-ligand interactions: A continuous differentiable function tuned to compute binding affinities. J. Comput. Aided Mol. Des. 1996, 10, 427–440. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.R.; Fogel, L.J.; Freer, S.T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: Conformationally flexible docking by evolutionary programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- Muegge, I.; Martin, Y.C. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. PMF scoring revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Krammer, A.; Kirchhoff, P.D.; Jiang, X.; Venkatachalam, C.M.; Waldman, M. LigScore: A novel scoring function for predicting binding affinities. J. Mol. Graph. Model. 2005, 23, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput. Aided Mol. Des. 1998, 12, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Neudert, G.; Klebe, G. DSX: A Knowledge-Based Scoring Function for the Assessment of Protein–Ligand Complexes. J. Chem. Inf. Model. 2011, 51, 2731–2745. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accelrys Software Inc. Discovery Studio Modeling Environment 2013; Accelrys Software Inc.: San Diego, CA, USA, 2013. [Google Scholar]
- The PyMOL Molecular Graphics System. Available online: www.pymol.org (accessed on 9 April 2018).
Sample Availability: Samples of the compounds F02, F07, F08, F09 and F13 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd No | Name | IC50 1 | SD 2 |
|---|---|---|---|
| fentanyl and acyclic derivative | |||
| F01 | fentanyl | 1.23 | 0.14 |
| F02 | N-[3-(Methyl-phenethylamino)propyl]-N-phenyl propionamide | 60.25 | 3.51 |
| shorter N-chain | |||
| F03 | benzylfentanyl | 489.7 | 28.6 |
| F04 | thienylfentanyl | 245.5 | 12.9 |
| substitution at the N-chain | |||
| F053 | α-methylfentanyl | 0.32 | 0.01 |
| F063 | β-hydroxyfentanyl | 2.81 | 0.13 |
| substitution at the ring of the N-phenethyl | |||
| F07 | 3″,4″-dimethoxyfentanyl | 977.2 | 43.11 |
| F08 | 3″,4″-dimethoxy-para-trifluoromethylfentanyl | >1000.0 | - |
| variations at the propionamide chain | |||
| F09 | cyclopropylfentanyl | 0.77 | 0.04 |
| F10 | ω-hydroxyfentanyl | 97.7 | 5.80 |
| F113 | ω-1-hydroxyfentanyl | 489.0 | 40.59 |
| para-substitution | |||
| F12 | para-fluorofentanyl | 0.48 | 0.03 |
| F13 | para-trifluoromethylfentanyl | 95.5 | 6.2 |
| ohmefentanyl (β-OH, 3-Me) | |||
| F143 | ohmefentanyl | 0.27 | 0.03 |
| 4-substitution | |||
| F15 | carfentanil | 0.19 | 0.01 |
| F16 | lofentanil | 0.21 | 0.01 |
| F17 | remifentanil | 0.60 | 0.08 |
| F18 | norcarfentanil | 295.1 | 1.3 |
| F19 | alfentanil | 38.9 | 2.8 |
| N-thioethyl | |||
| F203 | 3-methylothiofentanyl | 1.10 | 0.10 |
| F21 | sufentanil | 0.40 | 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.F.J.; Kosson, P.; Matalińska, J.; Roszkowski, P.; Czarnocki, Z.; Jarończyk, M.; Misicka, A.; Dobrowolski, J.C.; Sadlej, J. Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules 2019, 24, 740. https://doi.org/10.3390/molecules24040740
Lipiński PFJ, Kosson P, Matalińska J, Roszkowski P, Czarnocki Z, Jarończyk M, Misicka A, Dobrowolski JC, Sadlej J. Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules. 2019; 24(4):740. https://doi.org/10.3390/molecules24040740
Chicago/Turabian StyleLipiński, Piotr F. J., Piotr Kosson, Joanna Matalińska, Piotr Roszkowski, Zbigniew Czarnocki, Małgorzata Jarończyk, Aleksandra Misicka, Jan Cz. Dobrowolski, and Joanna Sadlej. 2019. "Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis" Molecules 24, no. 4: 740. https://doi.org/10.3390/molecules24040740
APA StyleLipiński, P. F. J., Kosson, P., Matalińska, J., Roszkowski, P., Czarnocki, Z., Jarończyk, M., Misicka, A., Dobrowolski, J. C., & Sadlej, J. (2019). Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules, 24(4), 740. https://doi.org/10.3390/molecules24040740