
Synthesis of Phthalides and ?,?-butenolides by Transition Metal-Catalyzed Activation of C—H Bonds


Abstract
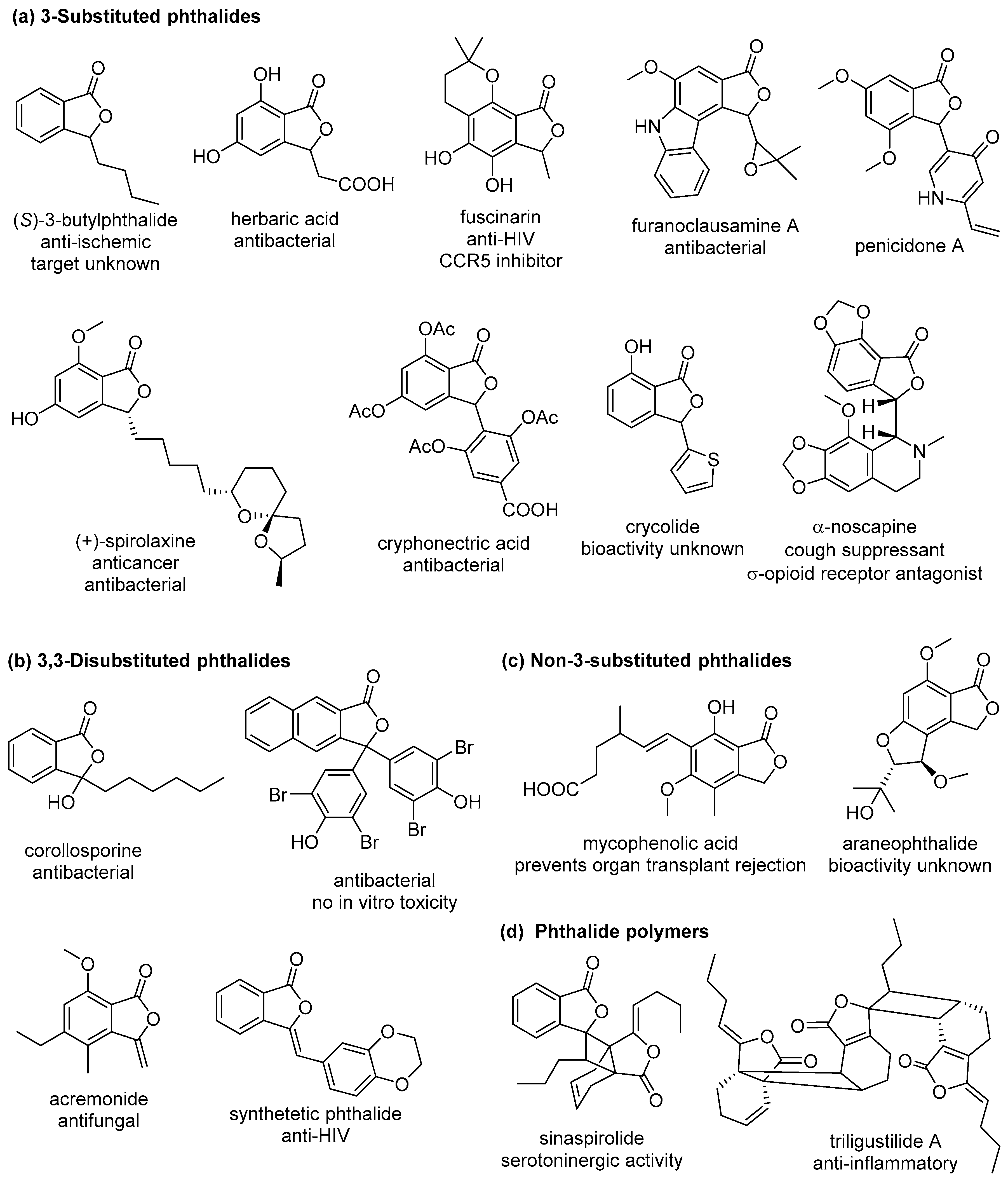
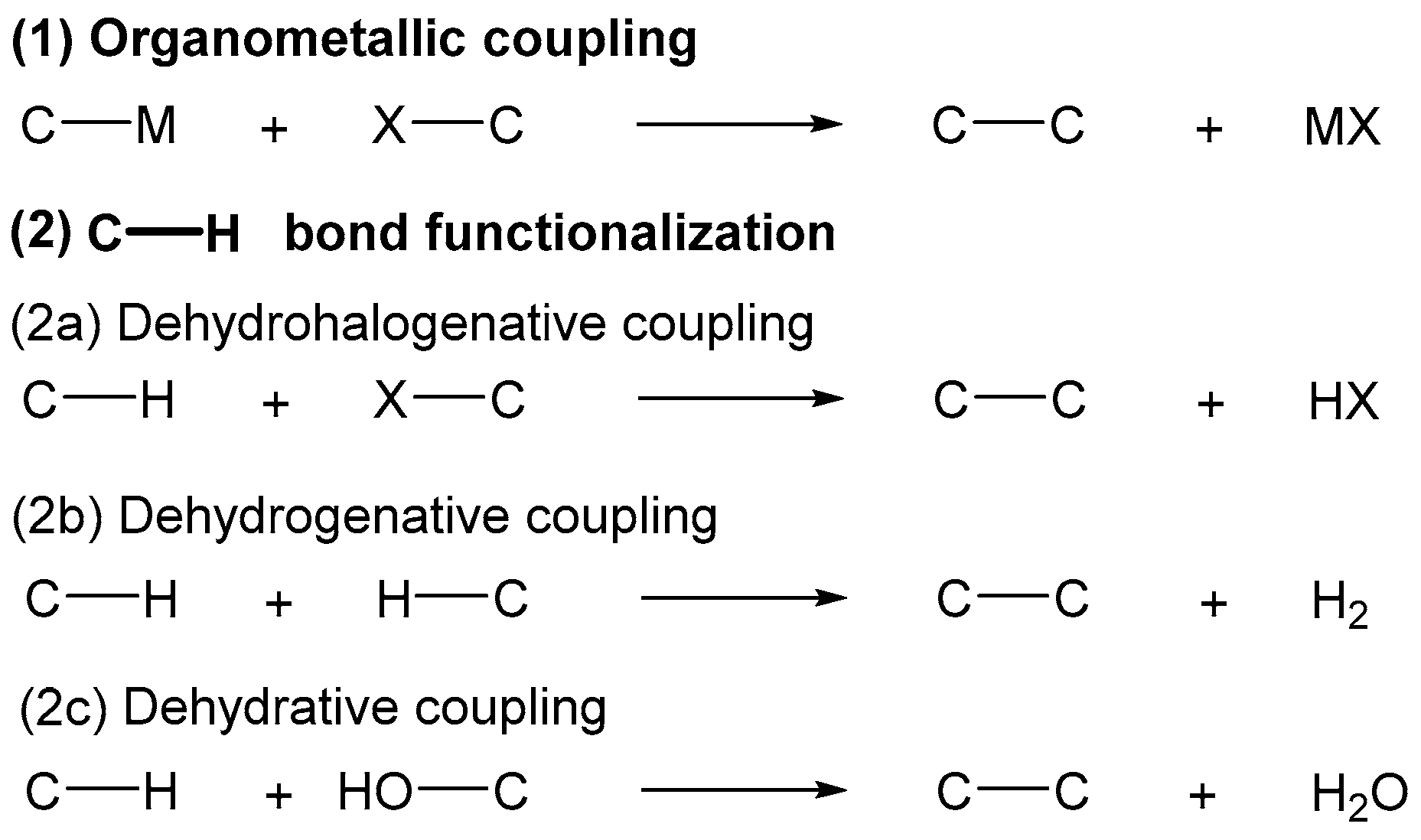
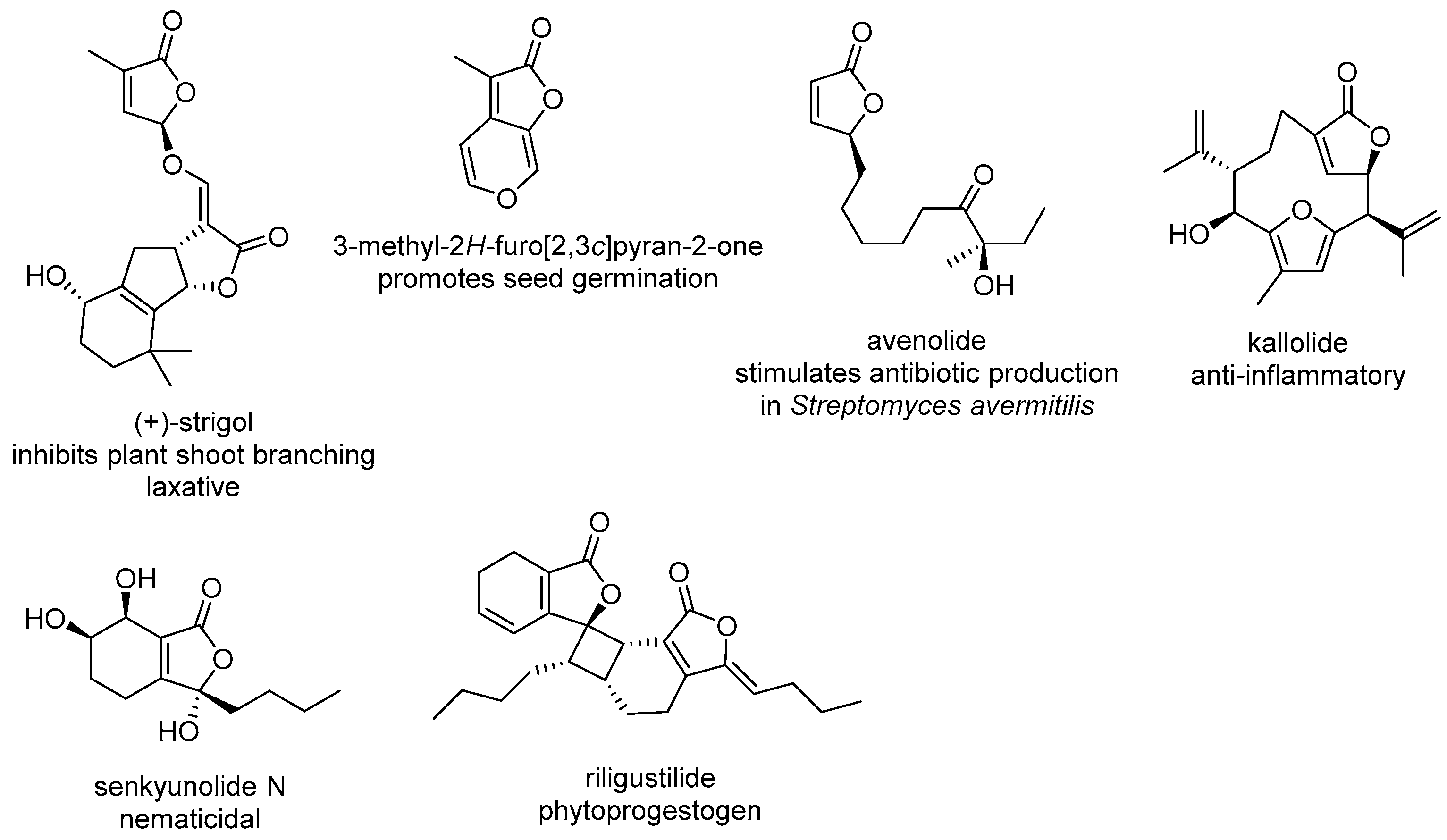
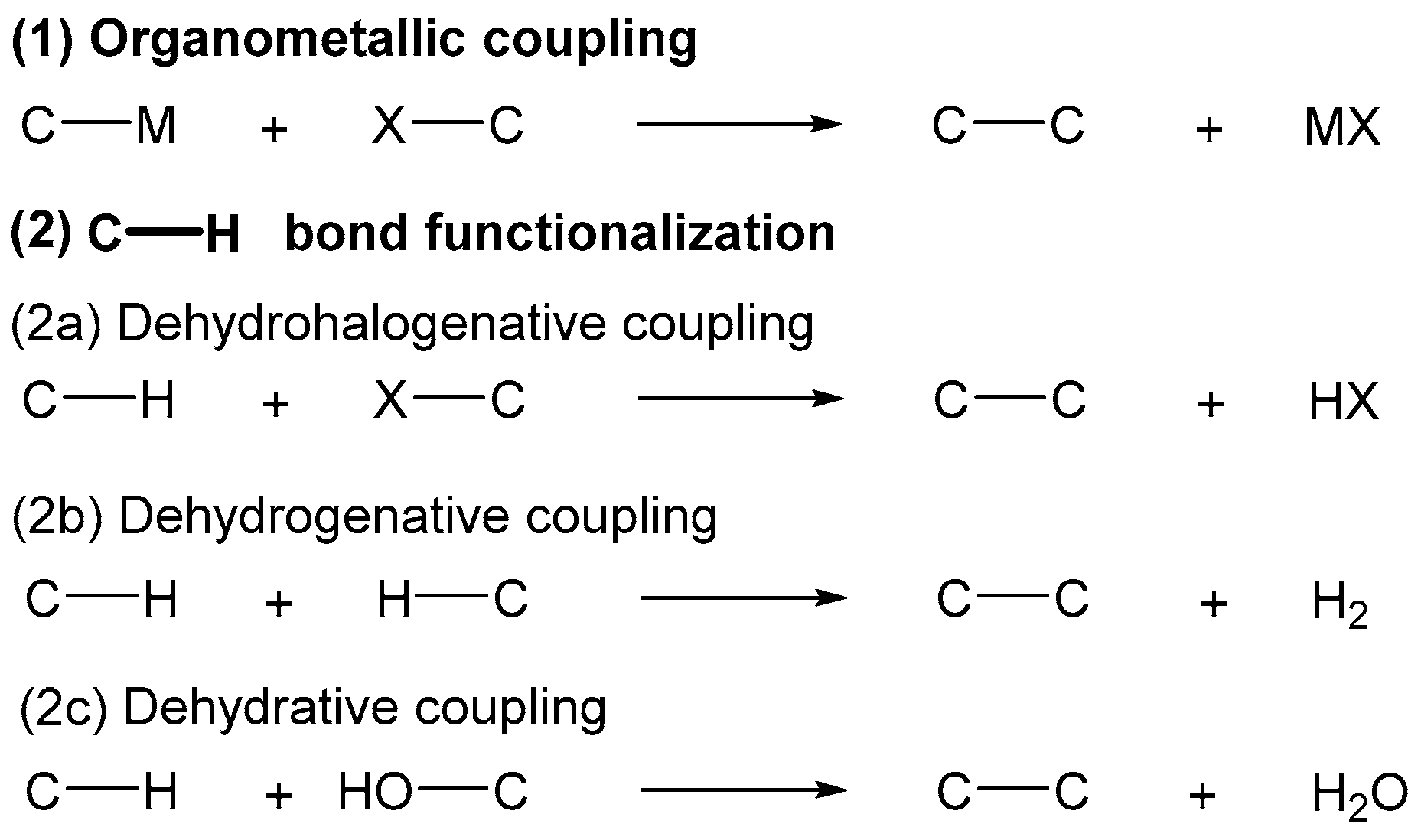
:1. Introduction
2. Phthalides
2.1. 3-Substituted Phthalides
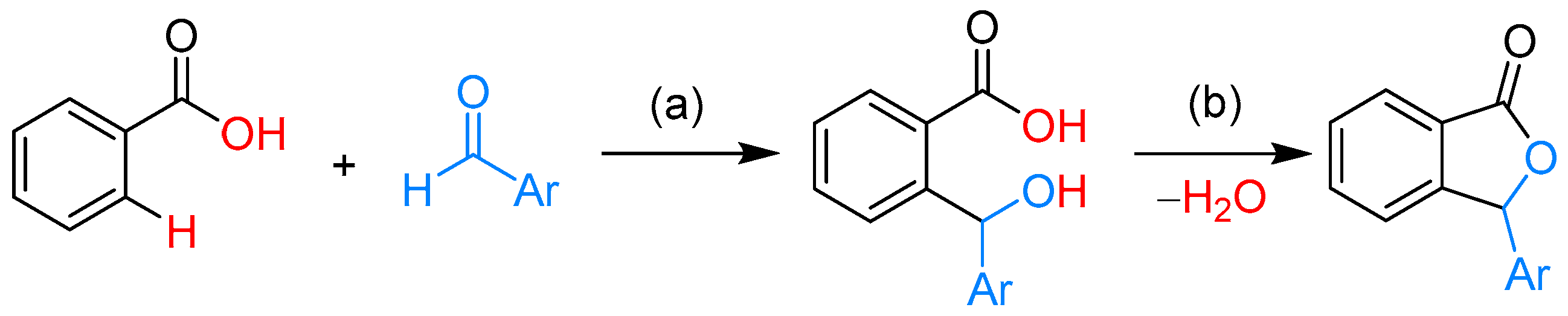
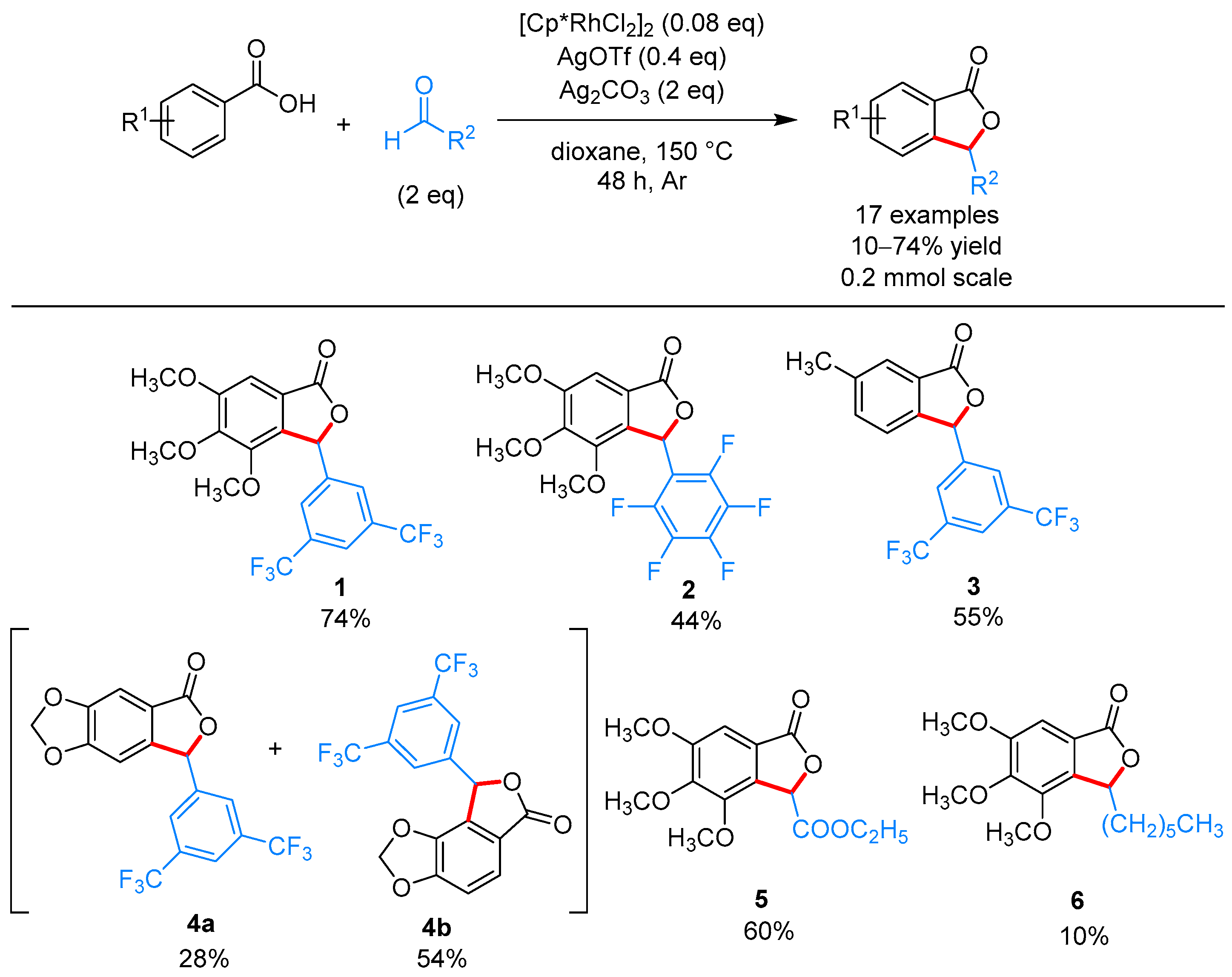
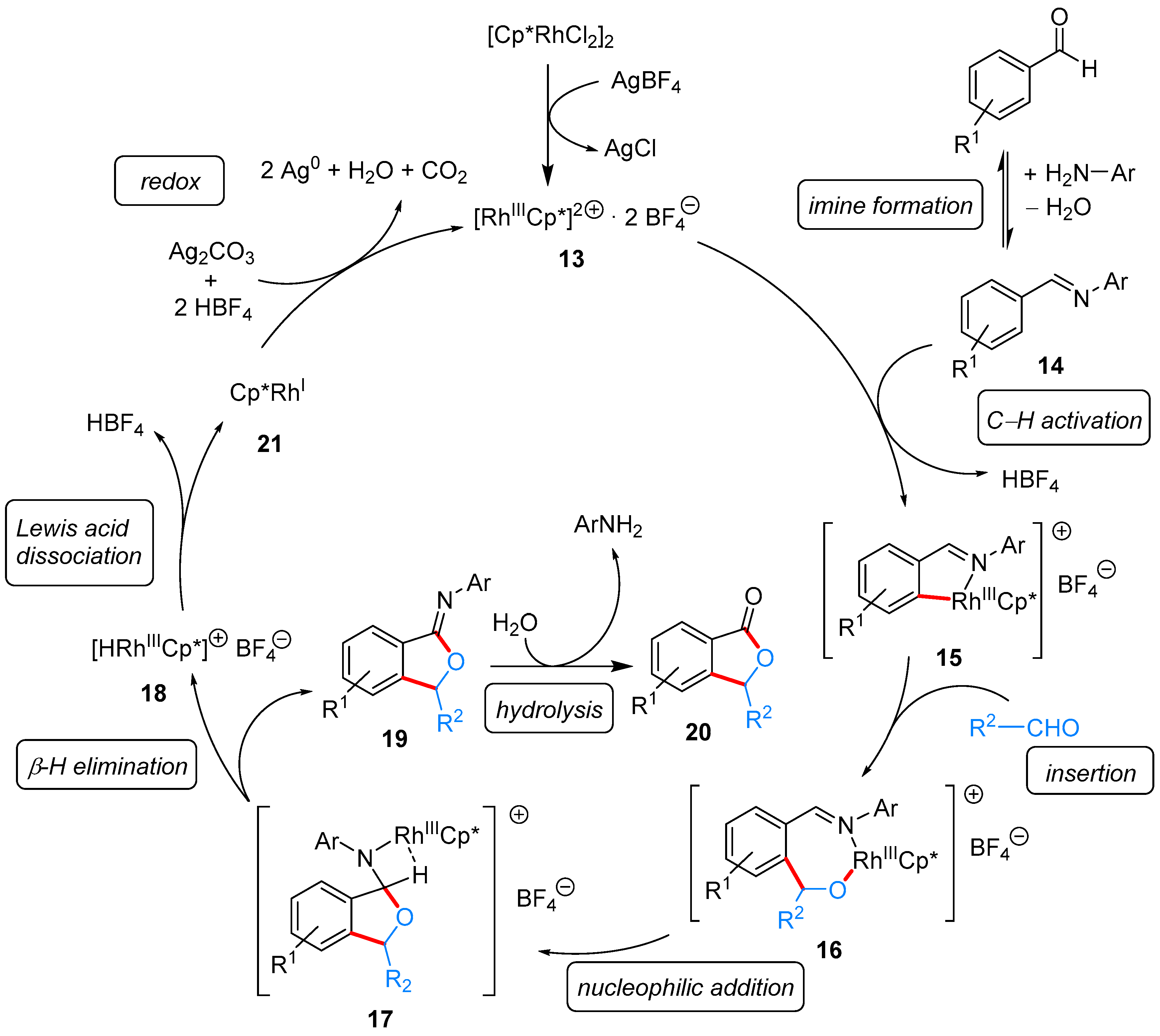
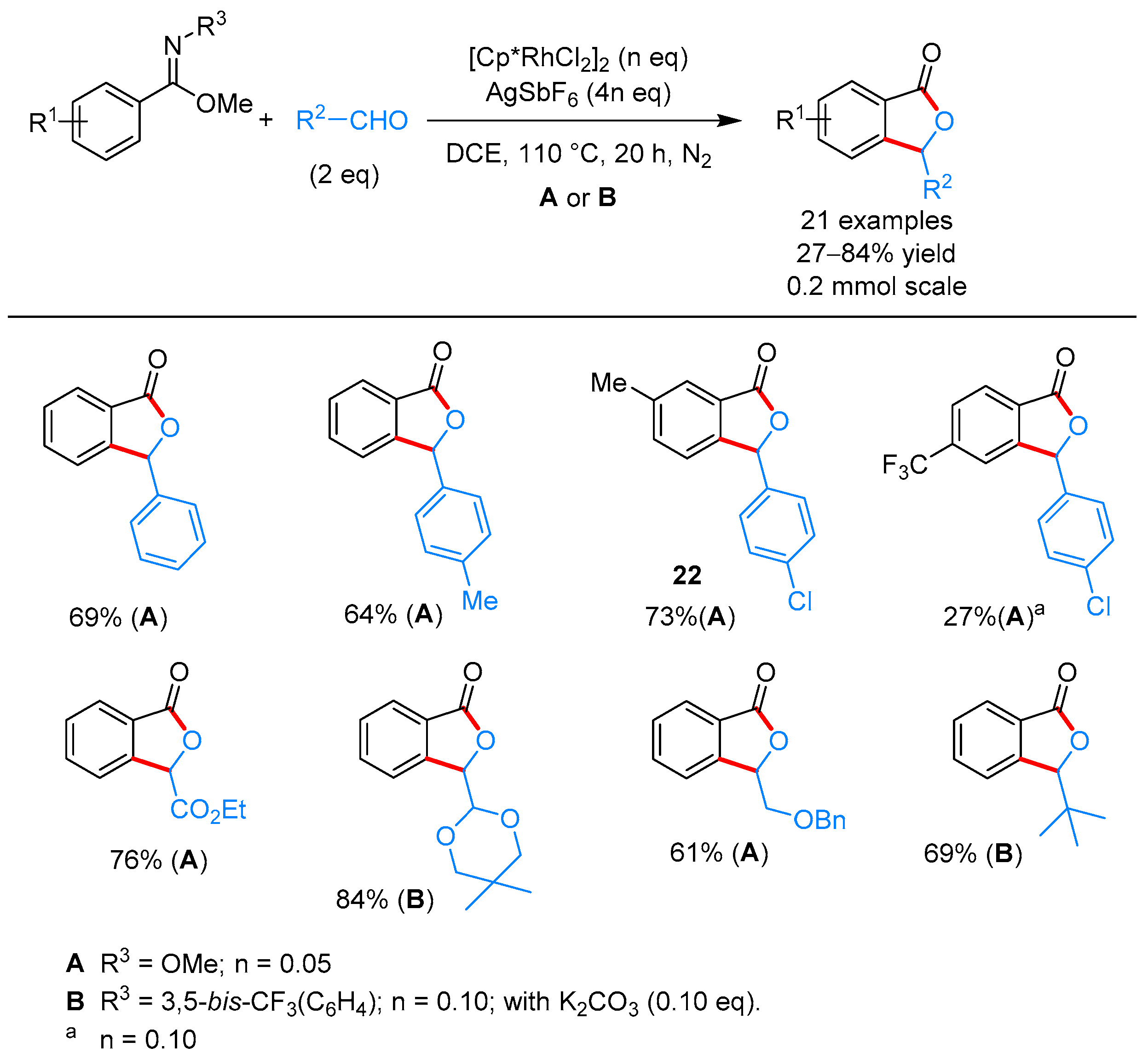
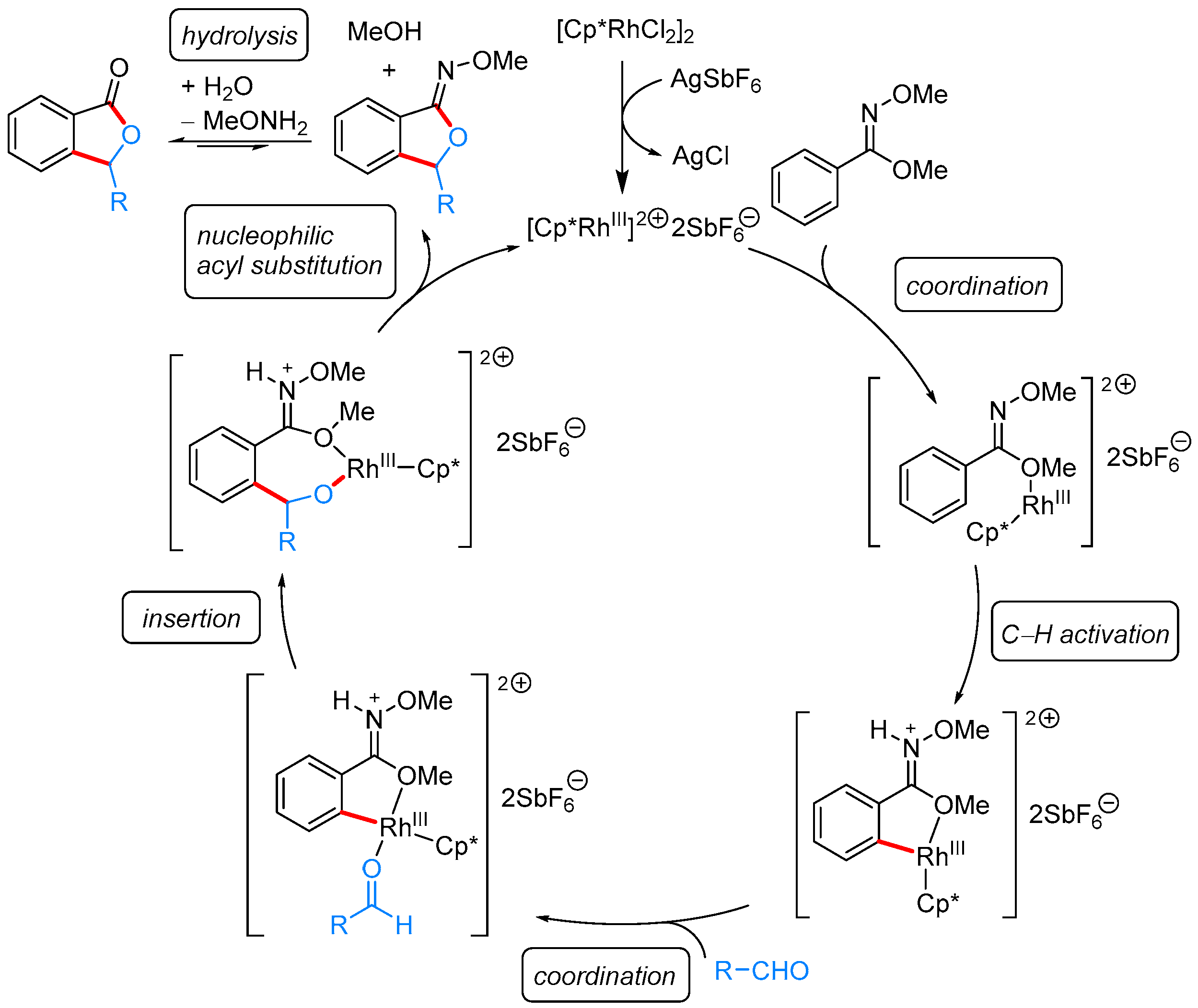
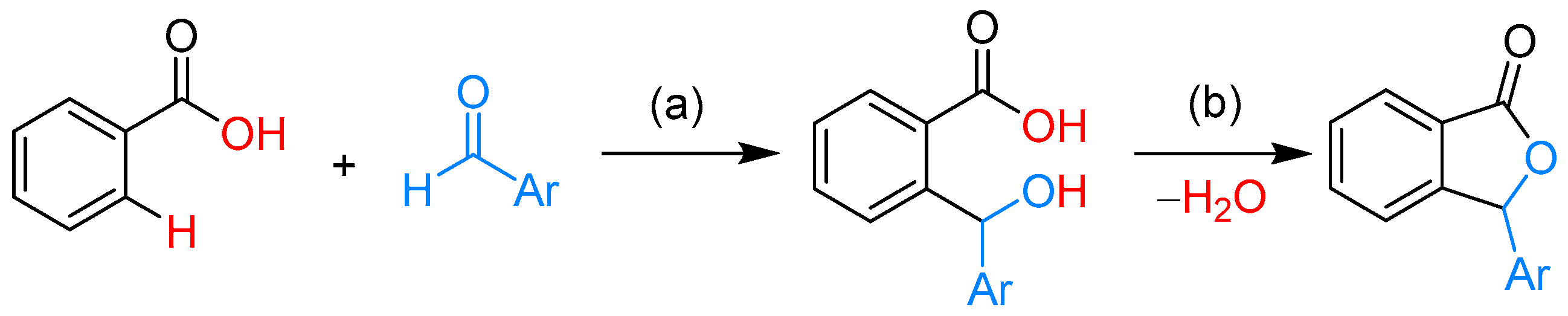
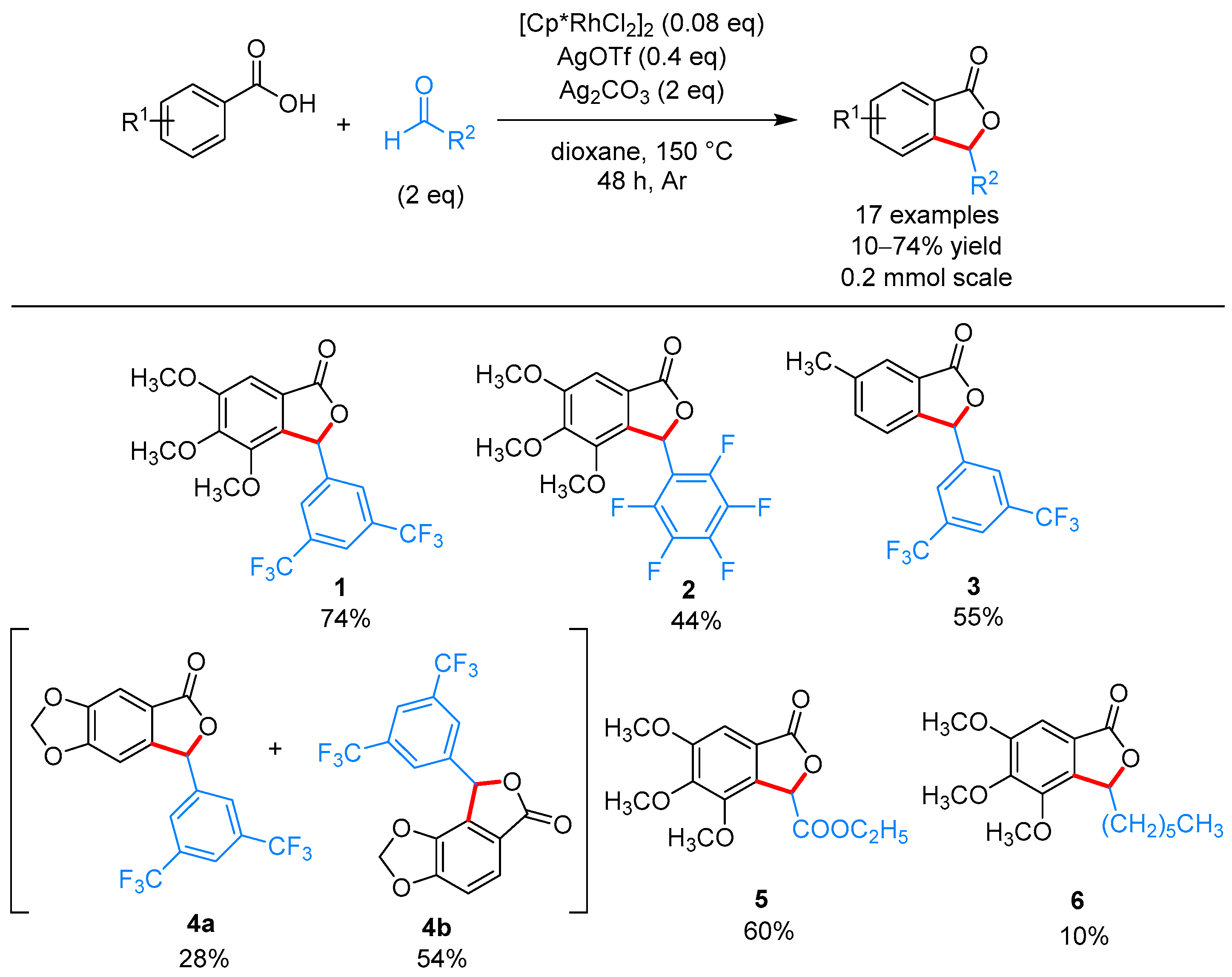
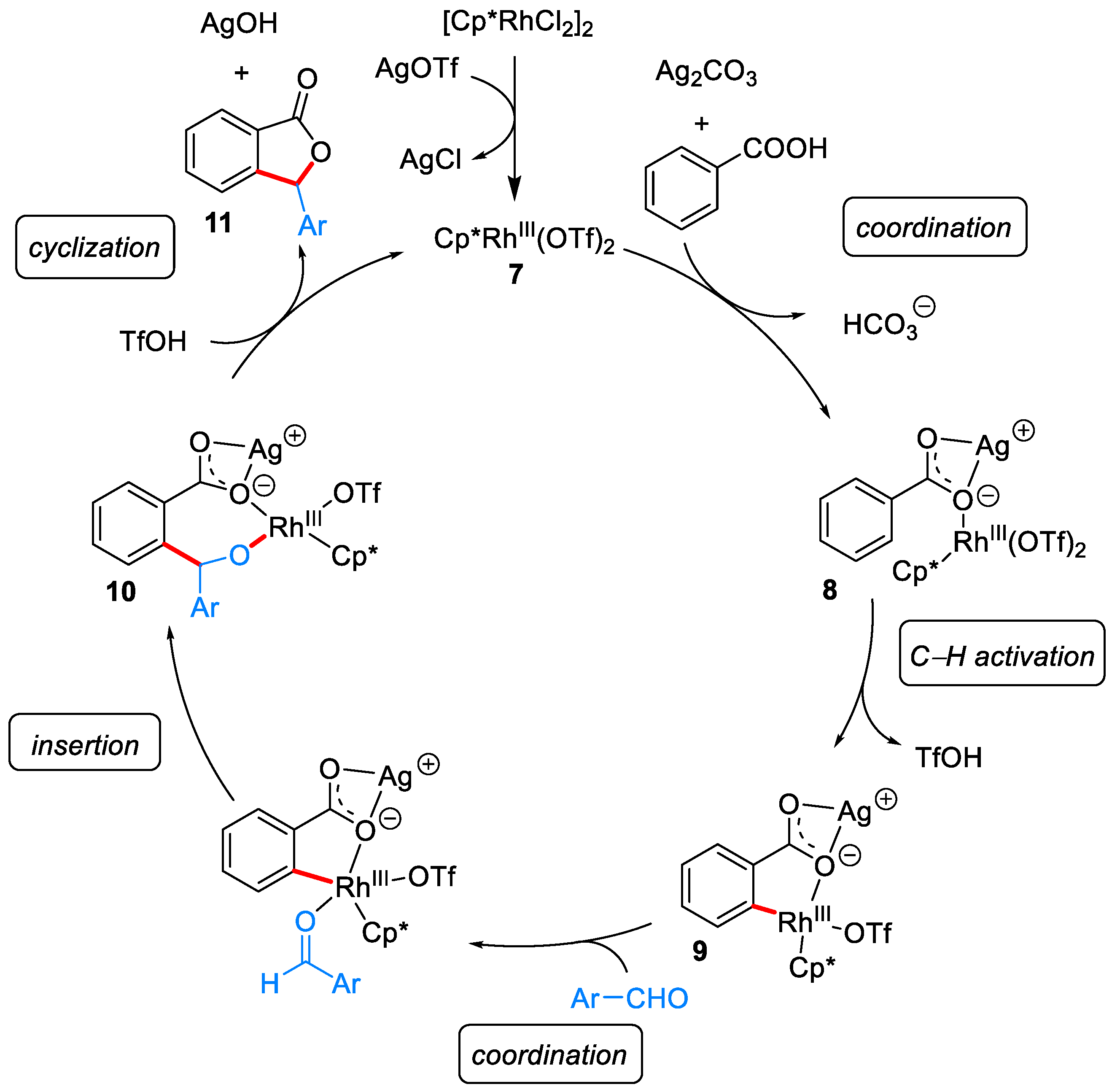
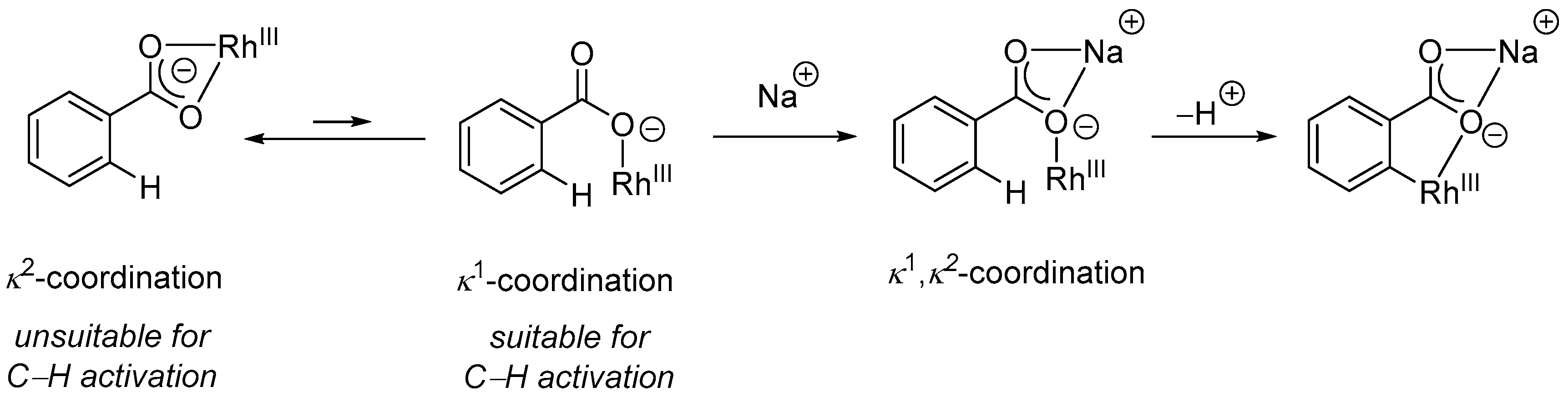
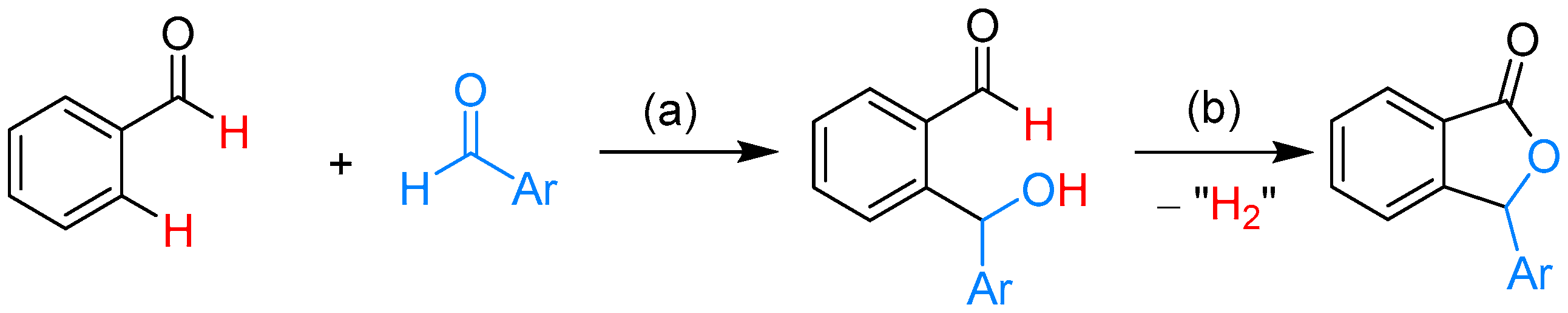
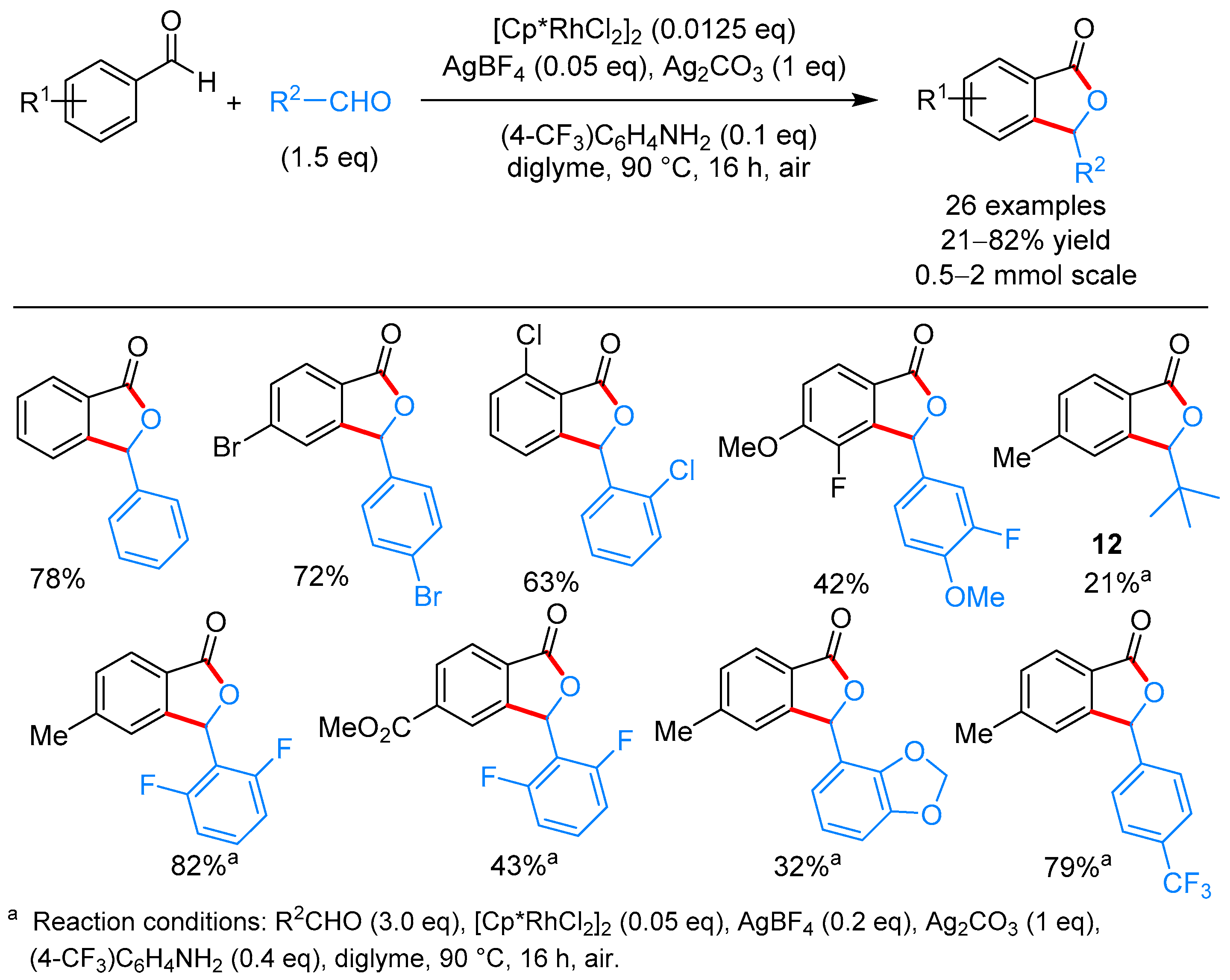
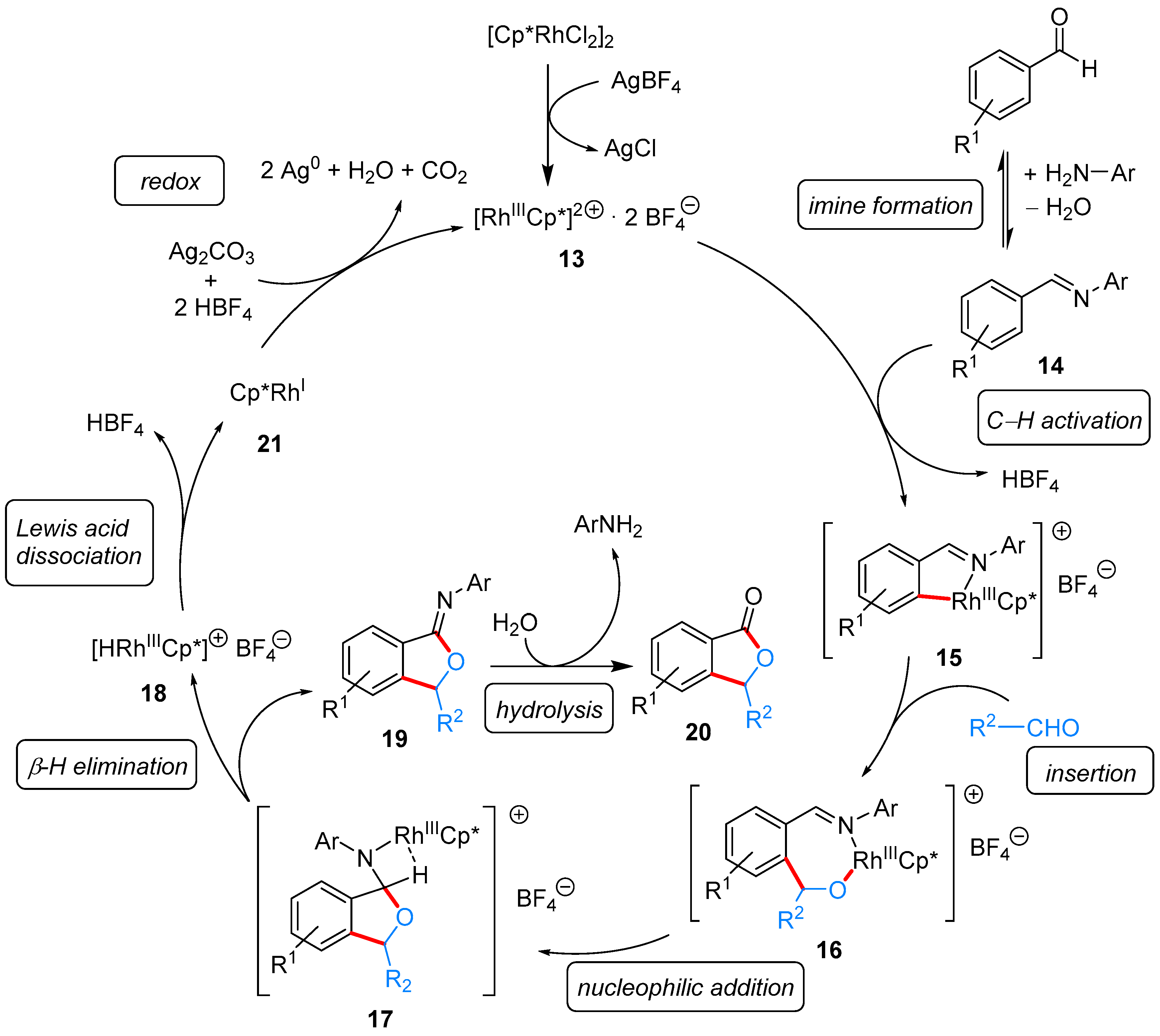
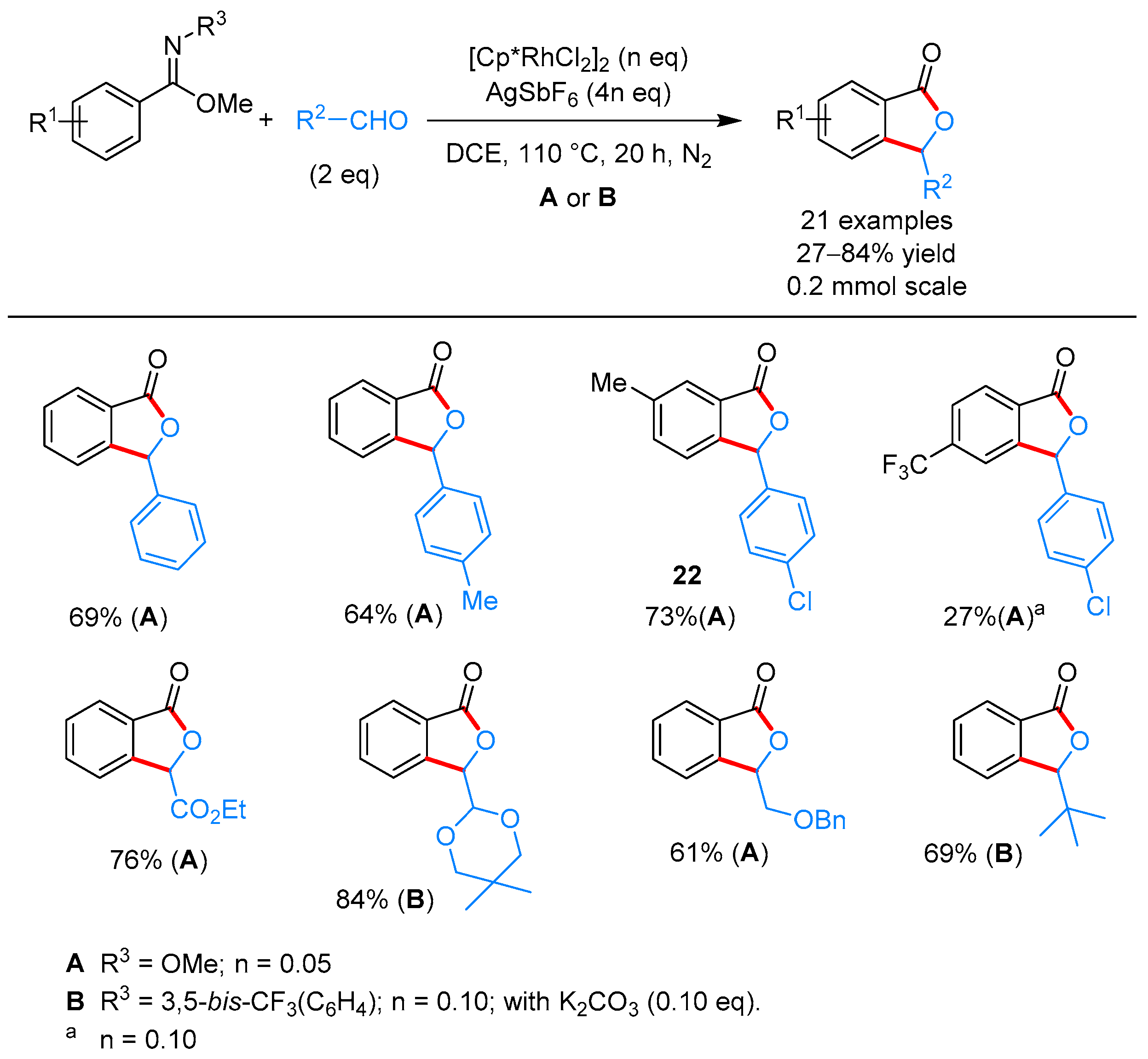
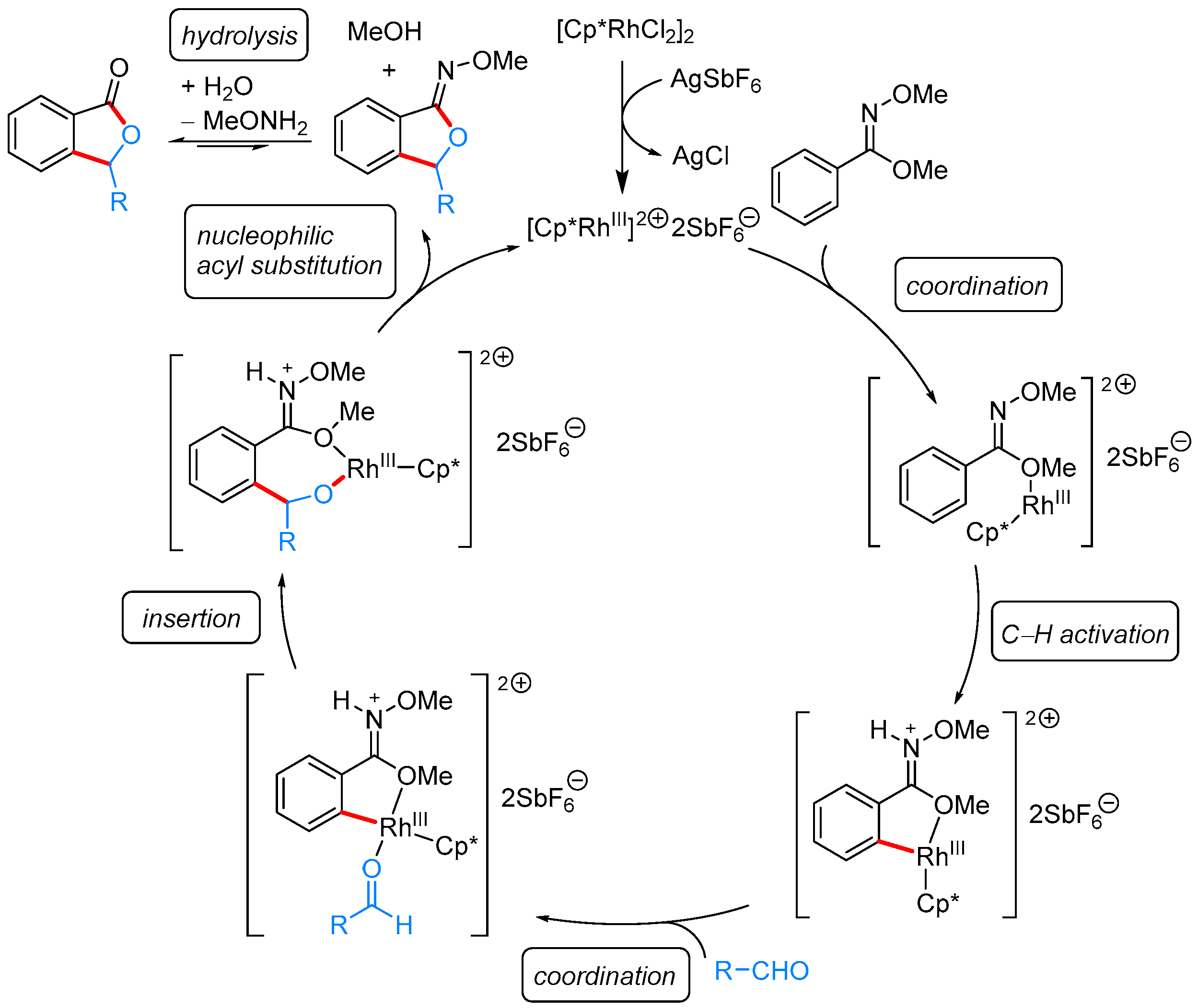
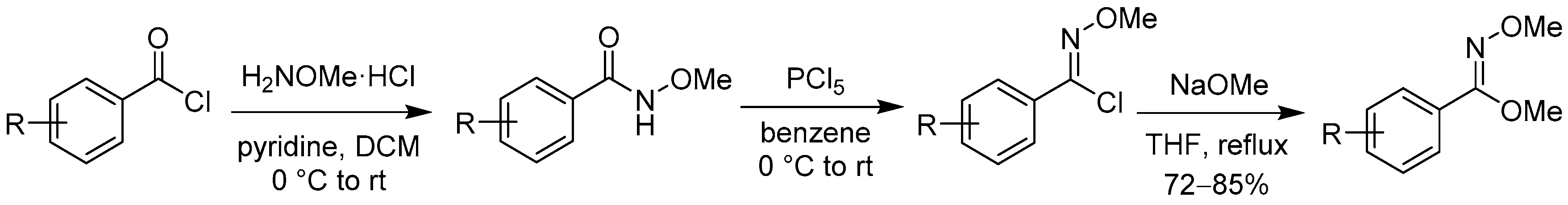
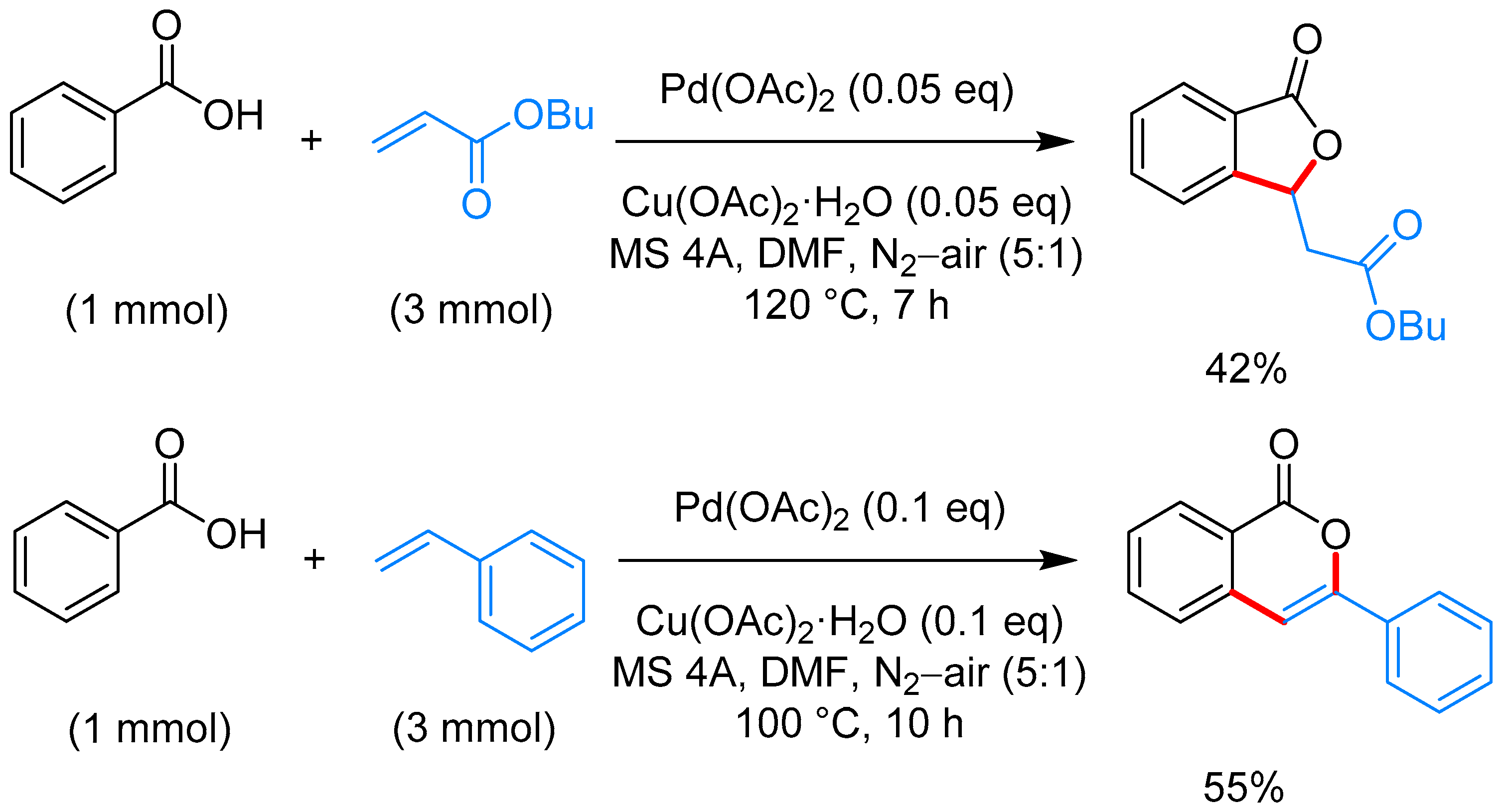
2.1.1. 3-Aryl Phthalides
2.1.2. 3-Alkyl Phthalides
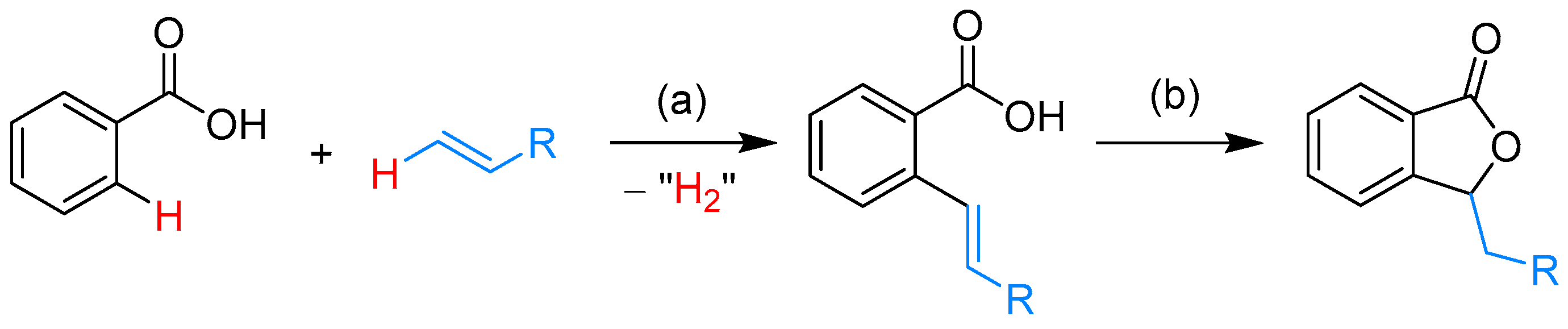
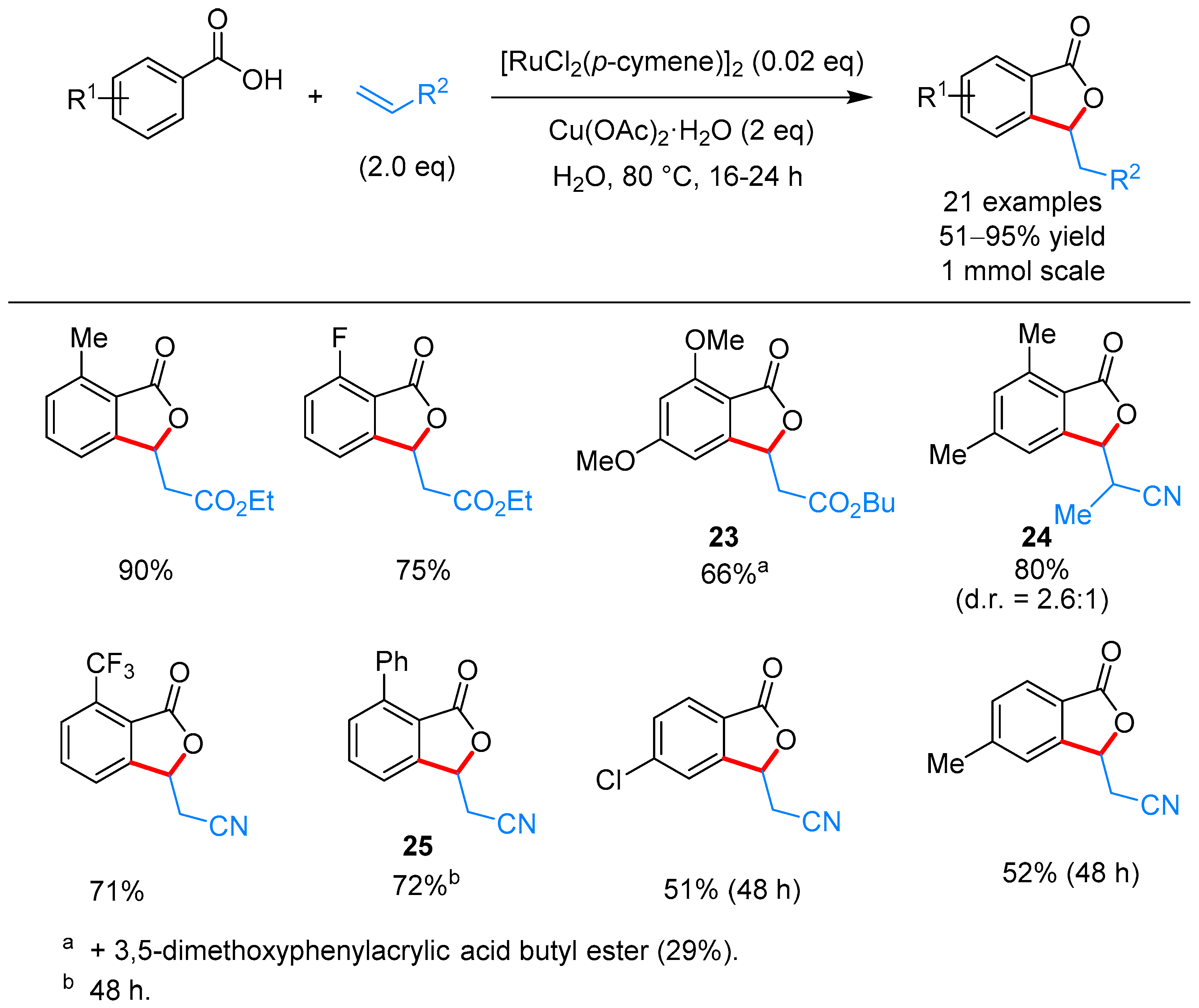
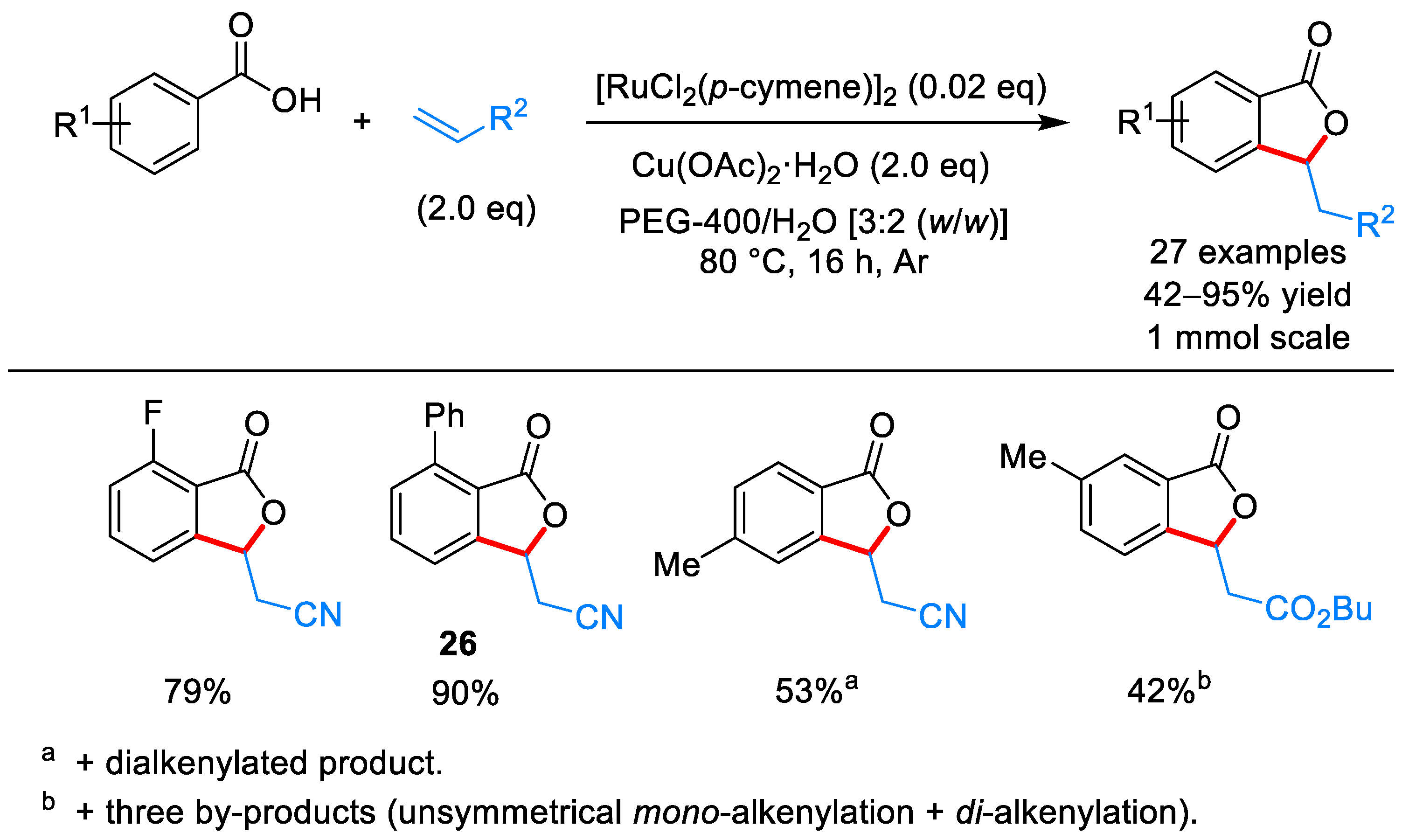
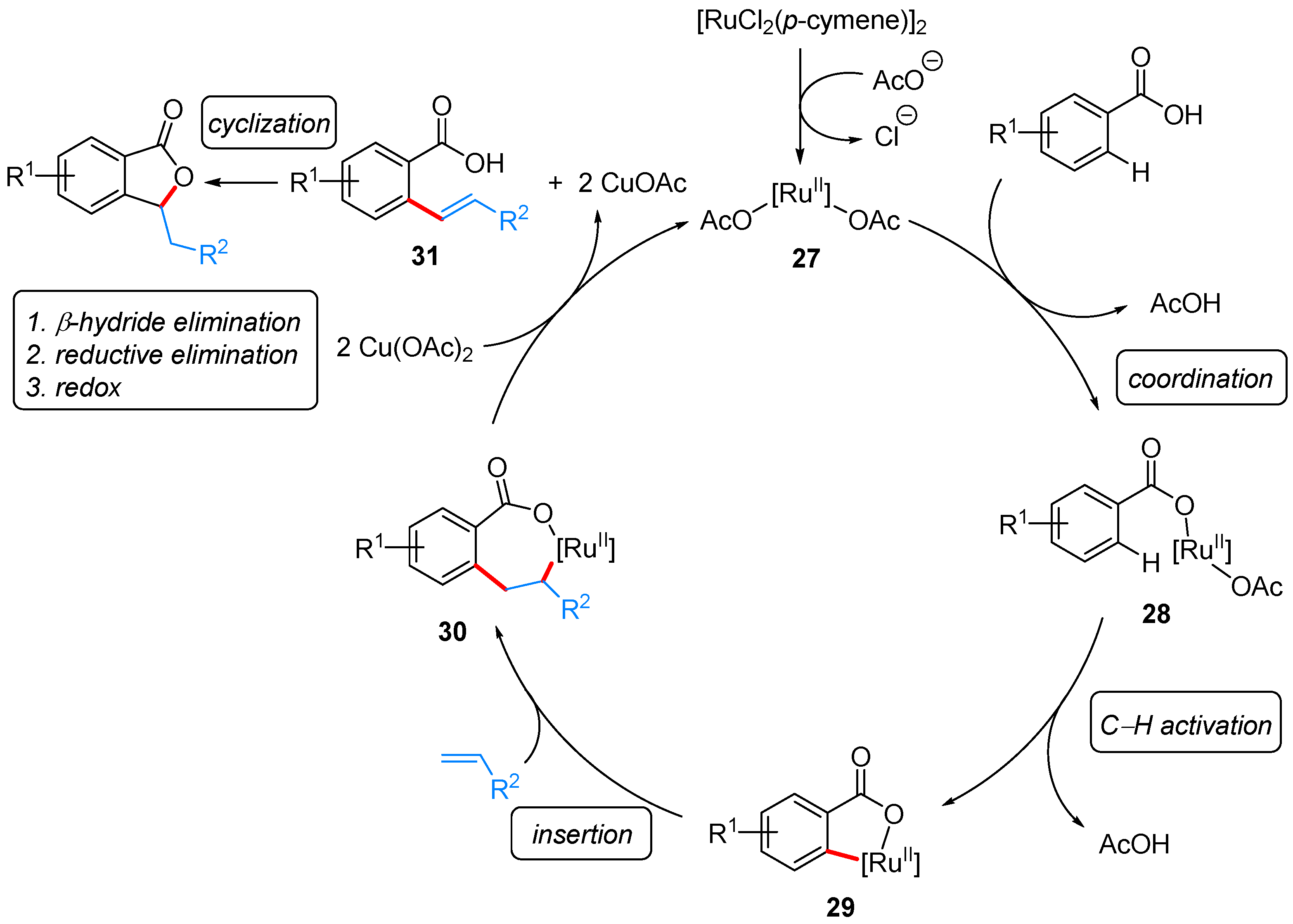
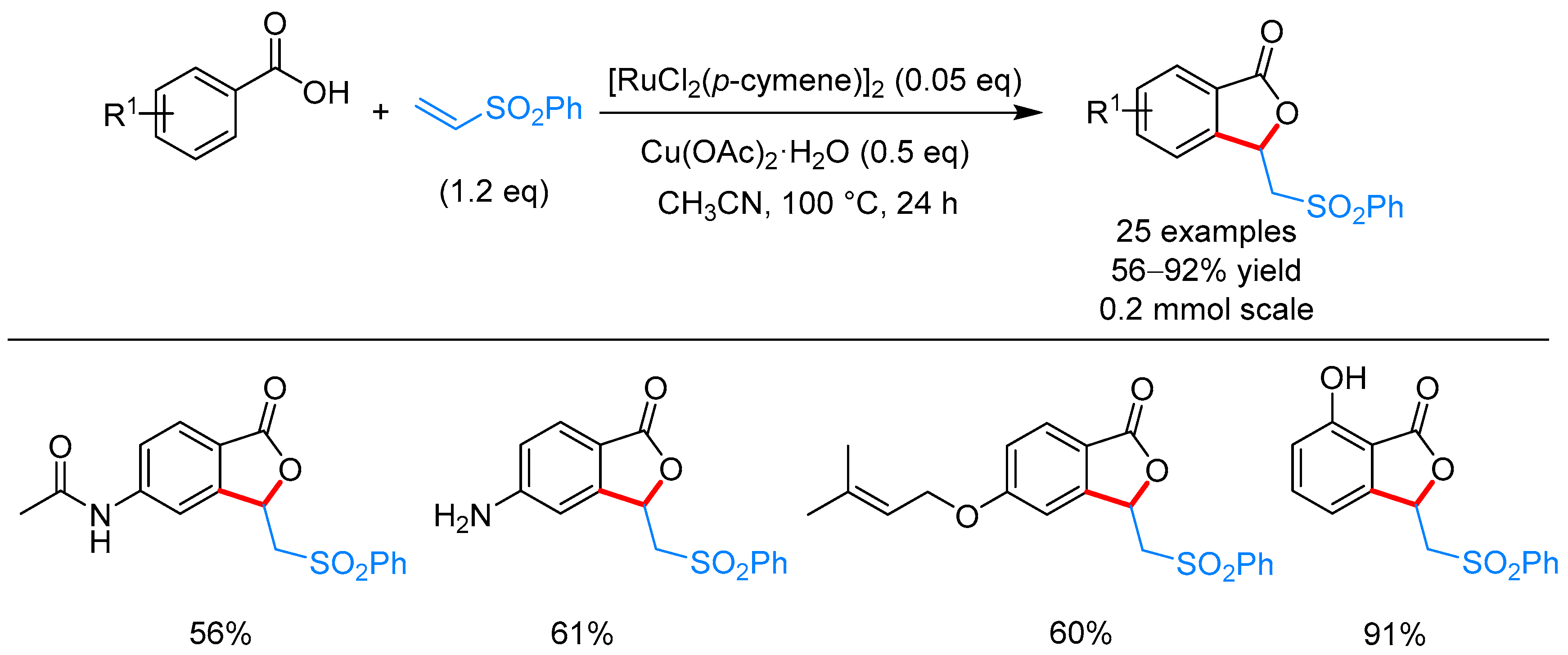
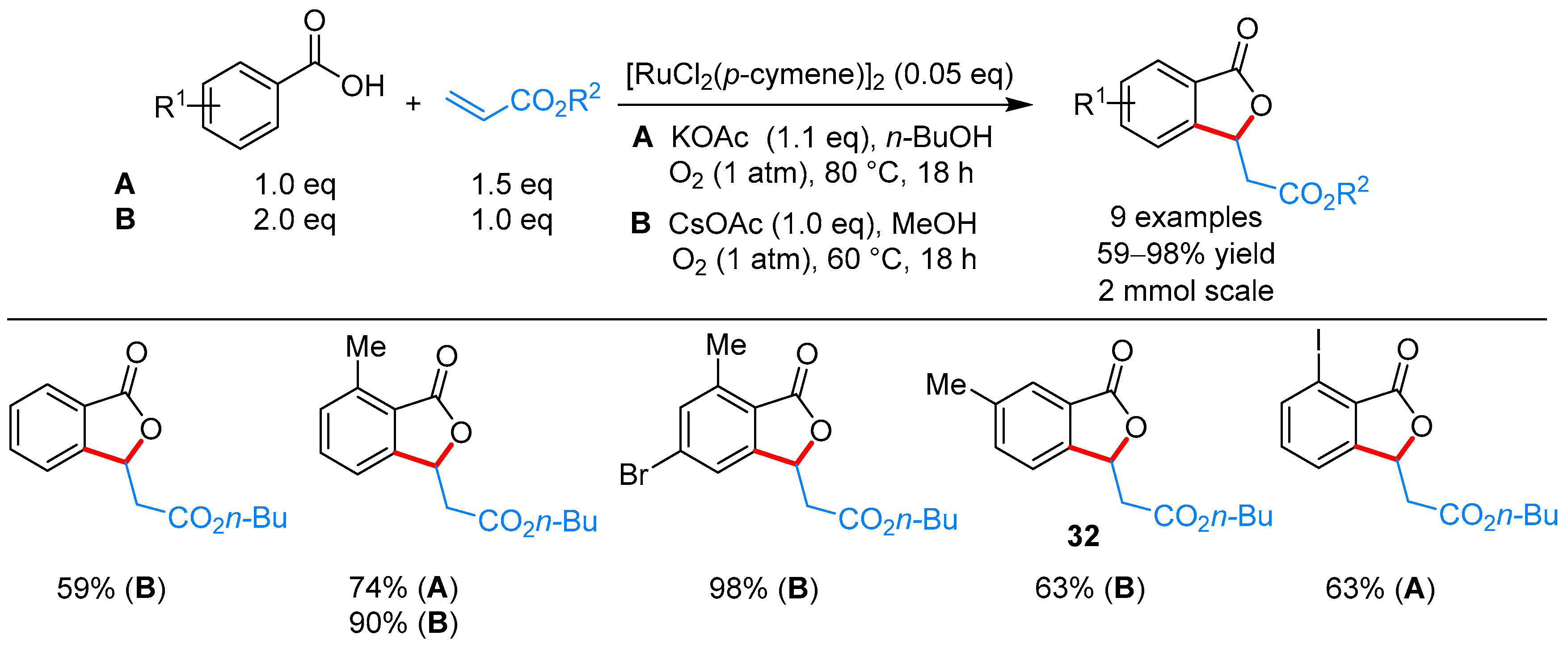
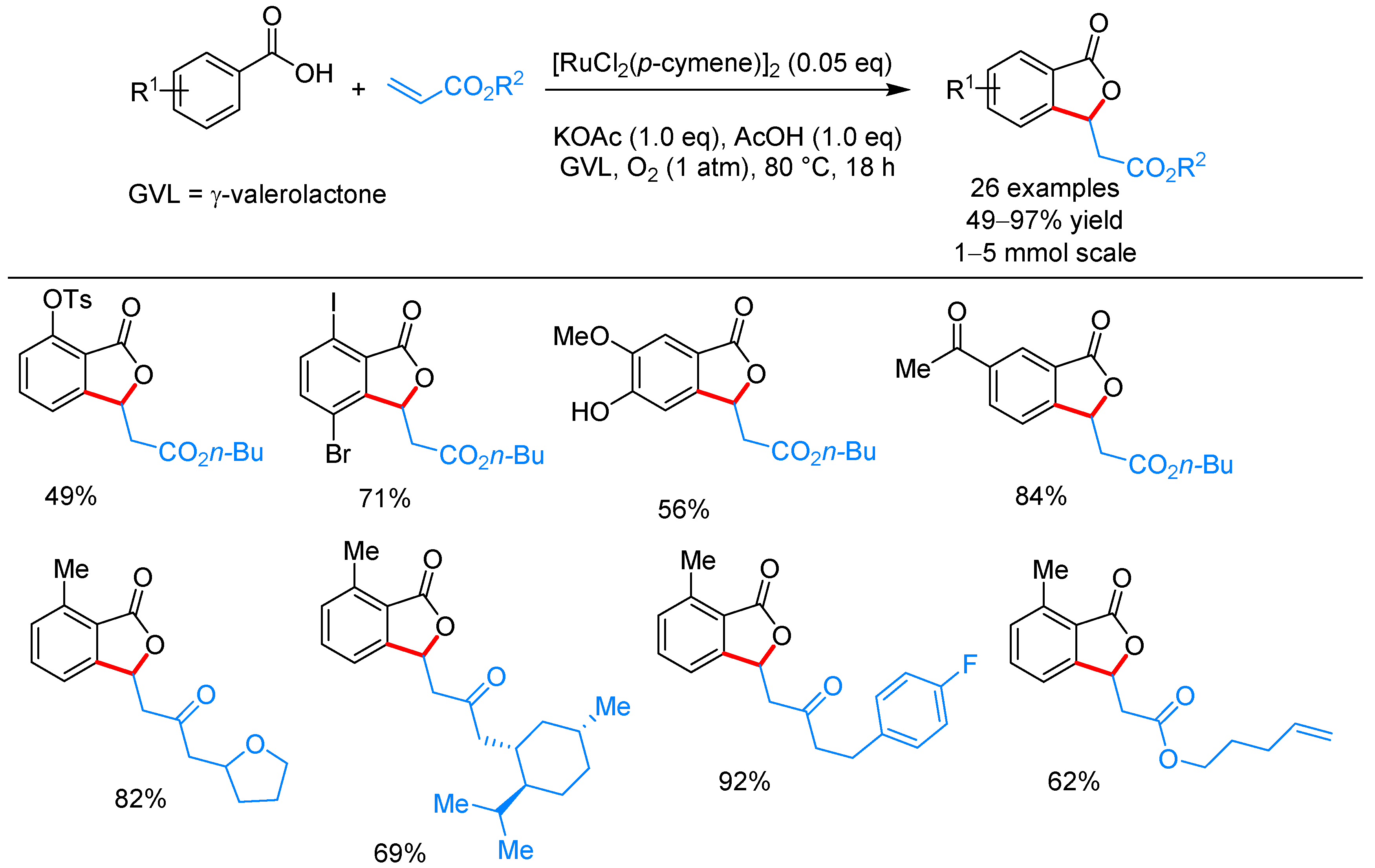
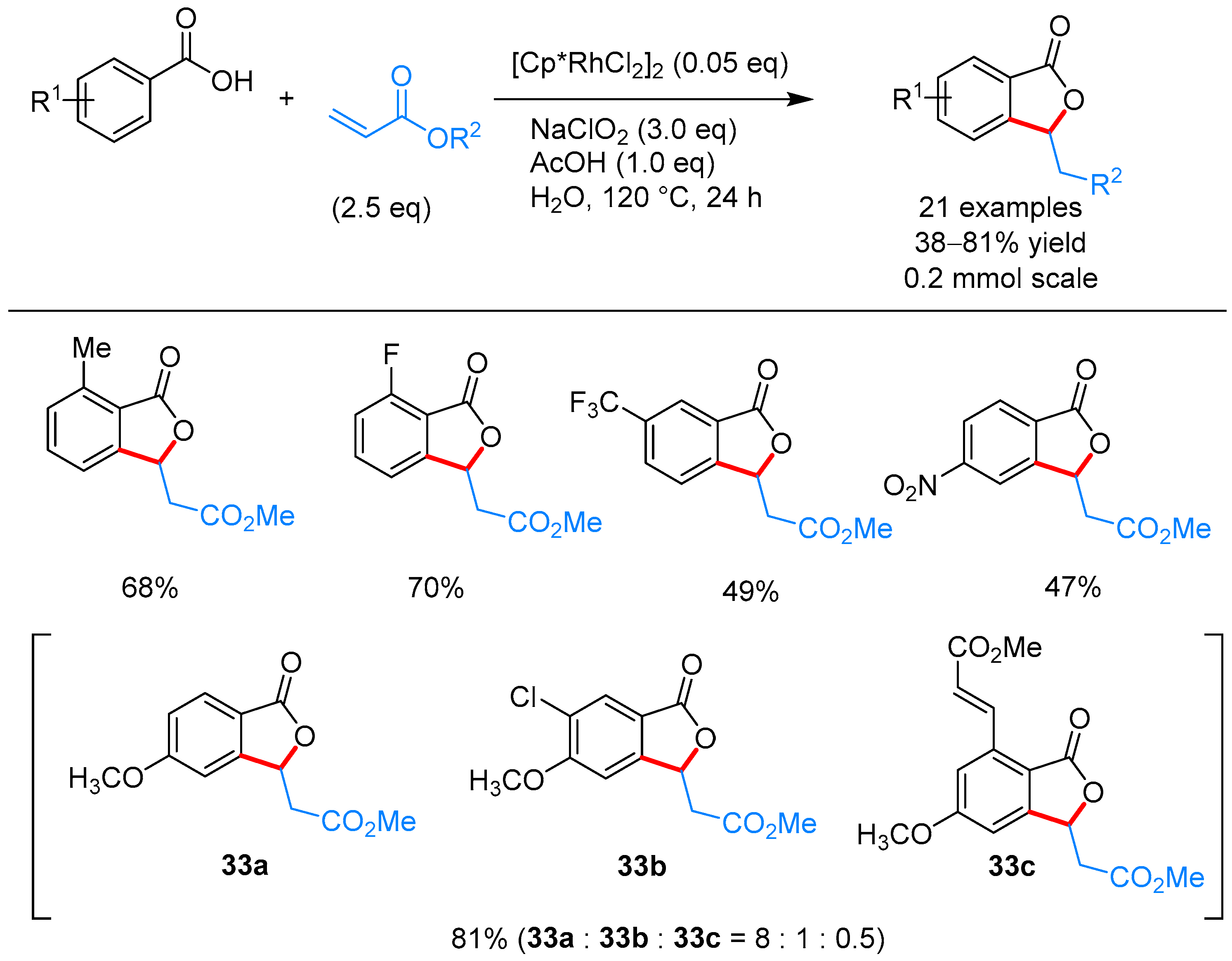
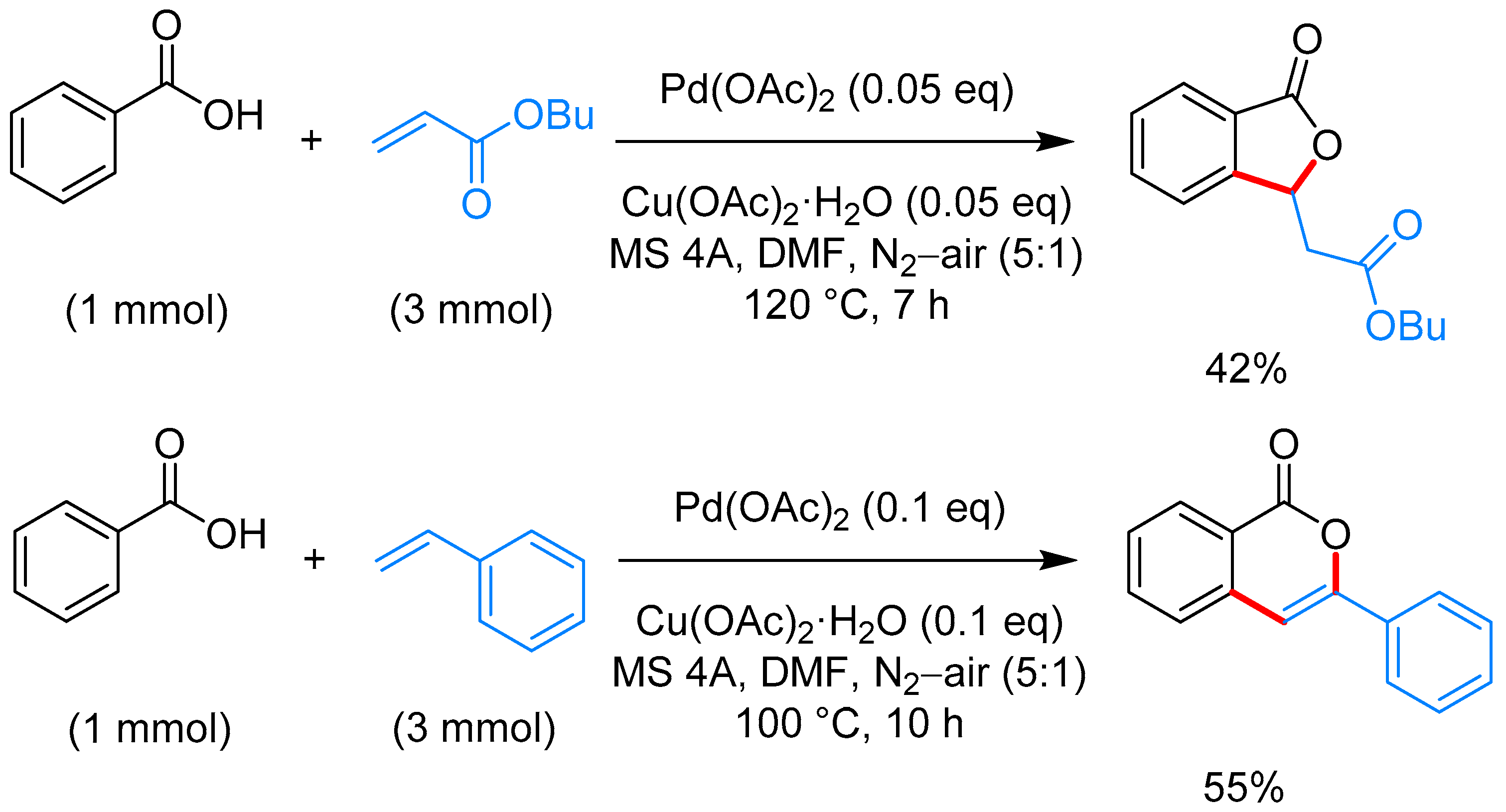
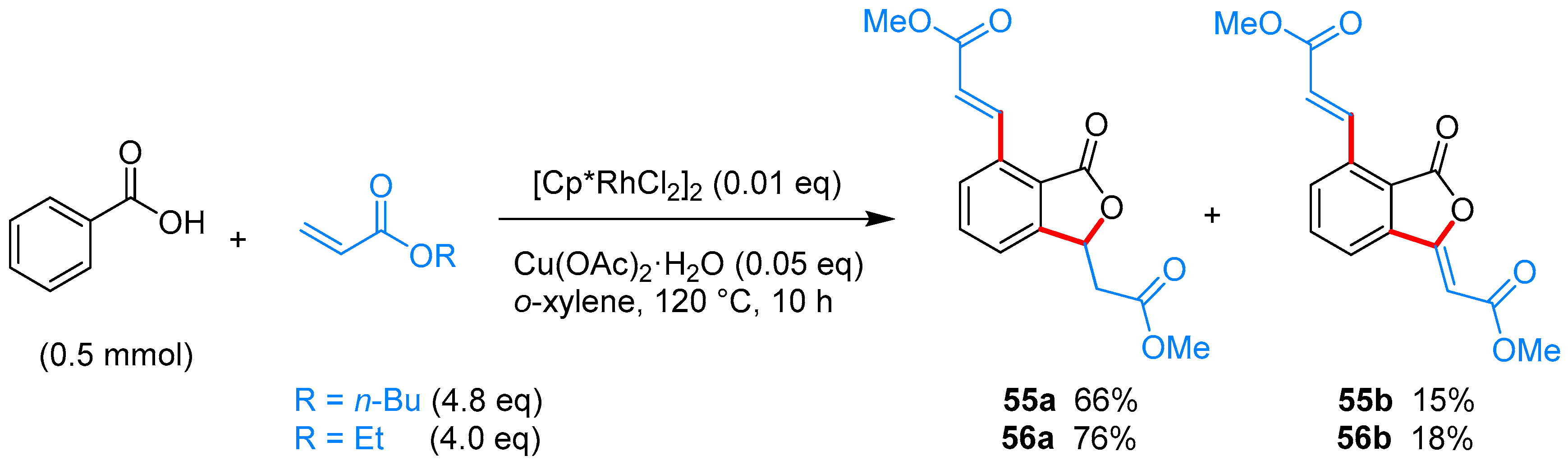
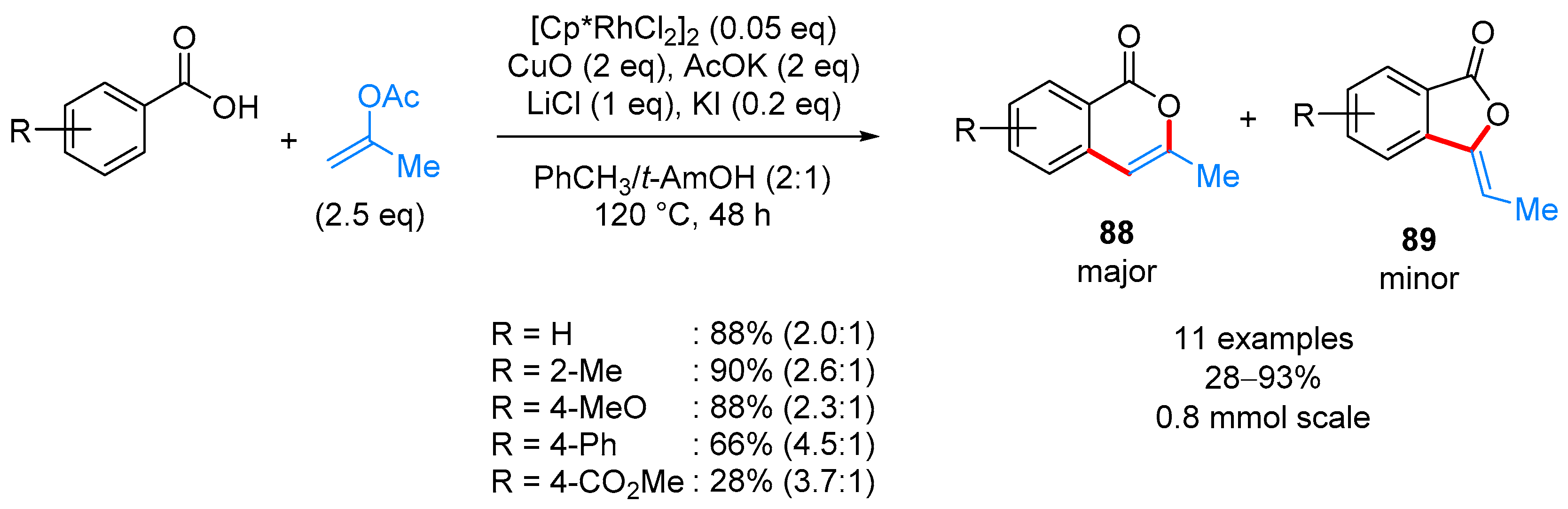
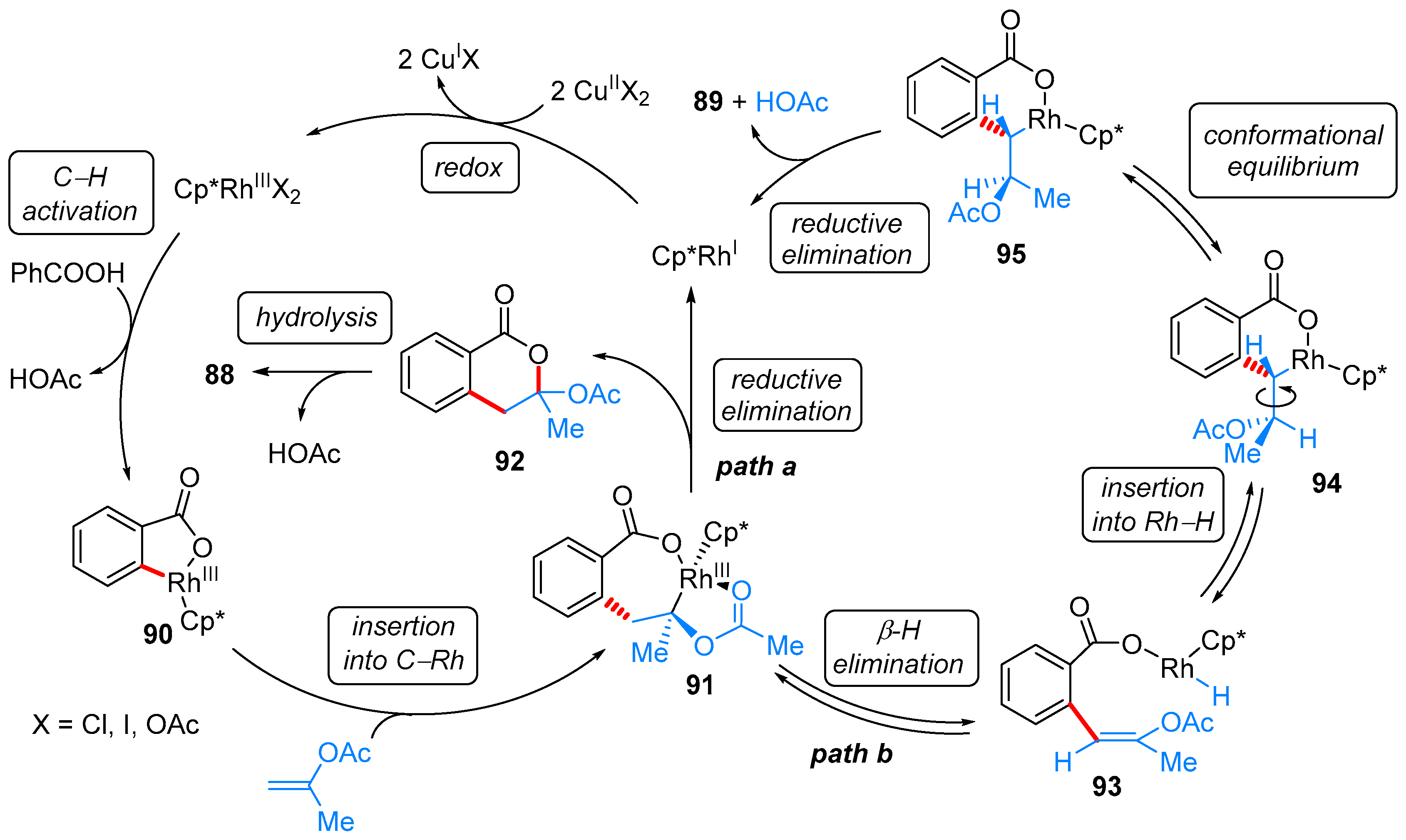
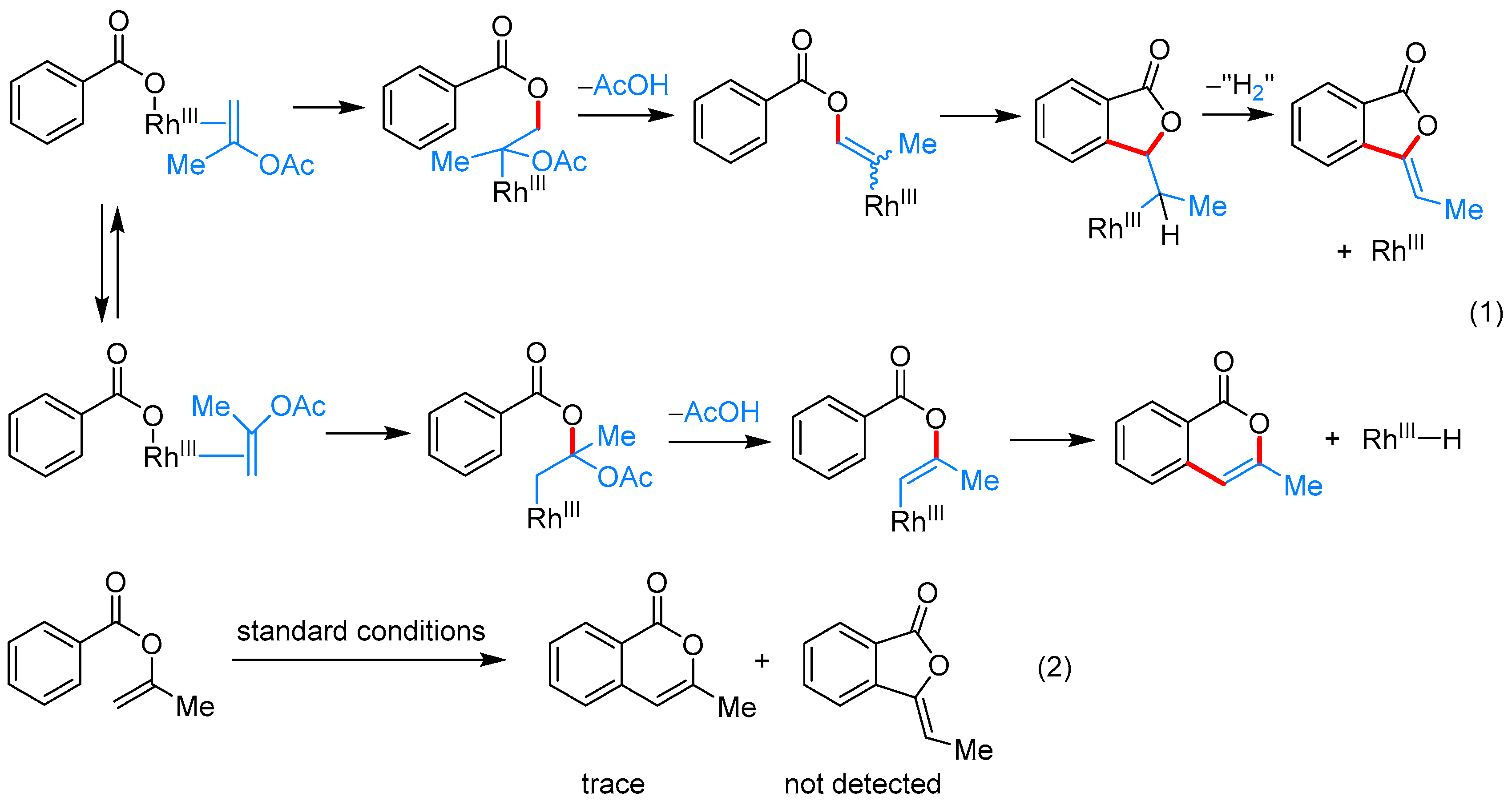
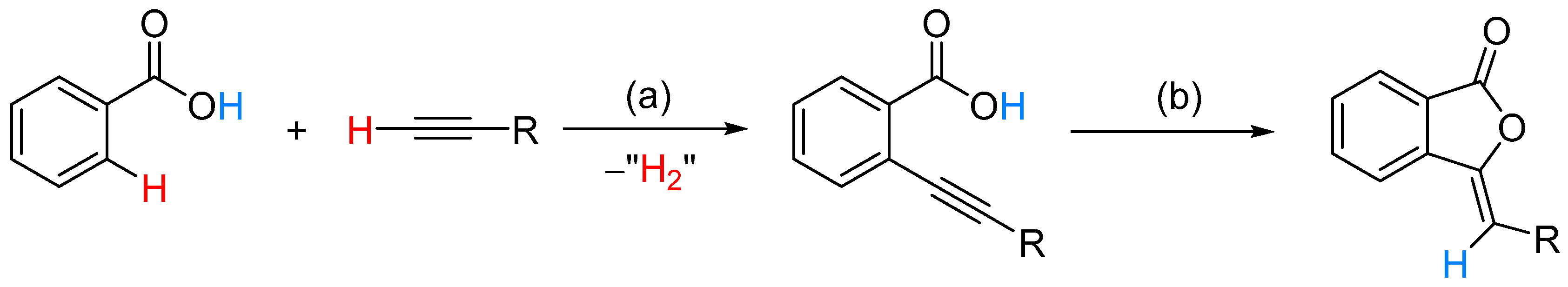
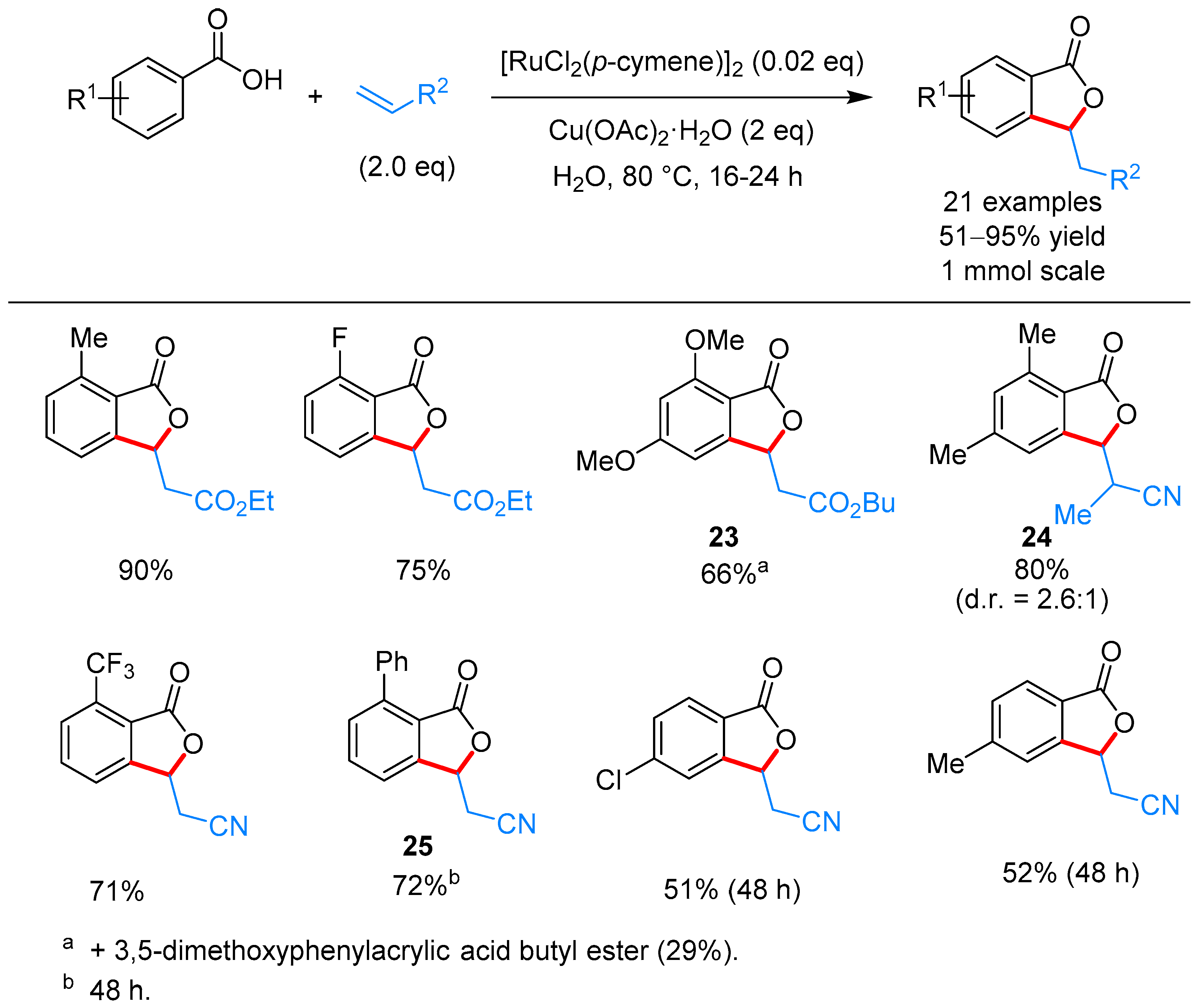
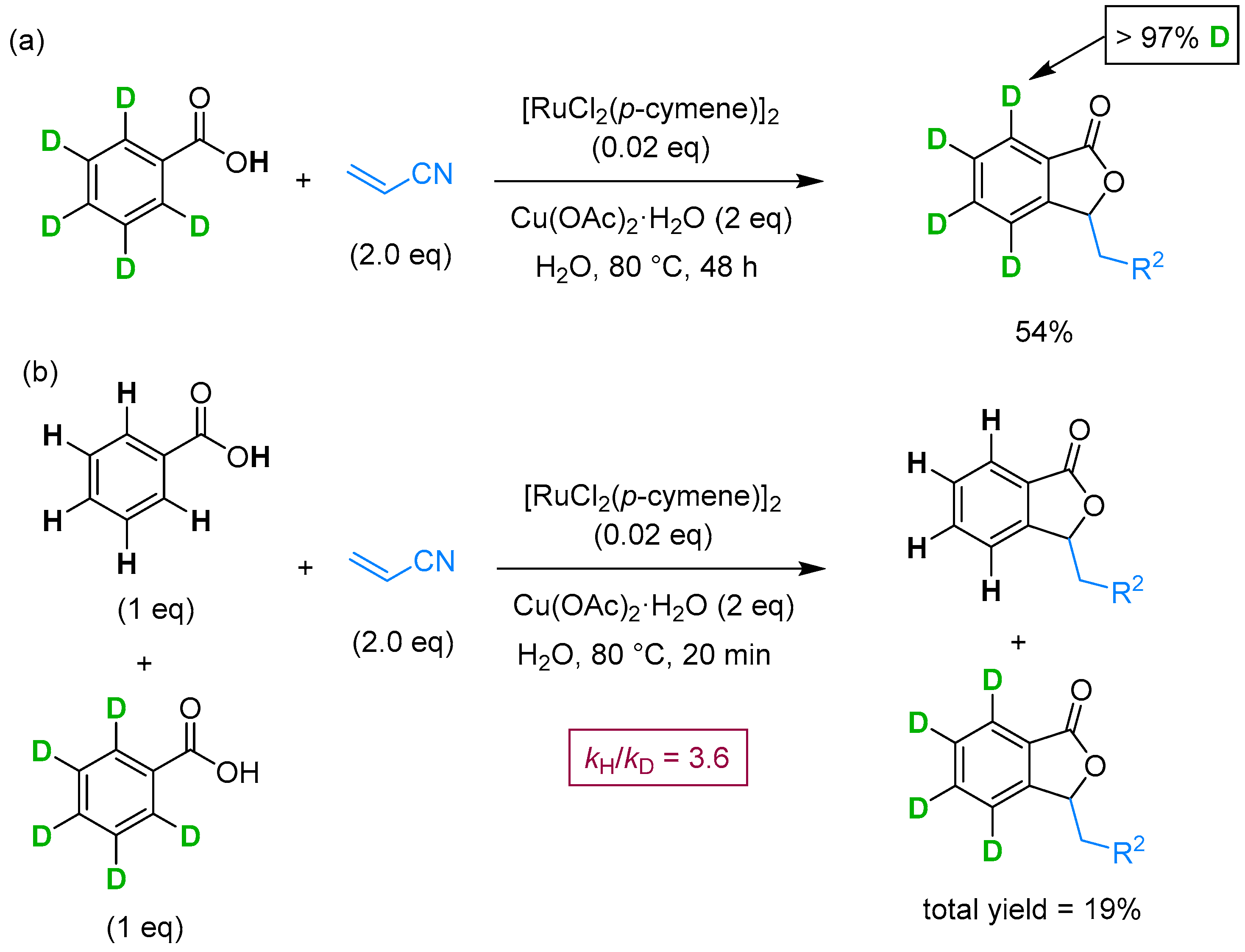
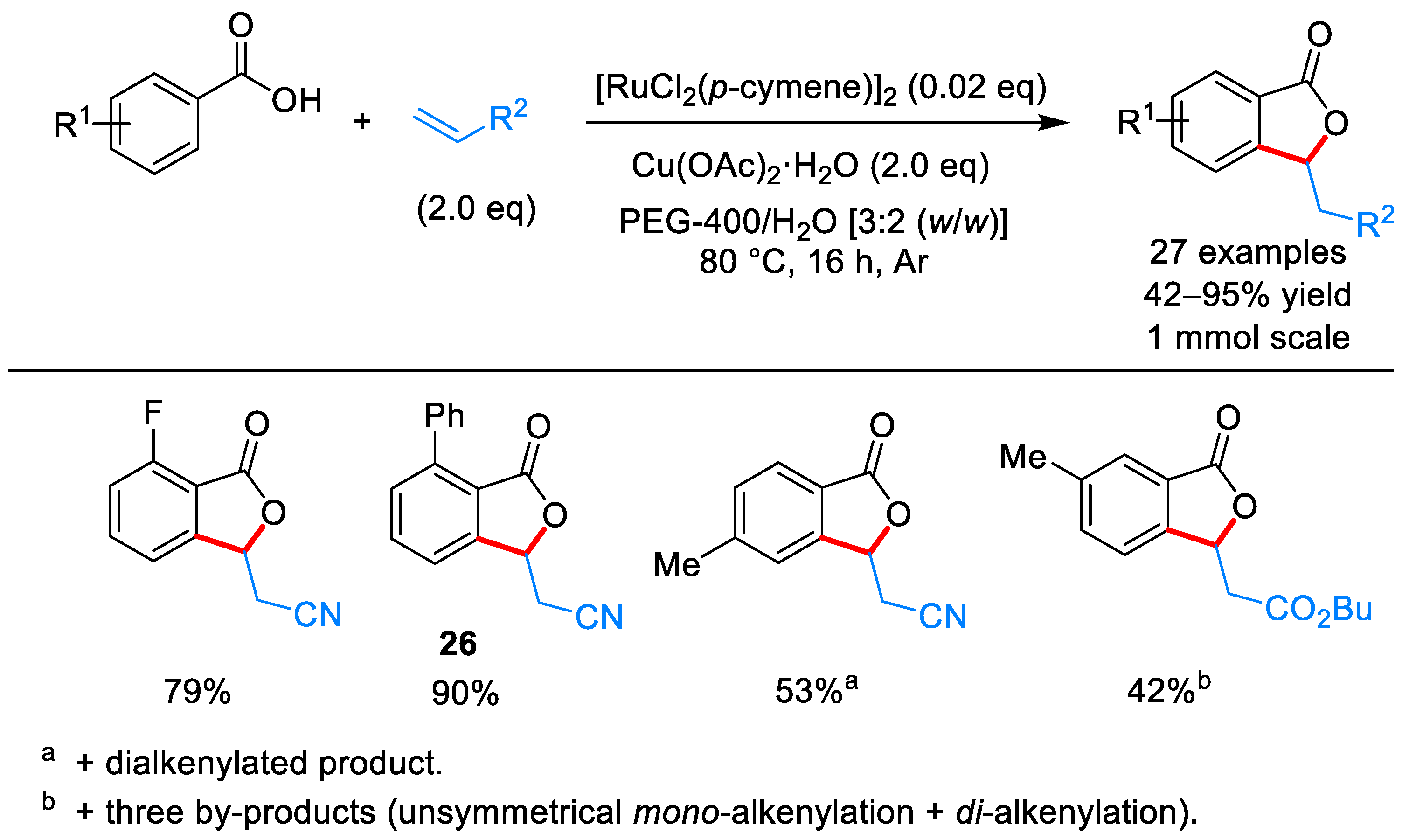
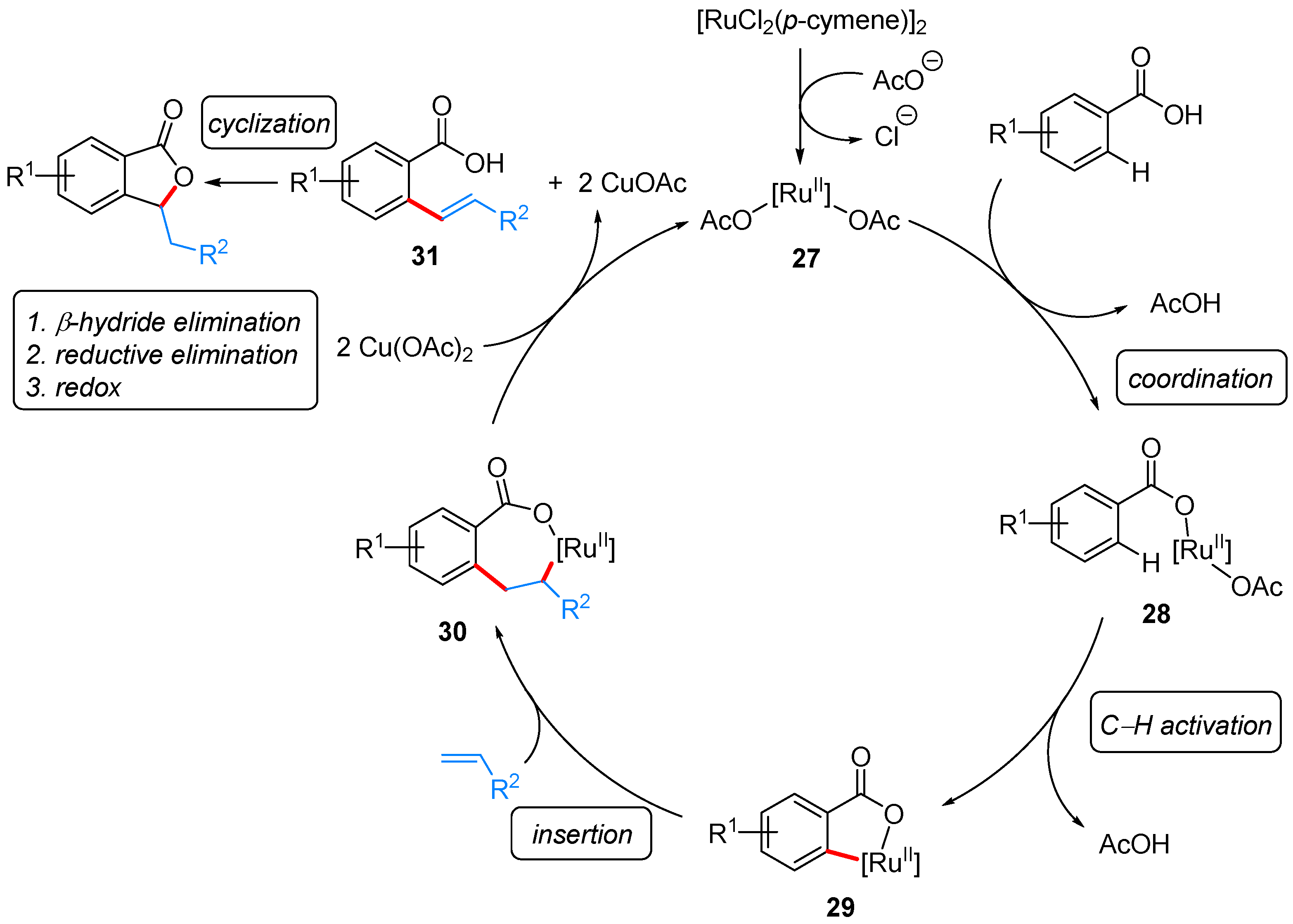
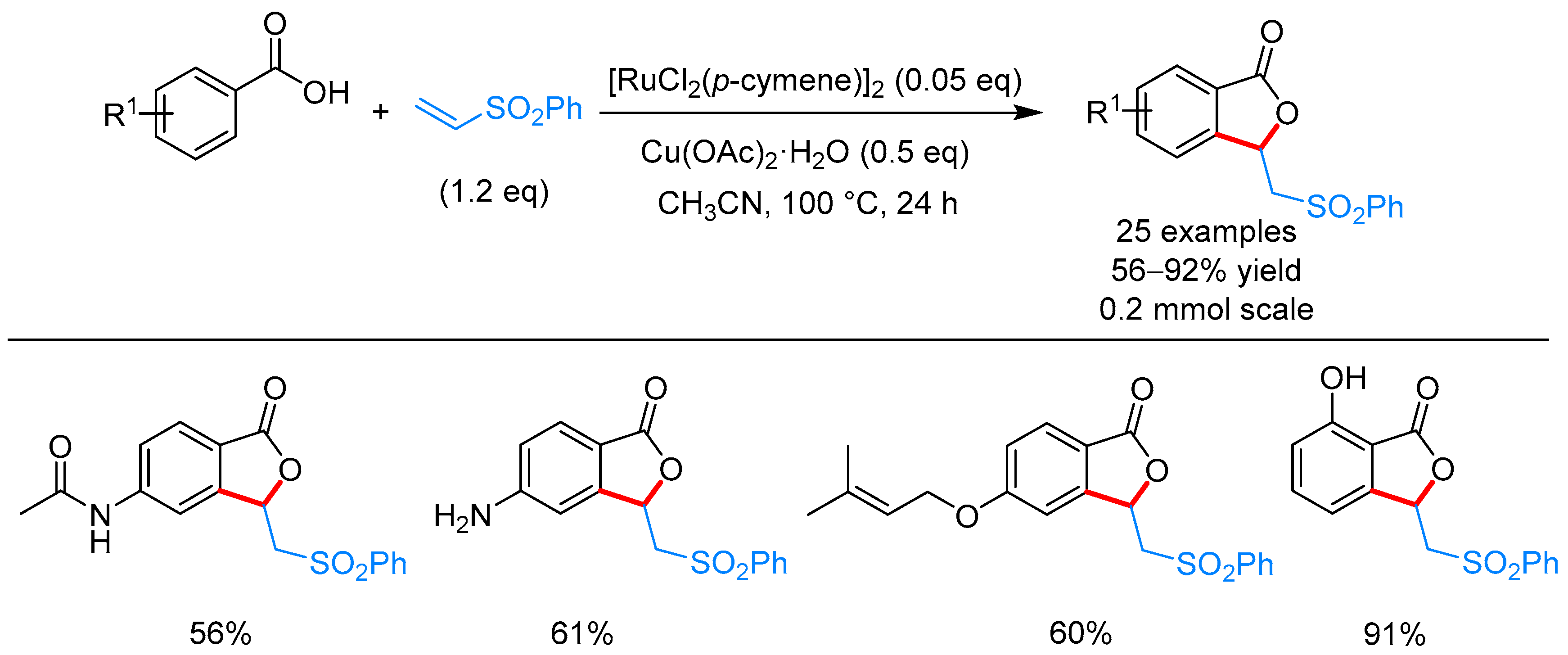
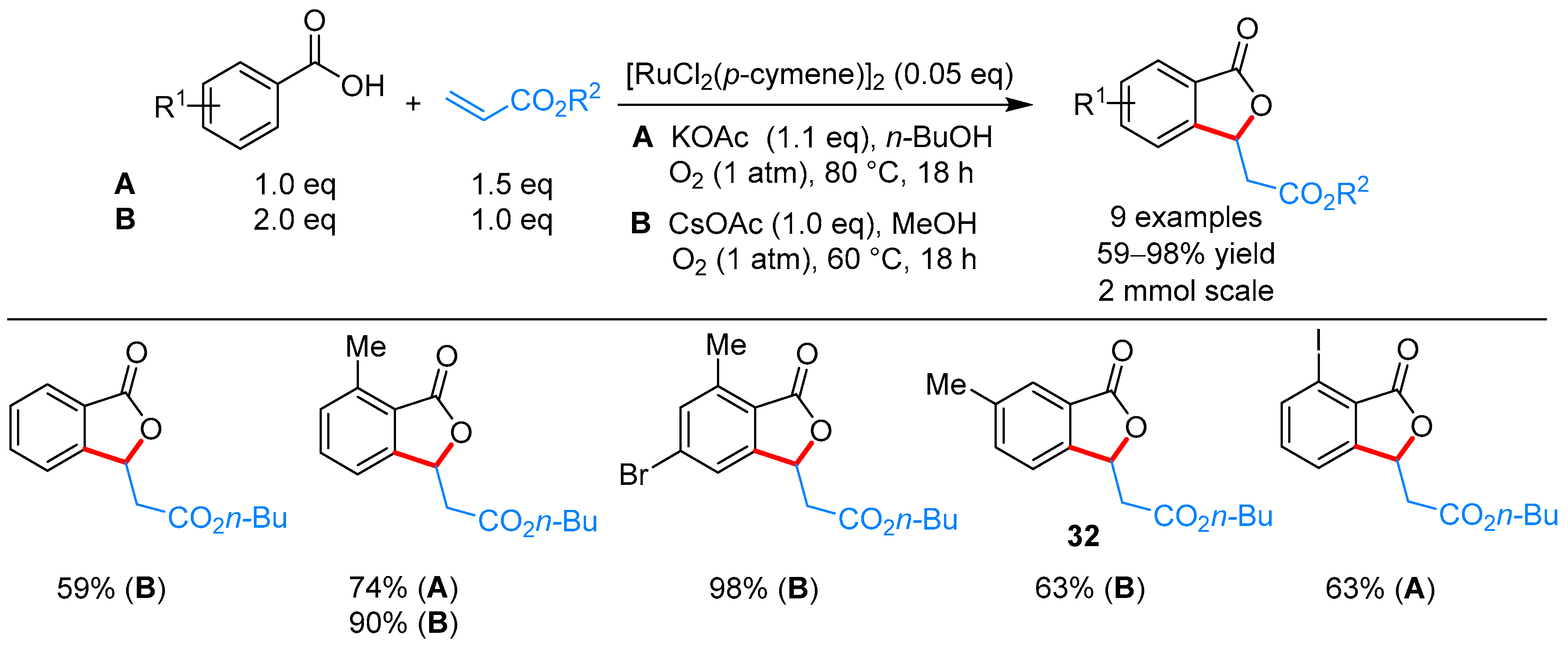
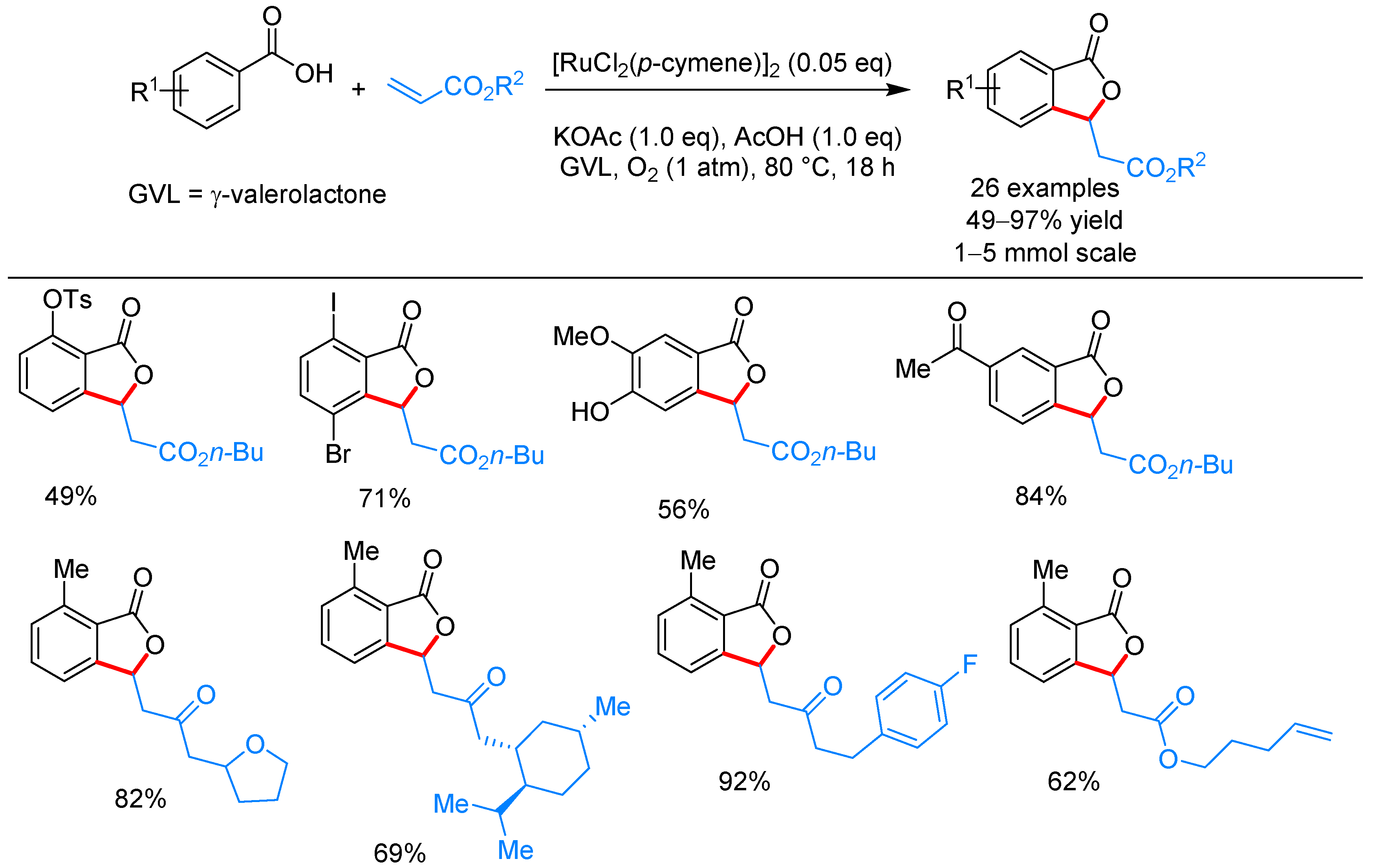
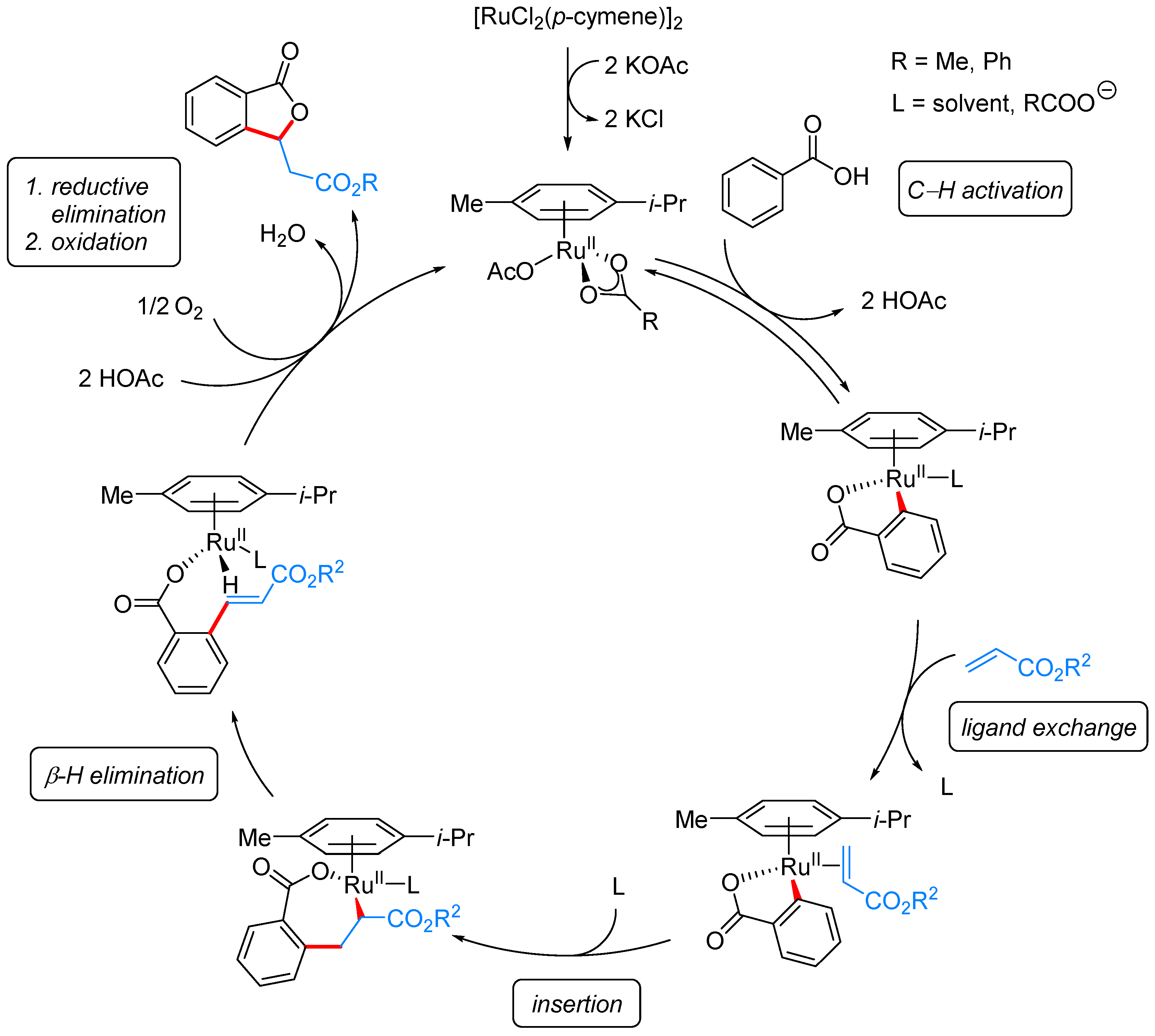
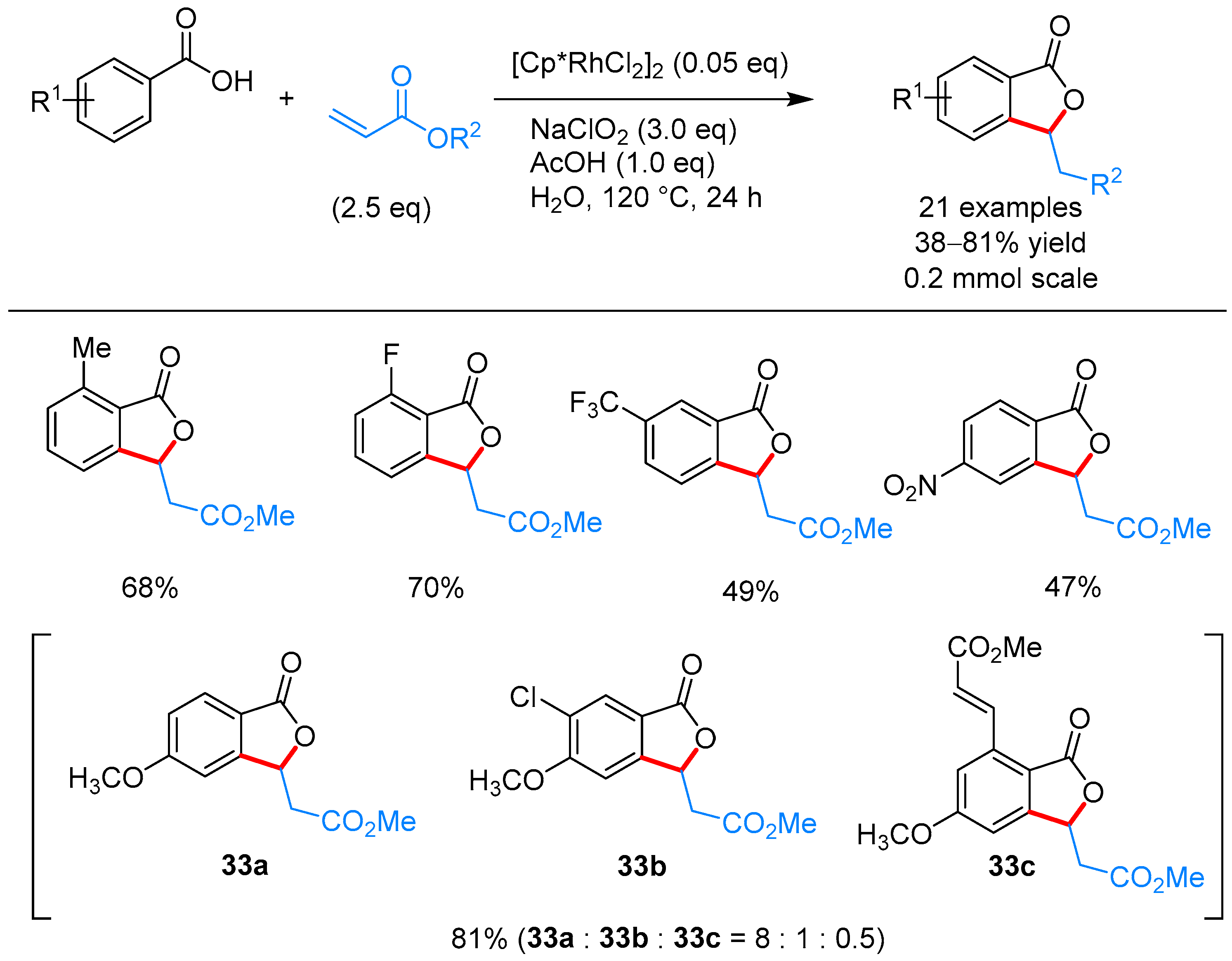
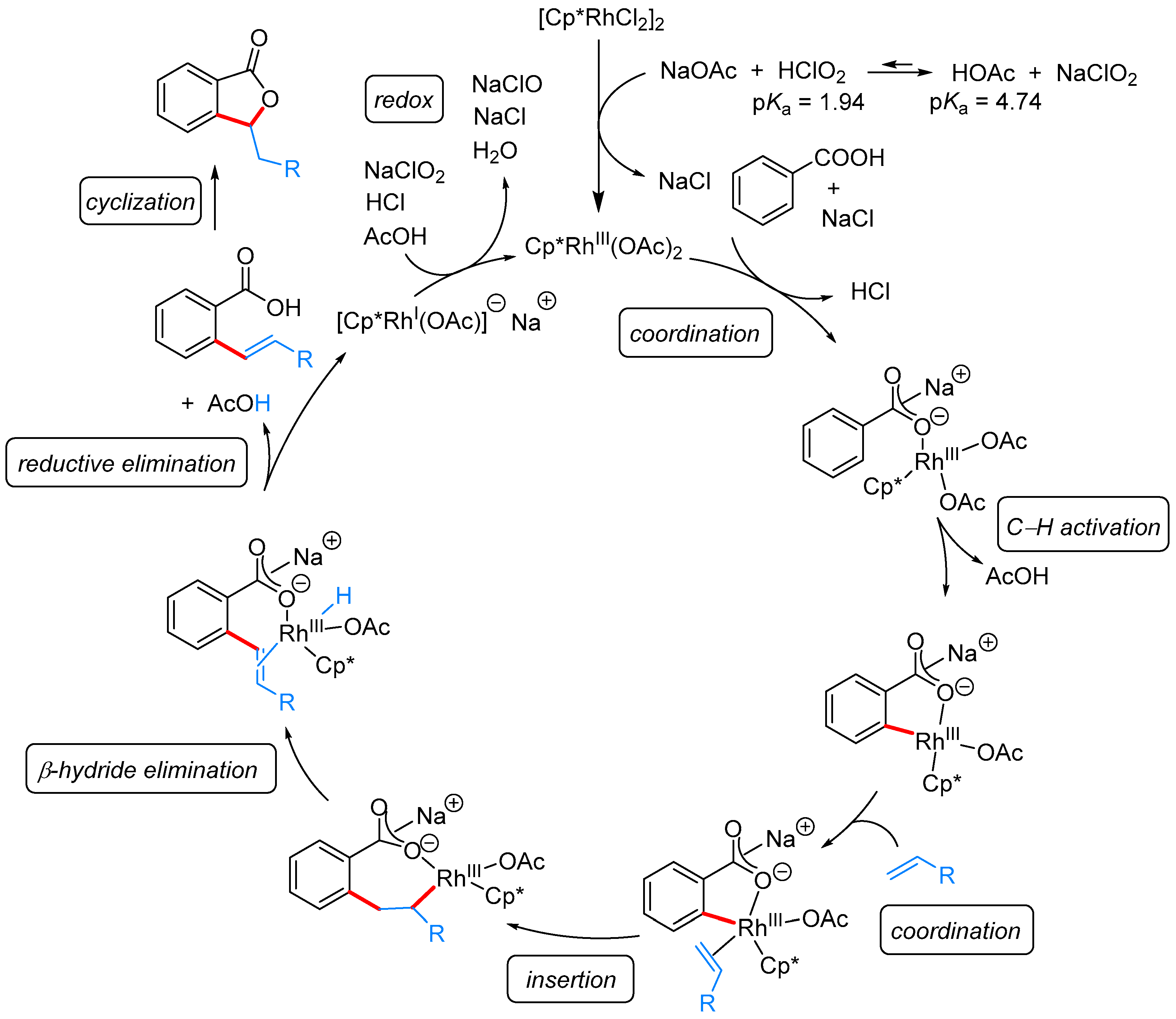
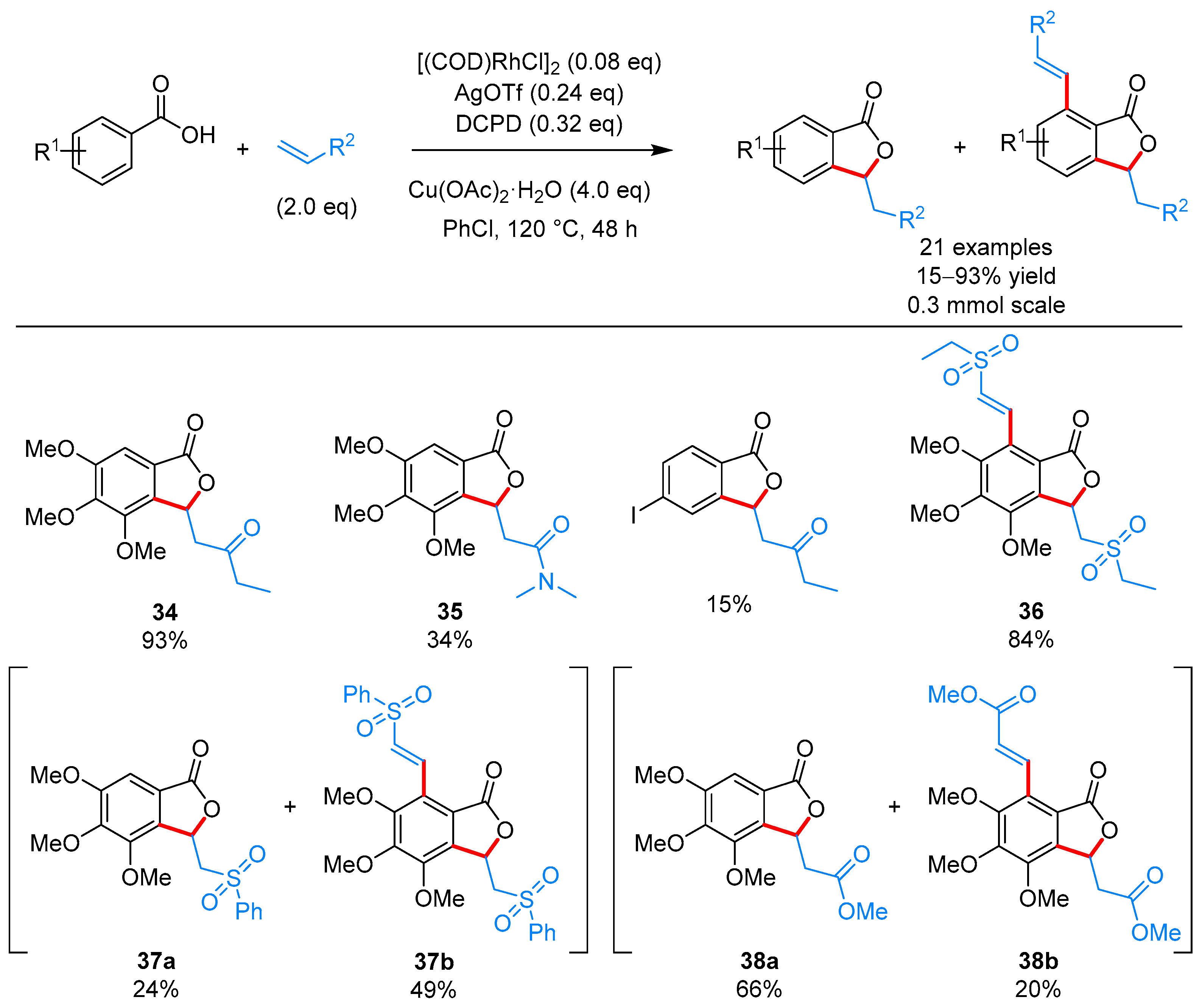
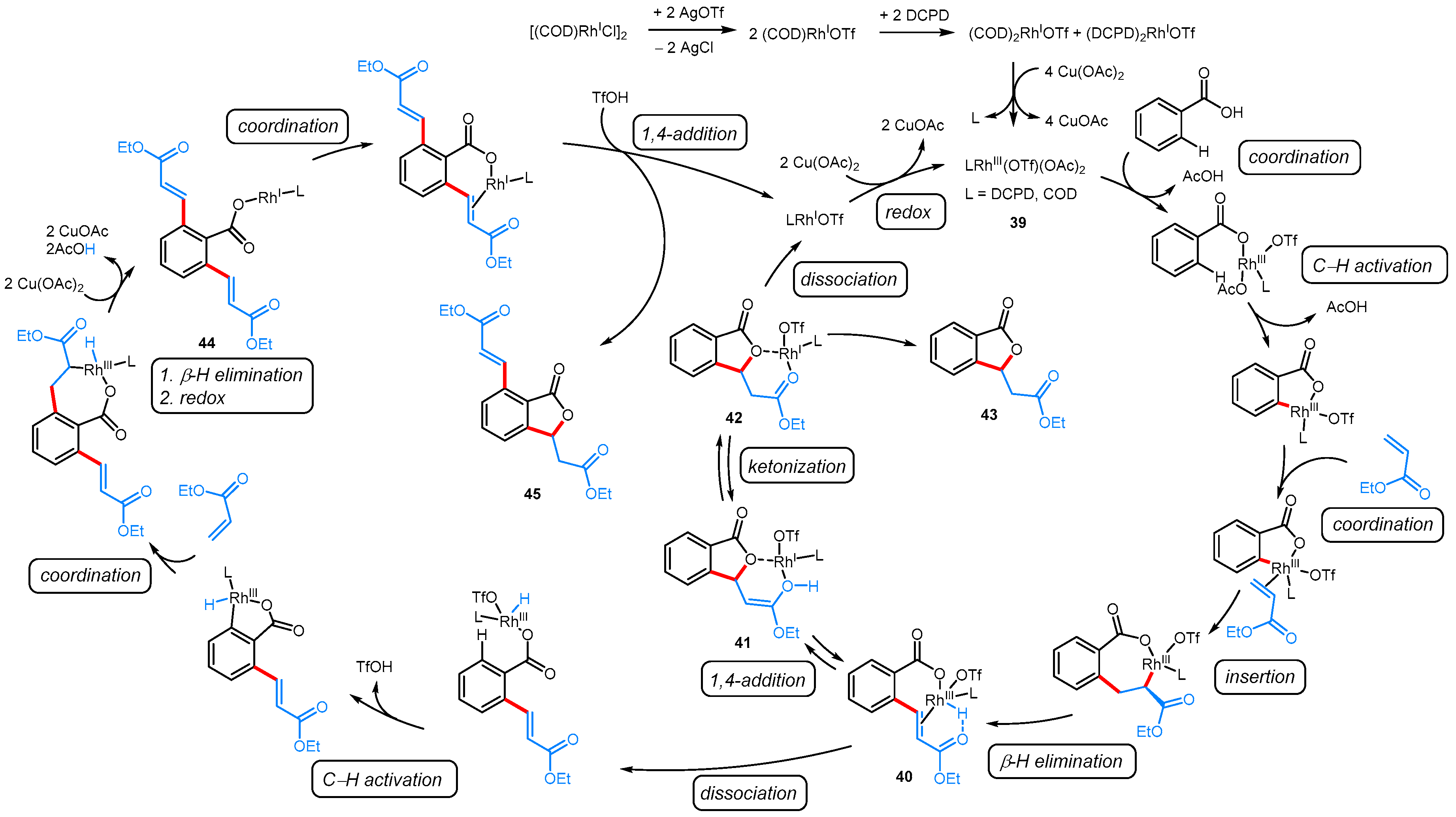
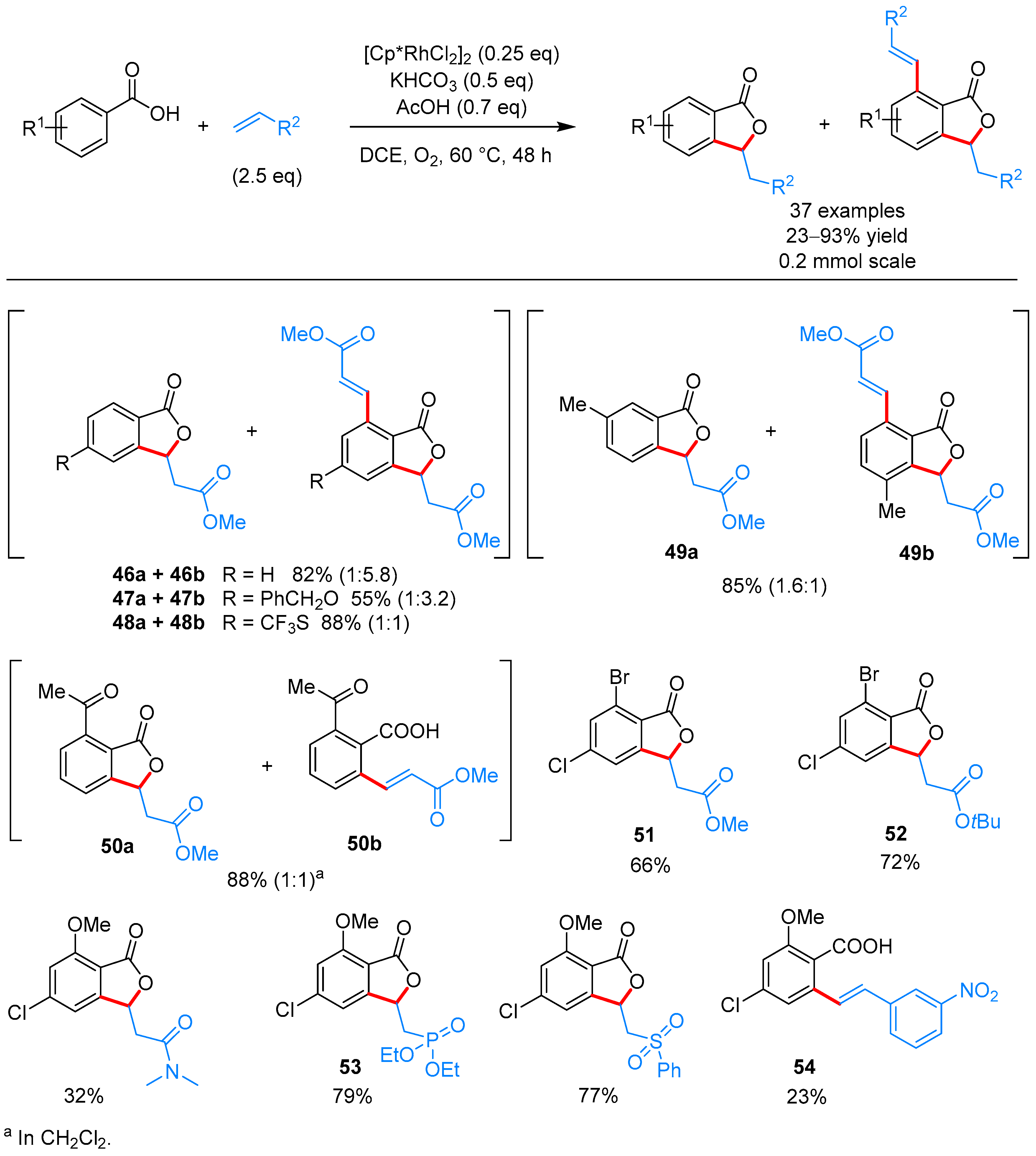
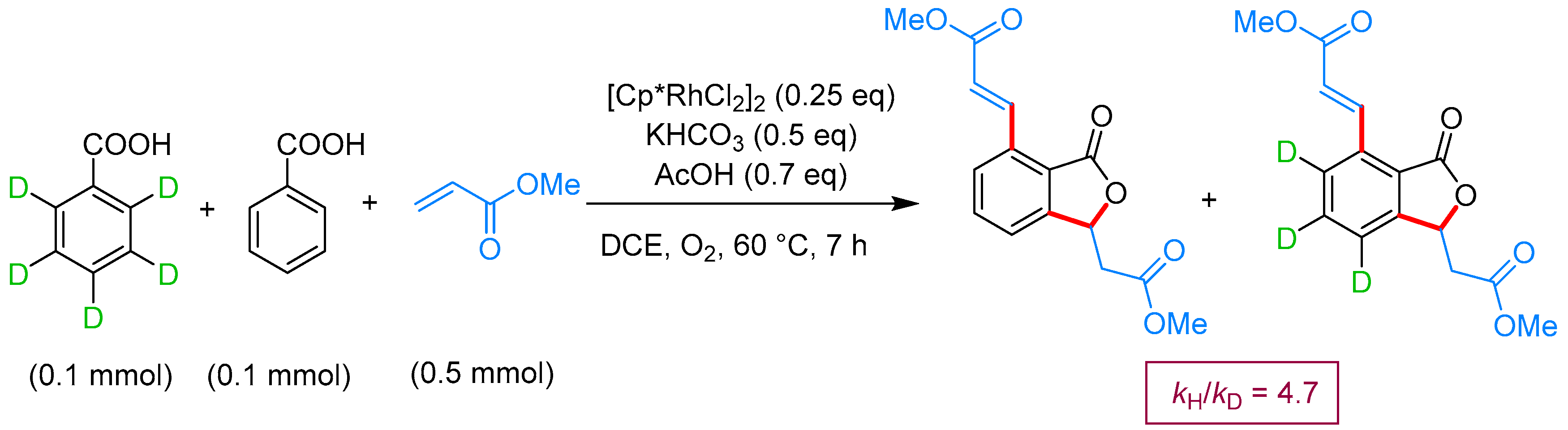
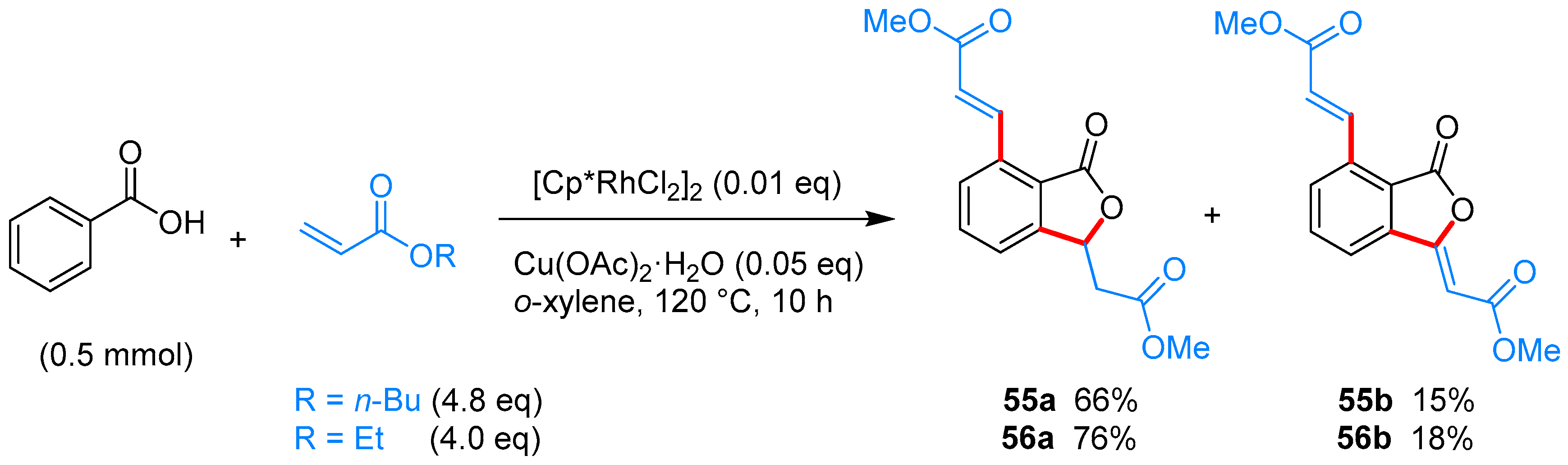
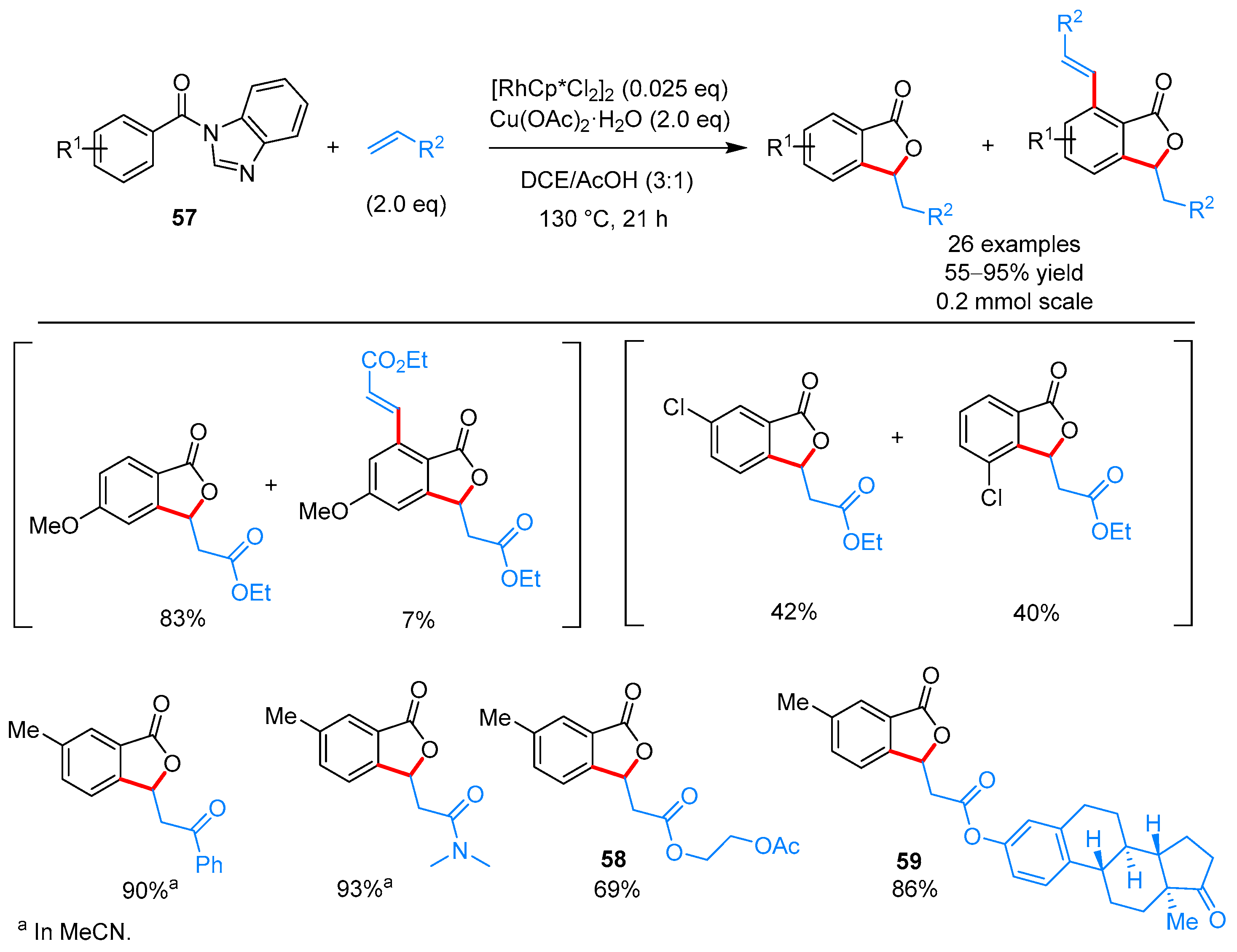
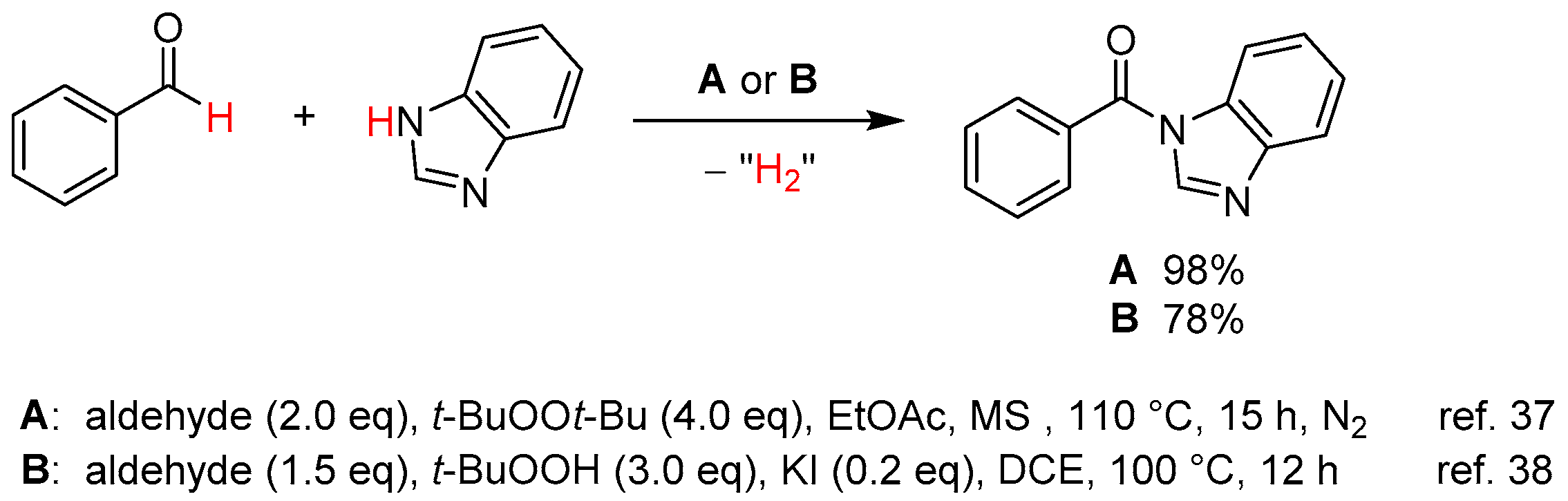
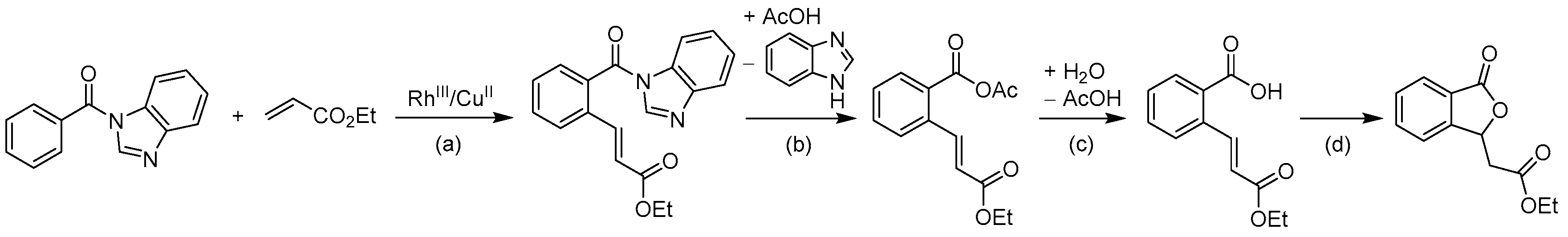
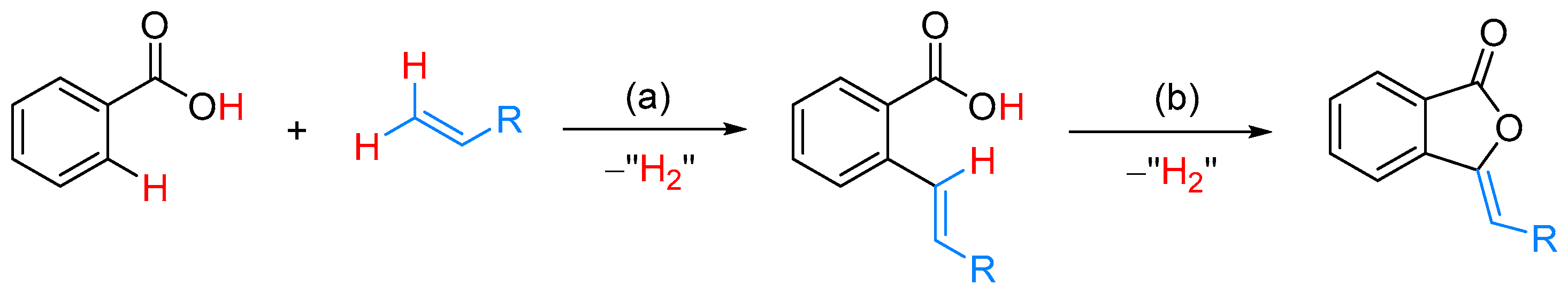
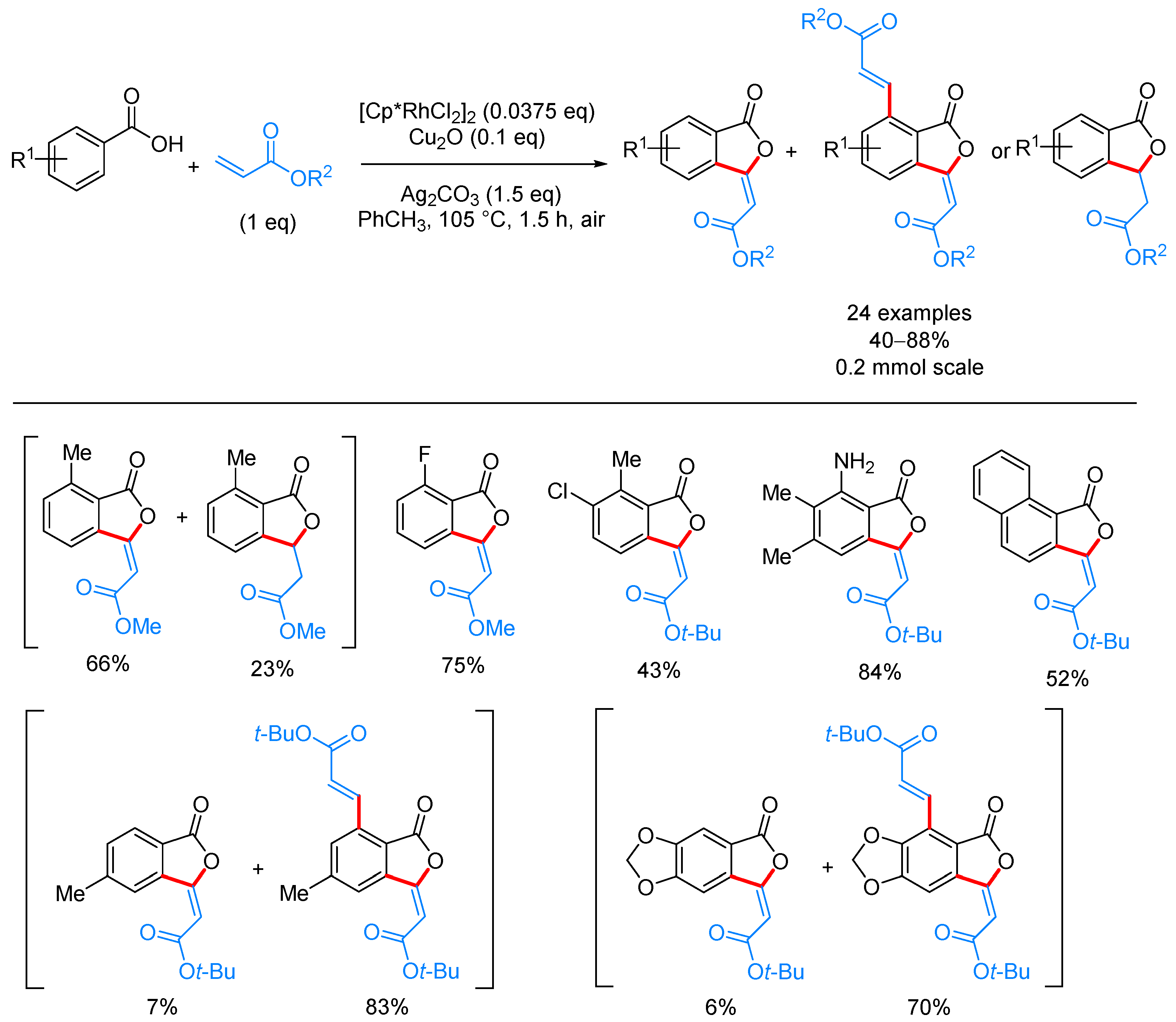
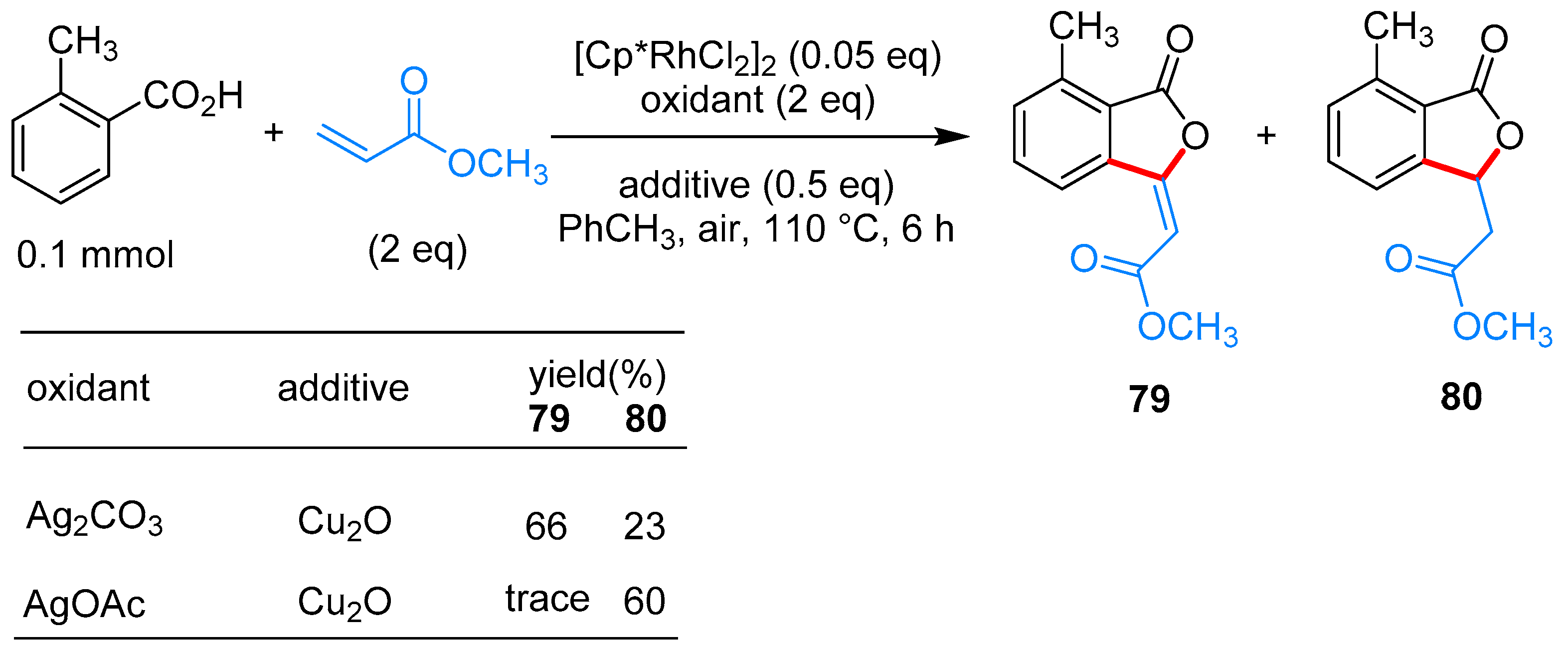
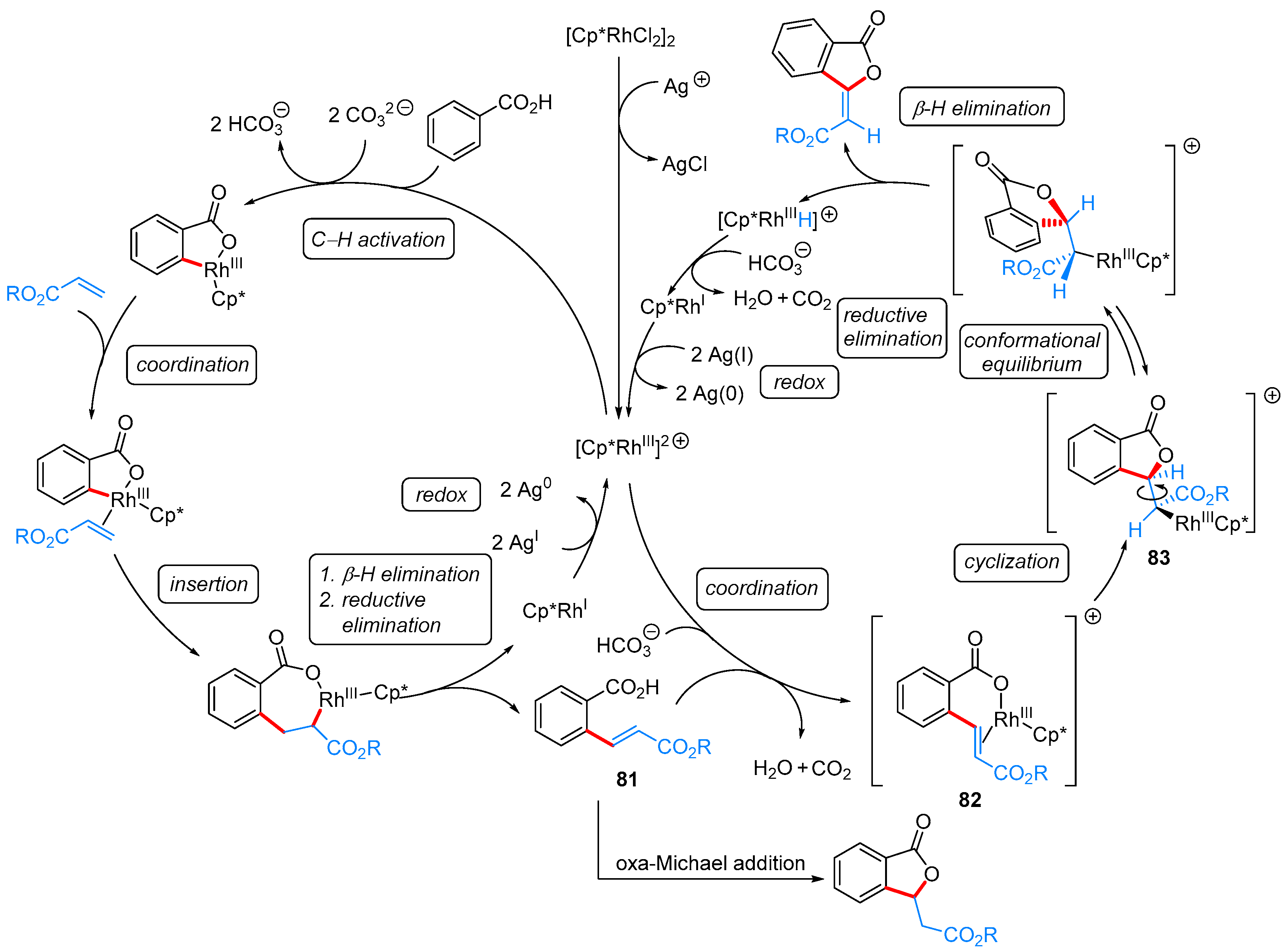
From Benzoic Acids and Electrophilic Alkenes
2.2. 3,3-Disubstituted Phthalides
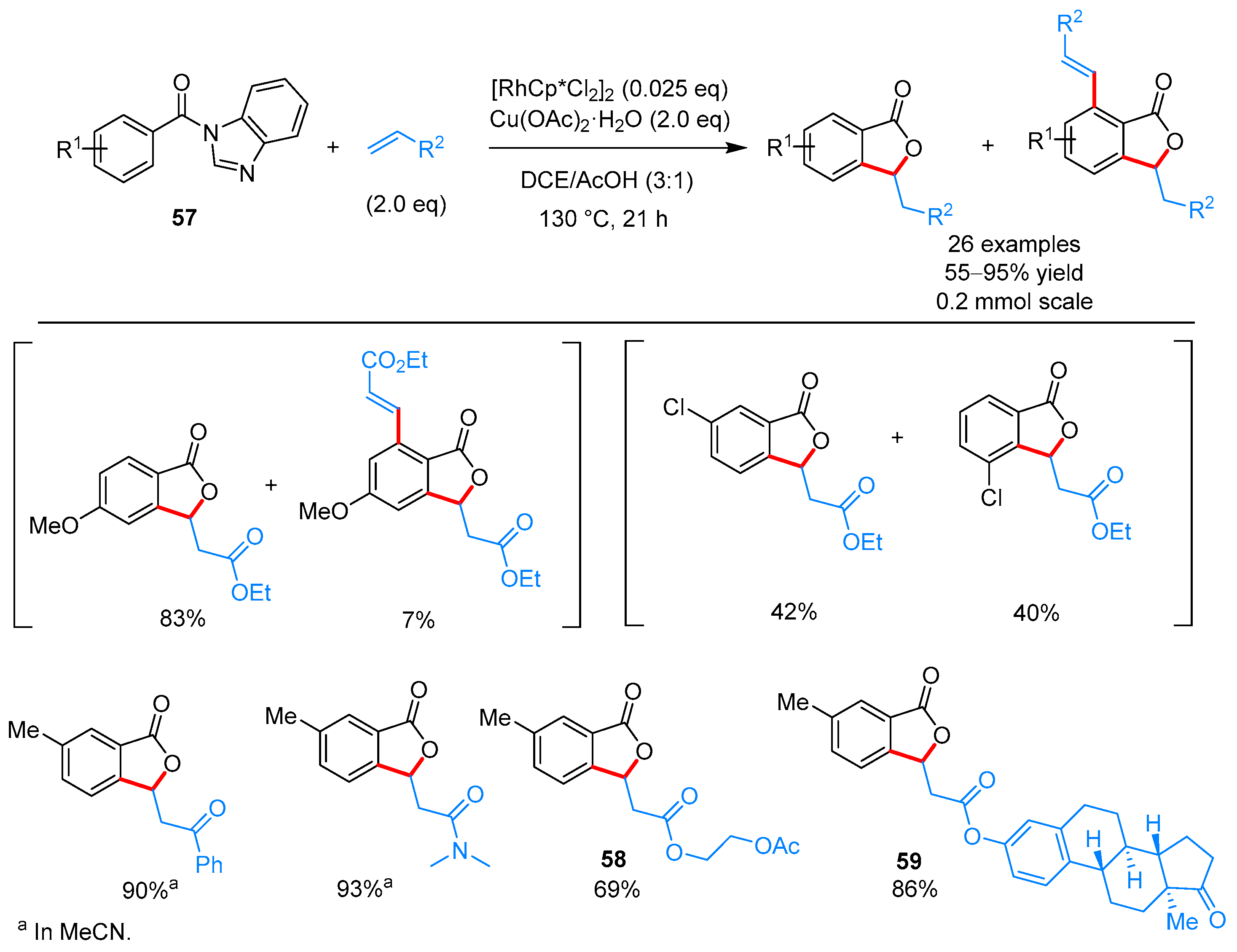
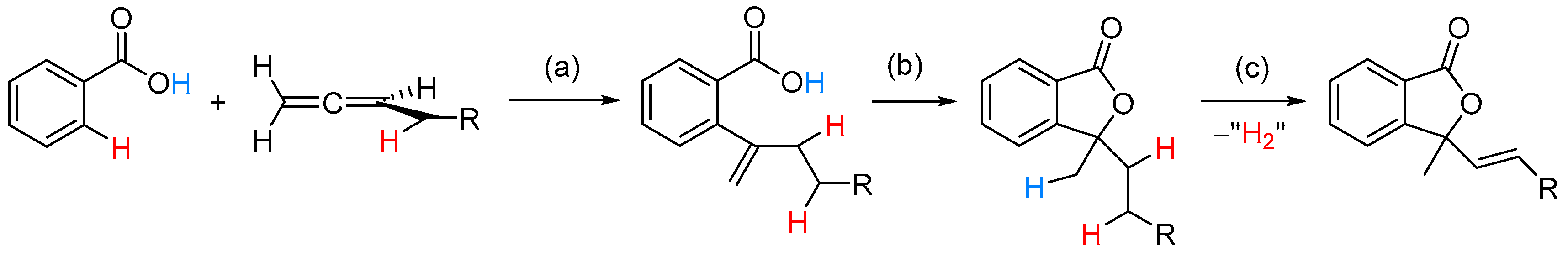
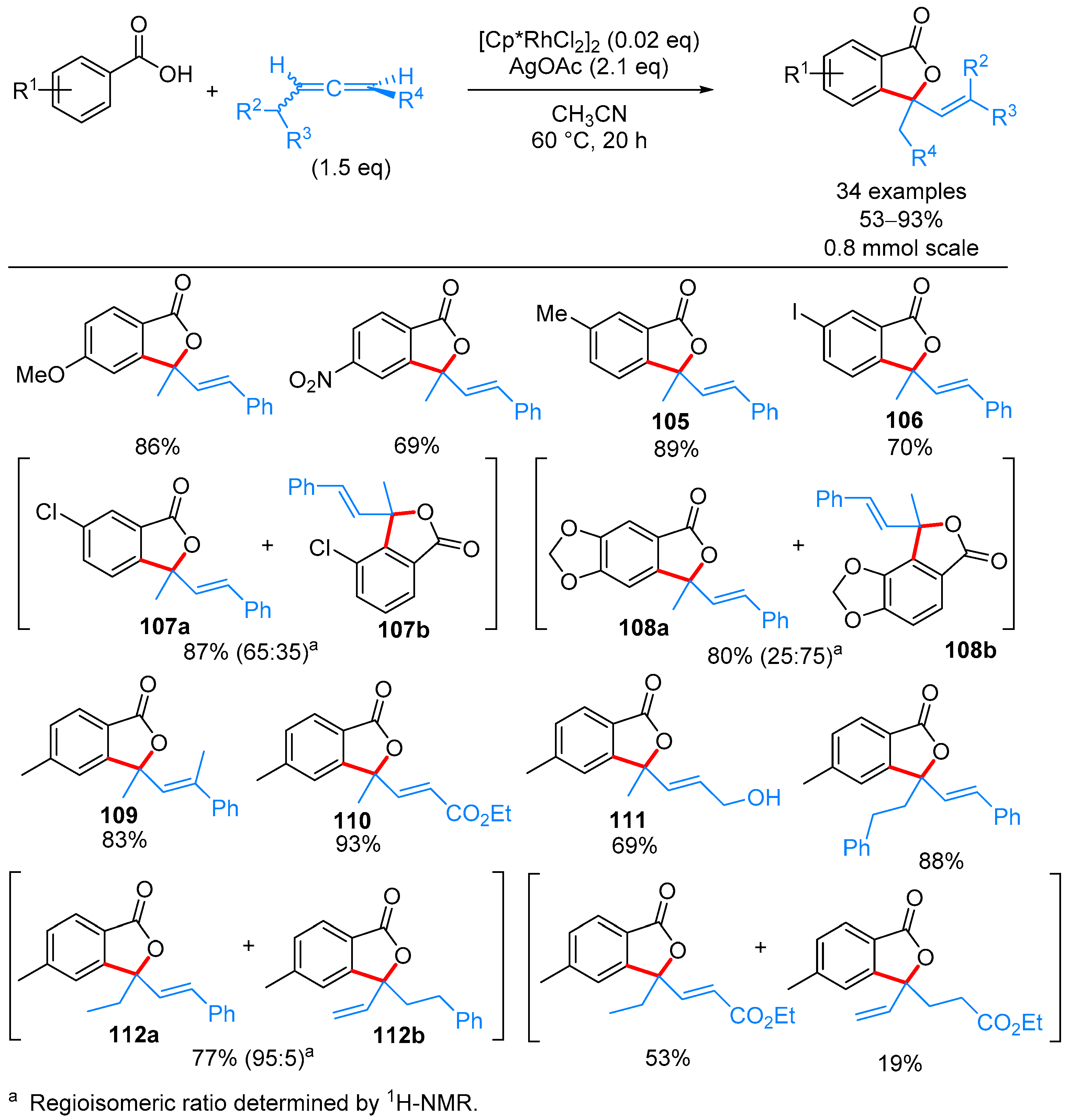
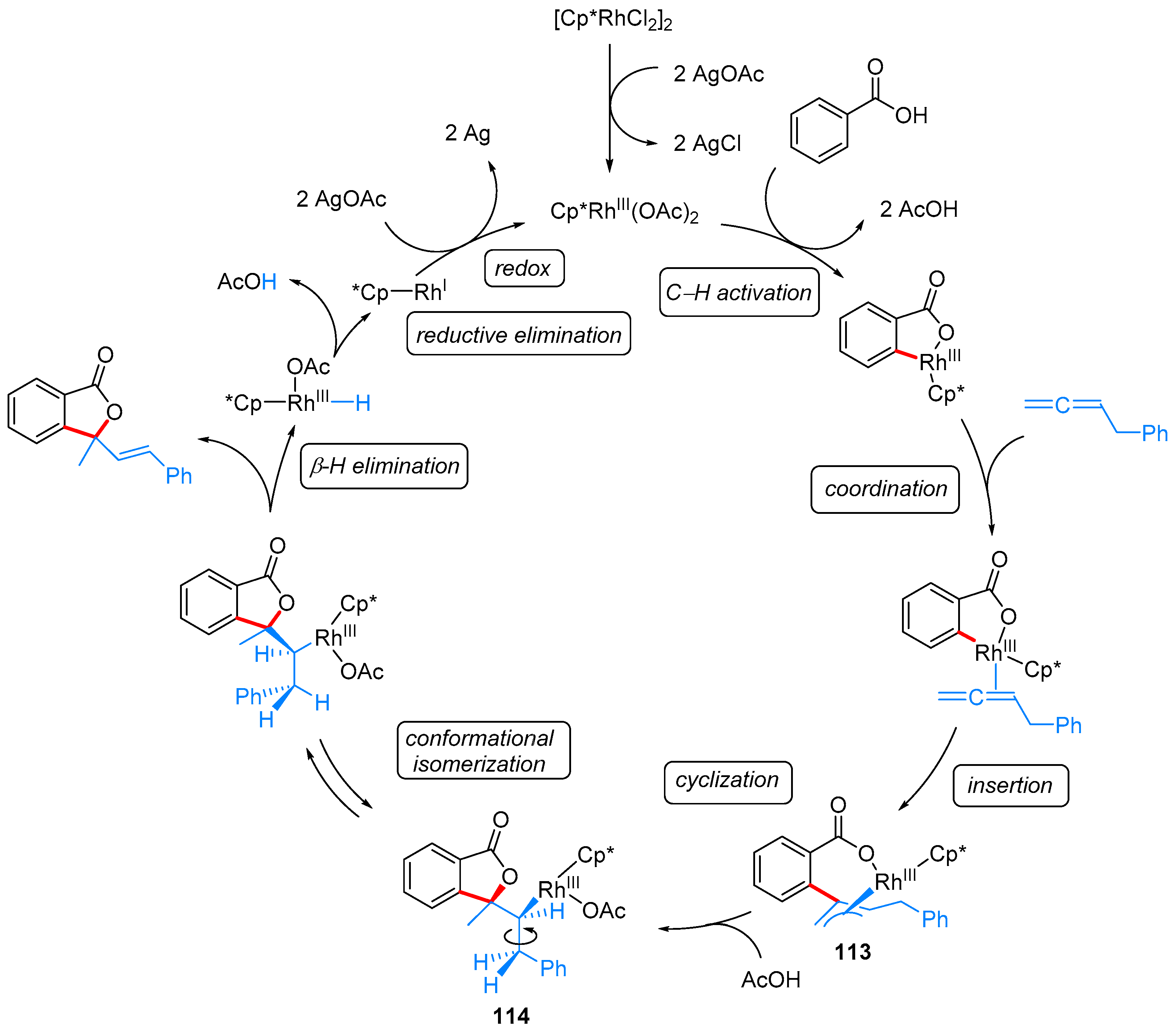
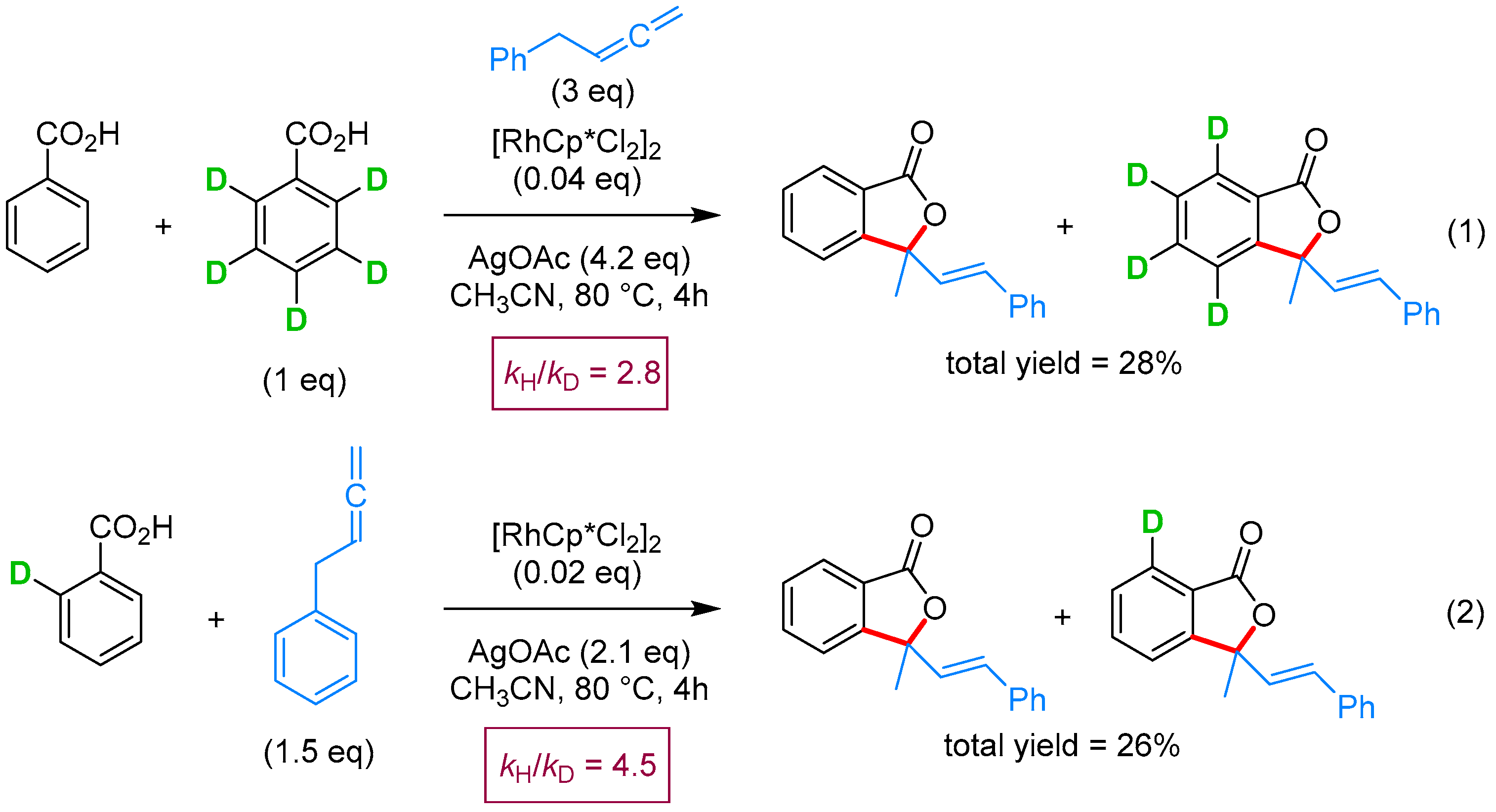
2.2.1. 3-Ylidene Phthalides
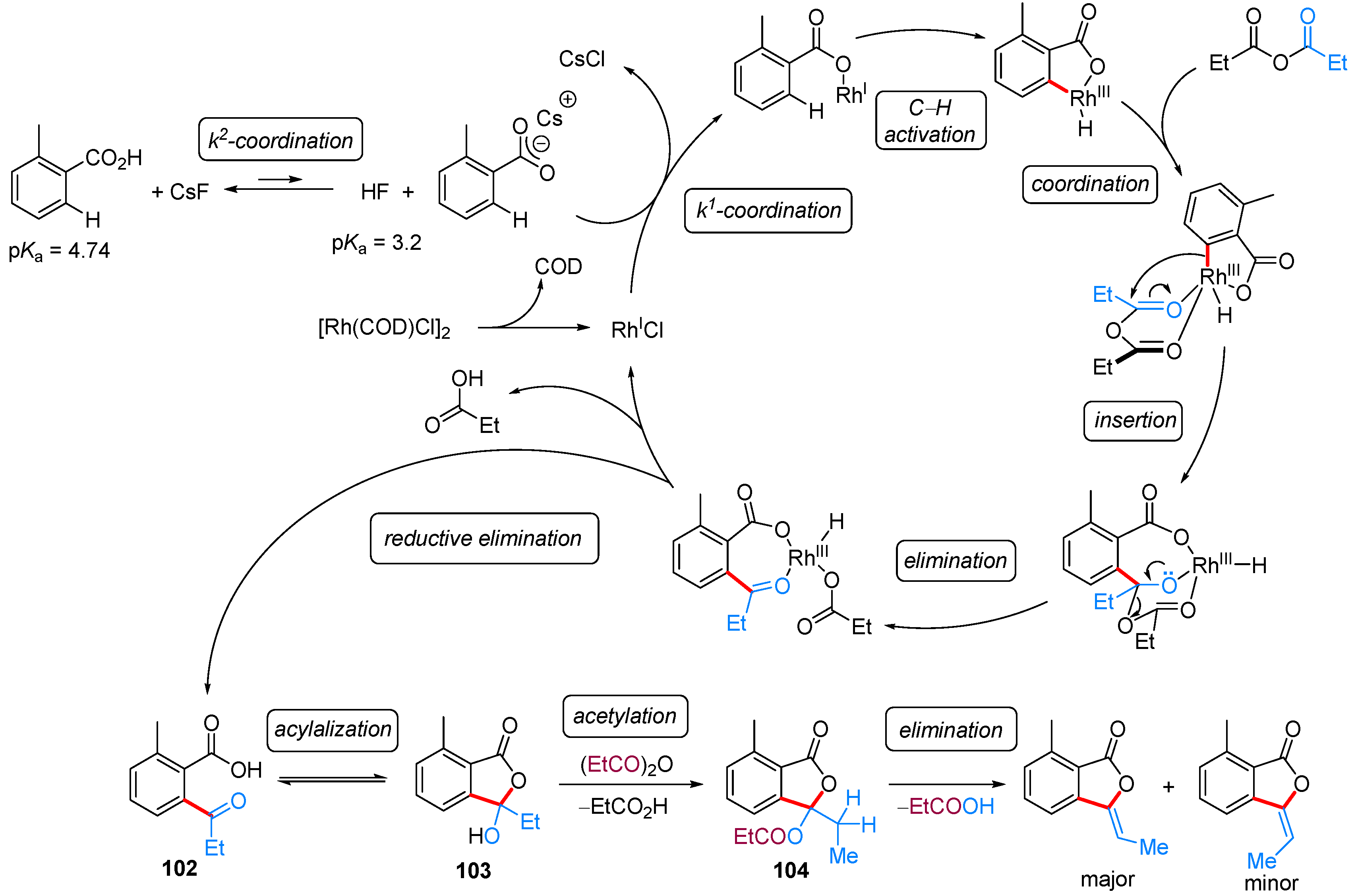
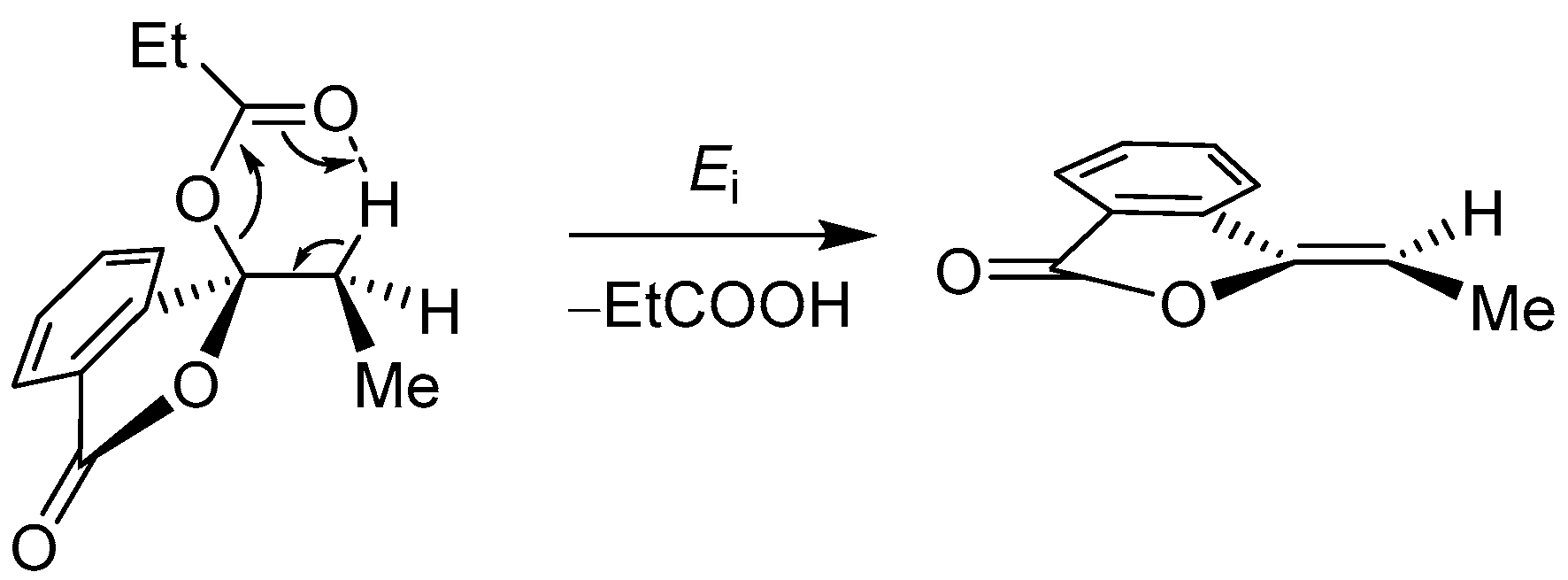
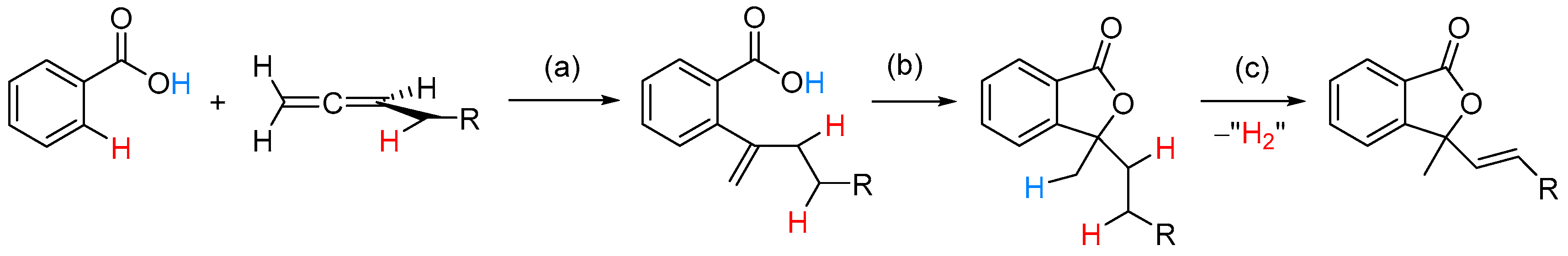
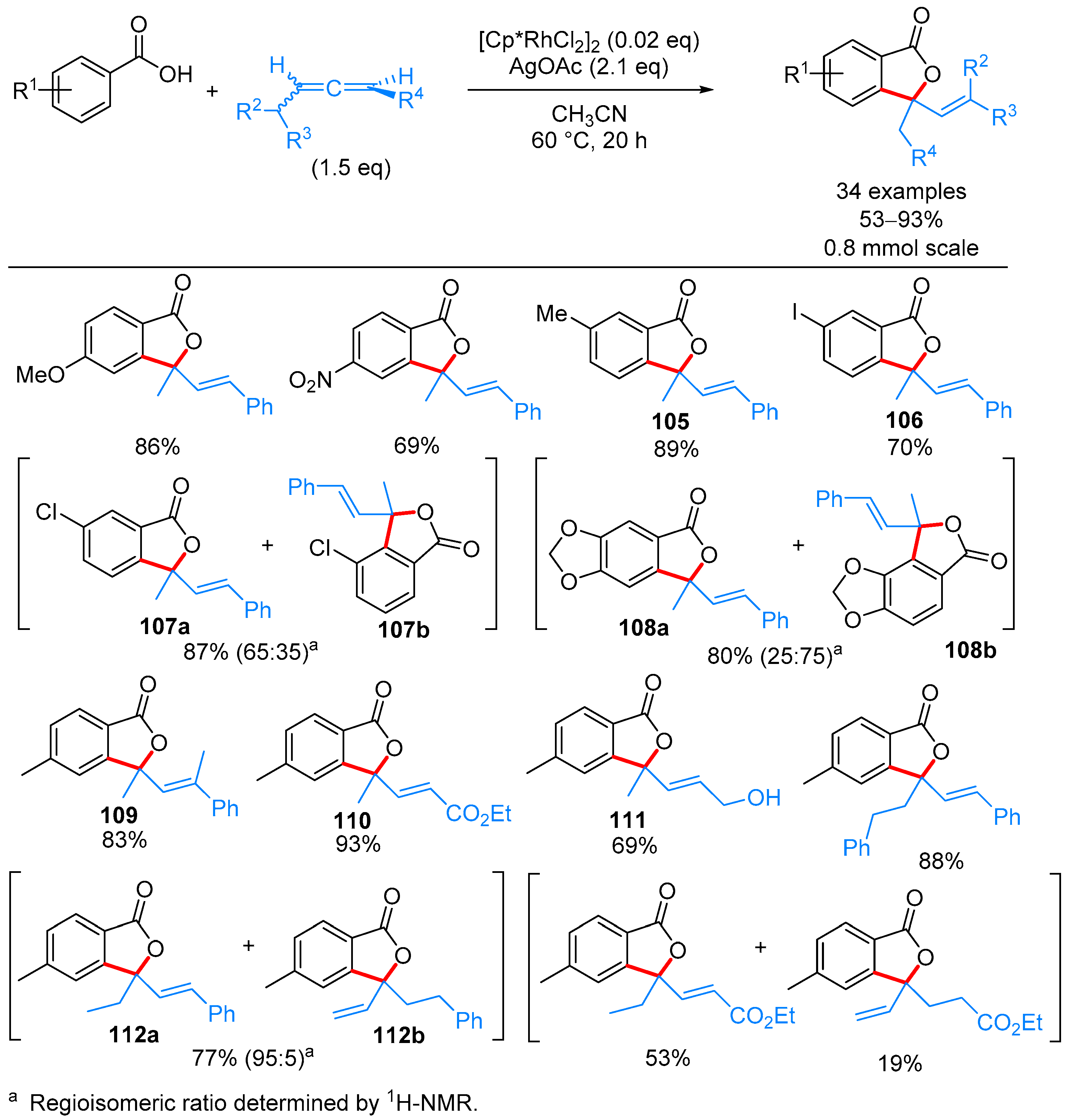
2.2.2. 3-Alkyl-3-Vinyl Phthalides
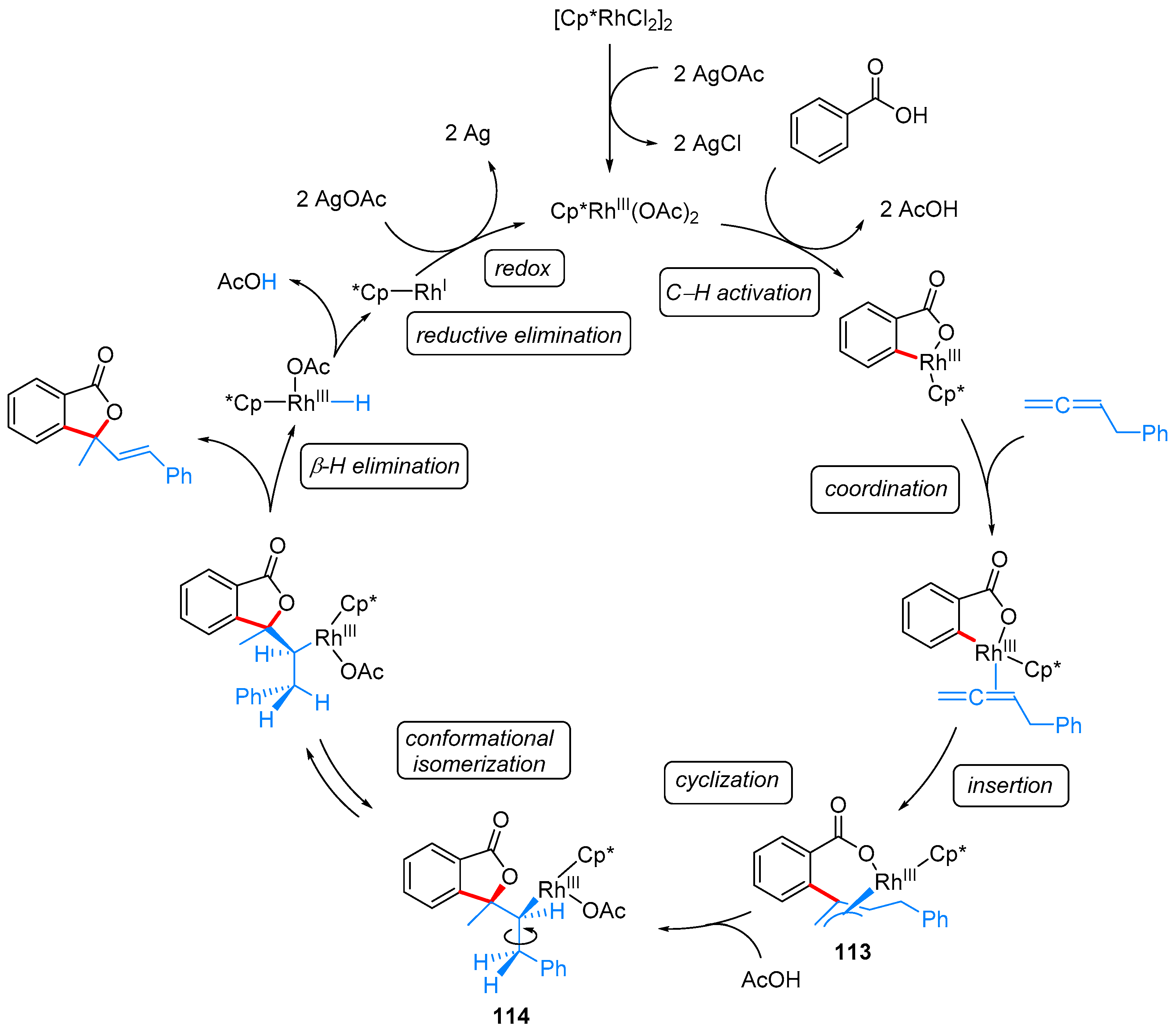
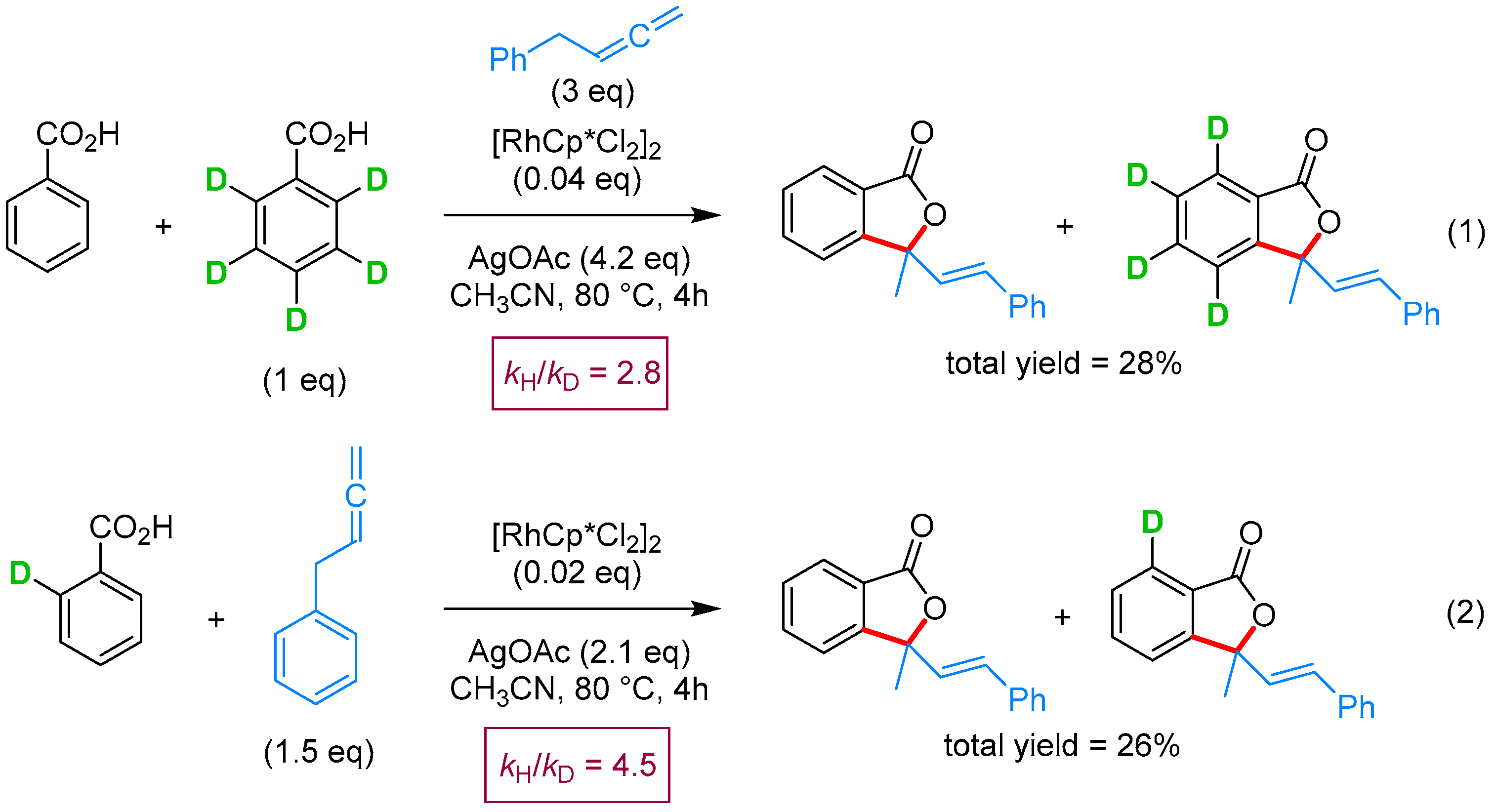
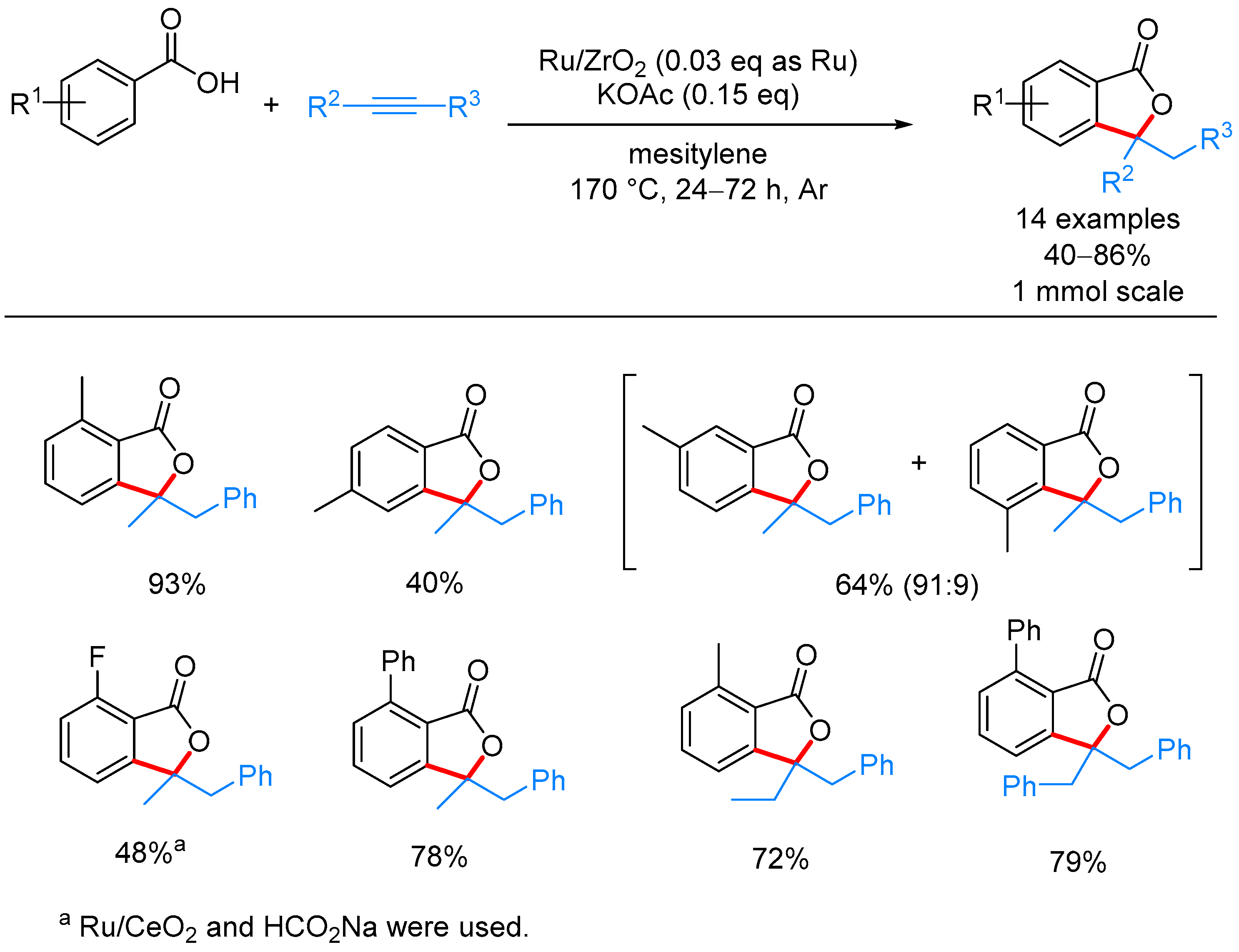
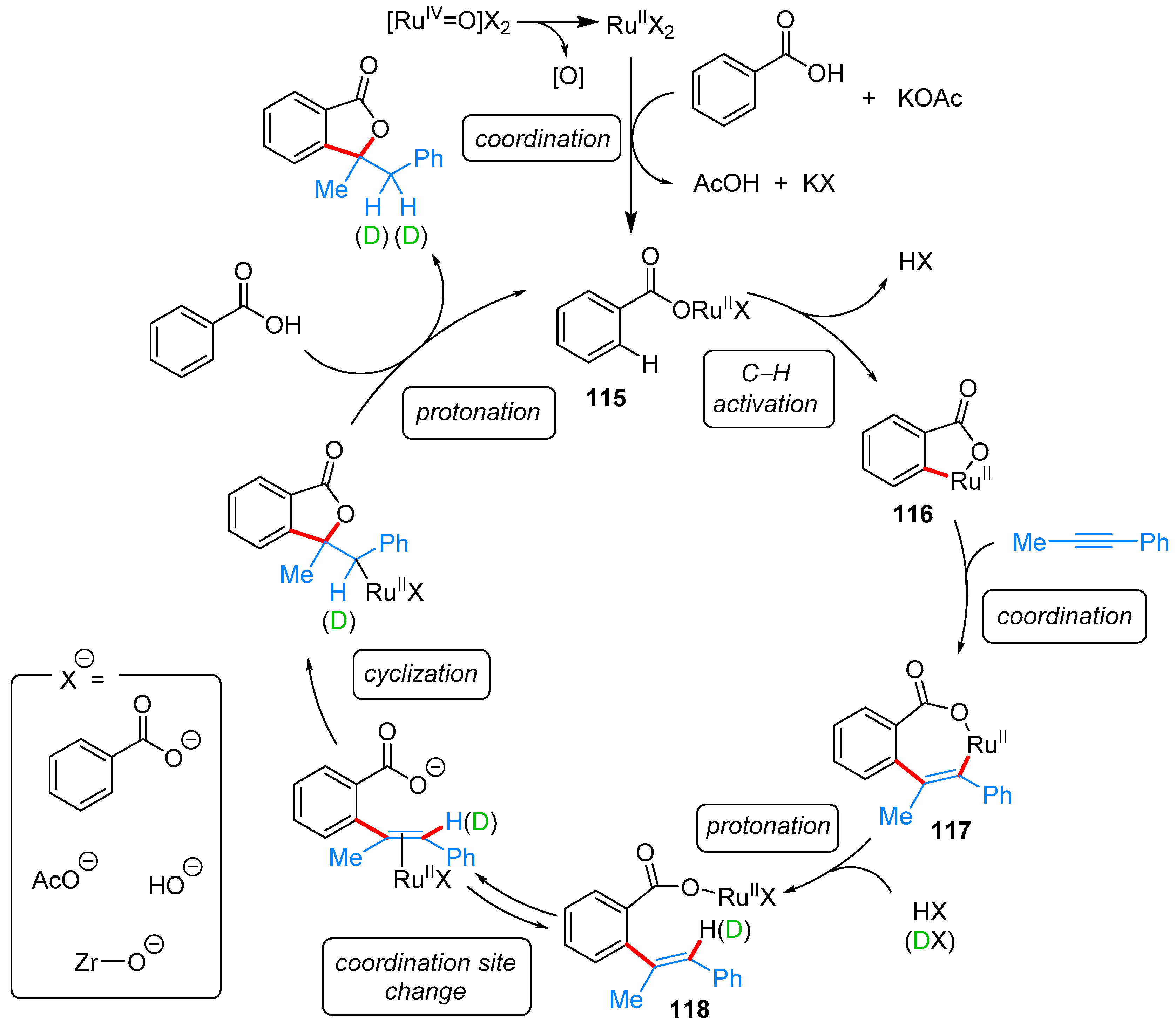
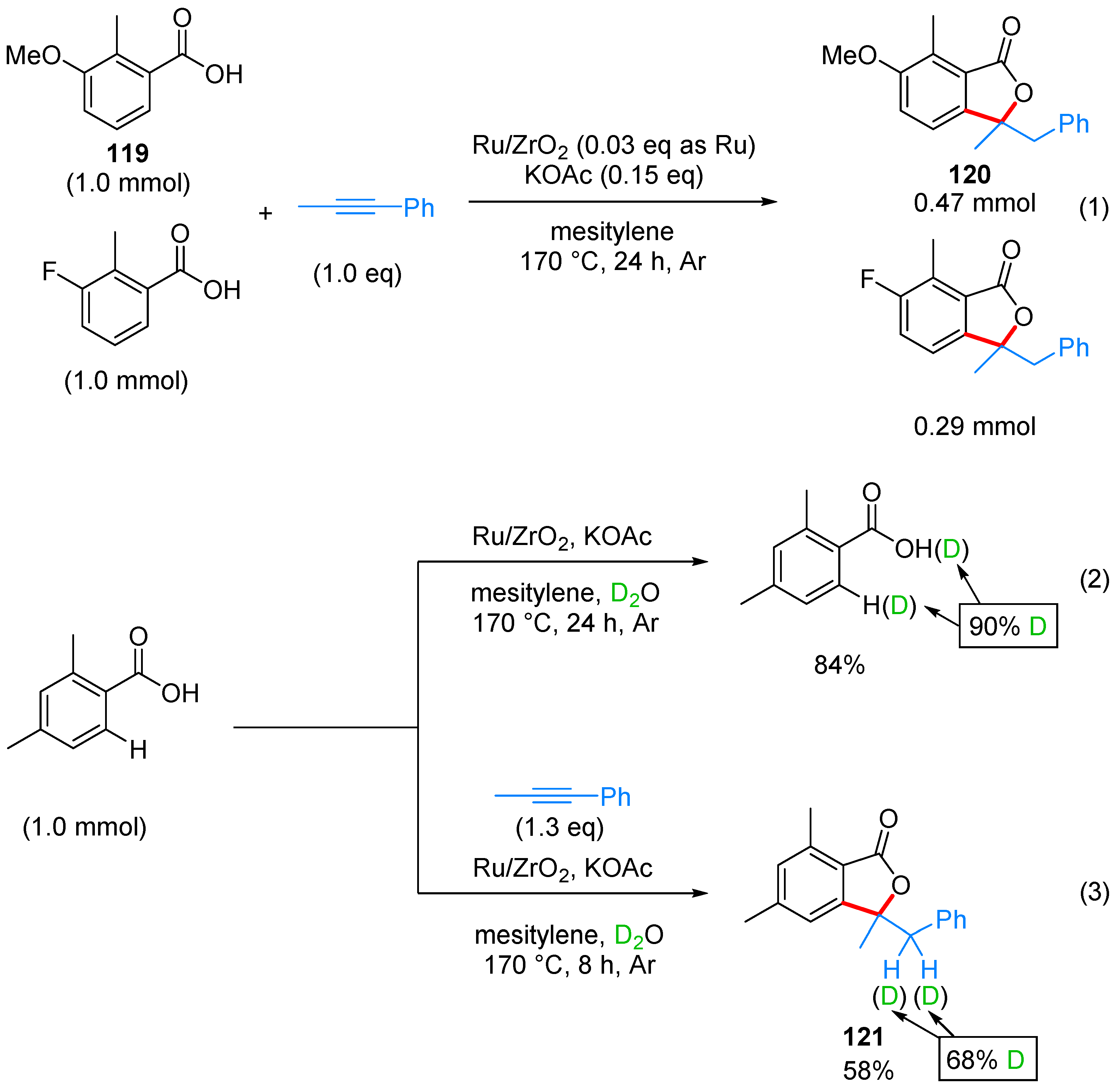
2.2.3. 3,3-Dialkyl Phthalides
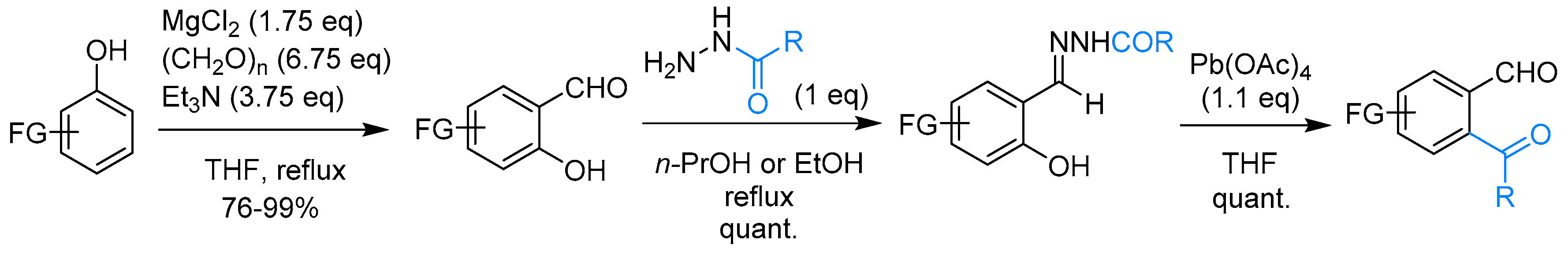
2.3. Unsubstituted Phthalide
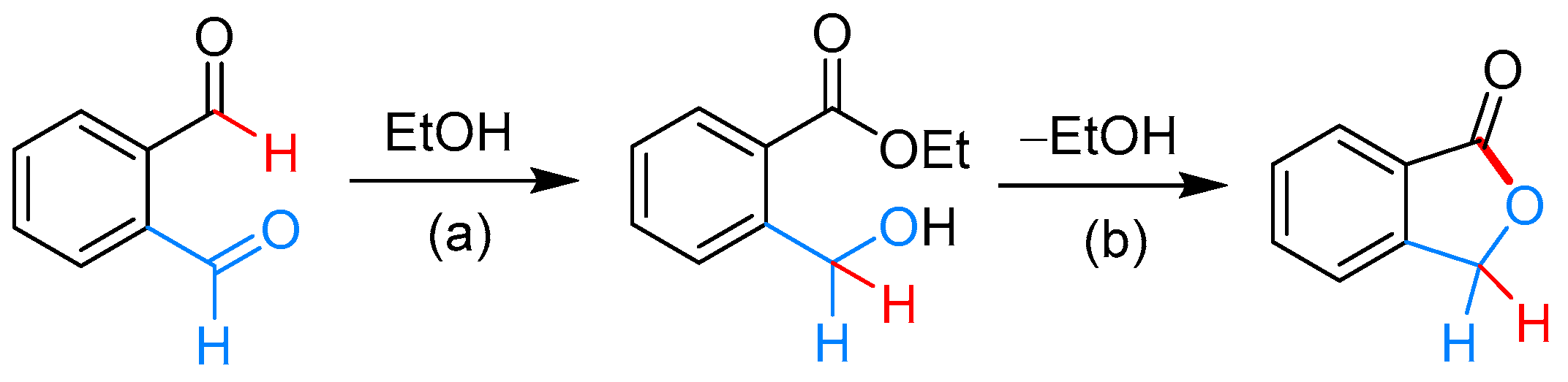
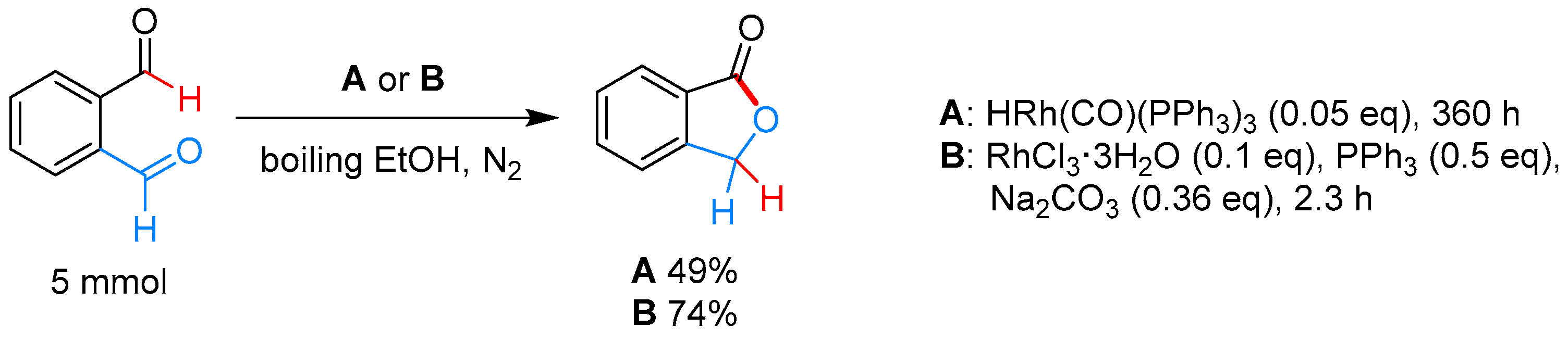
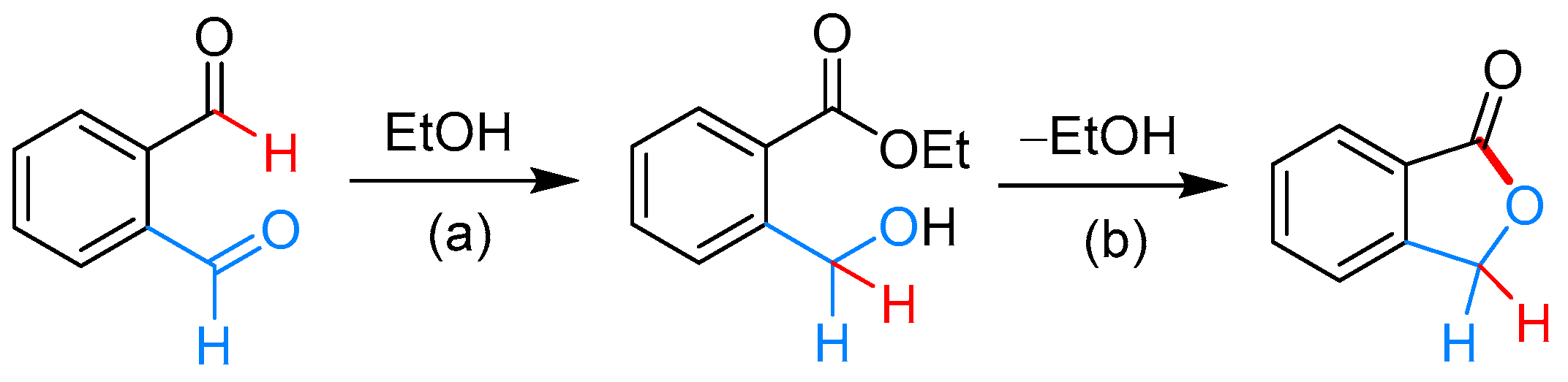
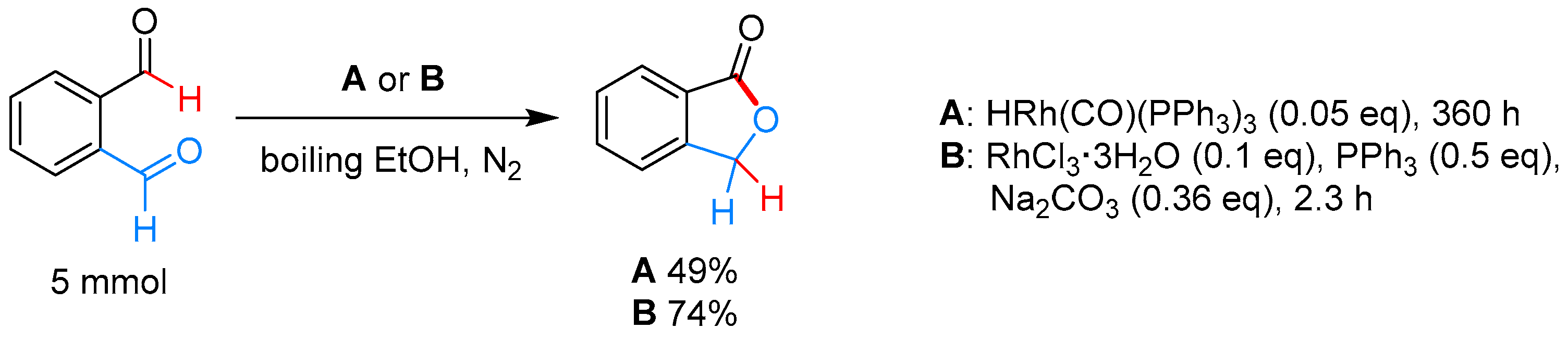
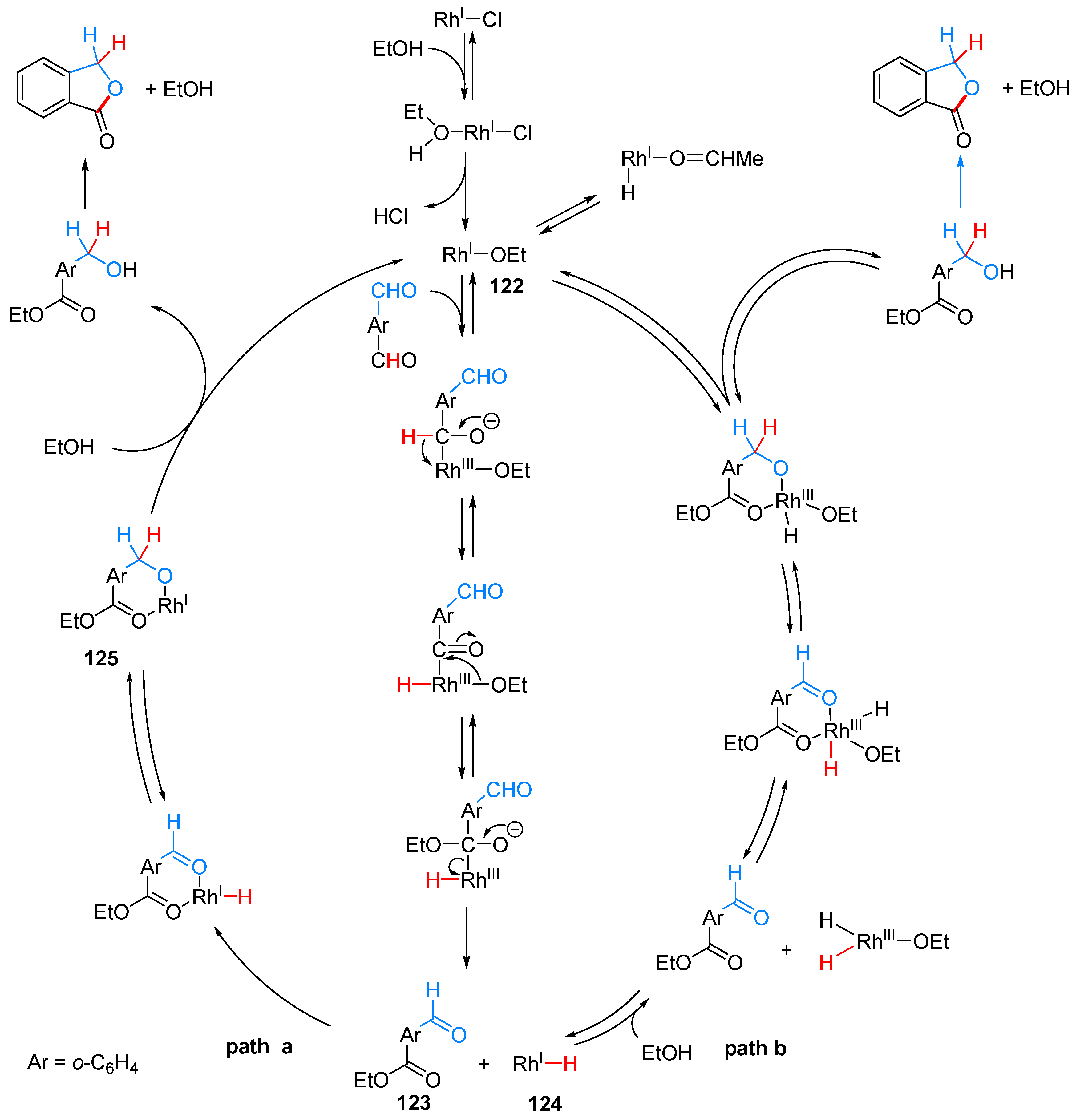
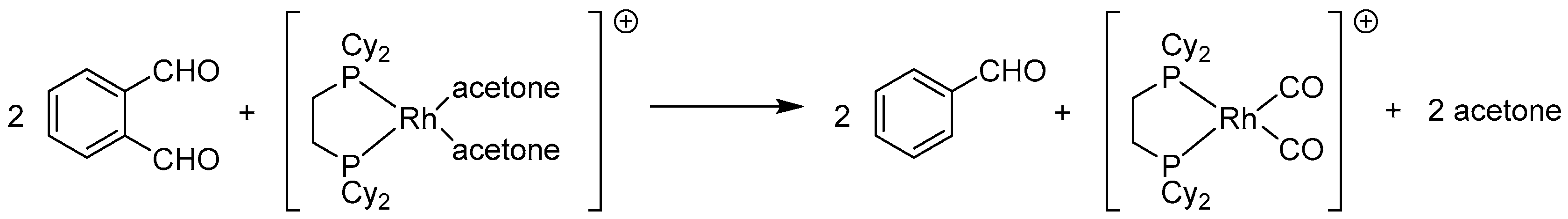
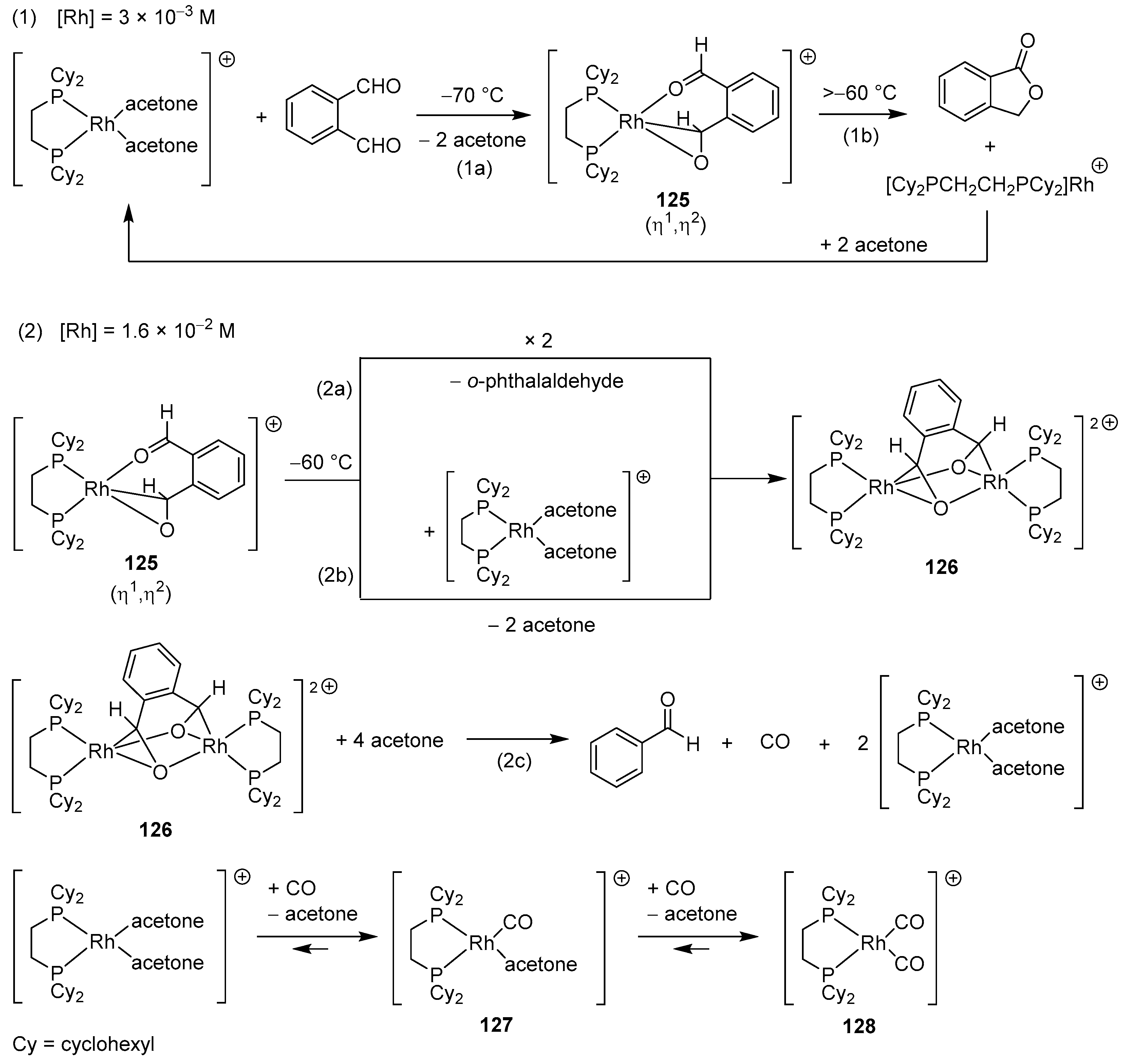
2.3.1. By Annulative Disproportionation of o-Phthalaldehyde
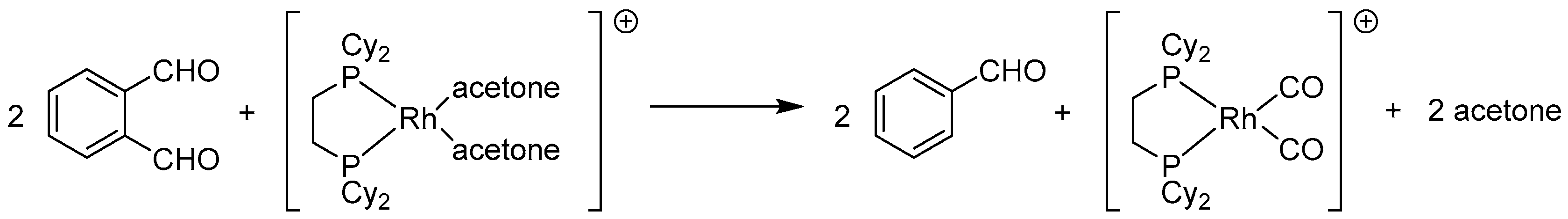
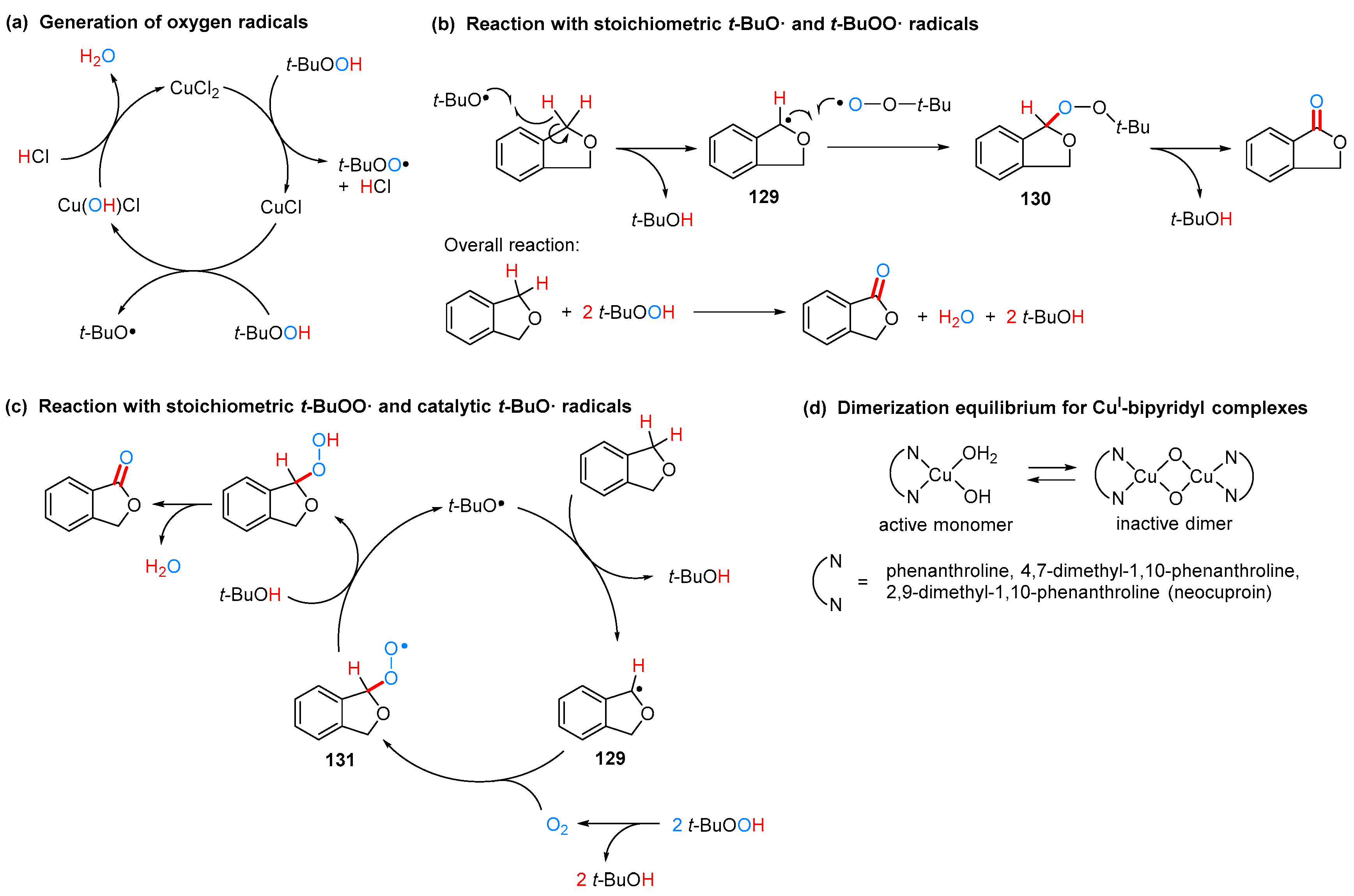
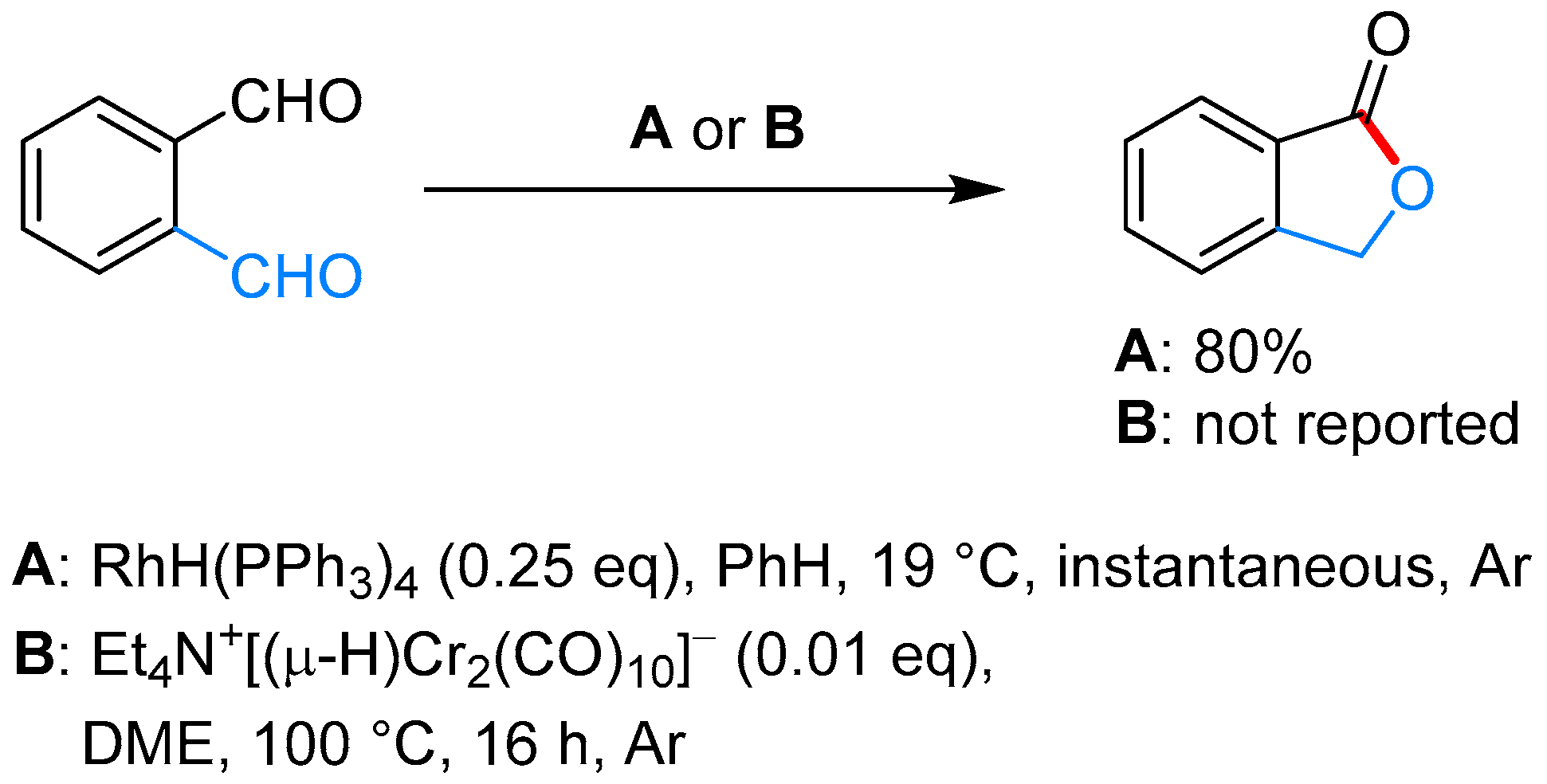
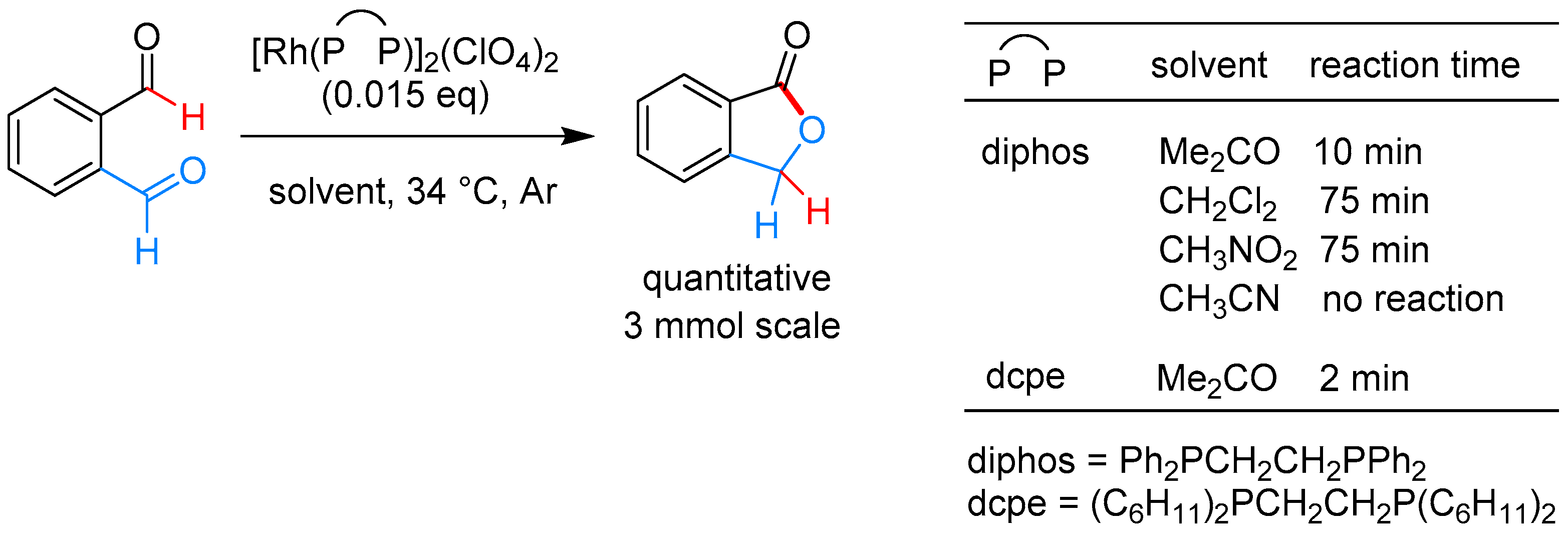
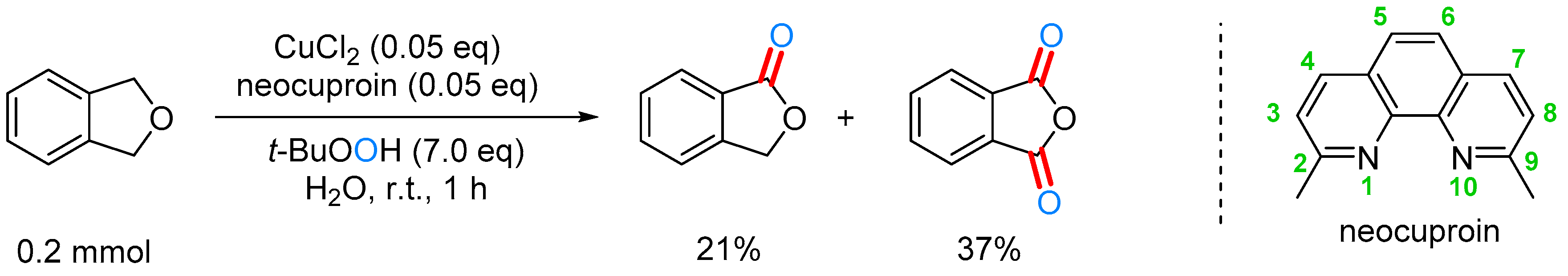
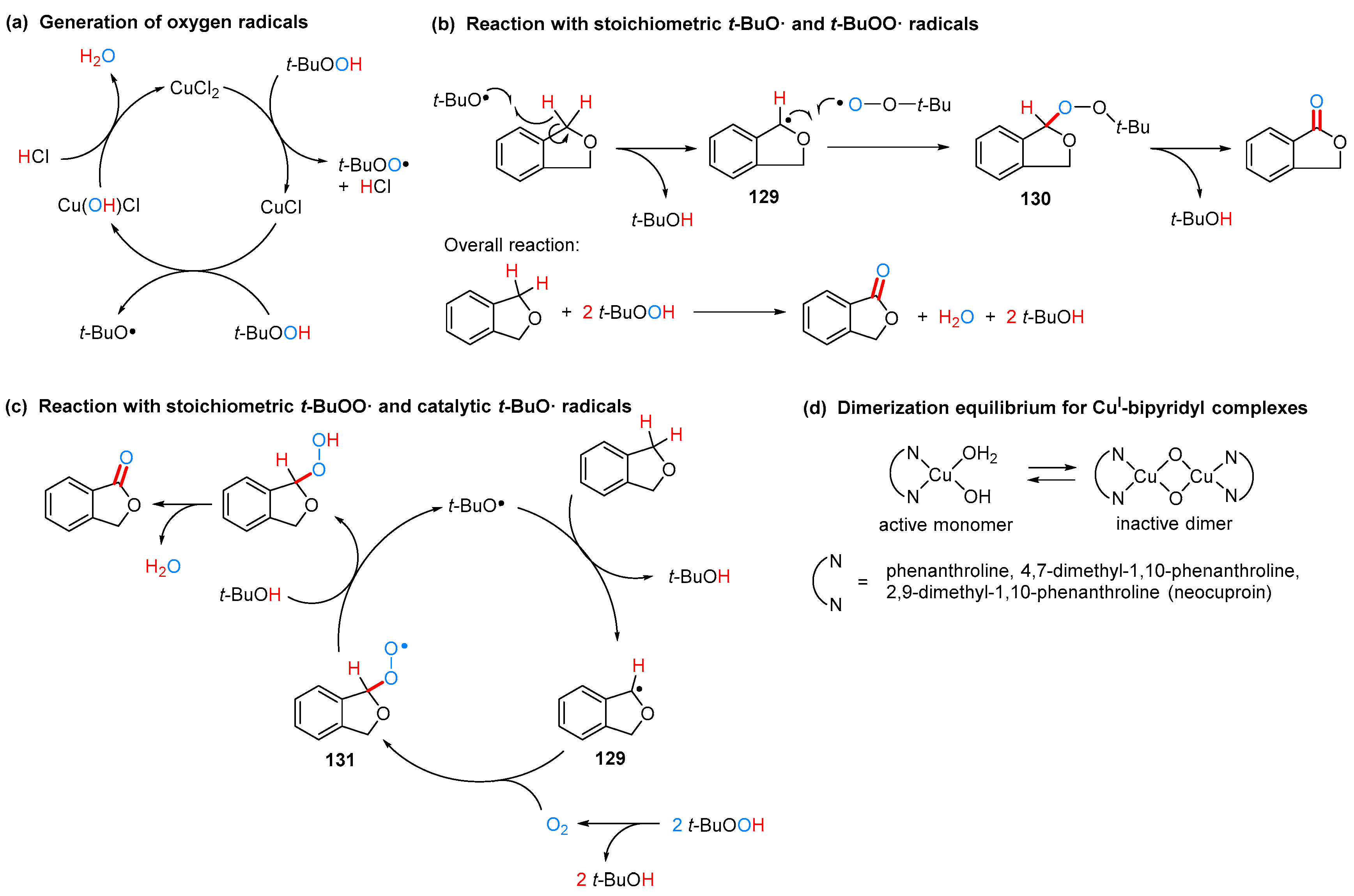
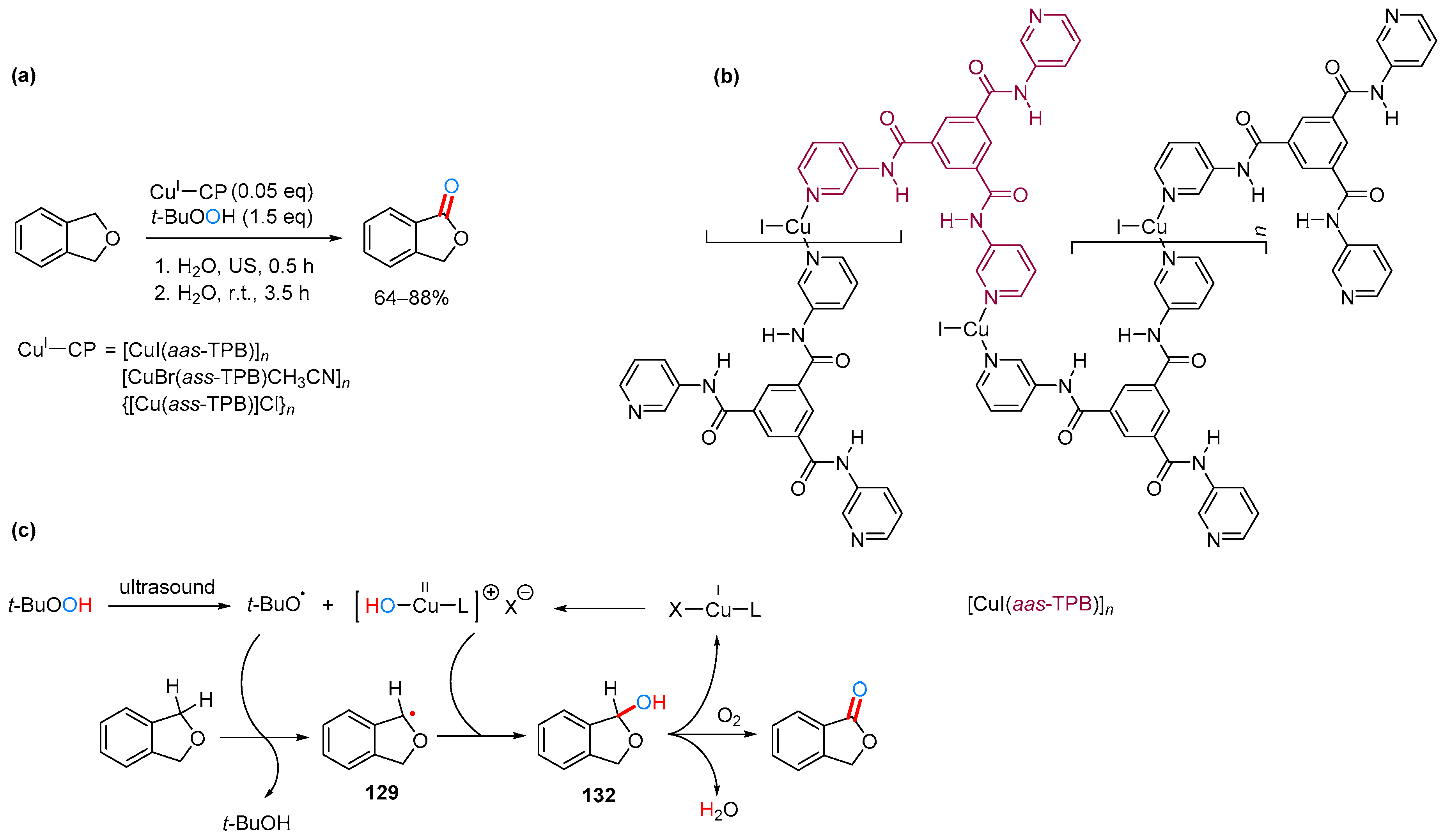
2.3.2. By Oxidation of 1,3-dihydroisobenzofuran in Homogeneous Phase
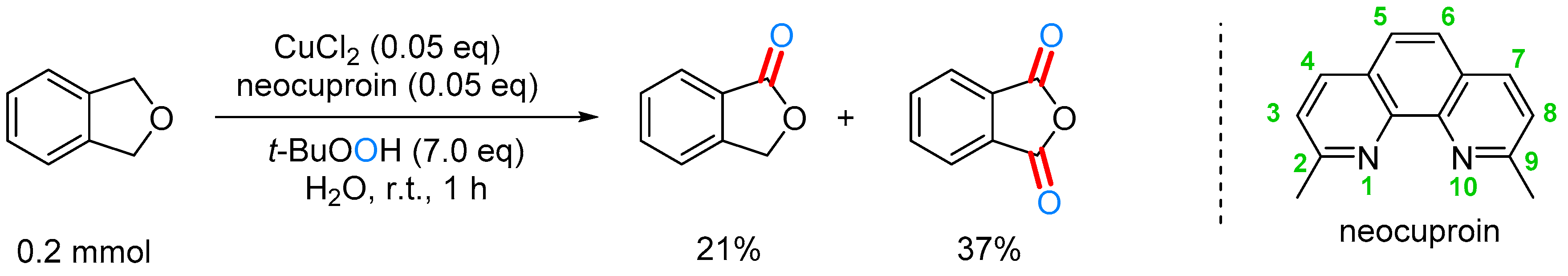
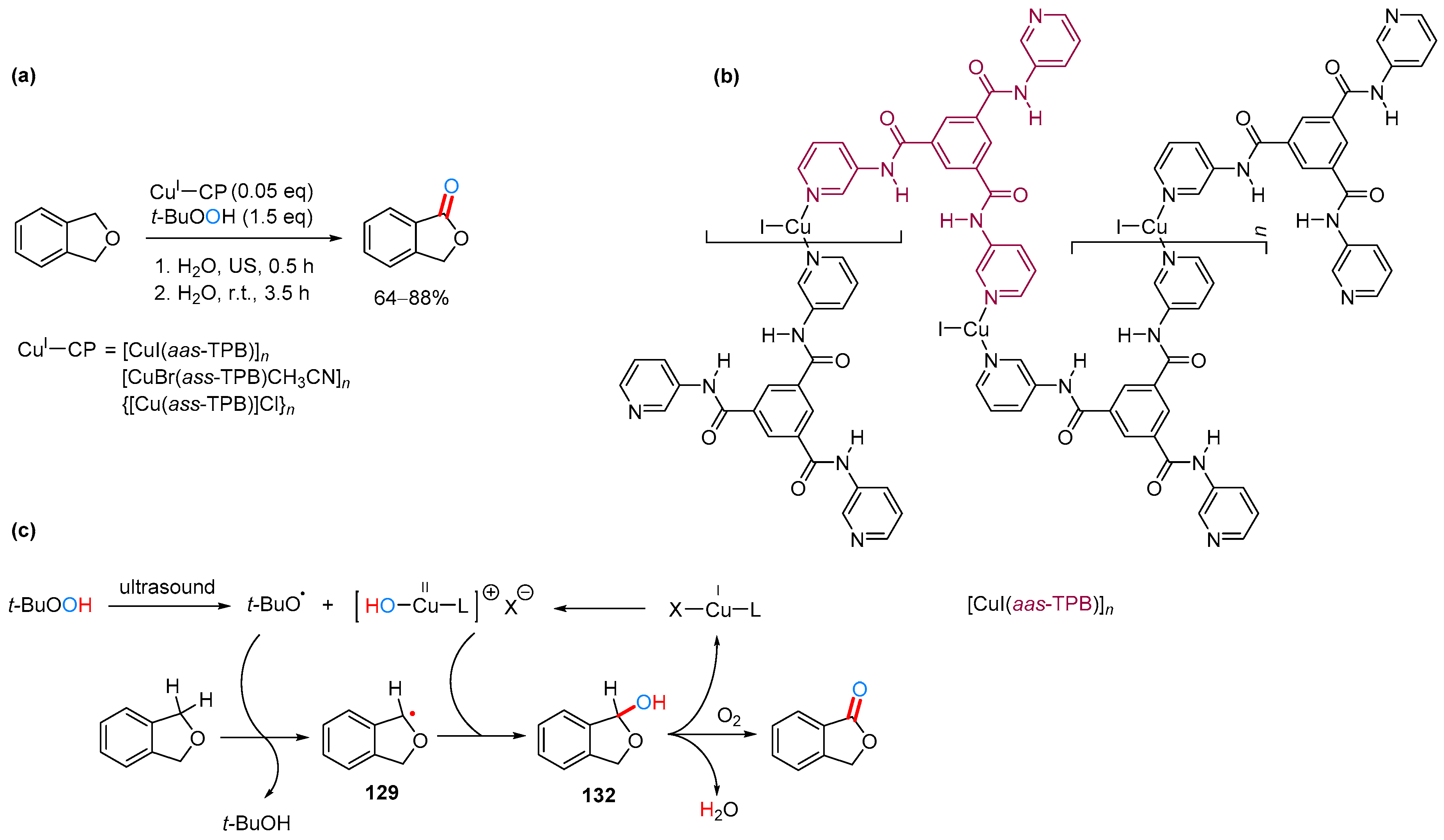
2.3.3. By Oxidation of 1,3-dihydroisobenzofuran in Heterogeneous Phase
3. α,β-Butenolides
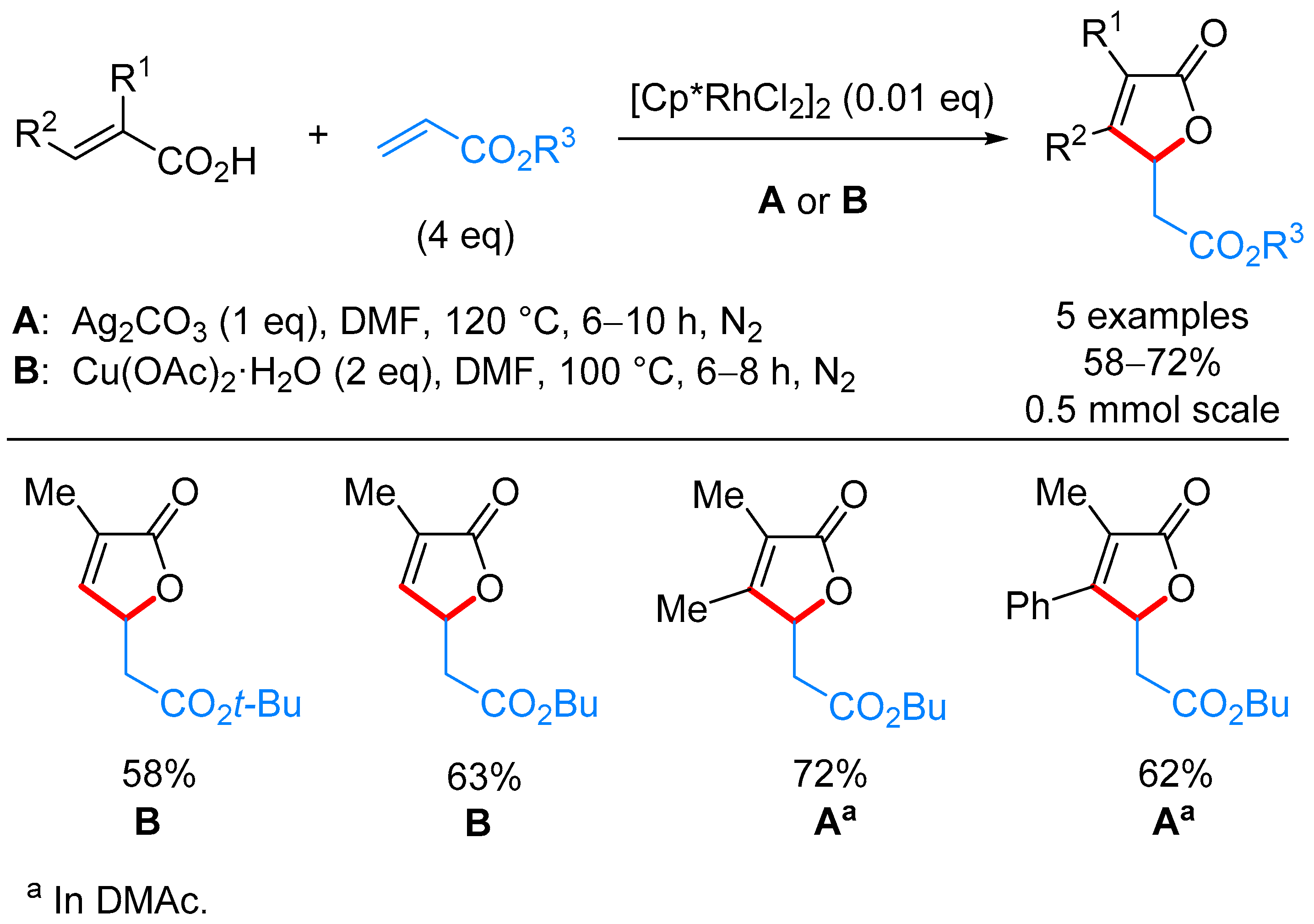
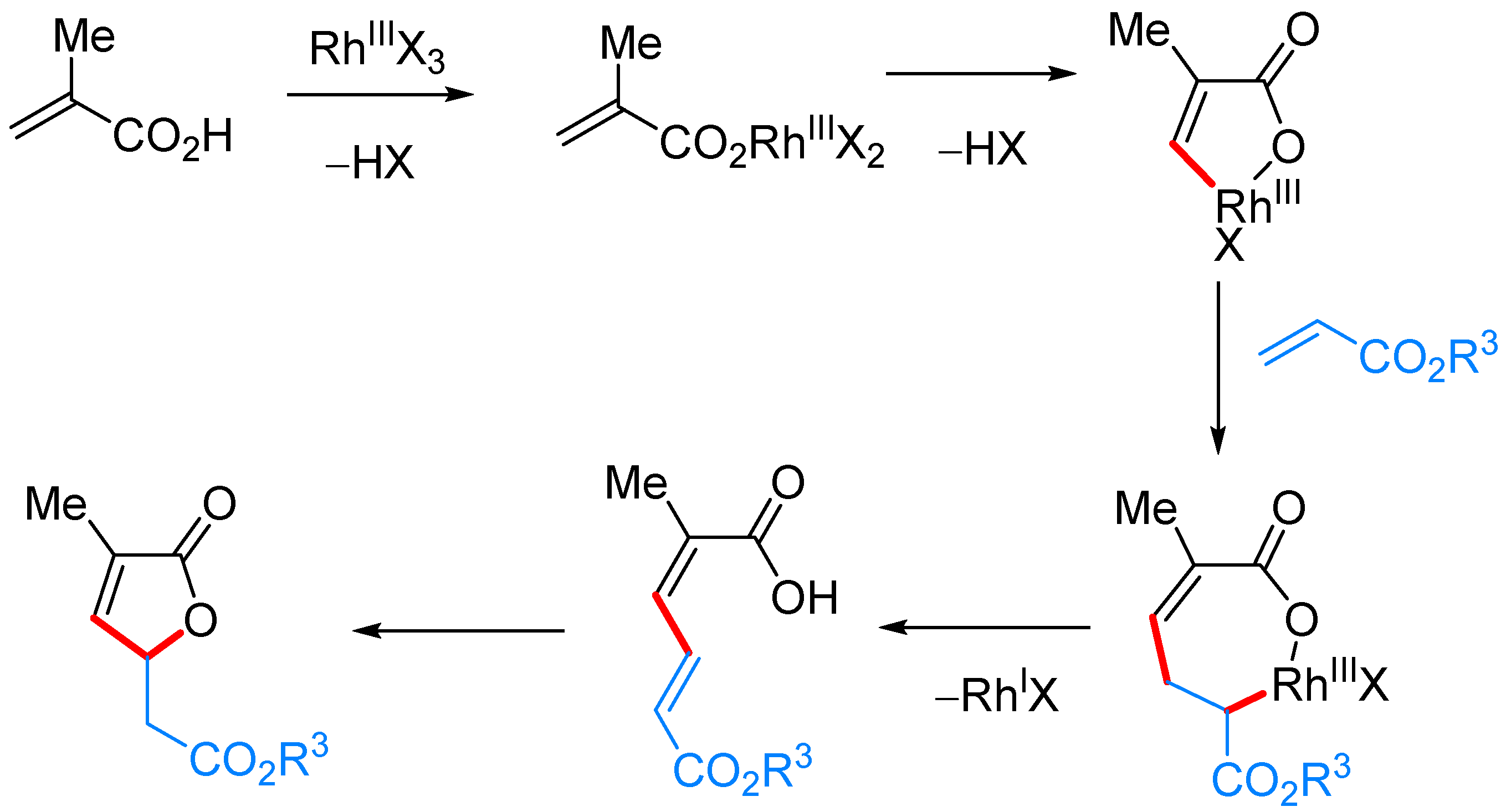
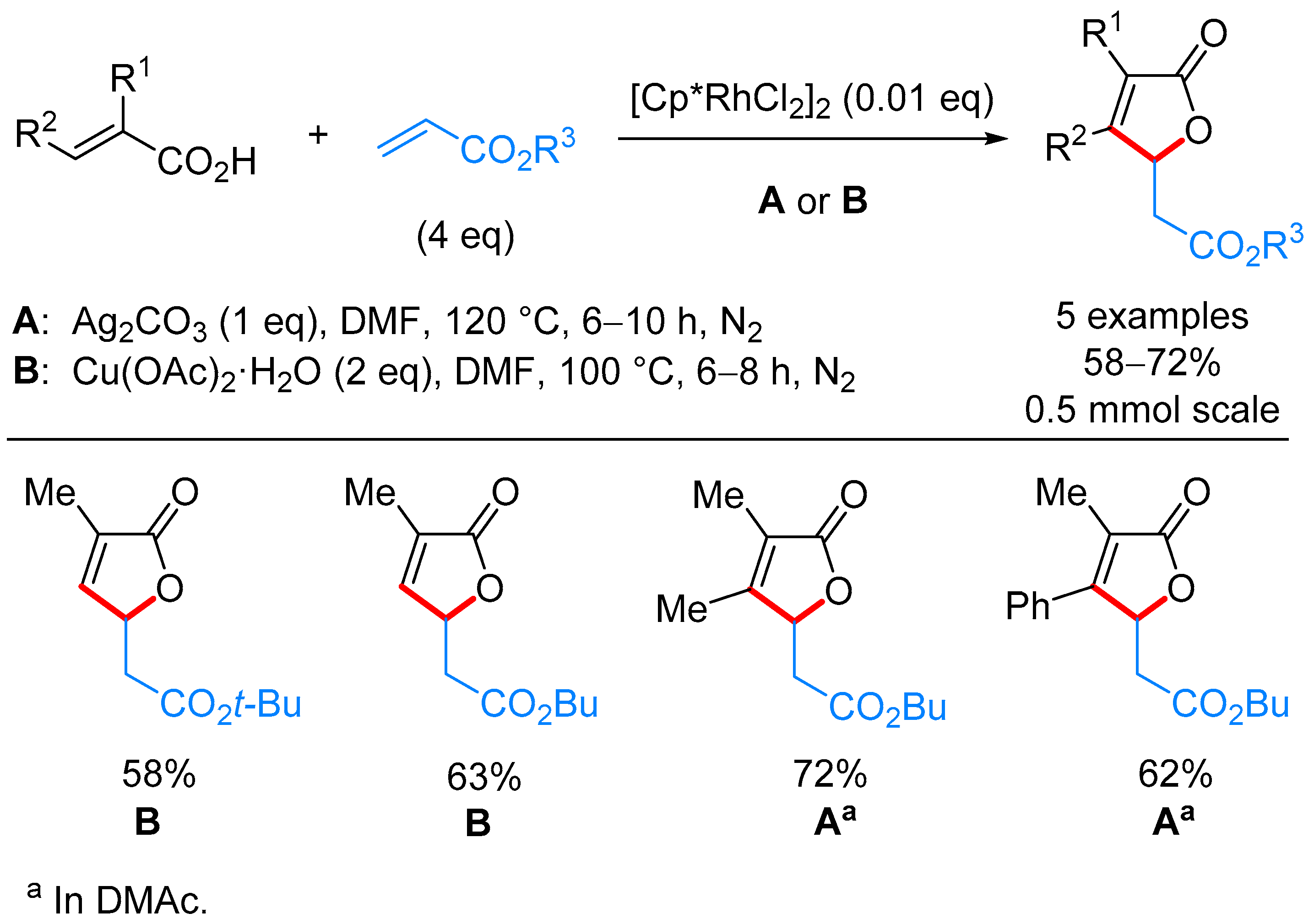
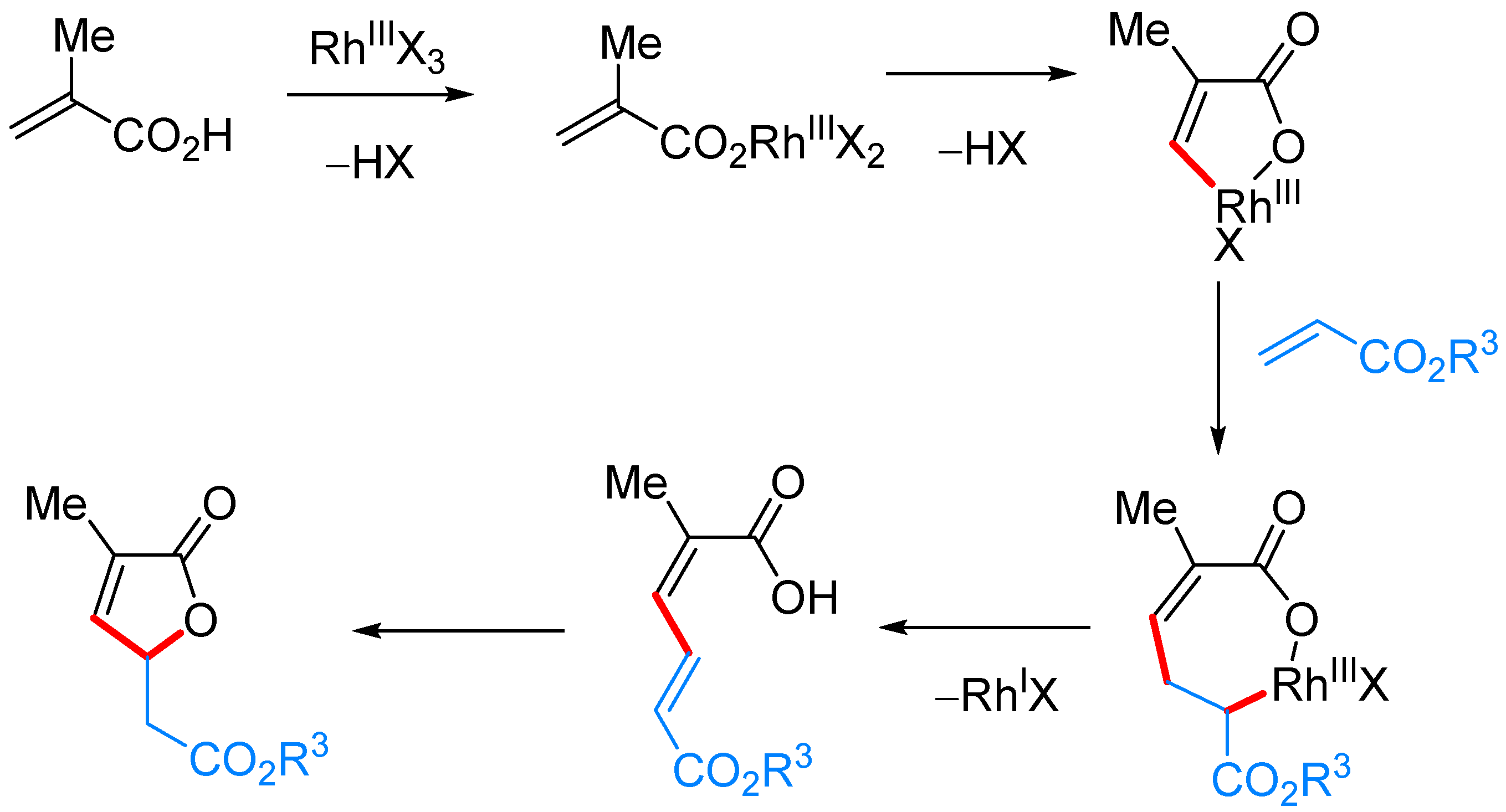
3.1. γ-Substituted α,β-butenolides
γ-Alkyl-α,β-butenolides
3.2. γ,γ-Disubstituted α,β-butenolides
3.2.1. γ-Alkyl-γ-vinyl-α,β-butenolides
3.2.2. γ-Alkylidenebutenolides
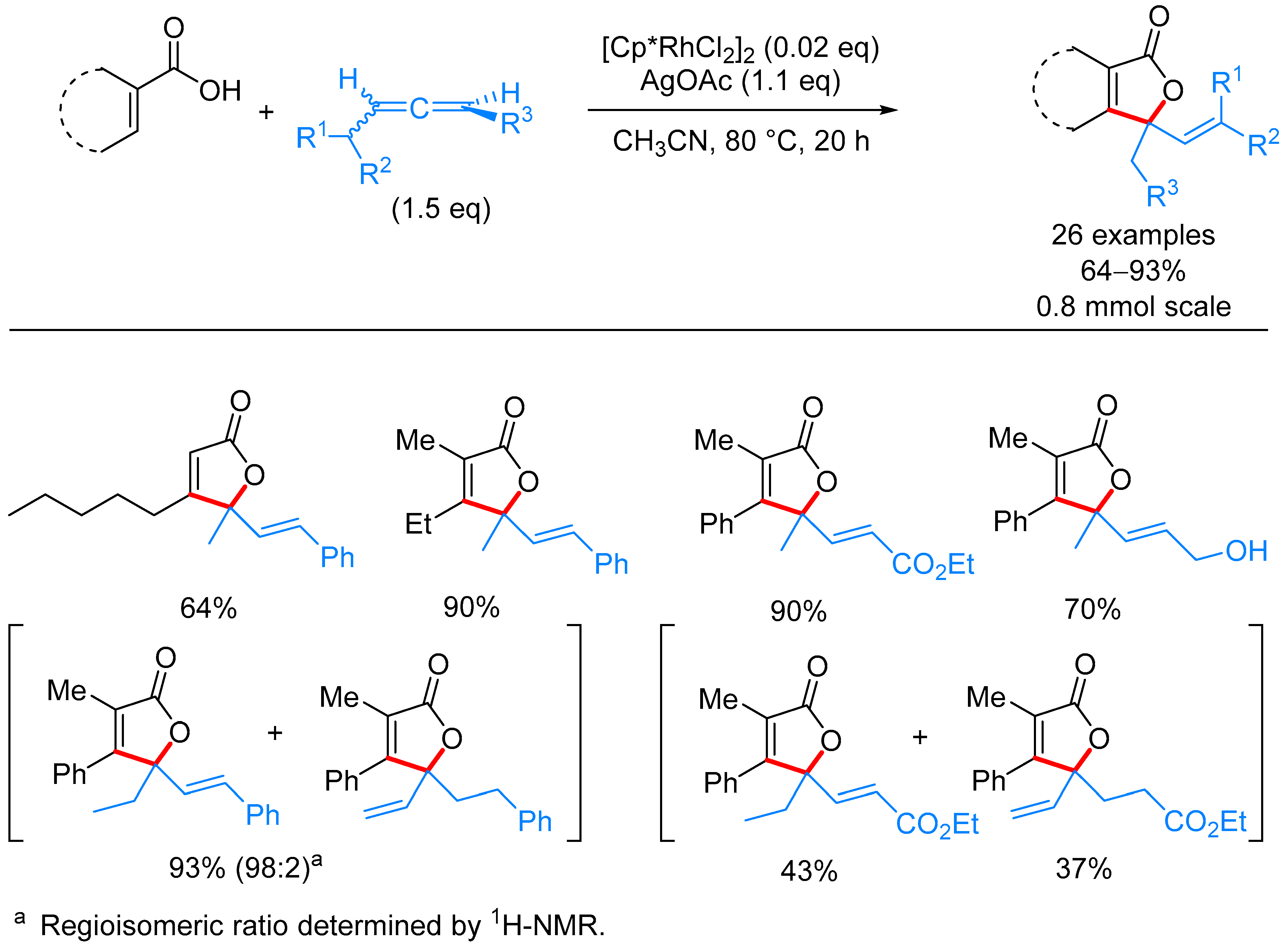
3.3. α,β-Disubstituted α,β-butenolides
4. Conclusions
- (1)
- All the reactions are catalyzed by a transition metal (usually RhI, RhIII, or RuII); most of them take place in homogeneous phase and follow an ionic mechanism.
- (2)
- Oxidative cyclizations require the use of an oxidant, either metallic (CuII or AgI) or organic (t-BuOOH); O2 can be used as the final oxidant in a few cases.
- (3)
- Phthalides are easier to obtain than α,β-butenolides due to the presence of phenyl ring, which assists the C—H activation step.
- (4)
- Several methodologies have been developed for the direct or semi-direct synthesis of 3-substituted phthalides; only a few are available for the preparation of 3,3-disubstituted phthalides and α,β-butenolides; none have been reported for the synthesis of spirophthalides or polymeric phthalides. Furthermore, there is no direct method available to introduce two fully saturated substituents in position 3. Phthalide disubstitution in position 3 through direct methods is made it difficult by the limited availability of unsaturated alkynes and allenes. Isolation from natural sources and classical multi-step synthesis are currently the only alternative for phtalides and butenolides that cannot be obtained by direct methods.
- (5)
- Despite the advantage provided in terms of sustainability, most methodologies still suffer from several limitations: (a) the use of expensive, non-recyclable catalysts (such as Rh or Pd), which compromises their application on a large scale; (b) the use of metal oxidants, which generate waste undermining the sustainability of the reaction; (c) the lack of enantioselectivity (except for ketone hydroacylation); and (d) the relatively small diversity of products. The reaction scope is quite large in terms of number (with over 20 examples of products reported in most cases), but the methodologies have usually been applied to the synthesis of simple structures rather than large drug-like molecules. Naturally occurring, reactive functional groups suitable for further functionalization, such as OH and NH2, are usually not tolerated on the coupling partners.
- (6)
- The development of new methods for the synthesis of heterocycles (including phthalides and butenolides) via C—H bond functionalization is made challenging by two aspects: (a) the large number of factors (substrate, catalyst, oxidant, additives, solvent, as well as reagents ratio and concentration) that affect chemo- and regio-selectivity and are difficult to control; and (b) the paucity of comprehensive mechanistic studies, which are necessary to fully rationalize the results. The available mechanistic studies mostly consist of deuterium-labelling experiments and control experiments that focus on the C—H bond activation step and the identification of some intermediates. The exact nature of the active species as well as the role of oxidant, additive, solvent, and metal ligand are still poorly understood despite the extensive screening that is performed to optimize the reaction conditions. In particular, it is unclear whether the metal oxidant may contribute to catalysis, especially considering the fact that it is used in large (100–1000-fold) excess with respect to catalyst. The isolation and characterization of reactive intermediates would shed light on the mechanism, helping the design of chiral ligands and the development of analogous reactions.
Acknowledgments
Conflicts of Interest
References
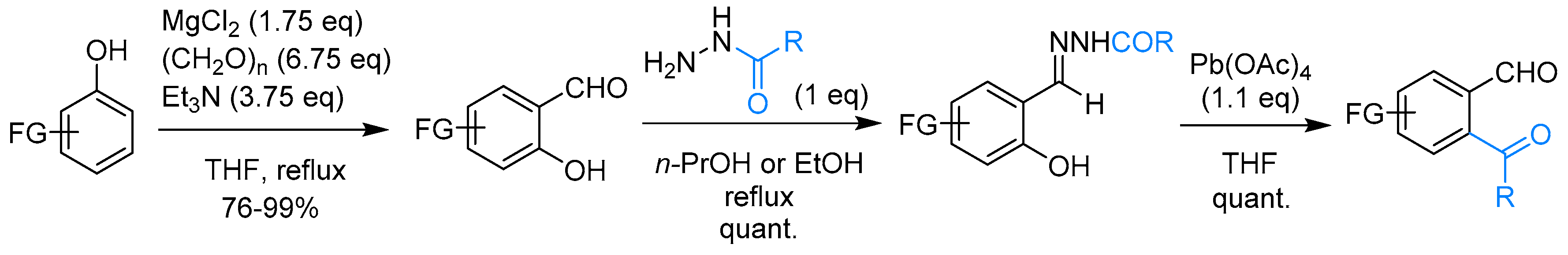
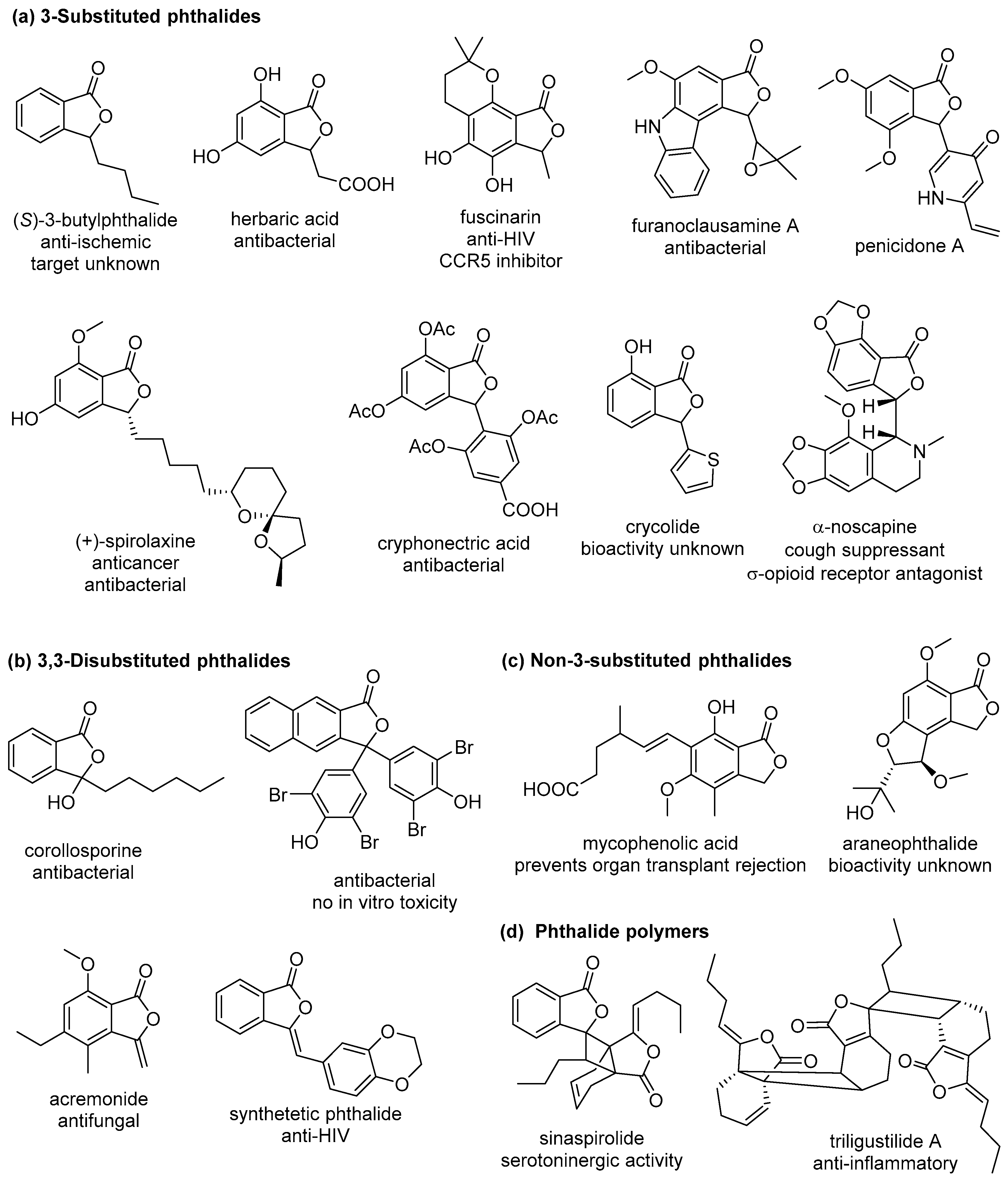
- León, A.; Del-Ángel, M.; Ávila, J.L.; Delgado, G. Phthalides: Distribution in Nature, Chemical Reactivity, Synthesis, and Biological Activity. In Progress in the Chemistry of Organic Natural Products 104; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J.I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 127–246. [Google Scholar]
- Lin, G.; Chan, S.S.-K.; Chung, H.-S.; Li, S.-L. Chemistry and biological activities of naturally occurring phthalides. Stud. Nat. Prod. Chem. 2005, 32, 611–669. [Google Scholar]
- Beck, J.J.; Chou, S.-C. The Structural Diversity of Phthalides from the Apiaceae. J. Nat. Prod. 2007, 70, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, C.; Gao, F.; Fu, Q.; Fu, C.; He, Y.; Zhang, J. A systematic review on the rhizome of Ligusticum chuanxiong Hort. (Chuanxiong). Food Chem. Toxicol. 2018, 119, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Fei, M.; Longjian, H.; Yong, Z.; Yuchen, P.; Changhong, X.; Yipu, F.; Xiaoliang, W.; Ying, P. Dl-3-n-Butylphthalide (NBP): A Promising Therapeutic Agent for Ischemic Stroke. CNS Neurol. Disord. Drug Targets 2018, 17, 338–347. [Google Scholar]
- Clinical Guidelines for Transplant Medications. BC Transplant, 25 June 2018; p. 103. Available online: http://www.transplant.bc.ca/Documents/Health%20Professionals/Clinical%20guidelines/Clinical%20Guidelines%20for%20Transplant%20Medications.pdf (accessed on 31 January 2019).
- Zou, J.; Chen, G.-D.; Zhao, H.; Huang, Y.; Luo, X.; Xu, W.; He, R.-R.; Hu, D.; Yao, X.-S.; Gao, H. Triligustilides A and B: Two Pairs of Phthalide Trimers from Angelica sinensis with a Complex Polycyclic Skeleton and Their Activities. Org. Lett. 2018, 20, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Giap, T.H.; Dung, N.A.; Thoa, H.T.; Dang, N.H.; Dat, N.T.; Hang, N.T.M.; Van Cuong, P.; Van Hung, N.; Van Minh, C.; Mishchenko, N.P.; et al. Phthalides and Other Metabolites from Roots of Ligusticum wallichii. Chem. Nat. Compd. 2018, 54, 34–37. [Google Scholar] [CrossRef]
- Zheng, N.; Yao, F.; Liang, X.; Liu, Q.; Xu, W.; Liang, Y.; Liu, X.; Li, J.; Yang, R. A new phthalide from the endophytic fungus Xylaria sp. GDG-102. Nat. Prod. Res. 2018, 32, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Fañanás-Mastral, M.; Feringa, B.L. Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones. Chem. Rev. 2017, 117, 10502–10566. [Google Scholar] [CrossRef] [PubMed]
- Waldie, T.; McCulloch, H.; Leyser, O. Strigolactones and the control of plant development: Lessons from shoot branching. Plant J. 2014, 79, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Hanada, A.; Yoshida, S.; Akiyama, K.; Arite, T.; Takeda-Kamiya, N.; Magome, H.; Kamiya, Y.; Shirasu, K.; Yoneyama, K.; et al. Inhibition of shoot branching by new terpenoid plant hormones. Nature 2008, 455, 195. [Google Scholar] [CrossRef] [PubMed]
- Public Assessment Report for STRIGOL Paediatric 6.86 g Powder for Oral Solution and STRIGOL 13.72 g Powder for Oral Solution. Medicines & Healthcare products Regulatory Agency; p. 24. Available online: http://www.mhra.gov.uk/home/groups/par/documents/websiteresources/con745788.pdf (accessed on 31 January 2019).
- Kitani, S.; Miyamoto, K.T.; Takamatsu, S.; Herawati, E.; Iguchi, H.; Nishitomi, K.; Uchida, M.; Nagamitsu, T.; Omura, S.; Ikeda, H.; et al. Avenolide, a Streptomyces hormone controlling antibiotic production in Streptomyces avermitilis. Proc. Natl. Acad. Sci. USA 2011, 108, 16410–16415. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, R.; Pahari, P.; Mal, D. Phthalides and Phthalans: Synthetic Methodologies and Their Applications in the Total Synthesis. Chem. Rev. 2014, 114, 6213–6284. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. (Ed.) C–H Bond Activation in Organic Synthesis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Li, C.-J. (Ed.) From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Kumar, R.; Van der Eycken, E.V. Recent approaches for C–C bond formation via direct dehydrative coupling strategies. Chem. Soc. Rev. 2013, 42, 1121–1146. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, C.-J. A novel rhodium-catalyzed cascade cyclization. Direct synthesis of 3-substituted phthalides from aldehydes and aromatic acids. Adv. Synth. Catal. 2012, 354, 2933–2938. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.W.; Juwaini, N.A.B.; Seayad, J. Rhodium(III)-Amine Dual Catalysis for the Oxidative Coupling of Aldehydes by Directed C–H Activation: Synthesis of Phthalides. Org. Lett. 2013, 15, 5166–5169. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Bergman, R.G.; Ellman, J.A. Rhodium(III)-catalyzed synthesis of phthalides by cascade addition and cyclization of benzimidates with aldehydes. Chem. Sci. 2012, 3, 3088–3092. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Pospech, J. Ruthenium-Catalyzed Oxidative C–H Bond Alkenylations in Water: Expedient Synthesis of Annulated Lactones. Org. Lett. 2011, 13, 4153–4155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, T.; Yan, T.; Cai, M. Recyclable and Reusable [RuCl2(p-cymene)]2/Cu(OAc)2/PEG-400/H2O System for Oxidative C–H Bond Alkenylations: Green Synthesis of Phthalides. J. Org. Chem. 2015, 80, 8849–8855. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Dana, S.; Chowdhury, D.; Baidya, M. RuII-Catalyzed Annulative Coupling of Benzoic Acids with Vinyl Sulfone via Weak Carboxylate-Assisted C–H Bond Activation. Asian J. Org. Chem. 2018, 7, 1302–1306. [Google Scholar] [CrossRef]
- Bechtoldt, A.; Tirler, C.; Raghuvanshi, K.; Warratz, S.; Kornhaaß, C.; Ackermann, L. Ruthenium Oxidase Catalysis for Site-Selective C–H Alkenylations with Ambient O2 as the Sole Oxidant. Angew. Chem. Int. Ed. 2016, 55, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Bechtoldt, A.; Baumert, M.E.; Vaccaro, L.; Ackermann, L. Ruthenium(II) oxidase catalysis for C–H alkenylations in biomass-derived γ-valerolactone. Green Chem. 2018, 20, 398–402. [Google Scholar] [CrossRef]
- Zhu, Y.-Q.; Li, J.-X.; Han, T.-F.; He, J.-L.; Zhu, K. Rh(III)-Catalyzed Regioselective Synthesis of Phthalides with Water as the Solvent. Eur. J. Org. Chem. 2017, 2017, 806–811. [Google Scholar] [CrossRef]
- Renzetti, A.; Nakazawa, H.; Li, C.-J. Rhodium-catalysed tandem dehydrogenative coupling–Michael addition: Direct synthesis of phthalides from benzoic acids and alkenes. RSC Adv. 2016, 6, 40626–40630. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhu, C.; Zhao, H.; Su, W. Rh(III)-Catalyzed C–H Olefination of Benzoic Acids under Mild Conditions using Oxygen as the Sole Oxidant. Chem. Asian J. 2016, 11, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gil, K.; Solans-Monfort, X.; Rodriguez-Santiago, L.; Sodupe, M.; Jaque, P. DFT Study on the Relative Stabilities of Substituted Ruthenacyclobutane Intermediates Involved in Olefin Cross-Metathesis Reactions and Their Interconversion Pathways. Organometallics 2014, 33, 6065–6075. [Google Scholar] [CrossRef]
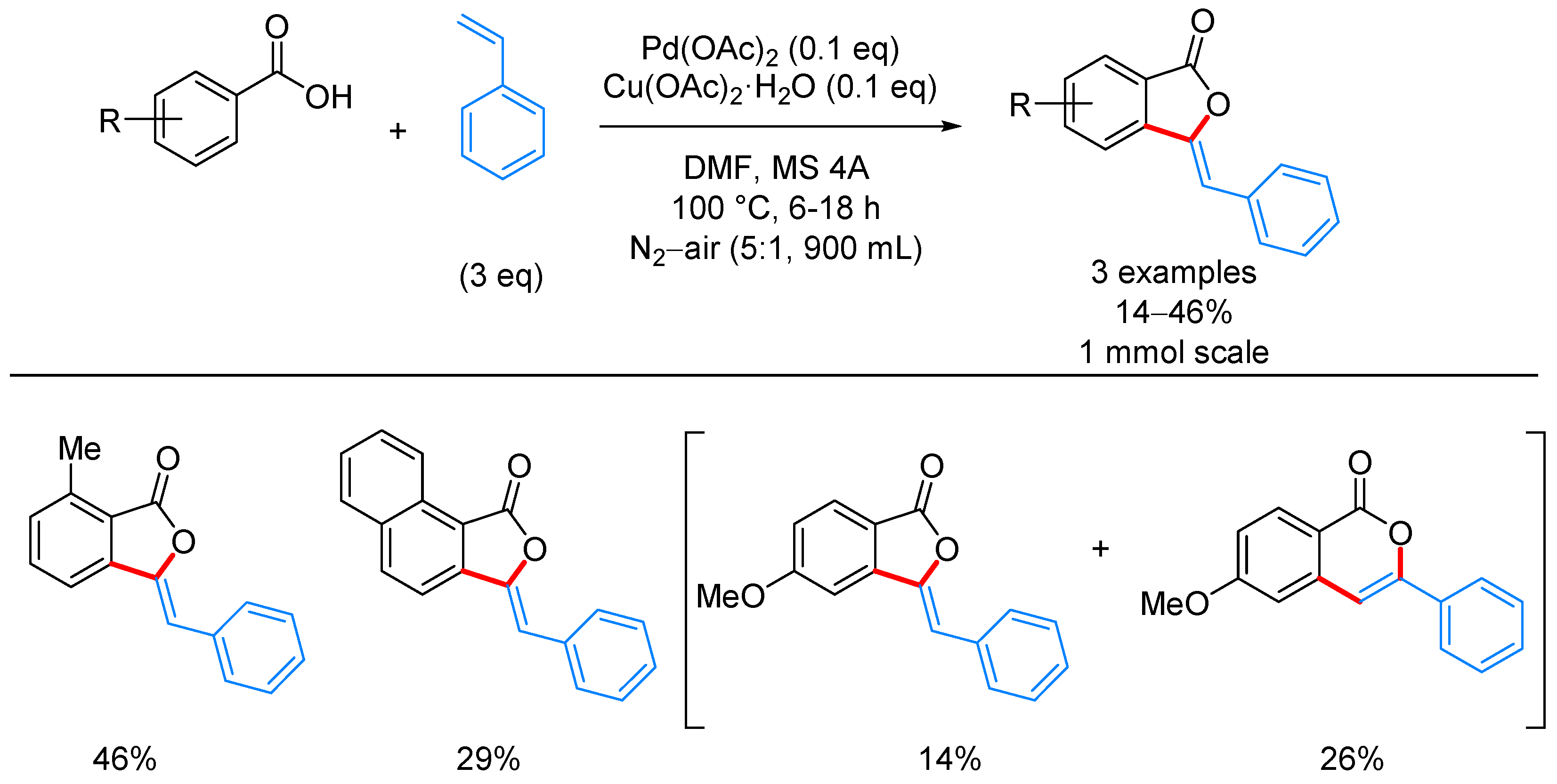
- Miura, M.; Tsuda, T.; Satoh, T.; Pivsa-Art, S.; Nomura, M. Oxidative Cross-Coupling of N-(2′-Phenylphenyl)benzene-sulfonamides or Benzoic and Naphthoic Acids with Alkenes Using a Palladium−Copper Catalyst System under Air. J. Org. Chem. 1998, 63, 5211–5215. [Google Scholar] [CrossRef]
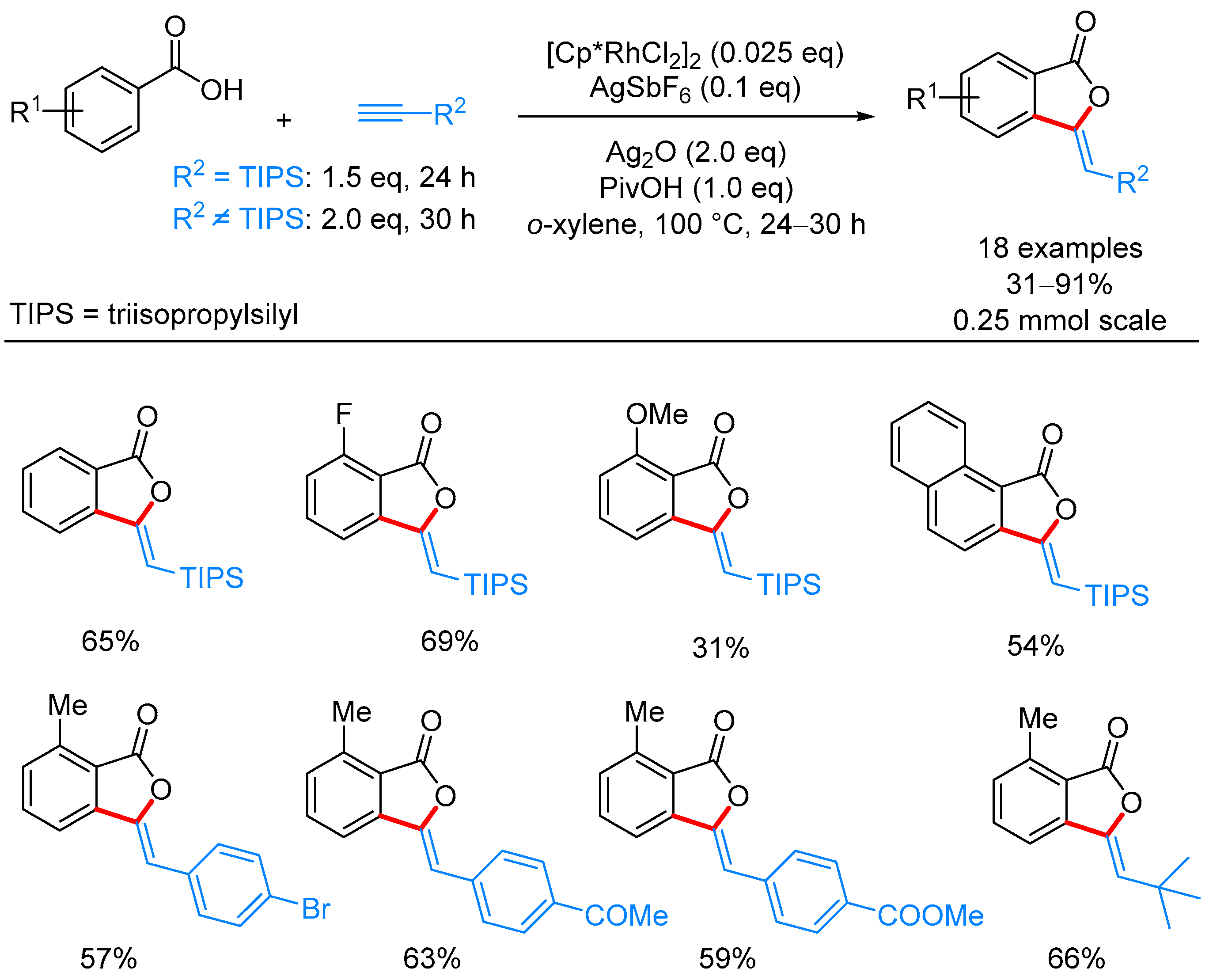
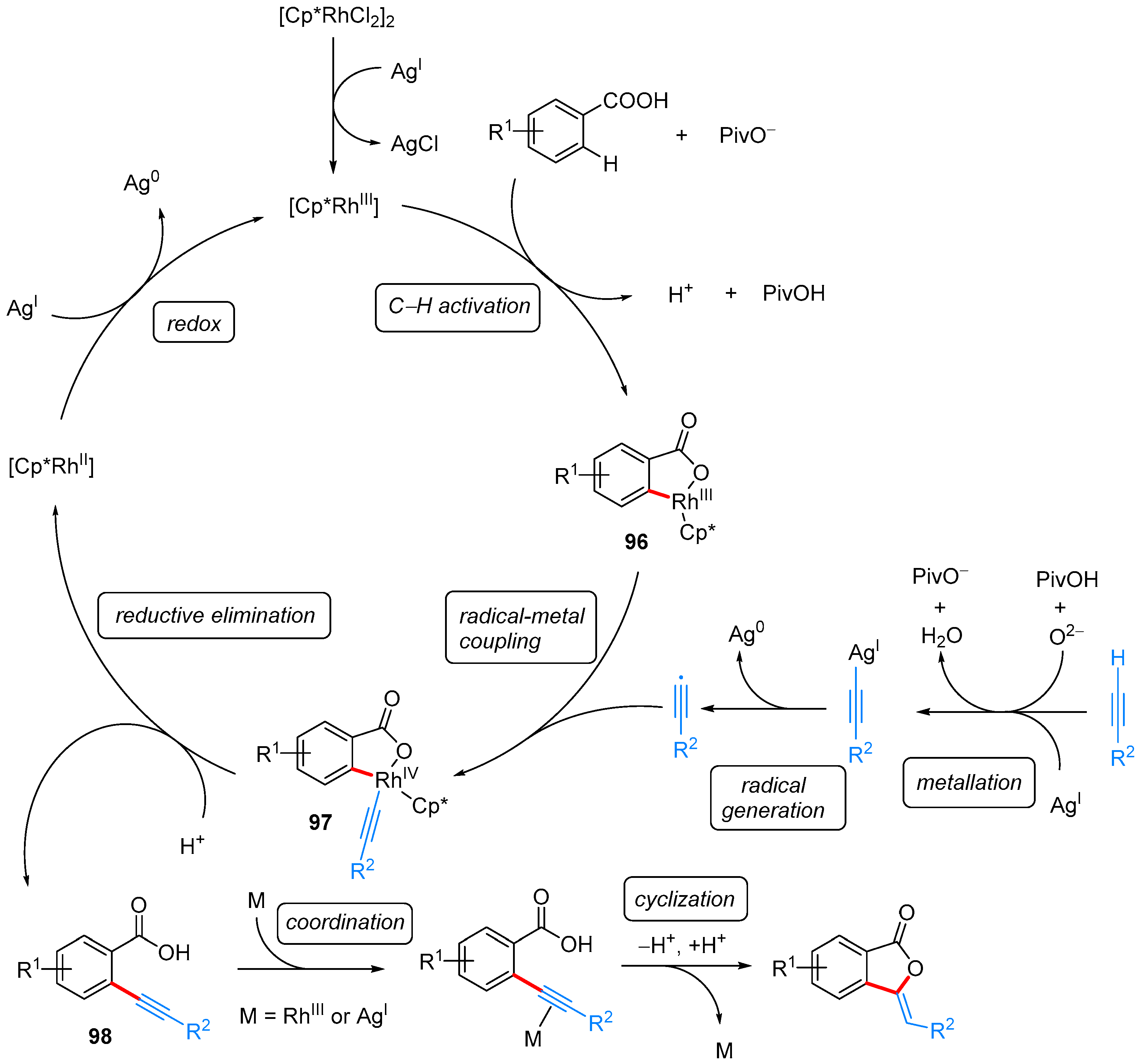
- Ueura, K.; Satoh, T.; Miura, M. An Efficient Waste-Free Oxidative Coupling via Regioselective C–H Bond Cleavage: Rh/Cu-Catalyzed Reaction of Benzoic Acids with Alkynes and Acrylates under Air. Org. Lett. 2007, 9, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Park, J.; Choi, M.; Sharma, S.; Jo, H.; Jeong, T.; Han, S.; Kim, S.; Kim, I.S. Synthesis of Phthalides through Tandem Rhodium-Catalyzed C–H Olefination and Annulation of Benzamides. Eur. J. Org. Chem. 2016, 2016, 3076–3083. [Google Scholar] [CrossRef]
- Li, B.-J.; Wang, H.-Y.; Zhu, Q.-L.; Shi, Z.-J. Rhodium/Copper-Catalyzed Annulation of Benzimides with Internal Alkynes: Indenone Synthesis through Sequential C–H and C–N Cleavage. Angew. Chem. Int. Ed. 2012, 51, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
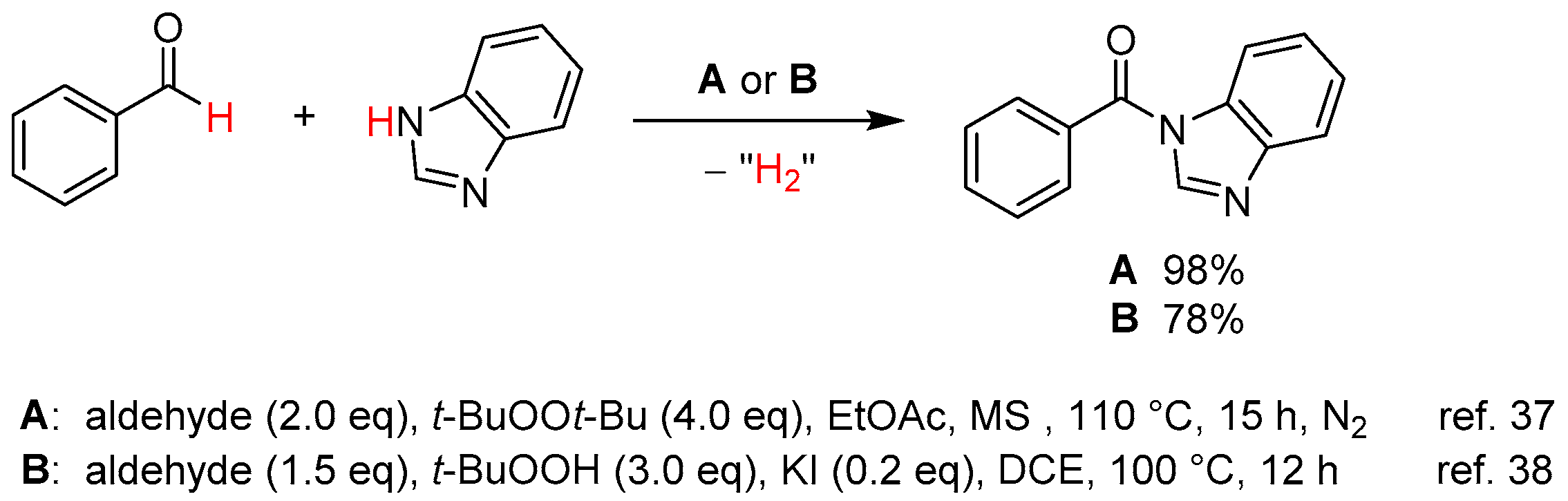
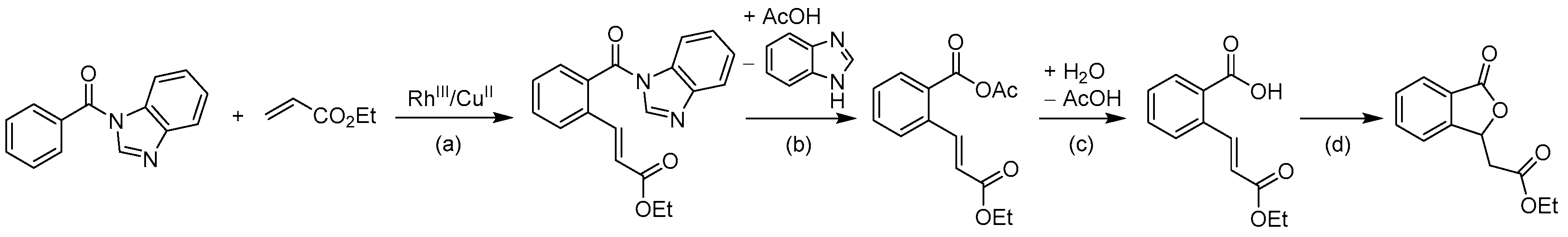
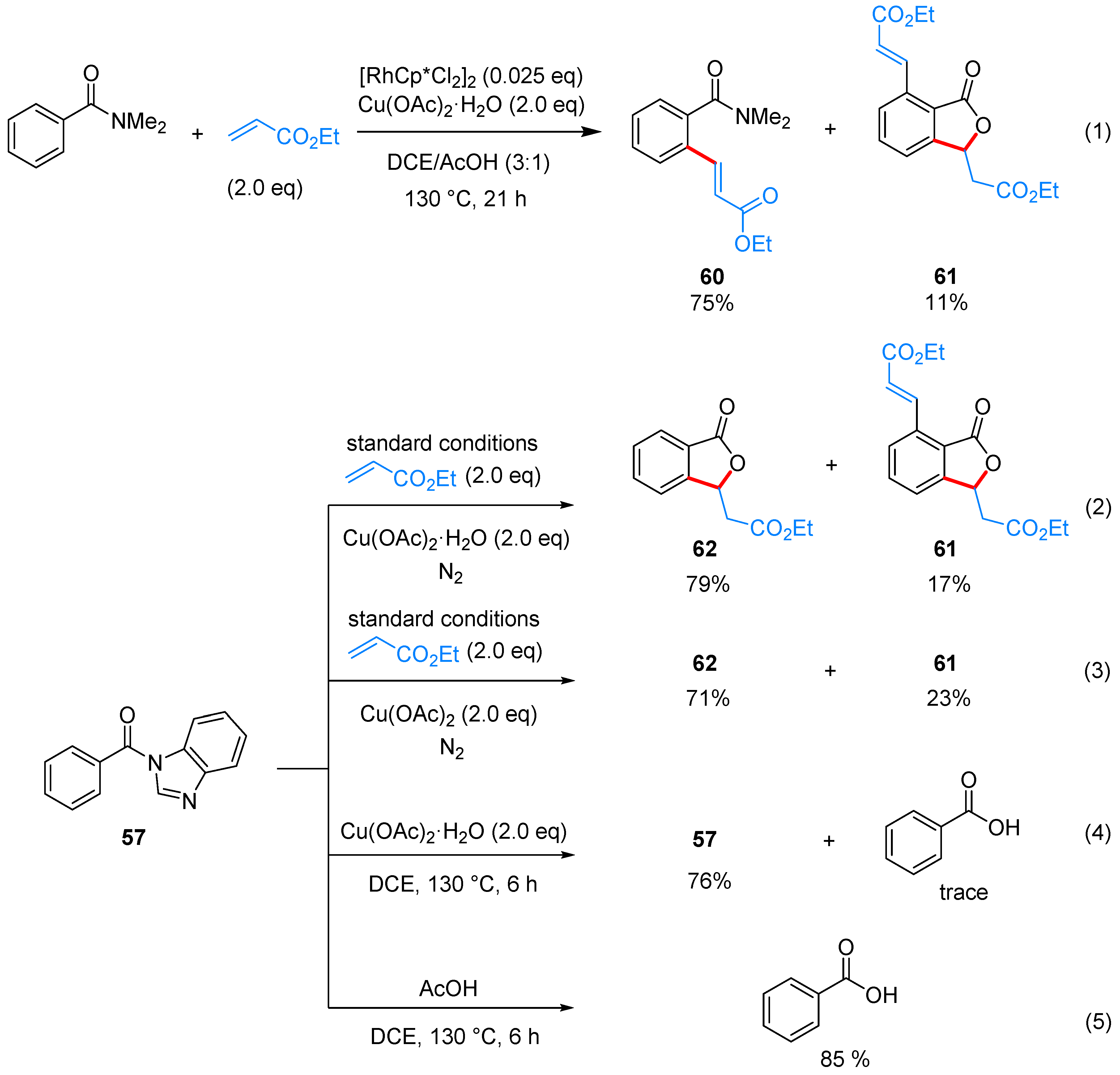
- Yu, L.; Wang, M.; Wang, L. Metal-free cross-dehydrogenative coupling of benzimidazoles with aldehydes to N-acylbenzimidazoles. Tetrahedron 2014, 70, 5391–5397. [Google Scholar] [CrossRef]
- Zhao, J.; Li, P.; Xia, C.; Li, F. Direct N-acylation of azoles via a metal-free catalyzed oxidative cross-coupling strategy. Chem. Commun. 2014, 50, 4751–4754. [Google Scholar] [CrossRef] [PubMed]
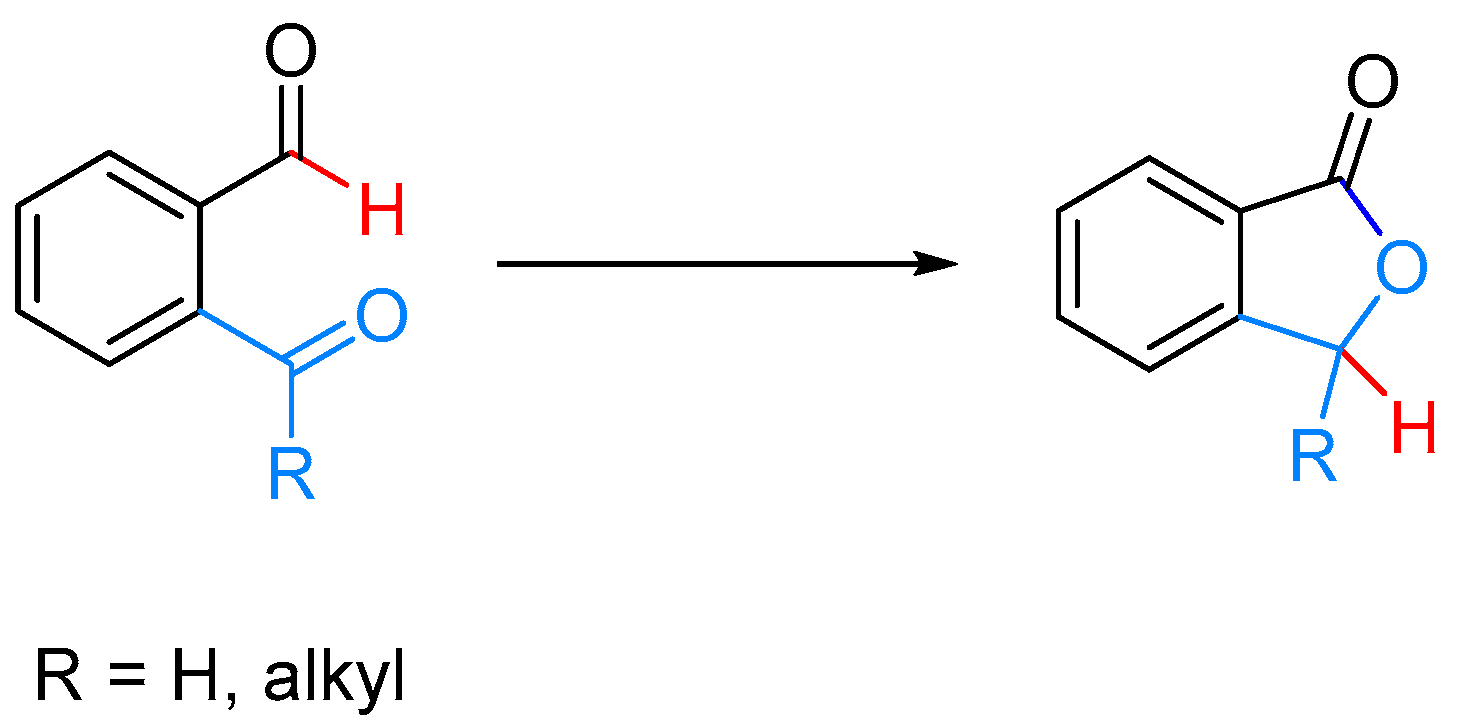
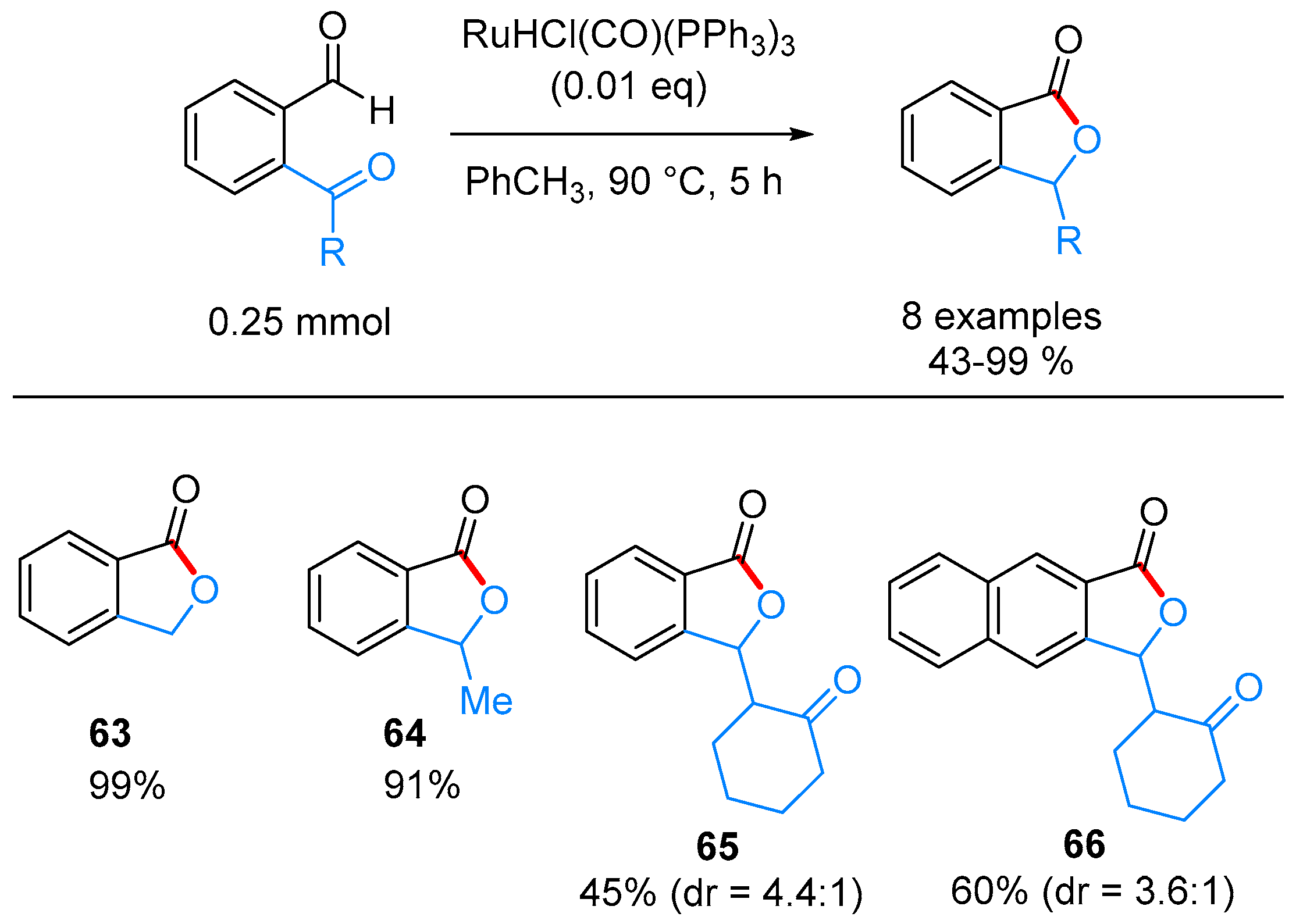
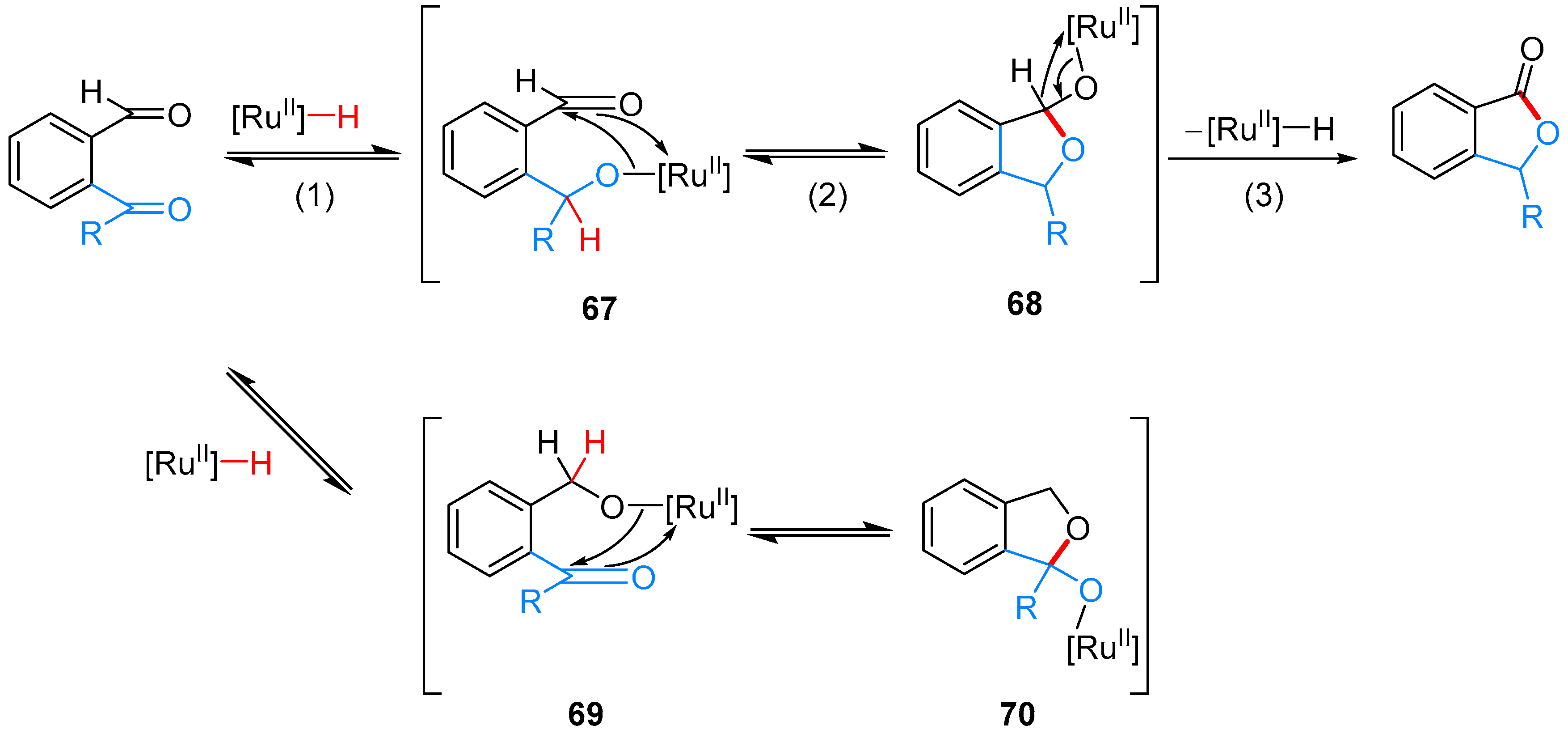
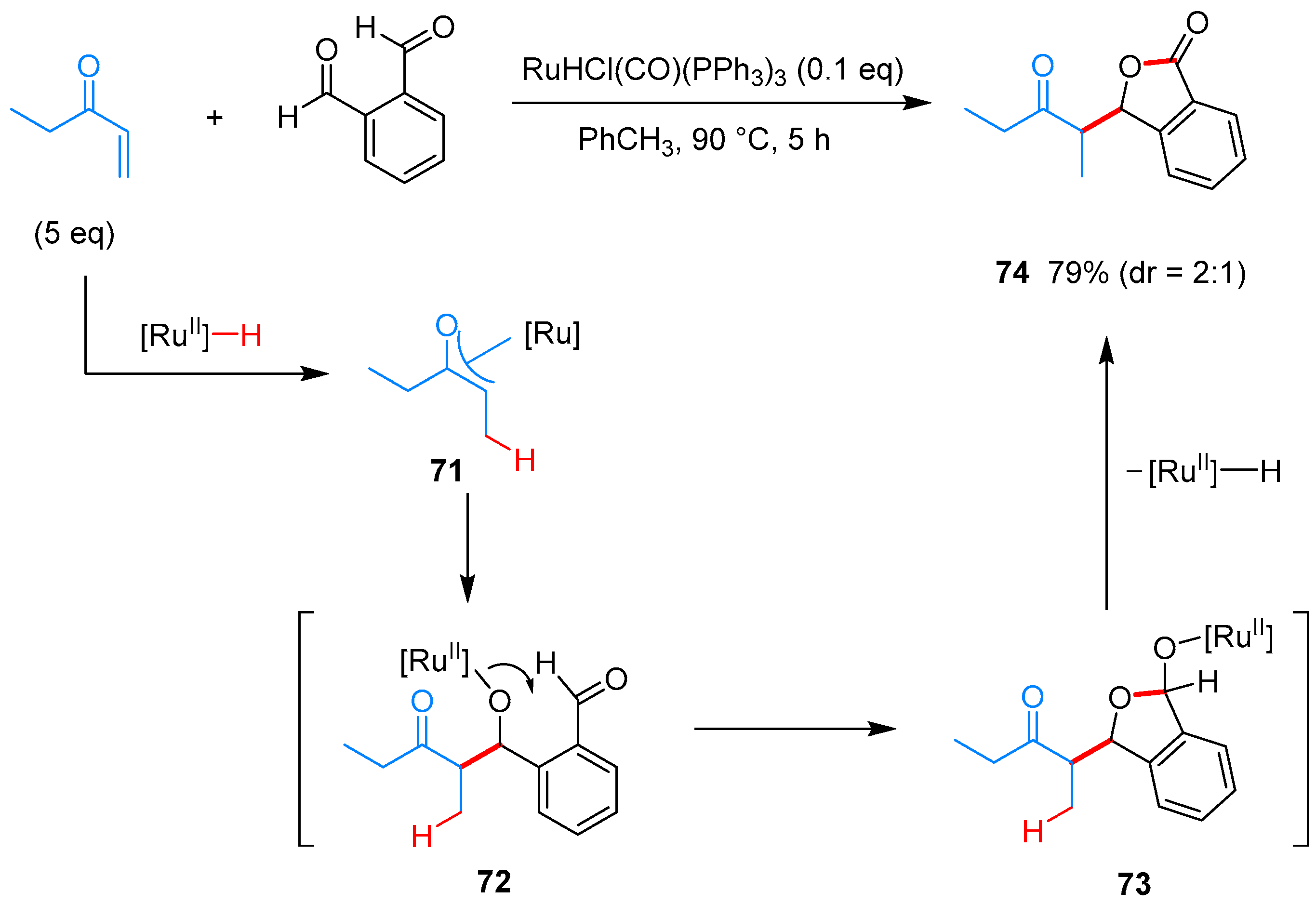
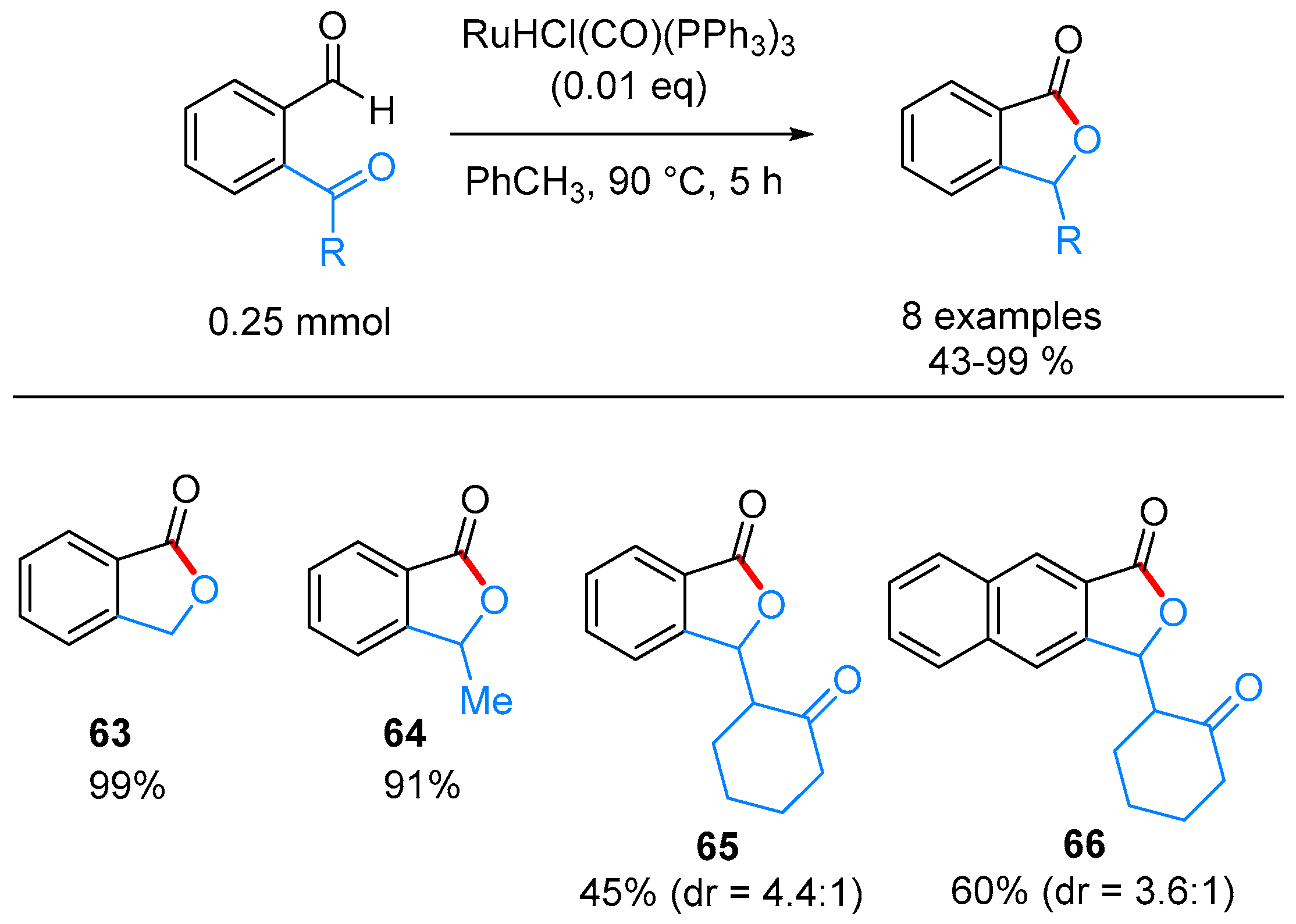
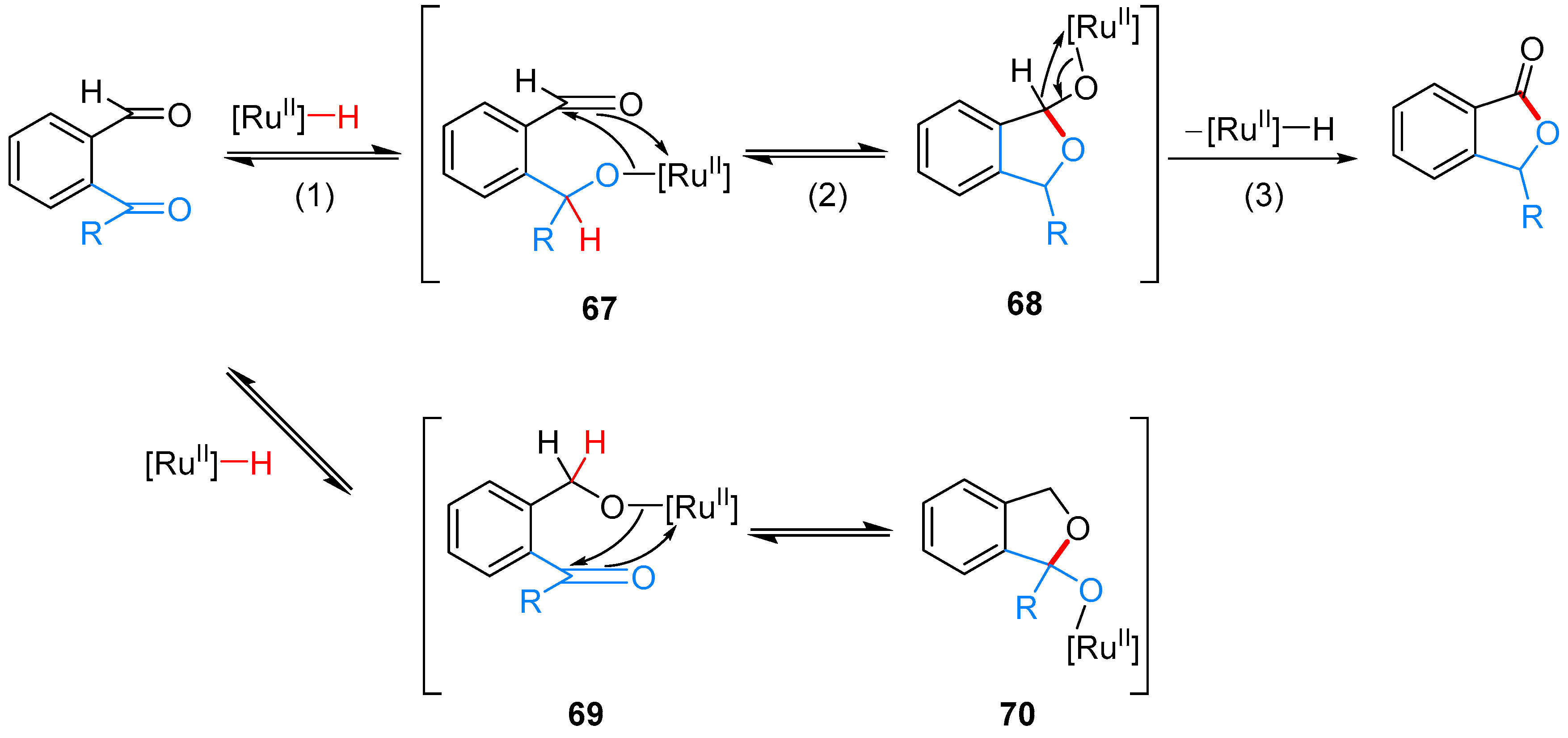
- Omura, S.; Fukuyama, T.; Murakami, Y.; Okamoto, H.; Ryu, I. Hydroruthenation triggered catalytic conversion of dialdehydes and keto aldehydes to lactones. Chem. Commun. 2009, 6741–6743. [Google Scholar] [CrossRef] [PubMed]
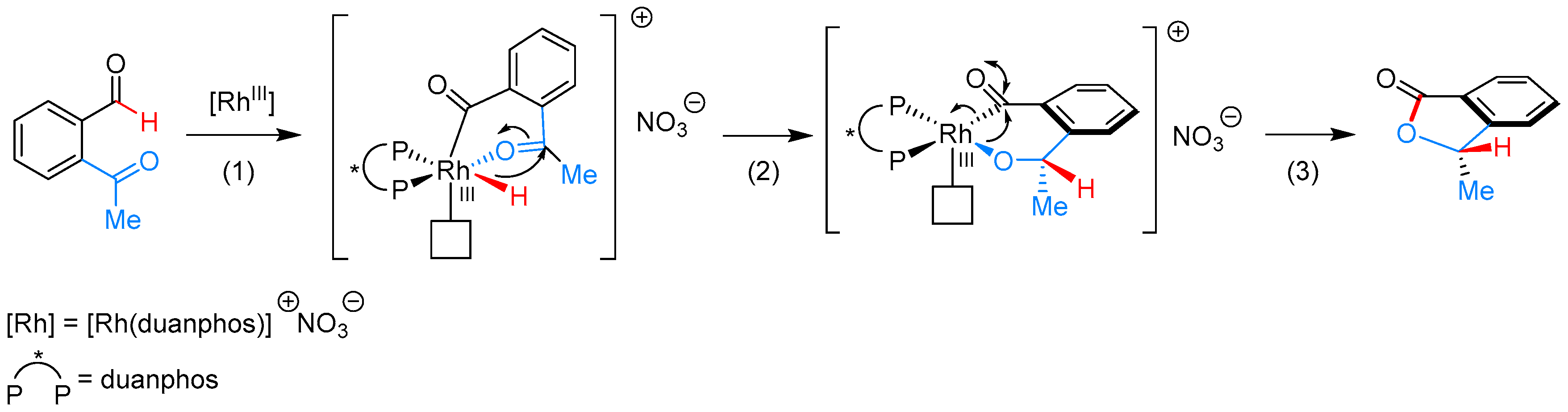
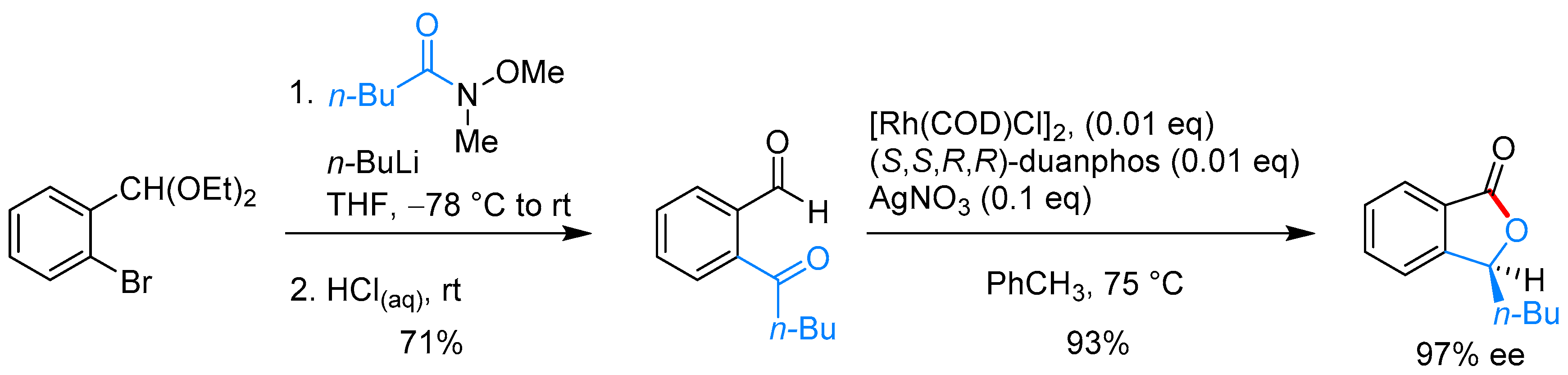
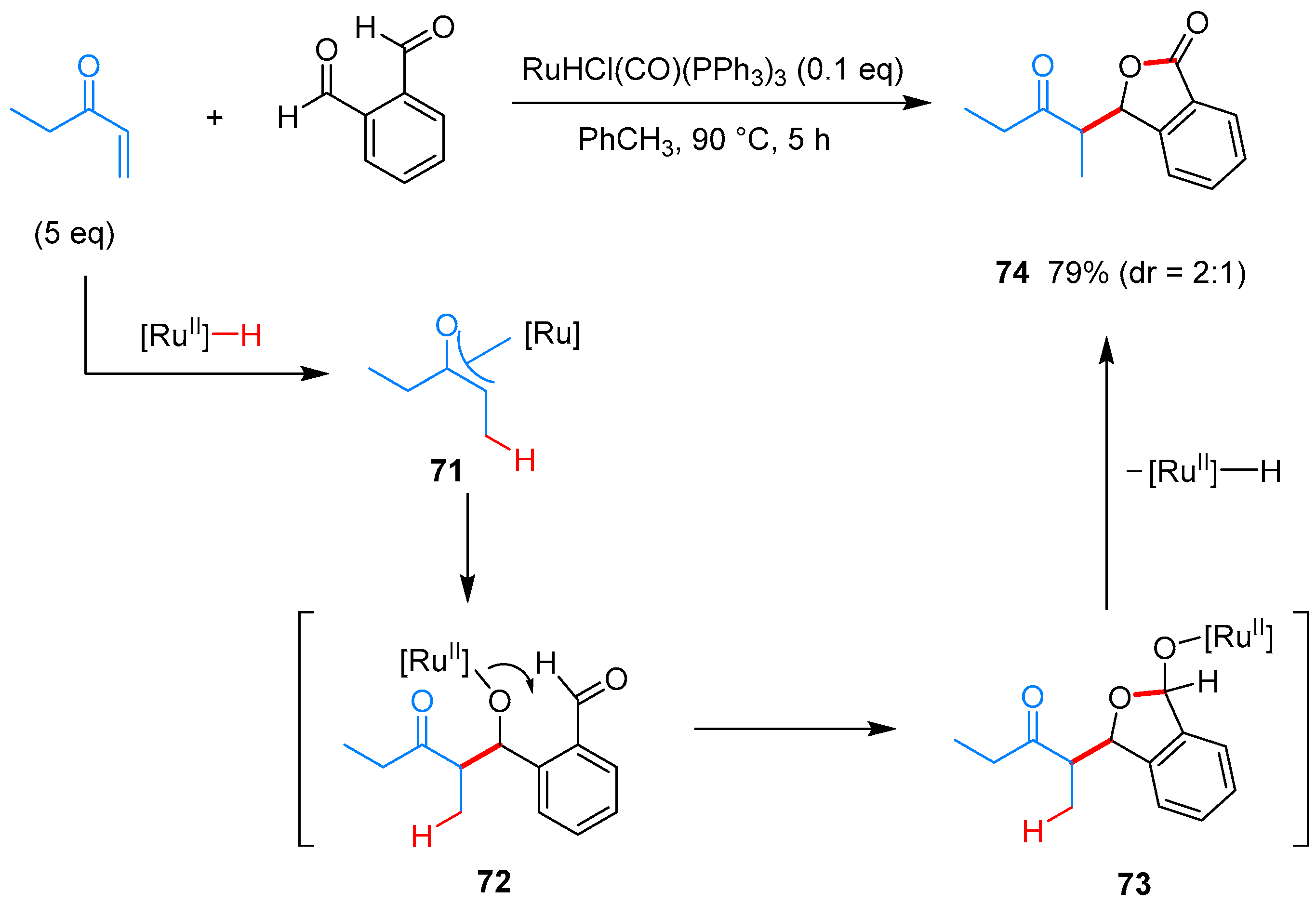
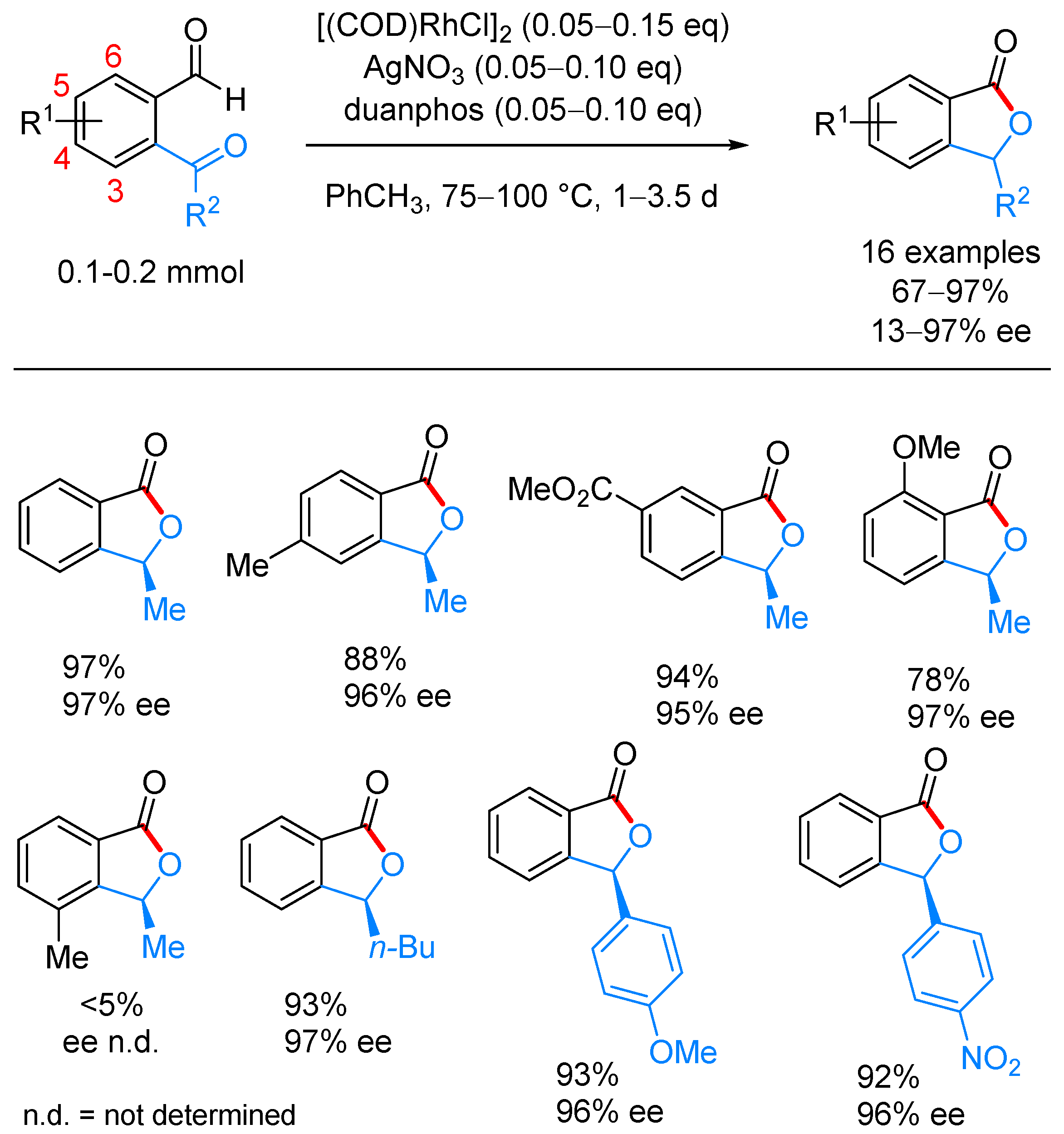
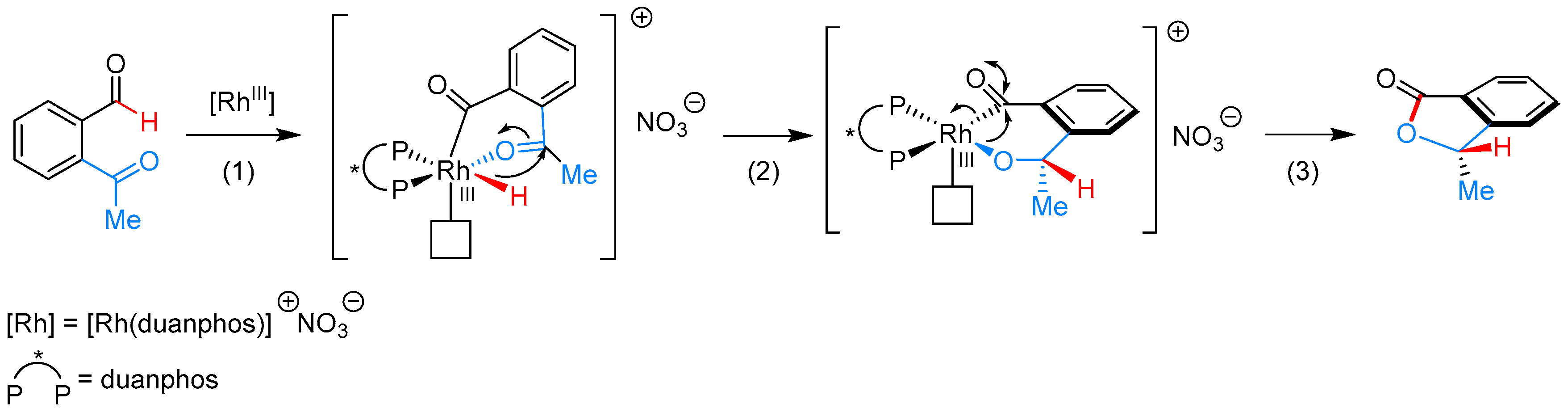
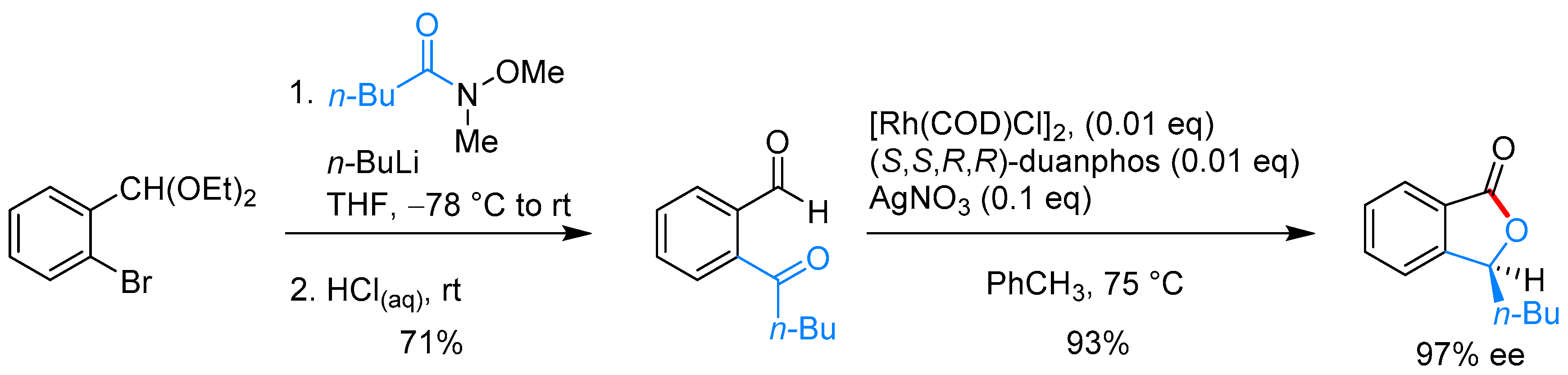
- Phan, D.H.T.; Kim, B.; Dong, V.M. Phthalides by Rhodium-Catalyzed Ketone Hydroacylation. J. Am. Chem. Soc. 2009, 131, 15608–15609. [Google Scholar] [CrossRef] [PubMed]
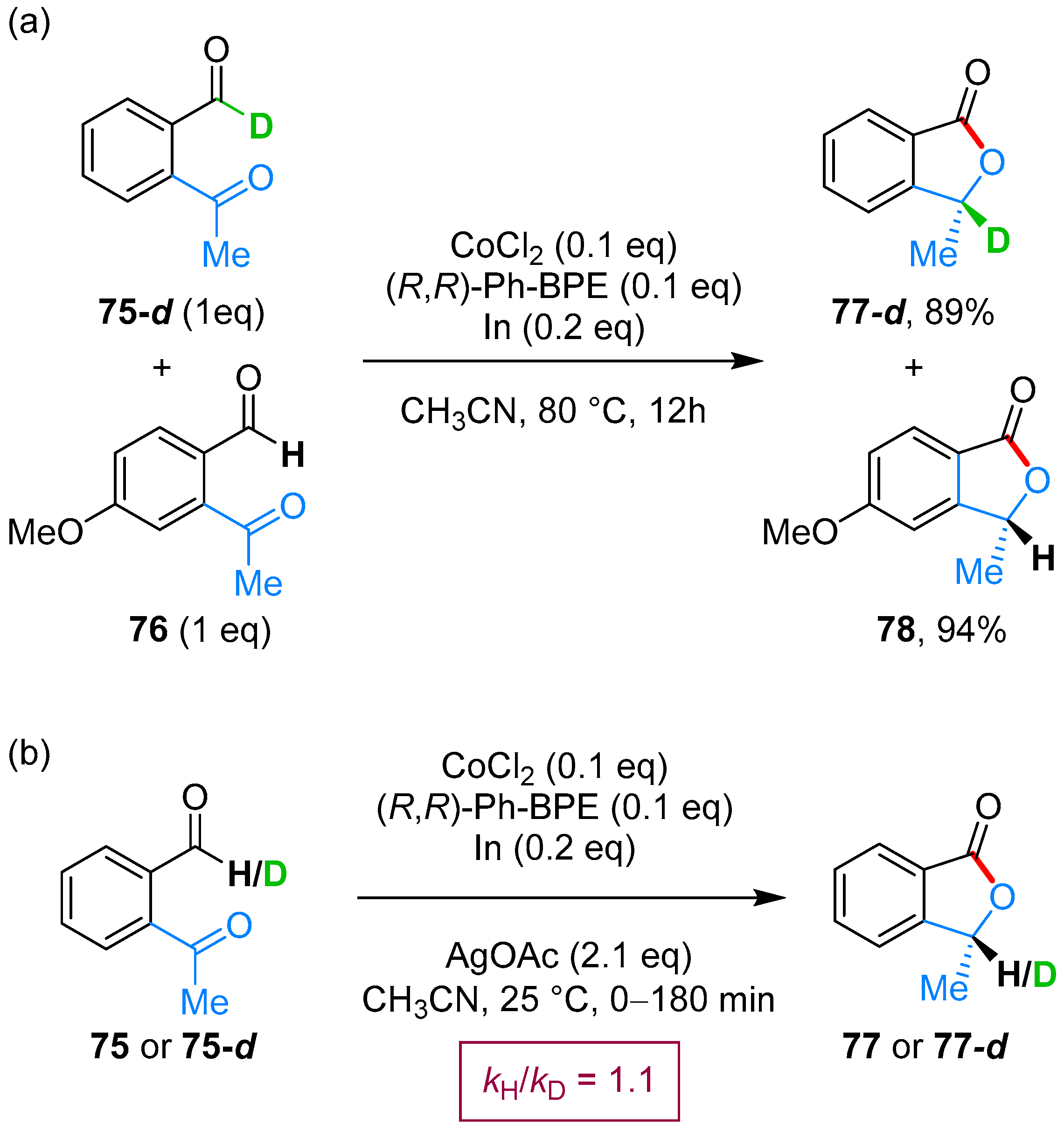
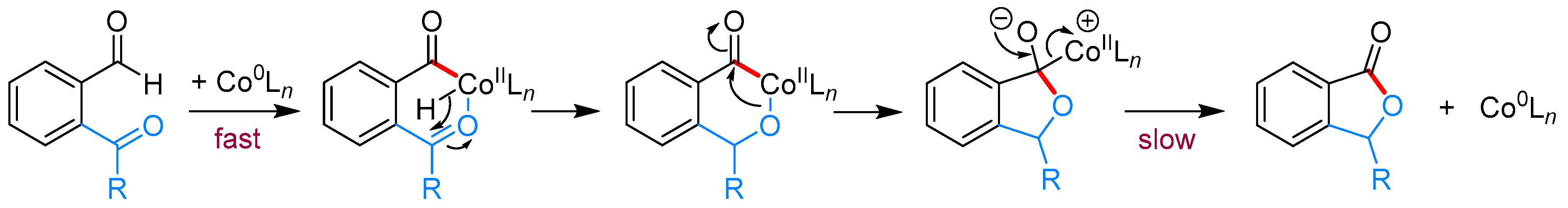
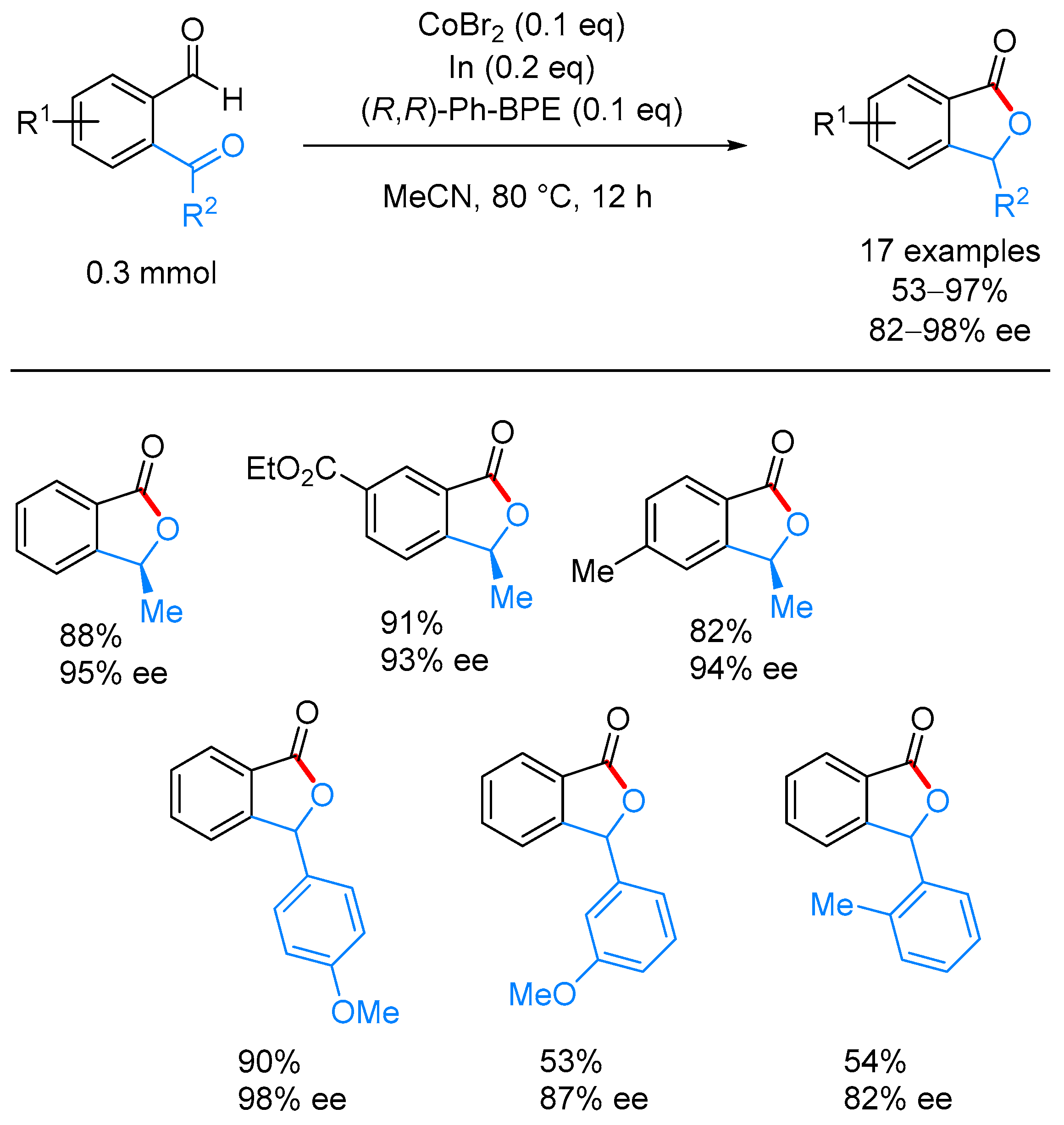
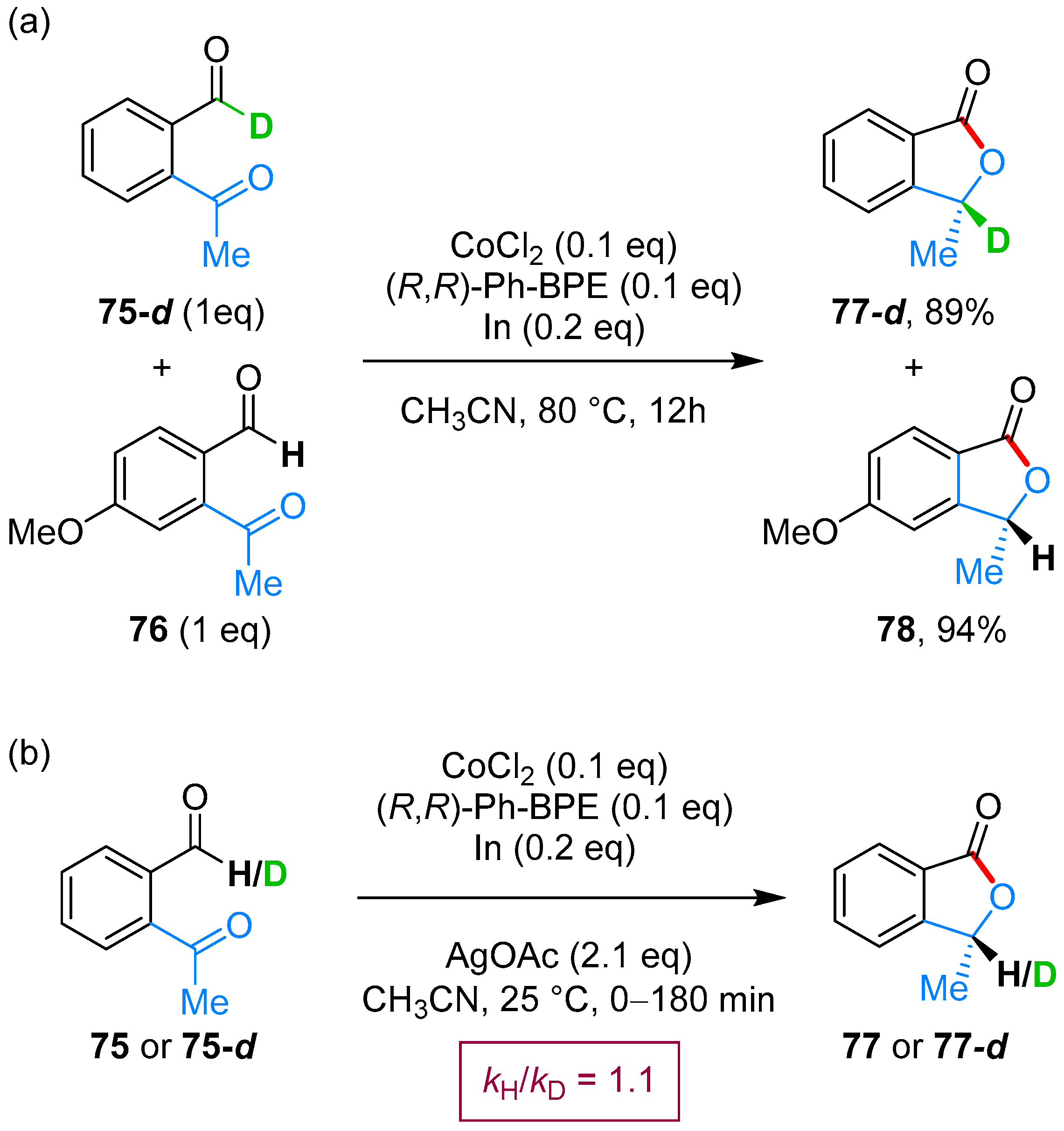
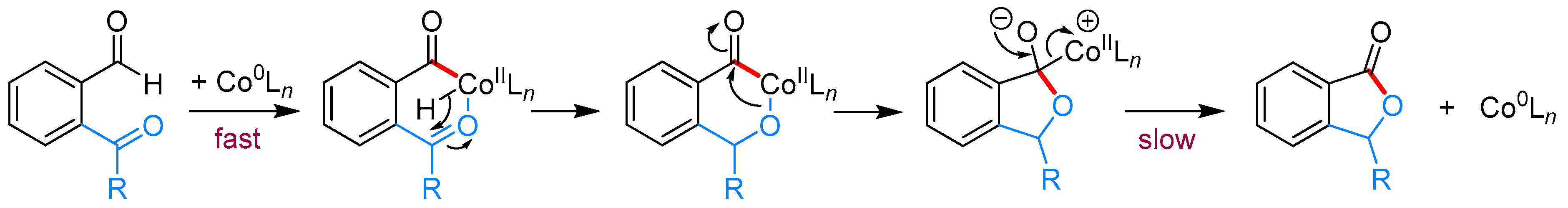
- Yang, J.; Yoshikai, N. Cobalt-Catalyzed Enantioselective Intramolecular Hydroacylation of Ketones and Olefins. J. Am. Chem. Soc. 2014, 136, 16748–16751. [Google Scholar] [CrossRef] [PubMed]
- Hiroshi, H.; Takashi, I.; Akio, Y. A New Tishchenko-Type Ester Formation Catalyzed by Rythenium Complexes. Chem. Lett. 1978, 7, 17–20. [Google Scholar]
- Takashi, I.; Hiroshi, H.; Yoshitaka, K.; Akio, Y. Selective Dimerization of Aldehydes to Esters Catalyzed by Hydridoruthenium Complexes. Bull. Chem. Soc. Jpn. 1982, 55, 504–512. [Google Scholar]
- Murahashi, S.; Naota, T.; Ito, K.; Maeda, Y.; Taki, H. Ruthenium-catalyzed oxidative transformation of alcohols and aldehydes to esters and lactones. J. Org. Chem. 1987, 52, 4319–4327. [Google Scholar] [CrossRef]
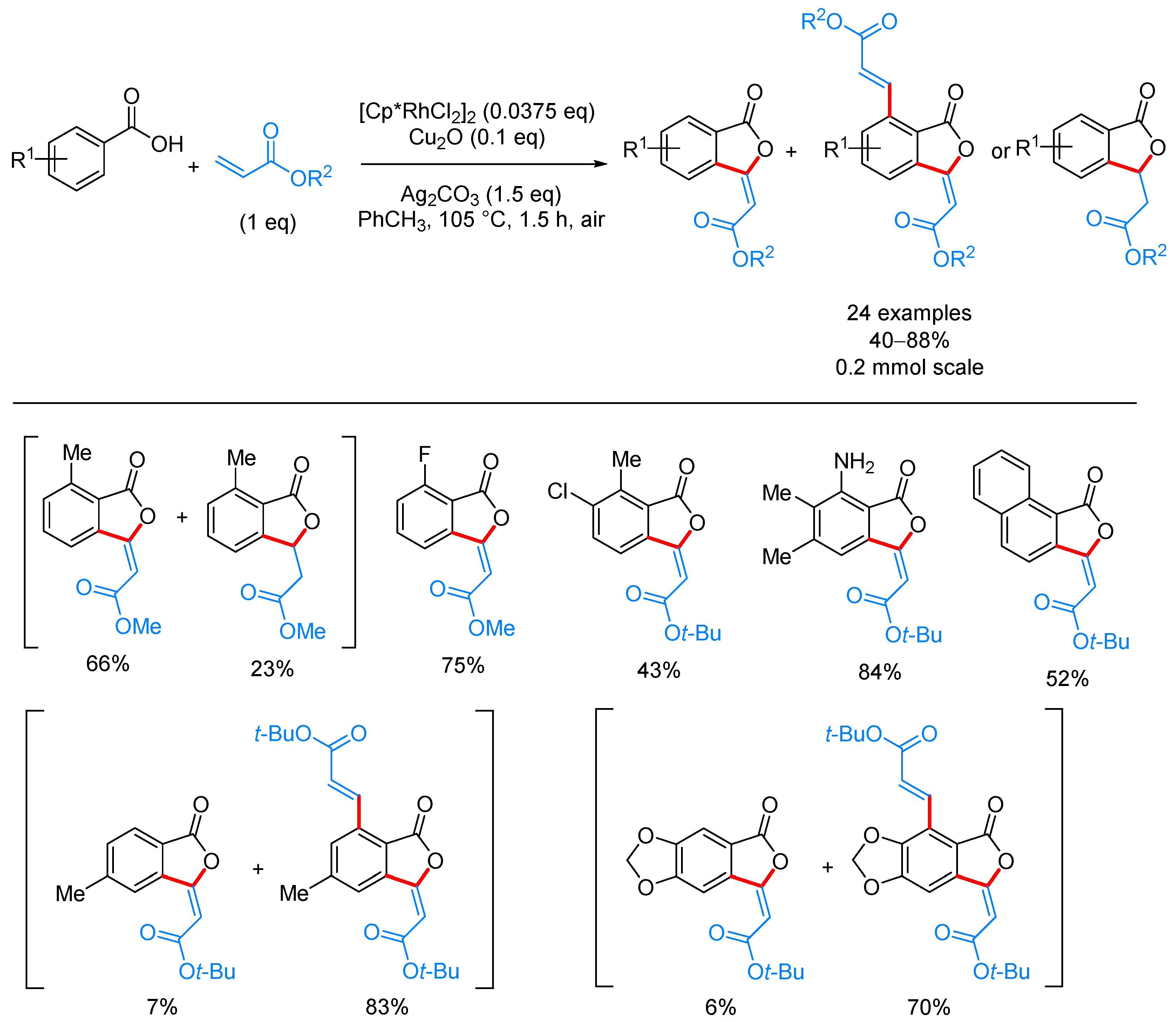
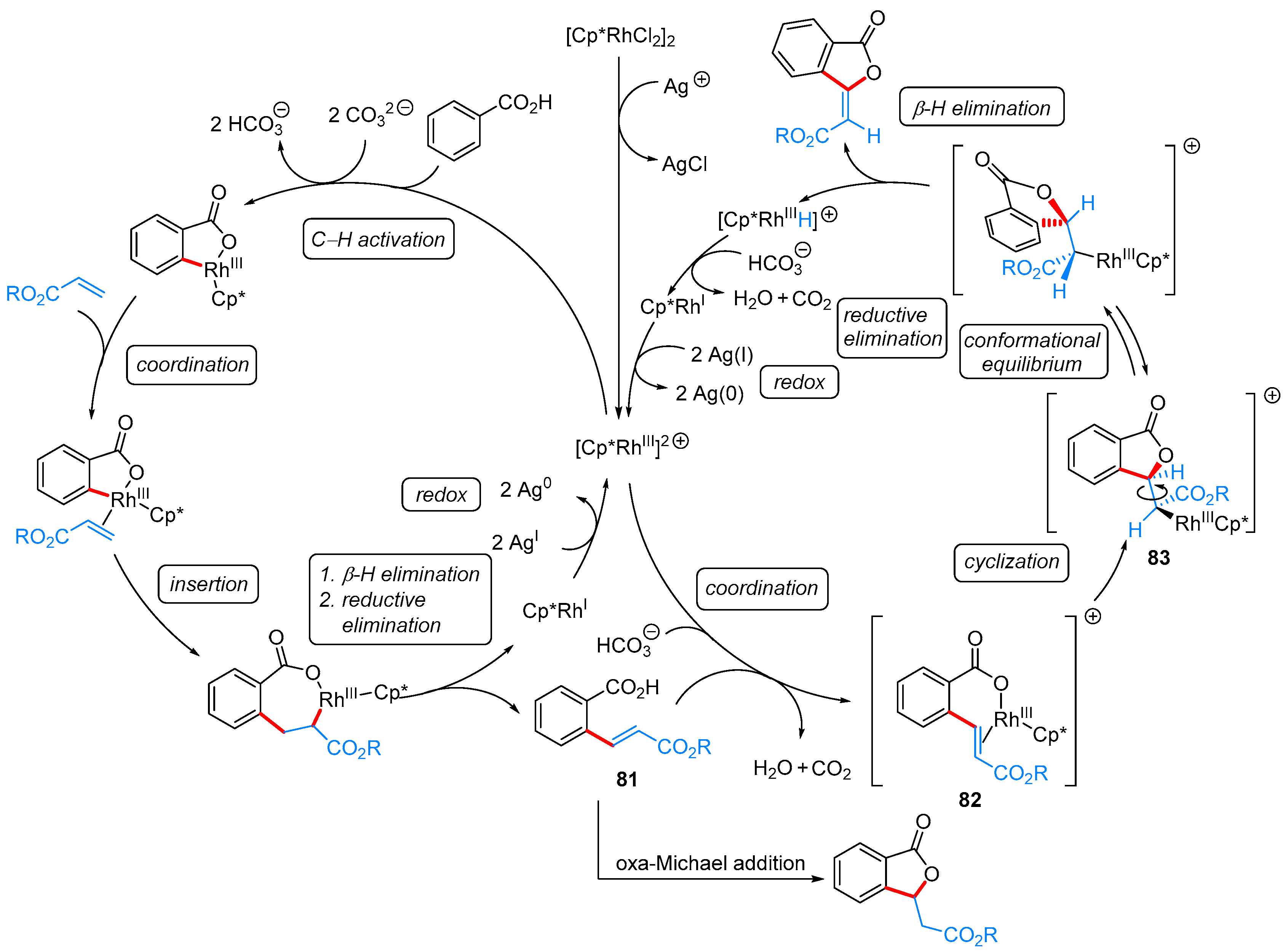
- Han, W.-J.; Pu, F.; Fan, J.; Liu, Z.-W.; Shi, X.-Y. Rhodium(III)-catalyzed tandem C–H olefination and oxidative cyclization of aromatic acids with acrylates for the synthesis of (E)-3-ylidenephthalides. Adv. Synth. Catal. 2017, 359, 3520–3525. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part 1: Fundamental principles. J. Chem. Ed. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part II: Underlying theories. J. Chem. Ed. 1968, 45, 643–648. [Google Scholar] [CrossRef]
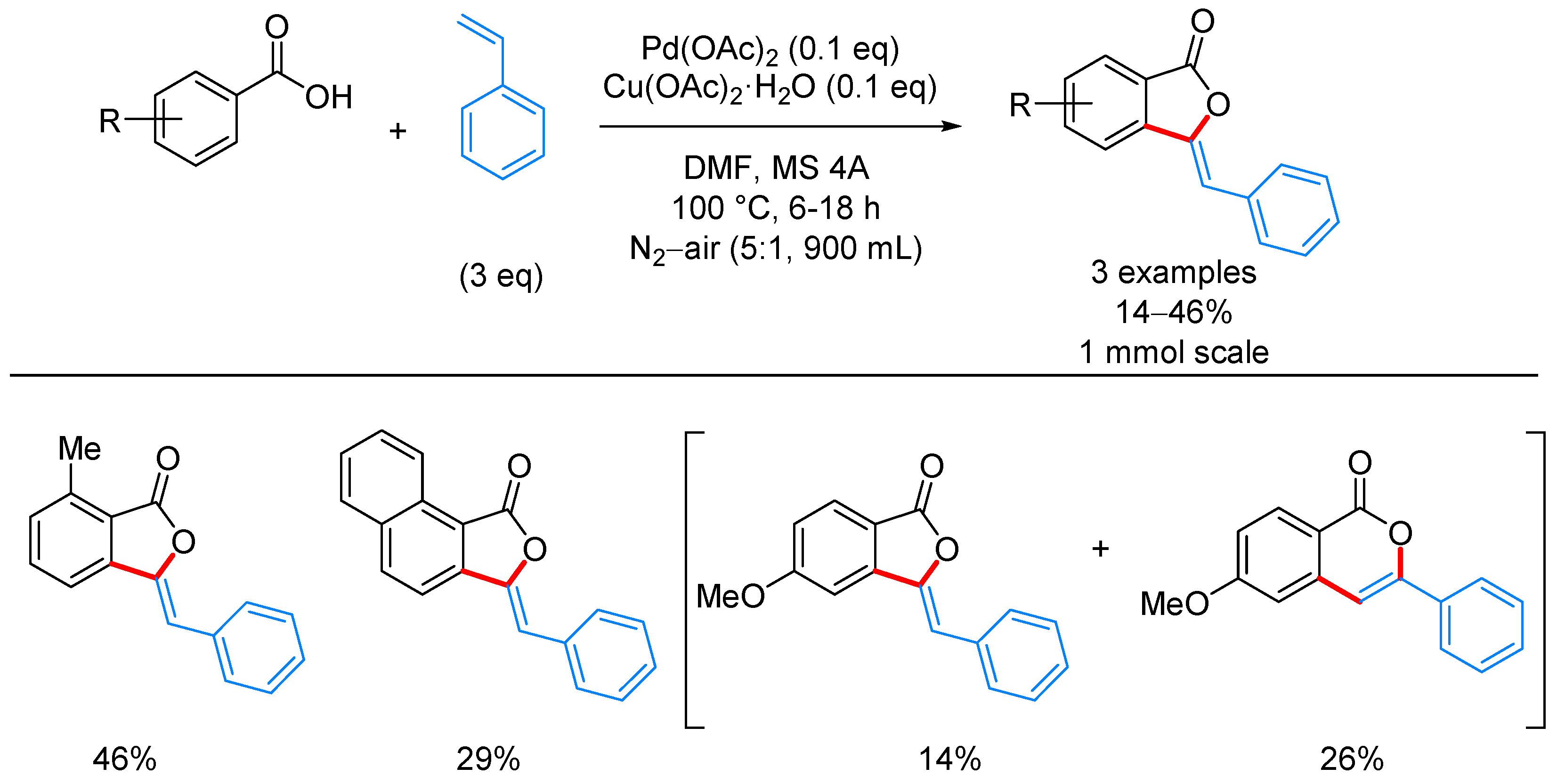
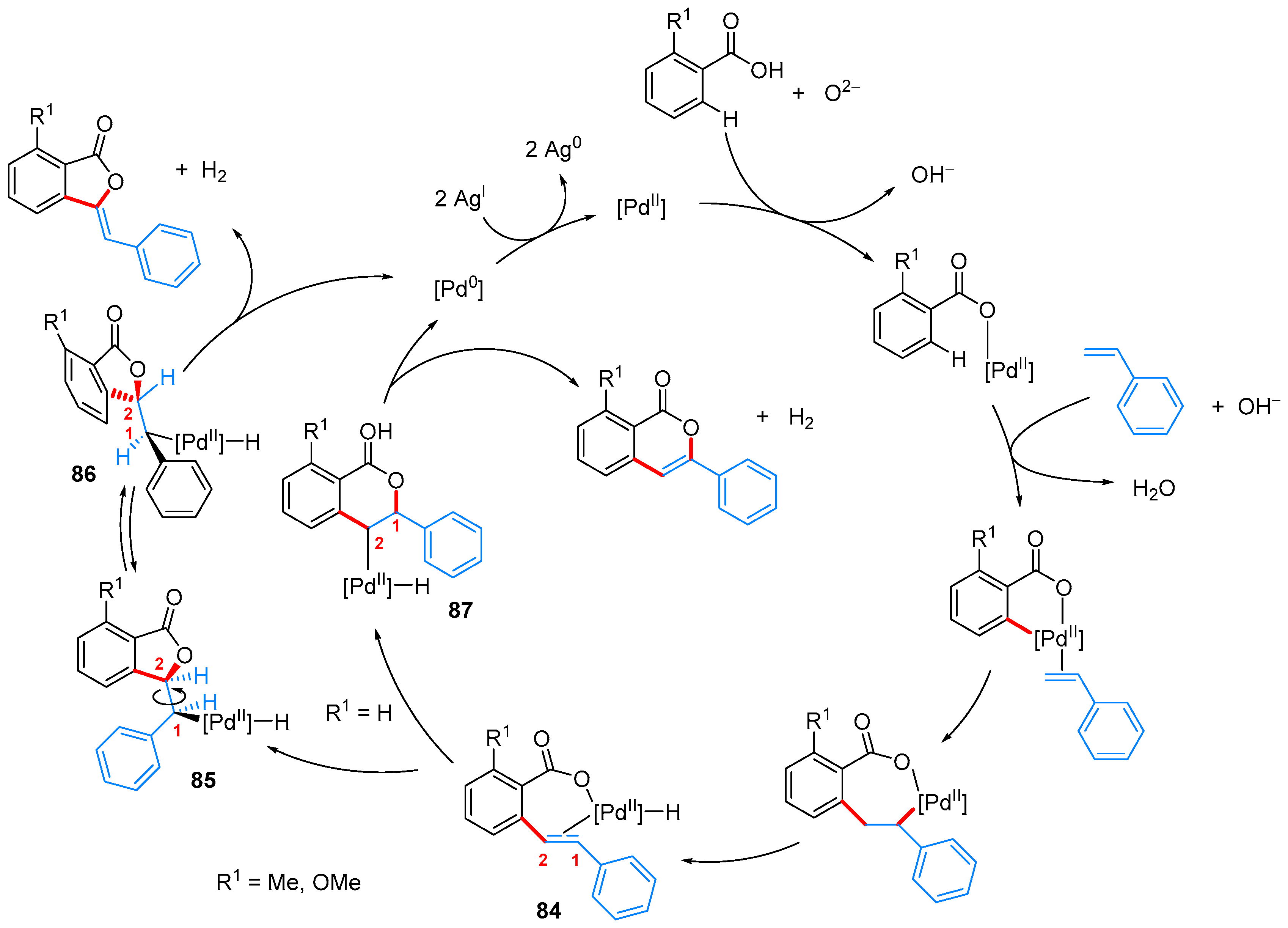
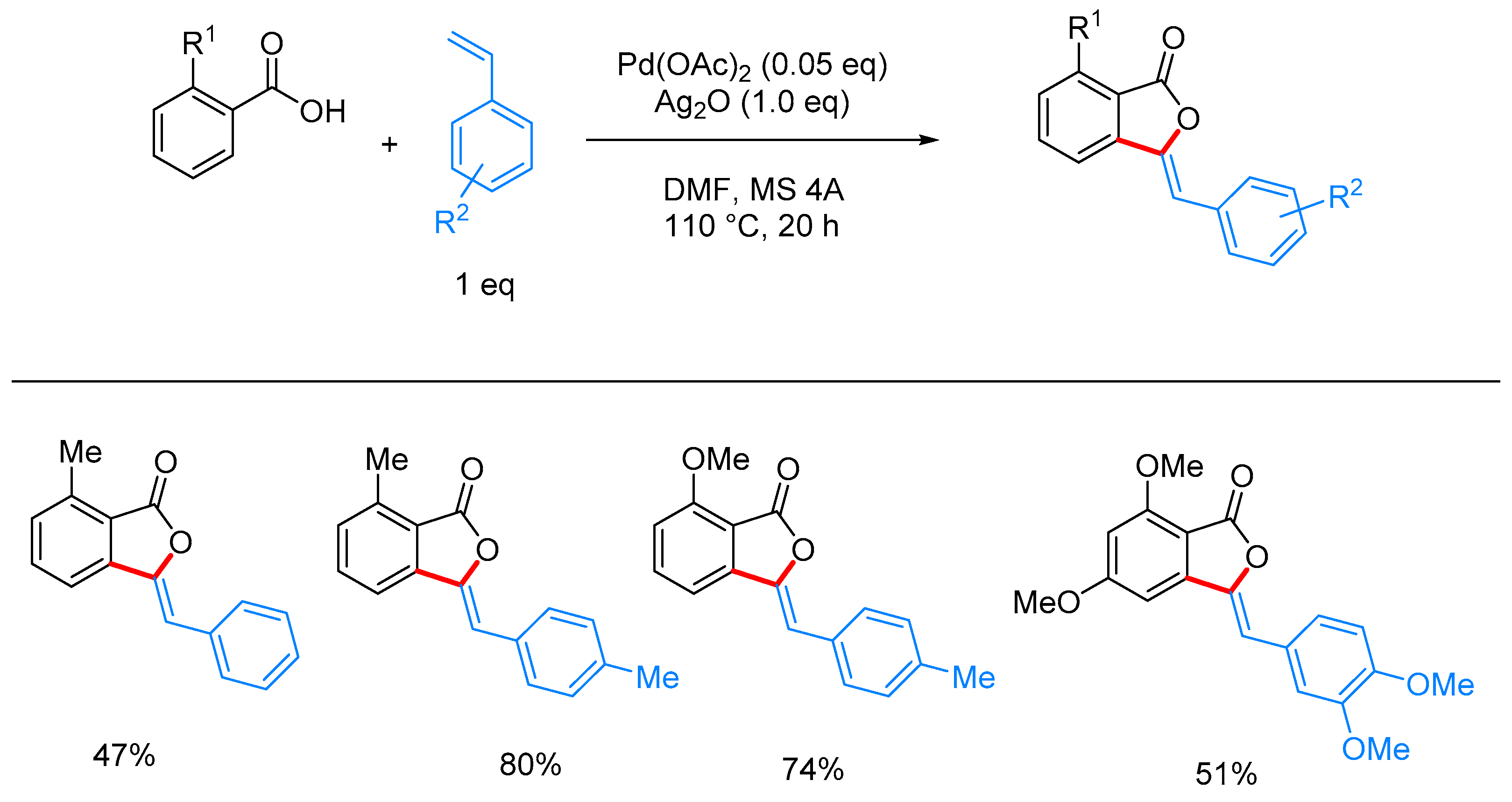
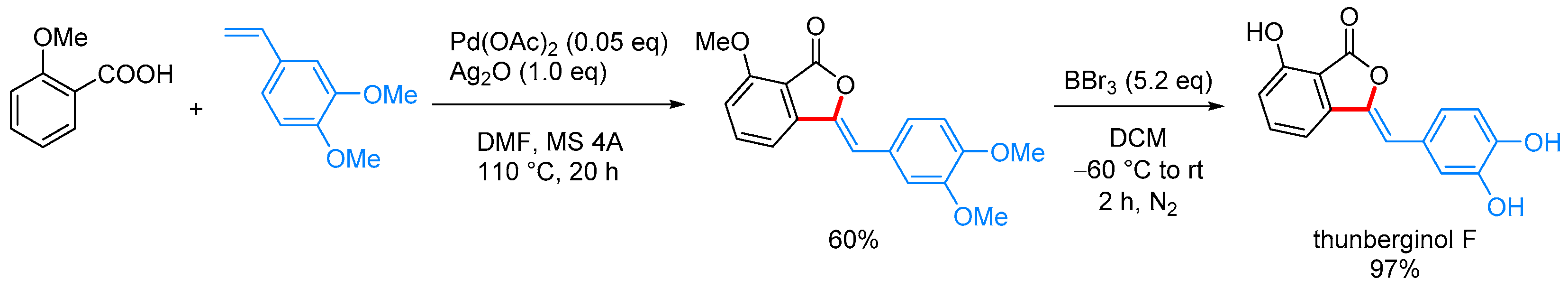
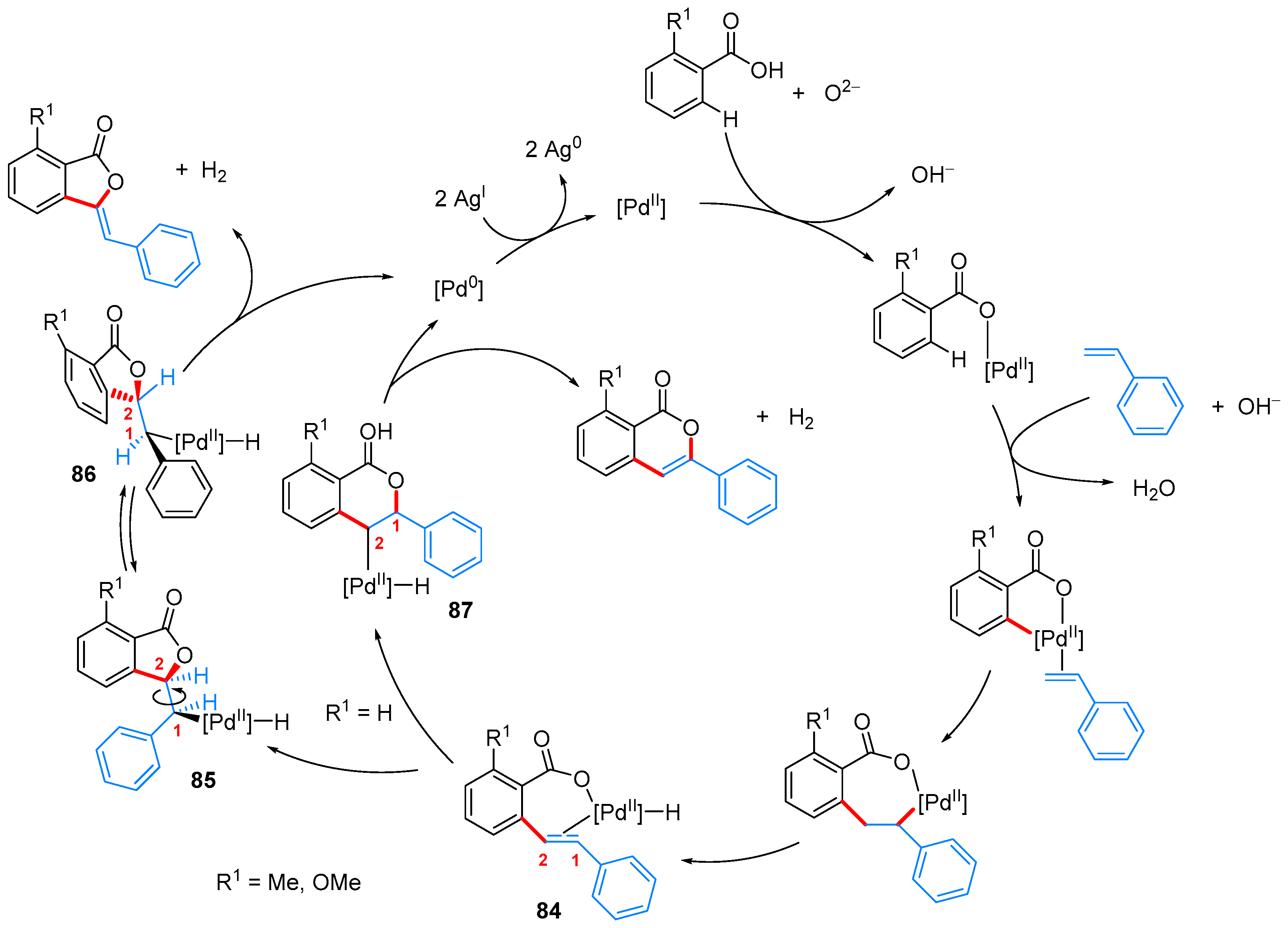
- Nandi, D.; Ghosh, D.; Chen, S.-J.; Kuo, B.-C.; Wang, N.M.; Lee, H.M. One-Step Synthesis of Isocoumarins and 3-Benzylidenephthalides via Ligandless Pd-Catalyzed Oxidative Coupling of Benzoic Acids and Vinylarenes. J. Org. Chem. 2013, 78, 3445–3451. [Google Scholar] [CrossRef] [PubMed]
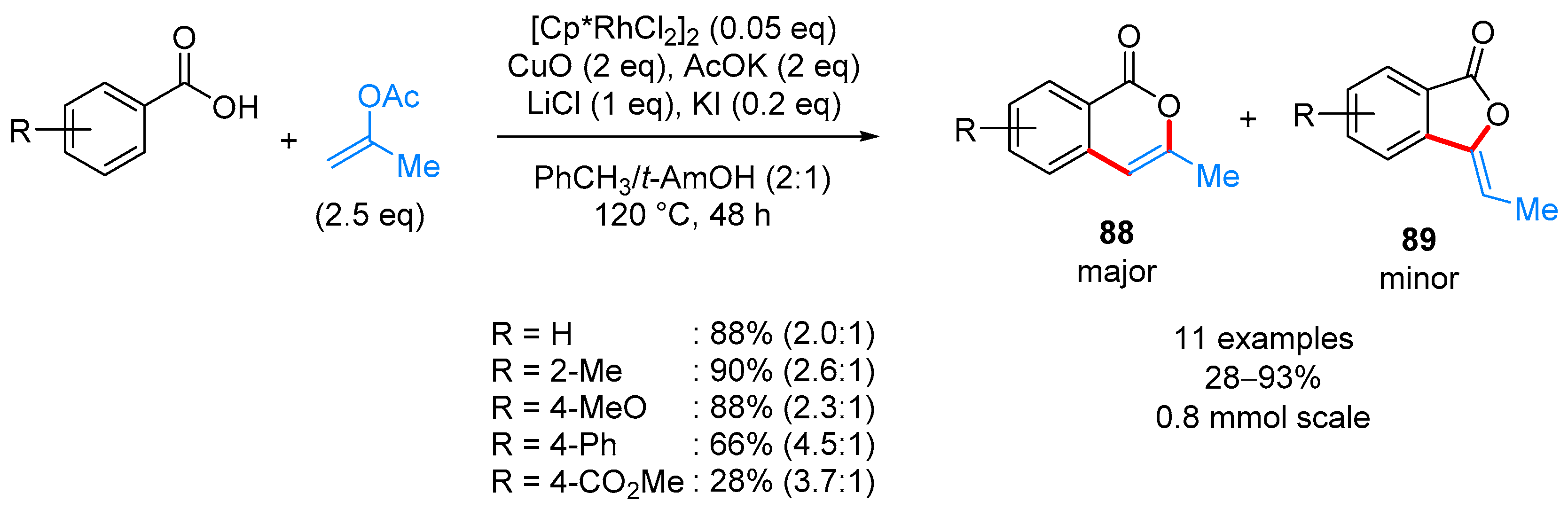
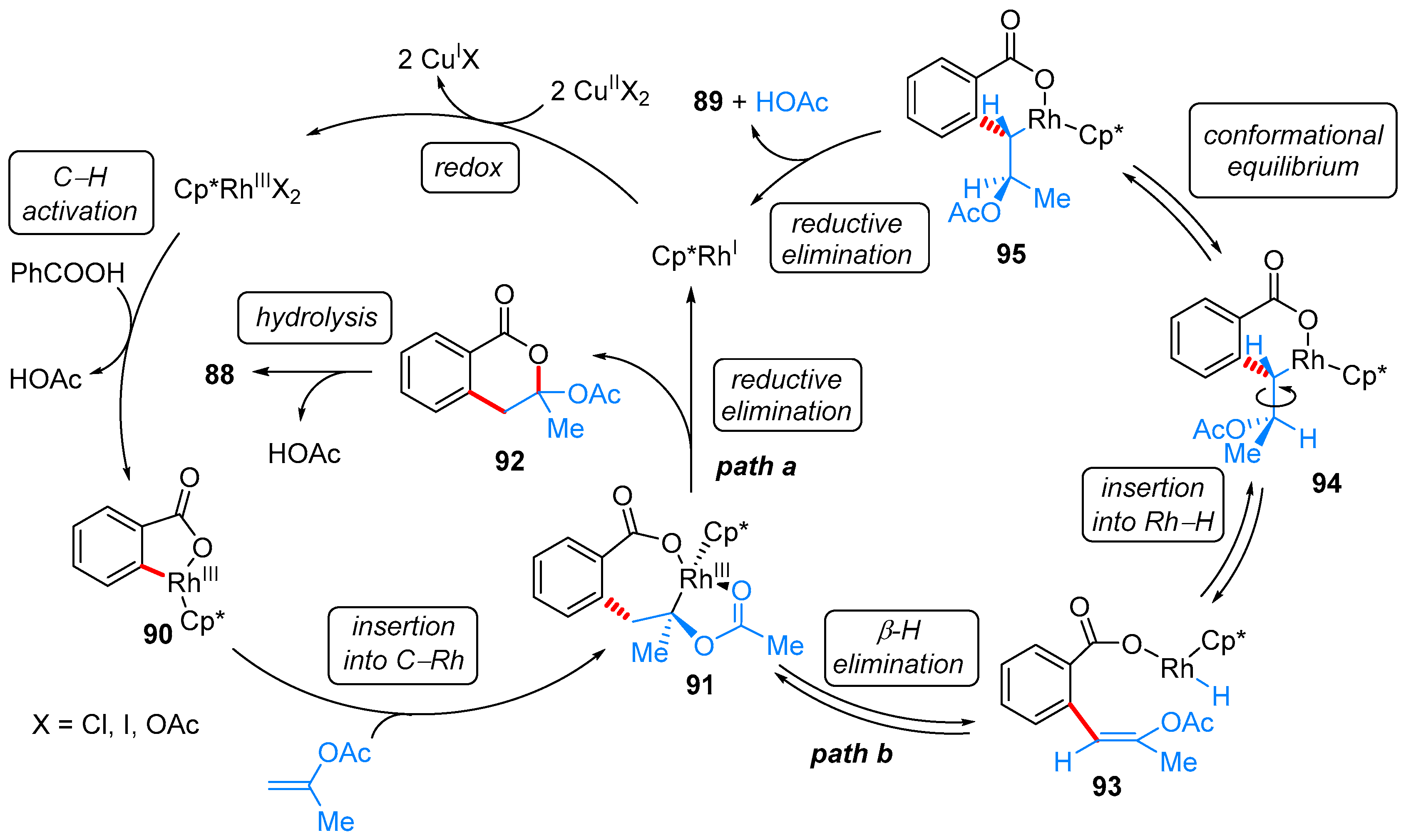
- Zhang, M.; Zhang, H.-J.; Han, T.; Ruan, W.; Wen, T.-B. Rh(III)-Catalyzed Oxidative Coupling of Benzoic Acids with Geminal-Substituted Vinyl Acetates: Synthesis of 3-Substituted Isocoumarins. J. Org. Chem. 2015, 80, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Shi, Y.; Wang, X.; Zhang, L.; Cheng, Y.; You, J. Rhodium-Catalyzed Oxidative Coupling of Benzoic Acids with Terminal Alkynes: An Efficient Access to 3-Ylidenephthalides. Organometallics 2016, 35, 1350–1353. [Google Scholar] [CrossRef]
- Danoun, G.; Mamone, P.; Goossen, L.J. One-Pot Synthesis of 3-Alkylidenephthalides from Benzoic Acids by a Rhodium-Catalyzed ortho-C-H Acylation Process. Chem. Eur. J. 2013, 19, 17287–17290. [Google Scholar] [CrossRef] [PubMed]
- Mamone, P.; Danoun, G.; Gooßen, L.J. Rhodium-Catalyzed ortho Acylation of Aromatic Carboxylic Acids. Angew. Chem. Int. Ed. 2013, 52, 6704–6708. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Rajamalli, P.; Cheng, C.-H. Rhodium(III)-Catalyzed [4+1] Annulation of Aromatic and Vinylic Carboxylic Acids with Allenes: An Efficient Method Towards Vinyl-Substituted Phthalides and 2-Furanones. Chem. Eur. J. 2015, 21, 9198–9203. [Google Scholar] [CrossRef] [PubMed]
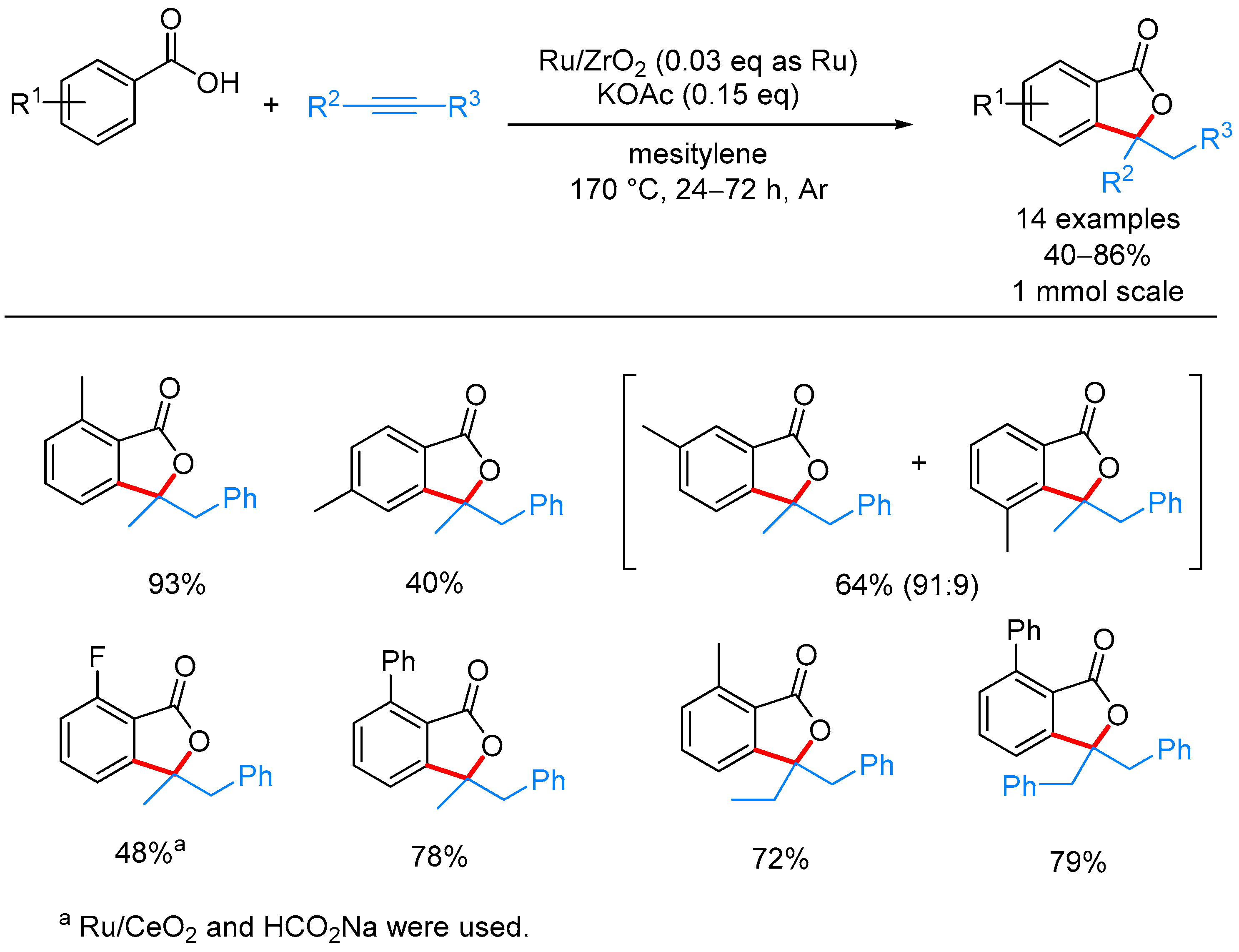
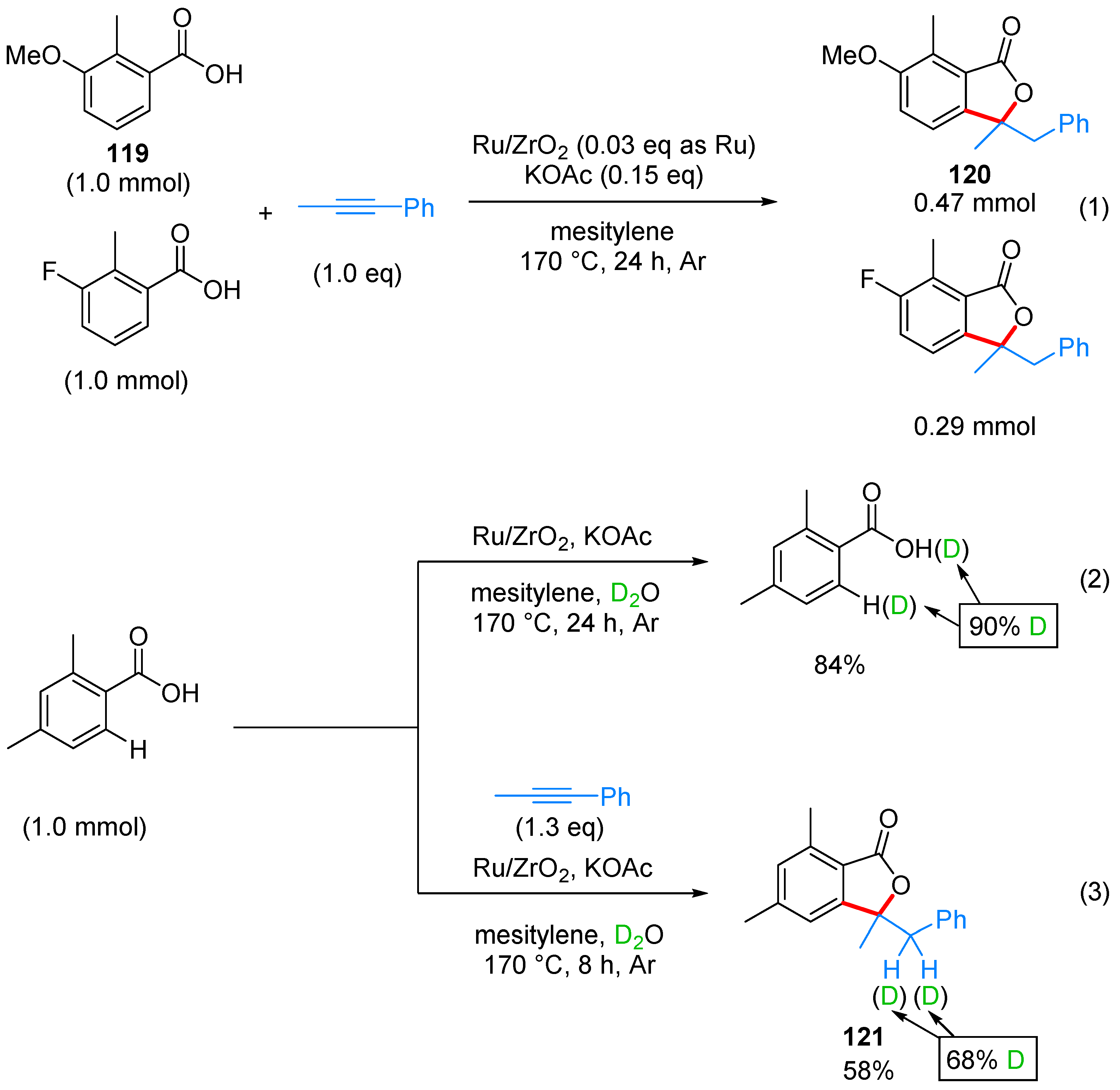
- Miura, H.; Tsutsui, K.; Wada, K.; Shishido, T. Coupling of carboxylic acids with internal alkynes by supported ruthenium catalysts: Direct and selective syntheses of multi-substituted phthalide derivatives. Chem. Commun. 2015, 51, 1654–1657. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.; Mitchell, T.R.B.; Sutthivaiyakit, S. Oxidation of alcohols by transition metal complexes—IV: The rhodium catalysed synthesis of esters from aldehydes and alcohols. Tetrahedron 1981, 37, 4313–4319. [Google Scholar] [CrossRef]
- Massoui, M.; Beaupère, D.; Nadjo, L.; Uzan, R. Homomolecular esterification of aldehydes catalyzed by hydridotetrakis(triphenylphosphine)rhodium(I). J. Organomet. Chem. 1983, 259, 345–353. [Google Scholar] [CrossRef]
- Boldrini, G.P.; Umani-Ronchi, A.; Panunzio, M. Chemistry of Alkali Metal Carbonylchromates. A New Method for the Selective Reduction of α,β-Unsaturated Carbonyl Compounds. Synthesis 1976, 1976, 596–598. [Google Scholar] [CrossRef]
- Fuchikami, T.; Ubukata, Y.; Tanaka, Y. Group 6 anionic μ-hydride complexes [HM2(CO)10]− (M = Cr, Mo, W): New catalysts for hydrogenation and hydrosilylation. Tetrahedron Lett. 1991, 32, 1199–1202. [Google Scholar] [CrossRef]
- Bergens, S.H.; Fairlie, D.P.; Bosnich, B. Homogeneous catalysis: Catalytic intramolecular conversion of 1,4-dialdehydes to γ-lactones. Organometallics 1990, 9, 566–571. [Google Scholar] [CrossRef]
- Hossain, M.M.; Shyu, S.-G. Biphasic copper-catalyzed C–H bond activation of arylalkanes to ketones with tert-butyl hydroperoxide in water at room temperature. Tetrahedron 2016, 72, 4252–4257. [Google Scholar] [CrossRef]
- Díaz-Torres, R.; Alvarez, S. Coordinating ability of anions and solvents towards transition metals and lanthanides. Dalton Trans. 2011, 40, 10742–10750. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, M.; Wang, Y.; Fan, H.; Wu, J.; Huang, C.; Hou, H. Cu(I) Coordination Polymers as the Green Heterogeneous Catalysts for Direct C—H Bonds Activation of Arylalkanes to Ketones in Water with Spatial Confinement Effect. Inorg. Chem. 2017, 56, 13329–13336. [Google Scholar] [CrossRef] [PubMed]
- Palmans, A.A.; Vekemans, J.J.; Spek, A. Hydrogen-bonded porous solid derived from trimesic amide. Chem. Commun. 1997, 22, 2247–2248. [Google Scholar] [CrossRef]
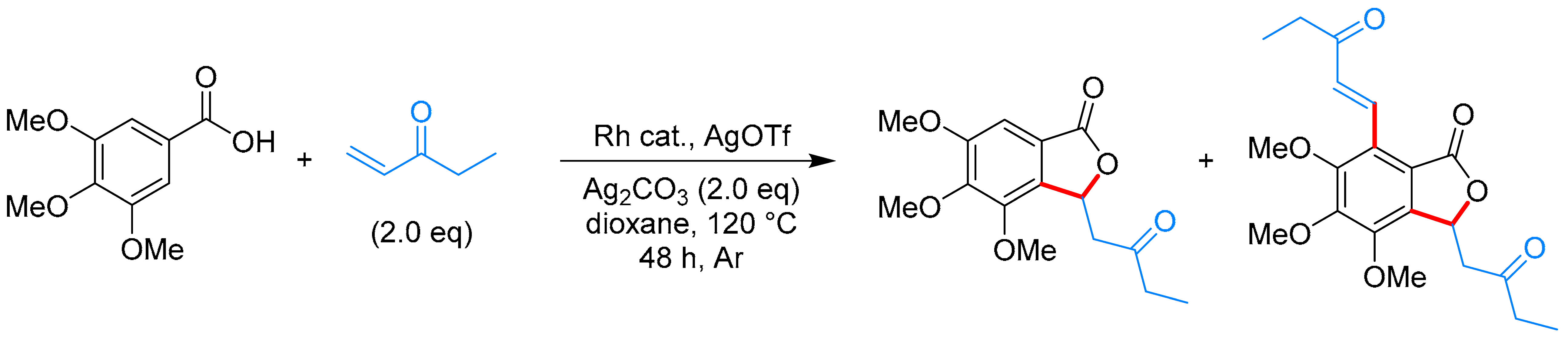
- Mochida, S.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of Functionalized α-Pyrone and Butenolide Derivatives by Rhodium-Catalyzed Oxidative Coupling of Substituted Acrylic Acids with Alkynes and Alkenes. J. Org. Chem. 2009, 74, 6295–6298. [Google Scholar] [CrossRef] [PubMed]
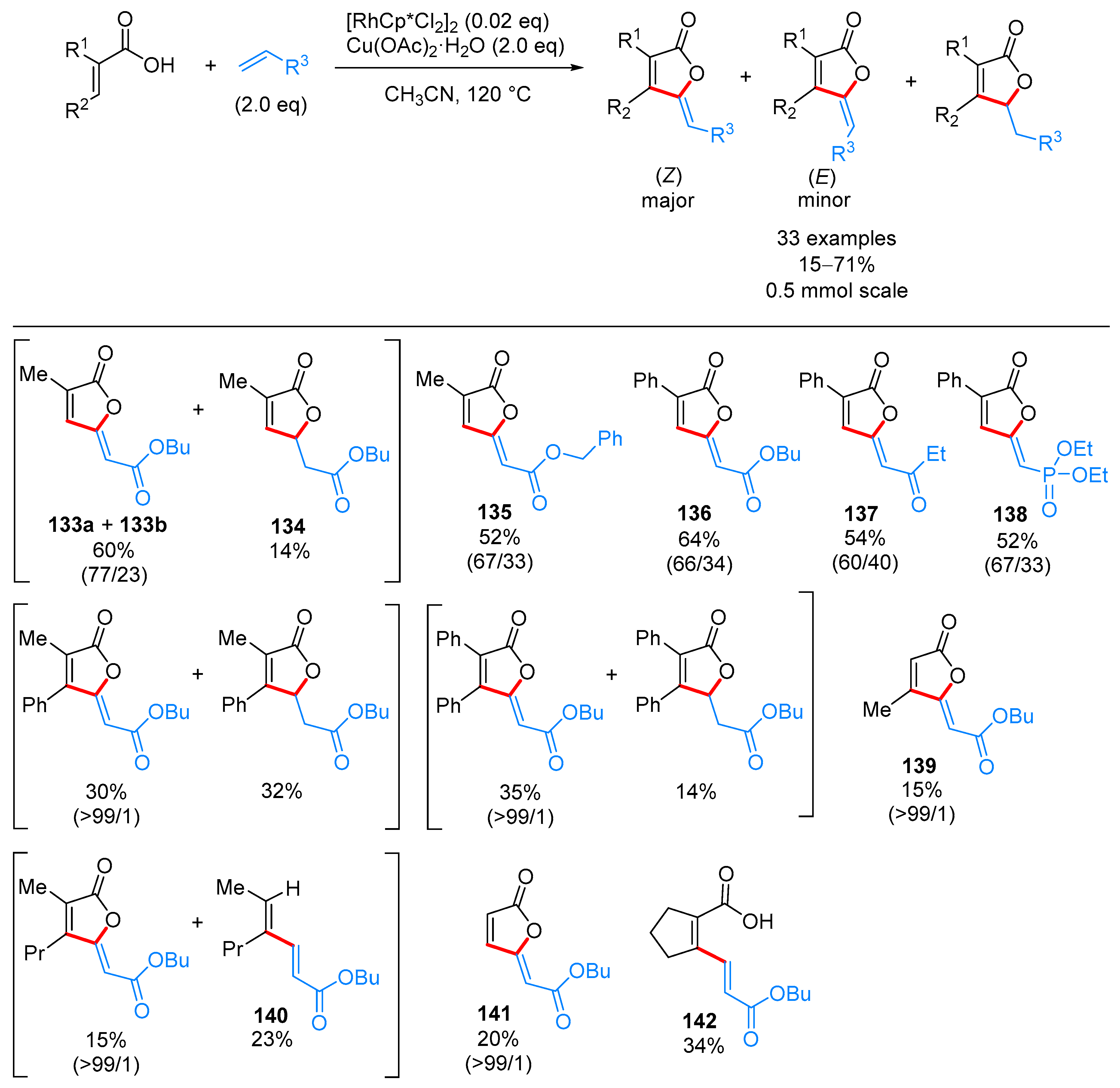
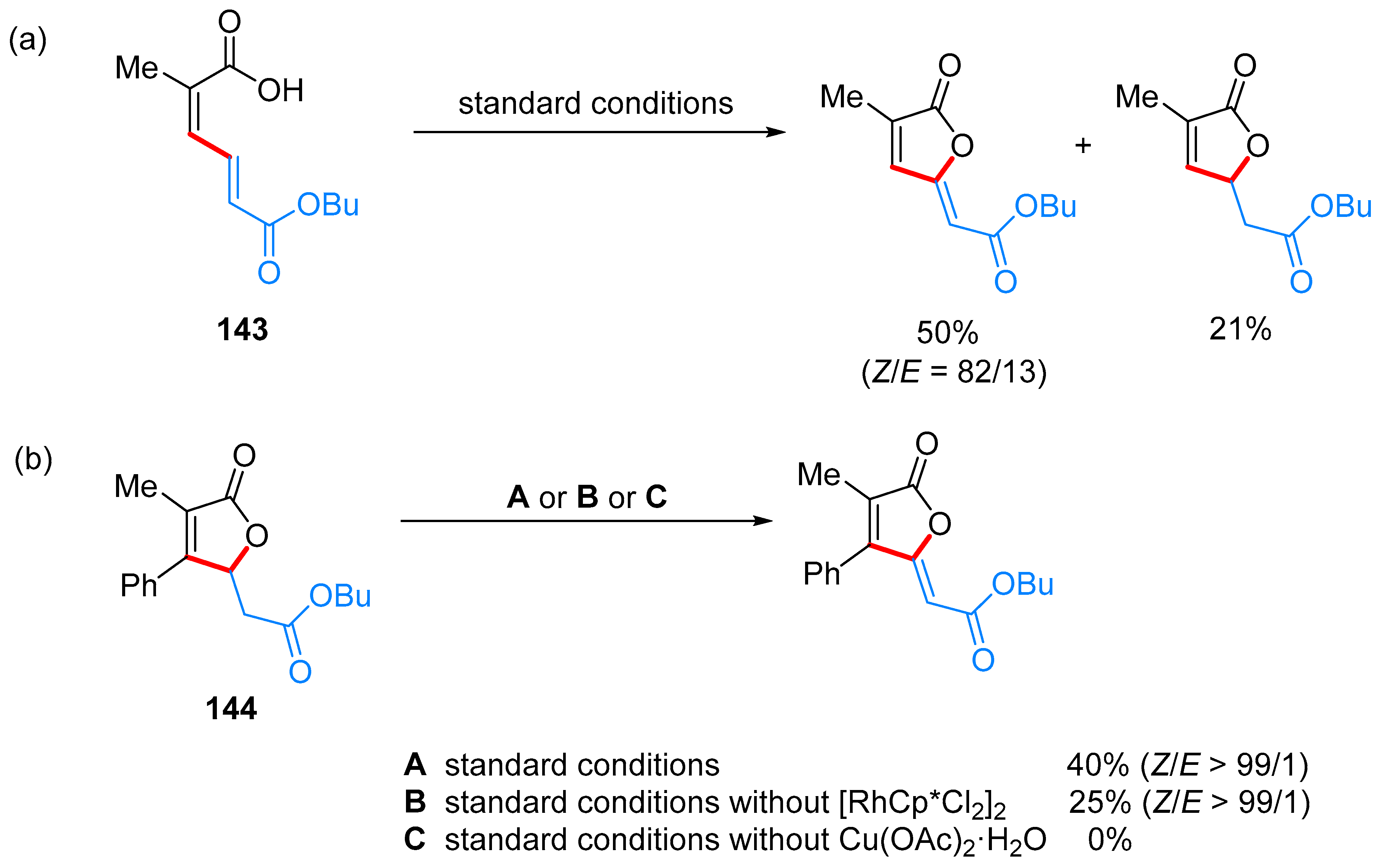
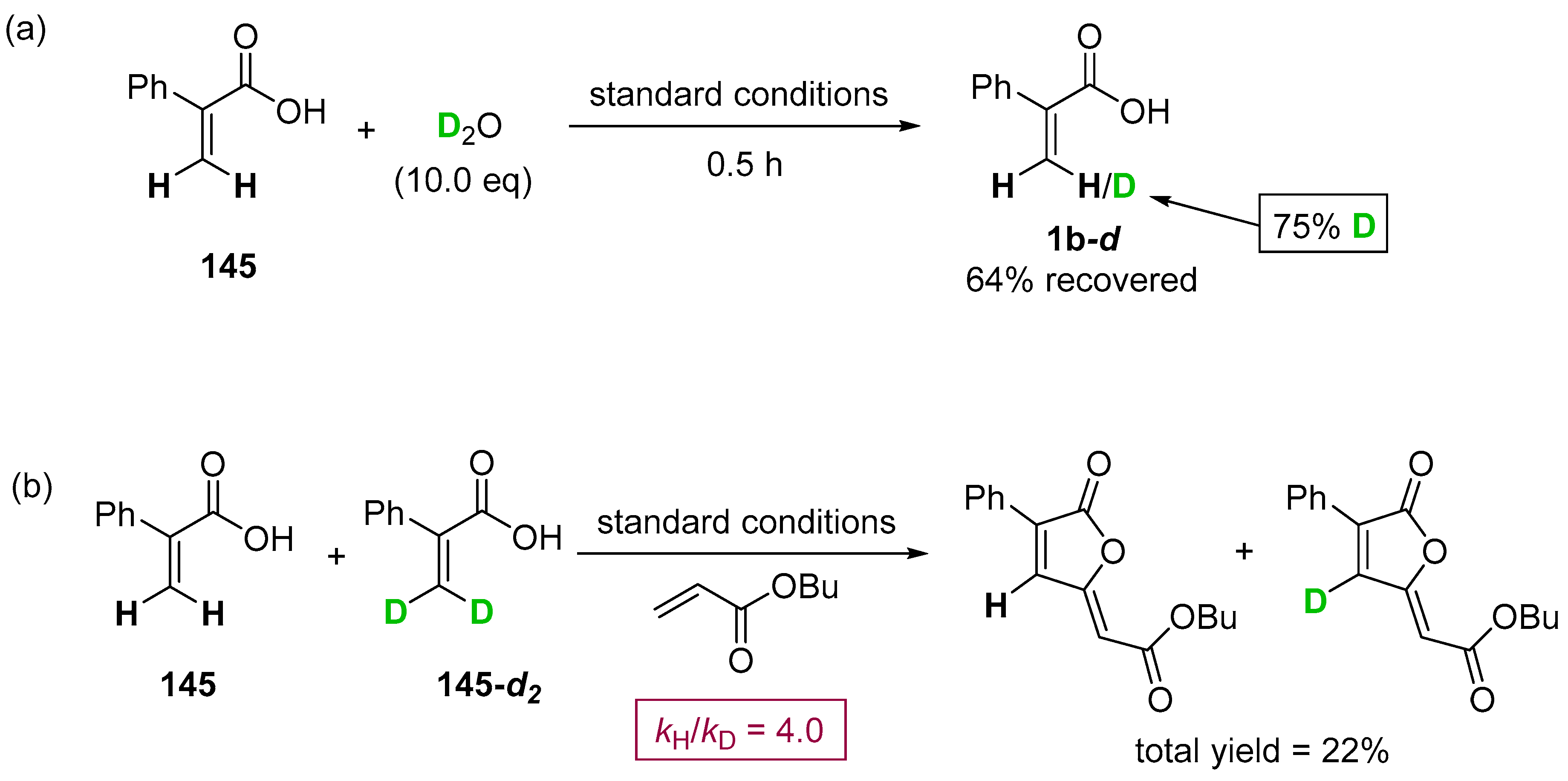
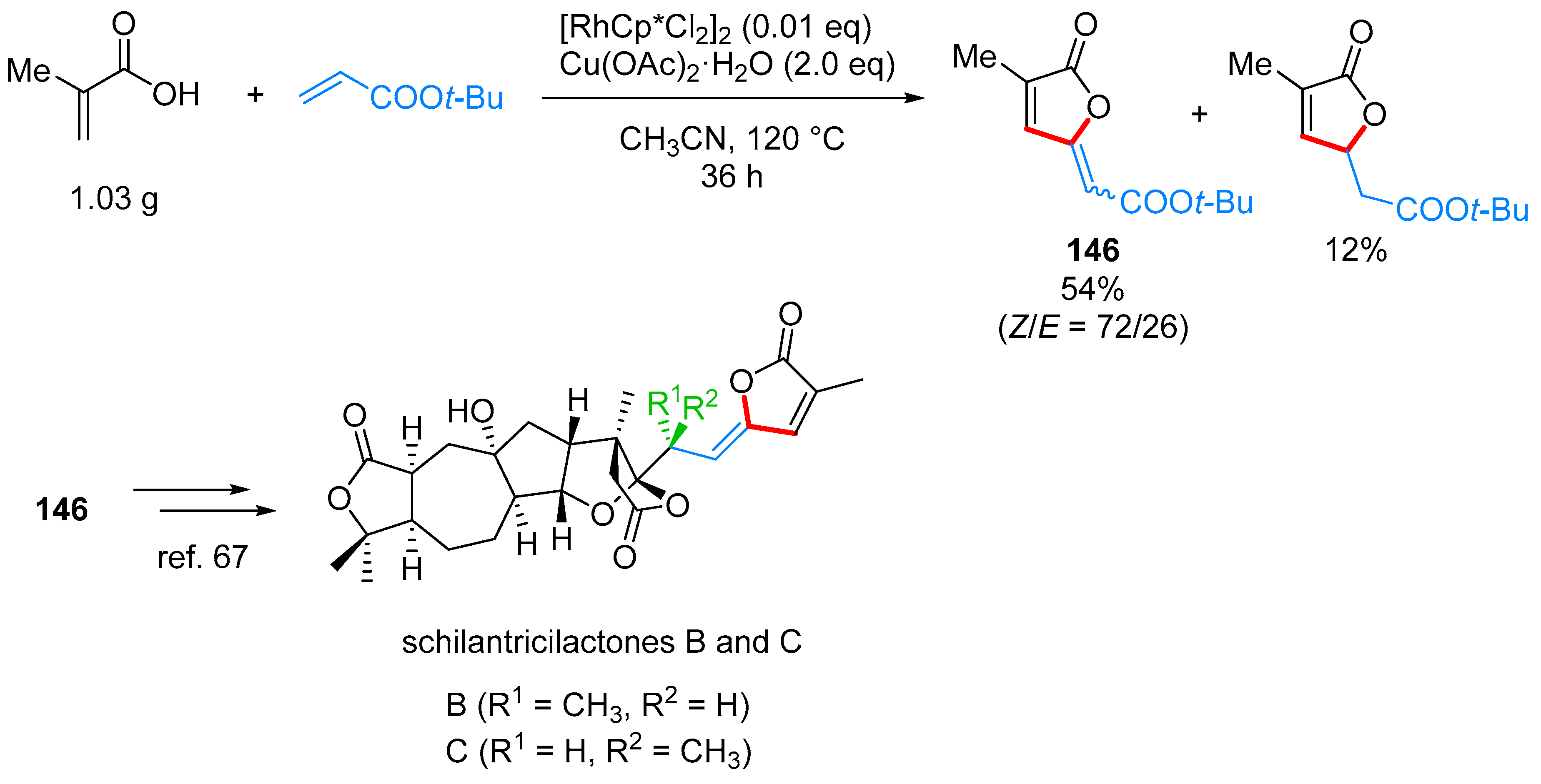
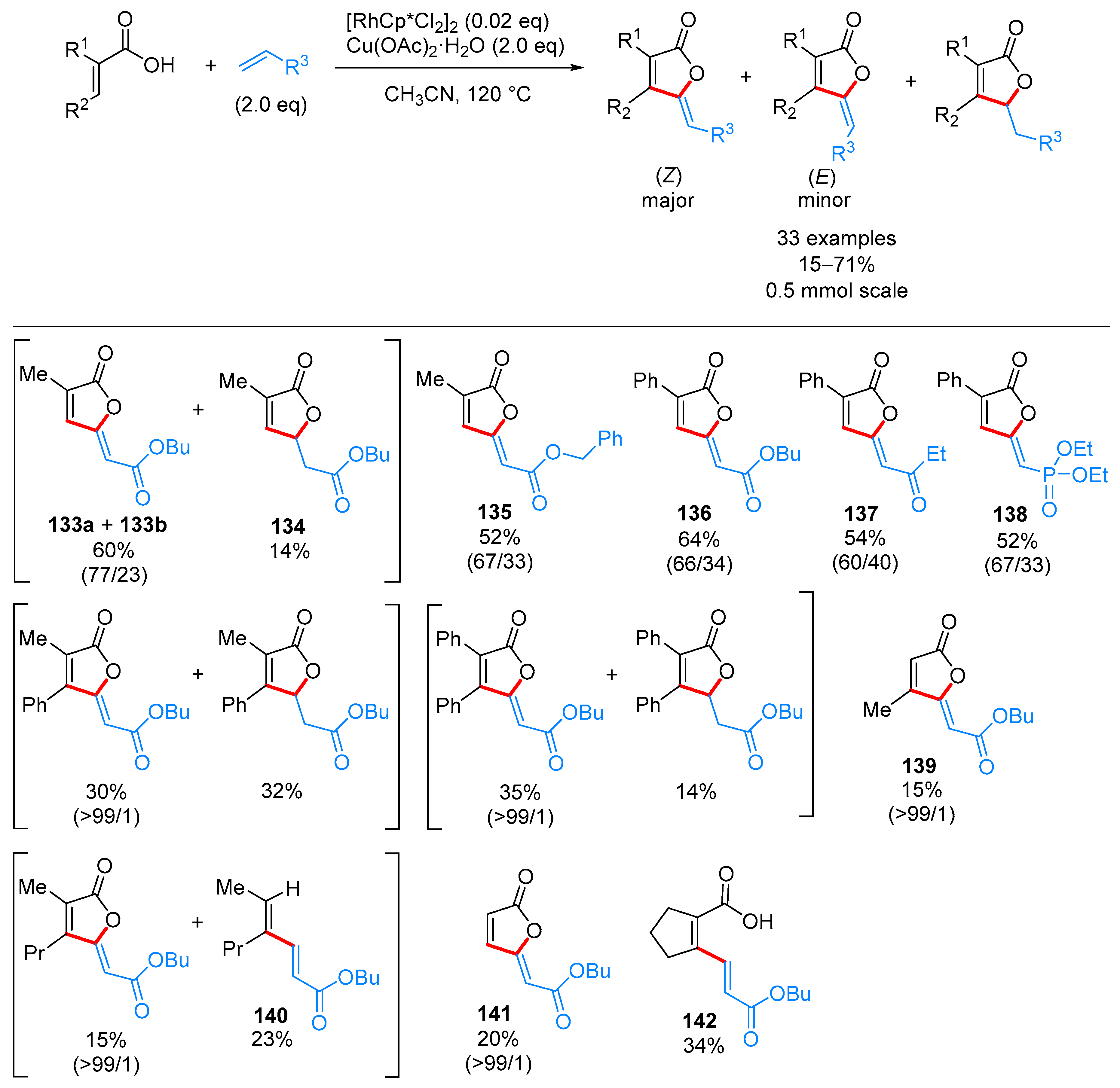
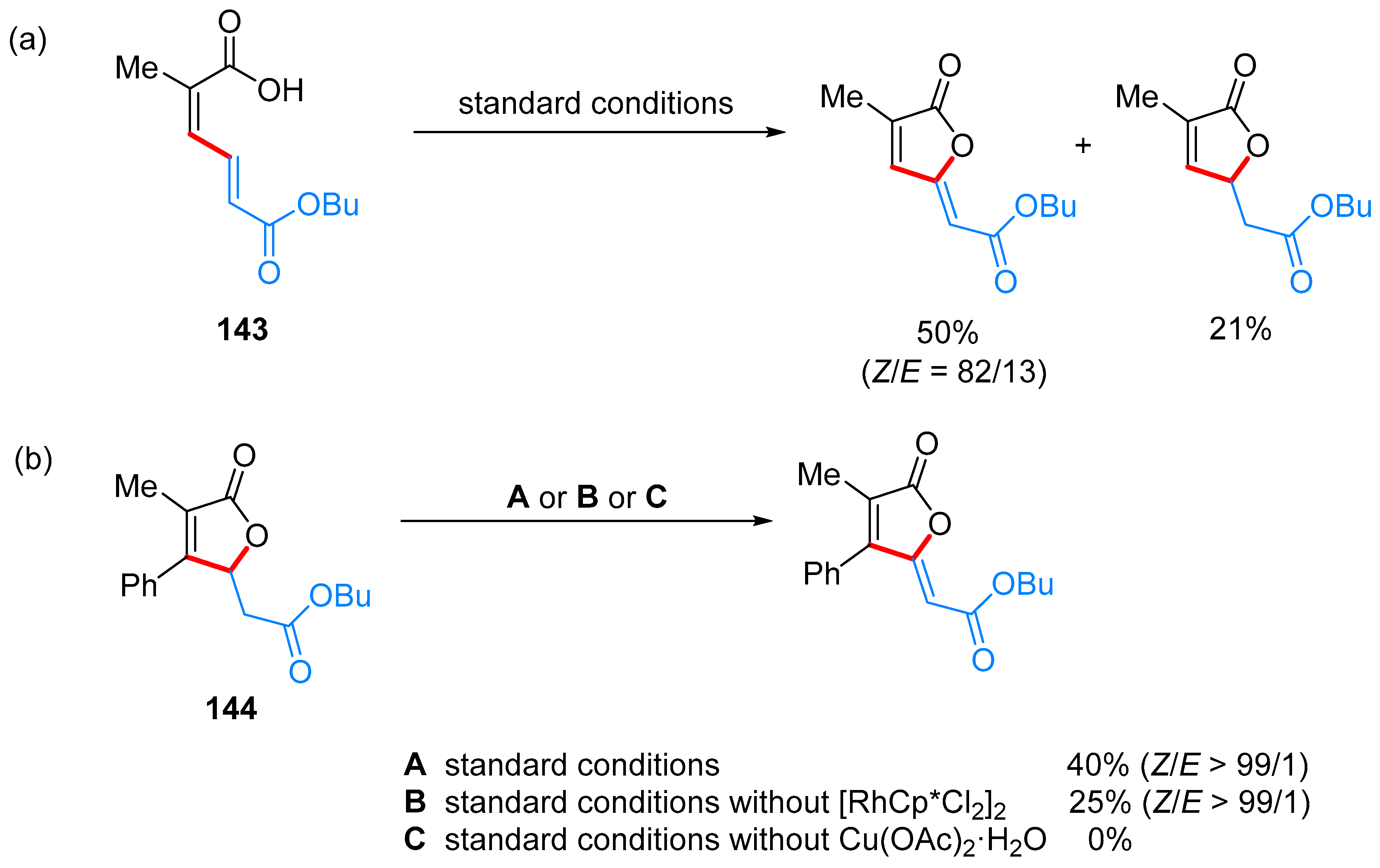
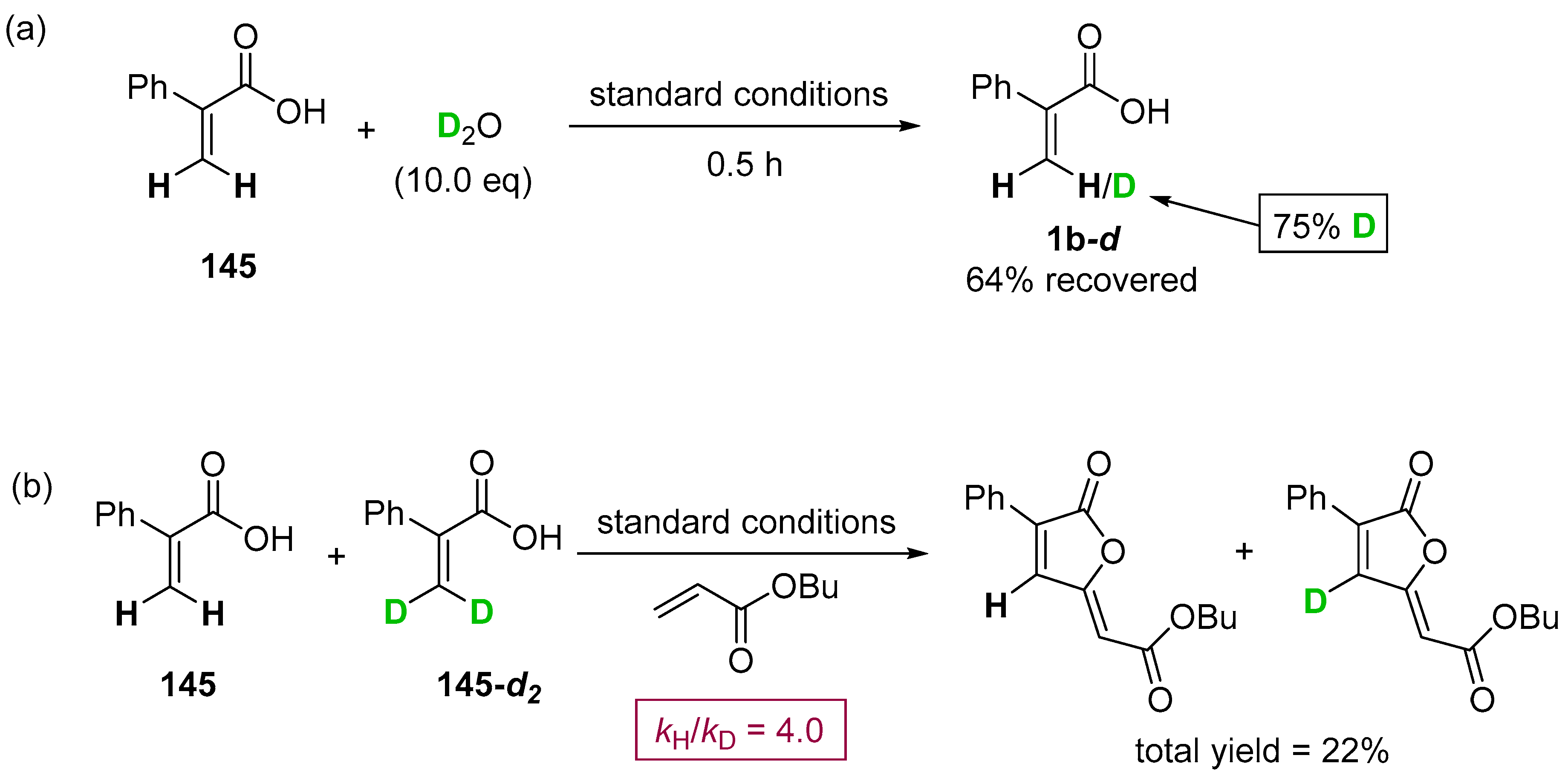
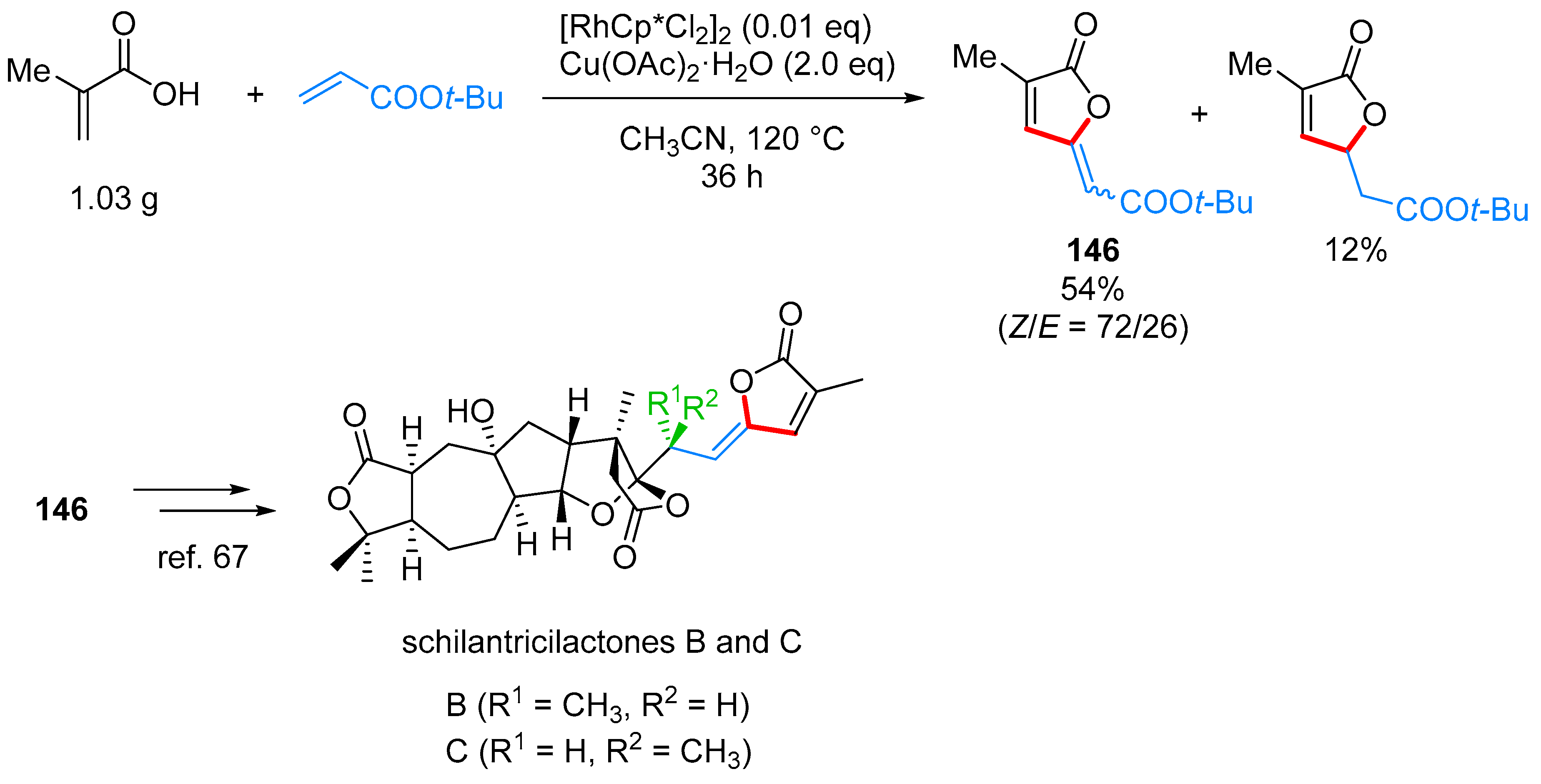
- Yu, C.; Zhang, J.; Zhong, G. One step synthesis of gamma-alkylidenebutenolides from simple vinyl carboxylic acids and alkenes. Chem. Commun. 2017, 53, 9902–9905. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Li, Y.; Tang, P. Total Synthesis of Schilancitrilactones B and C. Angew. Chem. Int. Ed. 2015, 54, 5732–5735. [Google Scholar] [CrossRef] [PubMed]
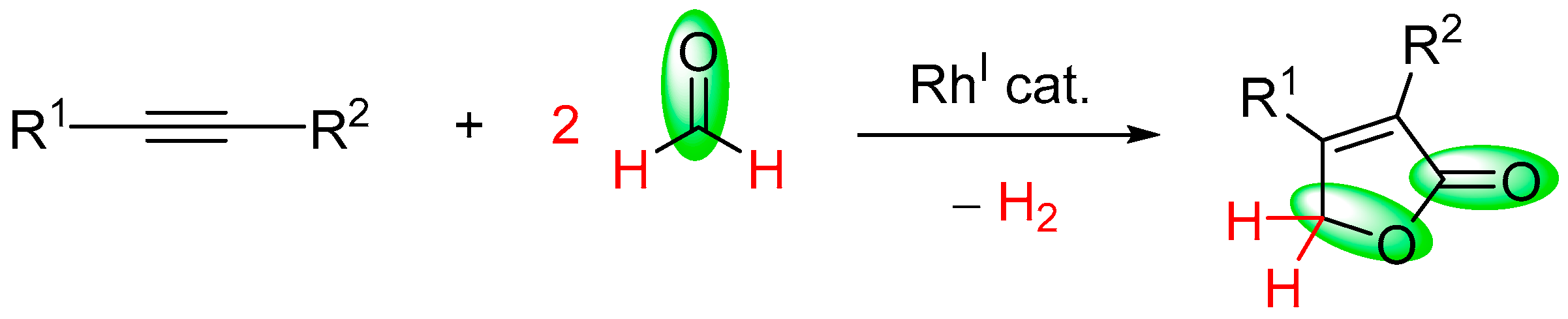
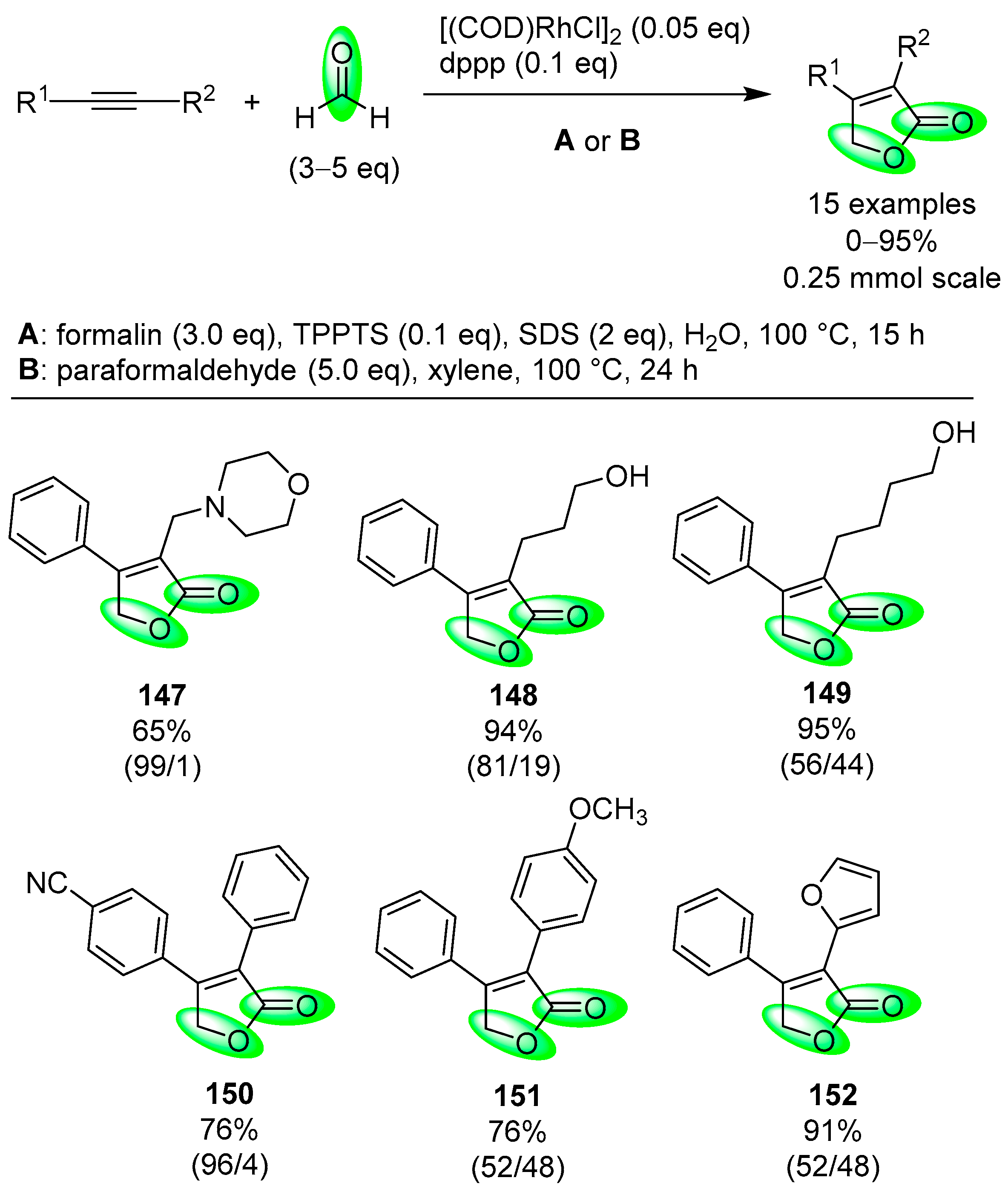
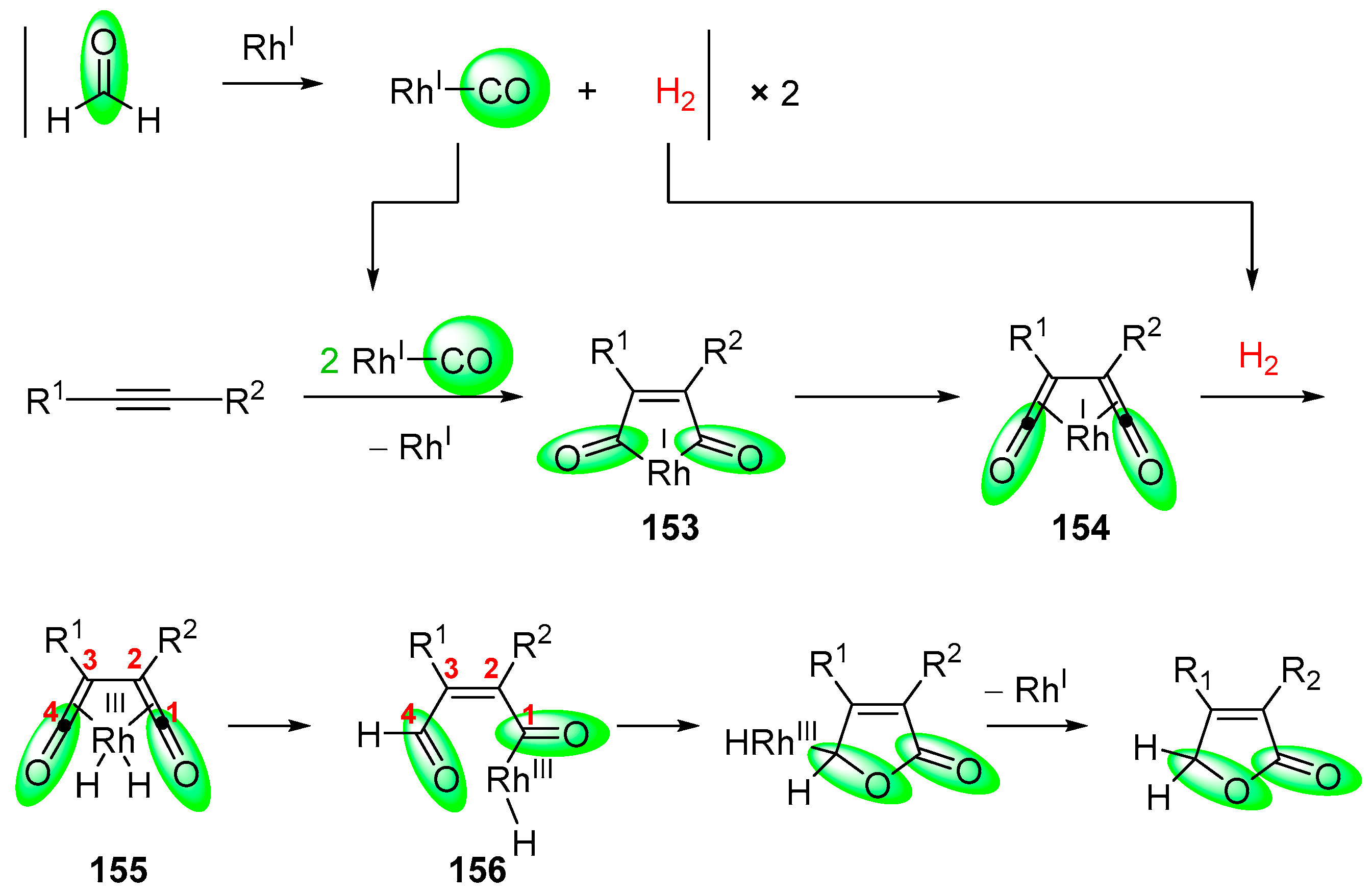
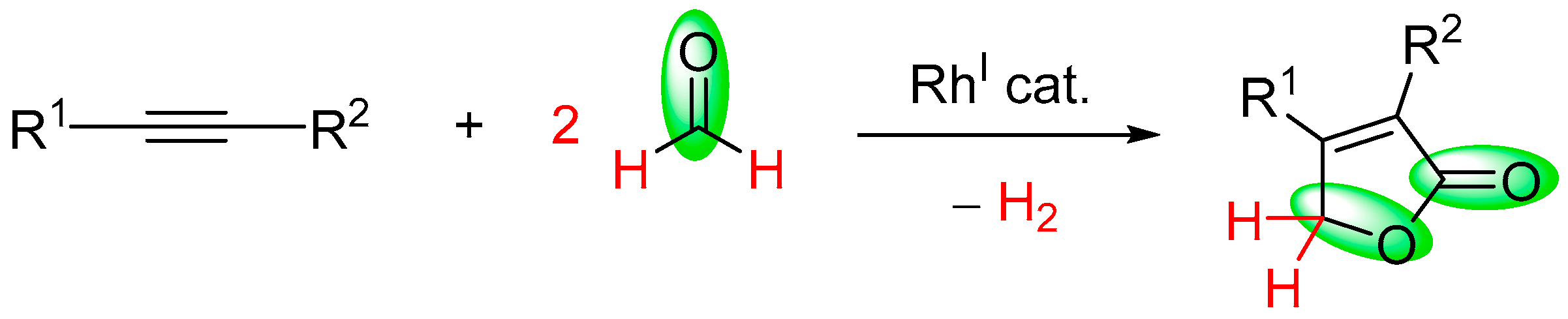
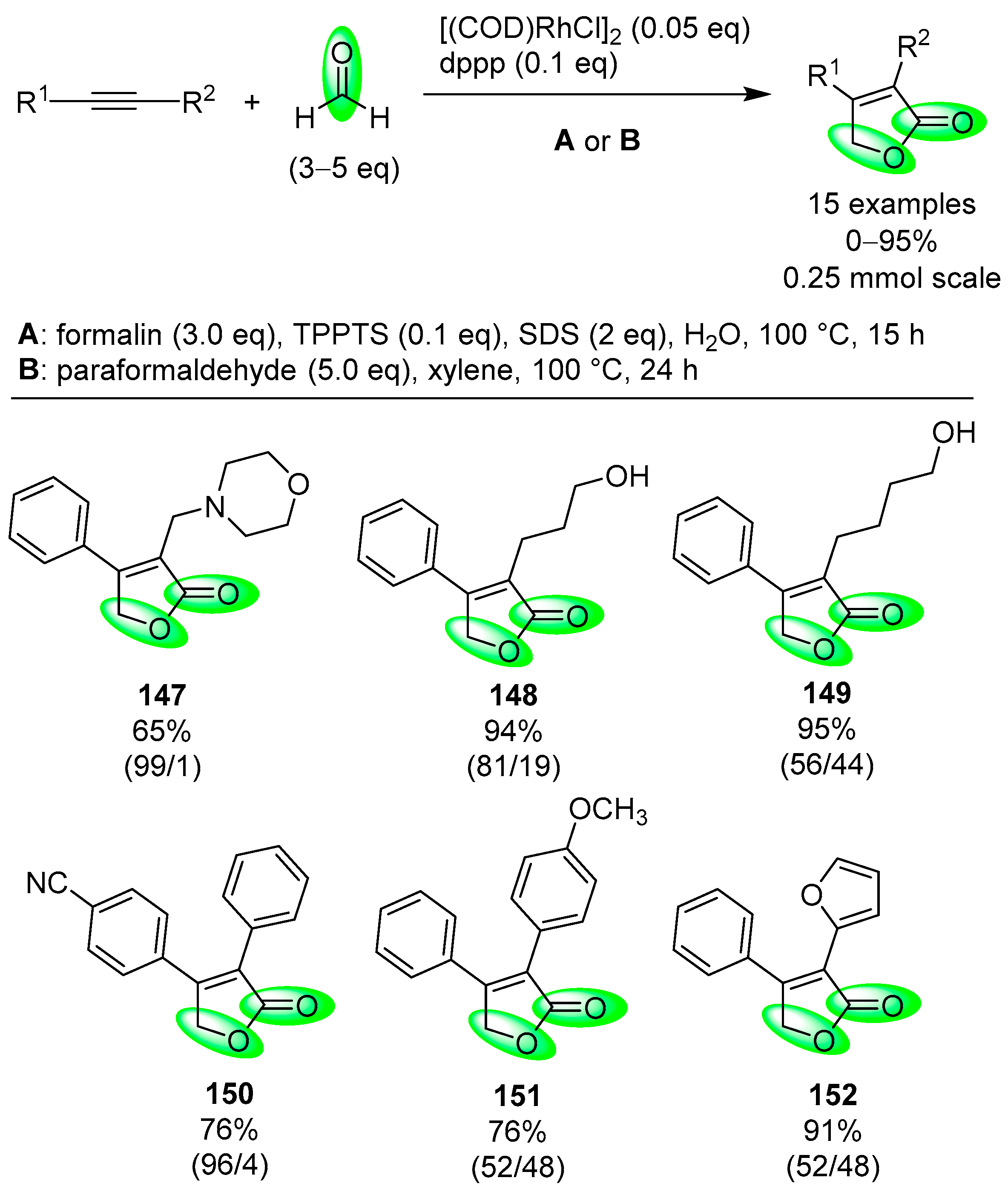
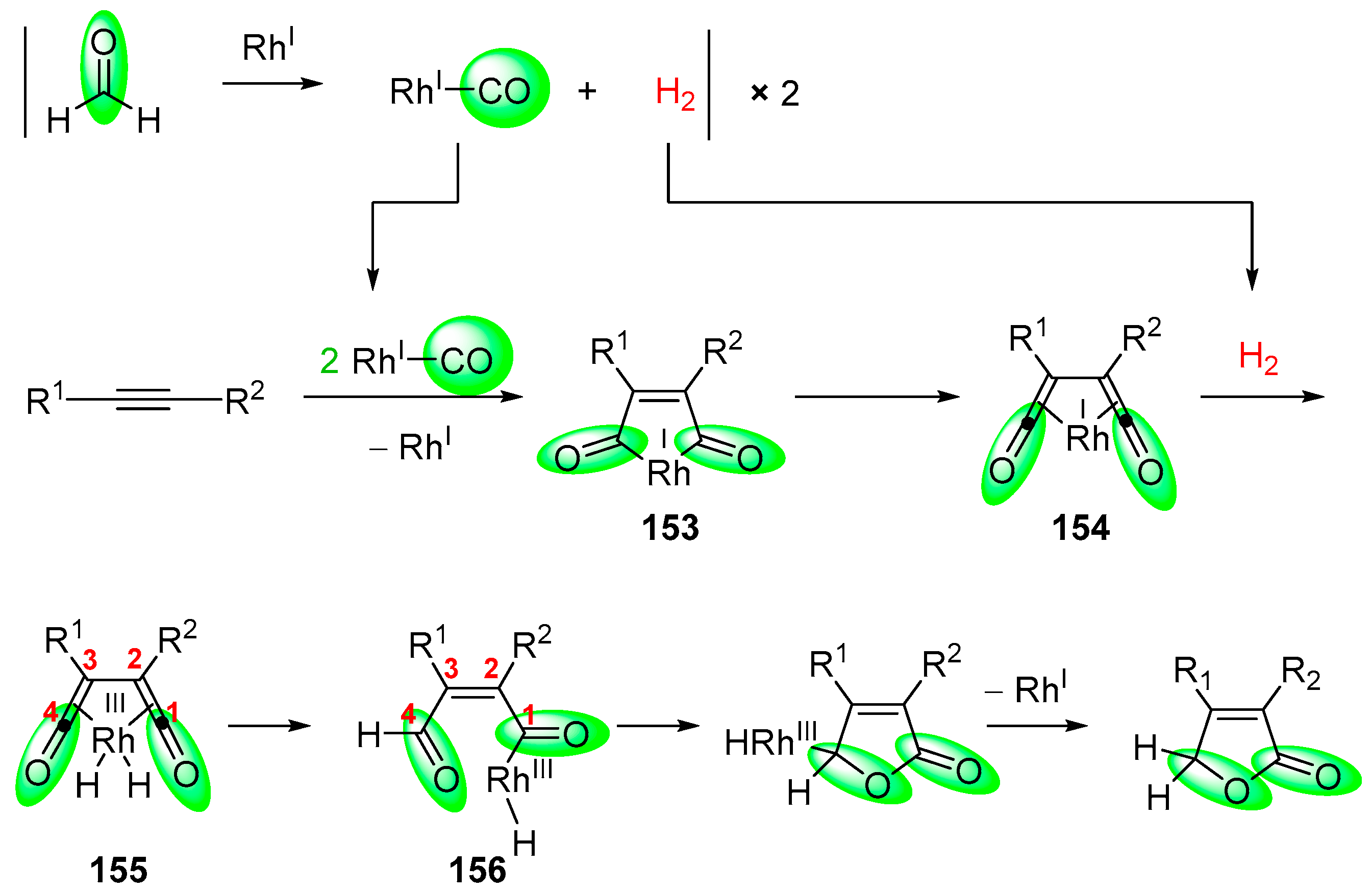
- Fuji, K.; Morimoto, T.; Tsutsumi, K.; Kakiuchi, K. Rh(I)-catalyzed CO gas-free cyclohydrocarbonylation of alkynes with formaldehyde to α,β-butenolides. Chem. Commun. 2005, 26, 3295–3297. [Google Scholar] [CrossRef] [PubMed]

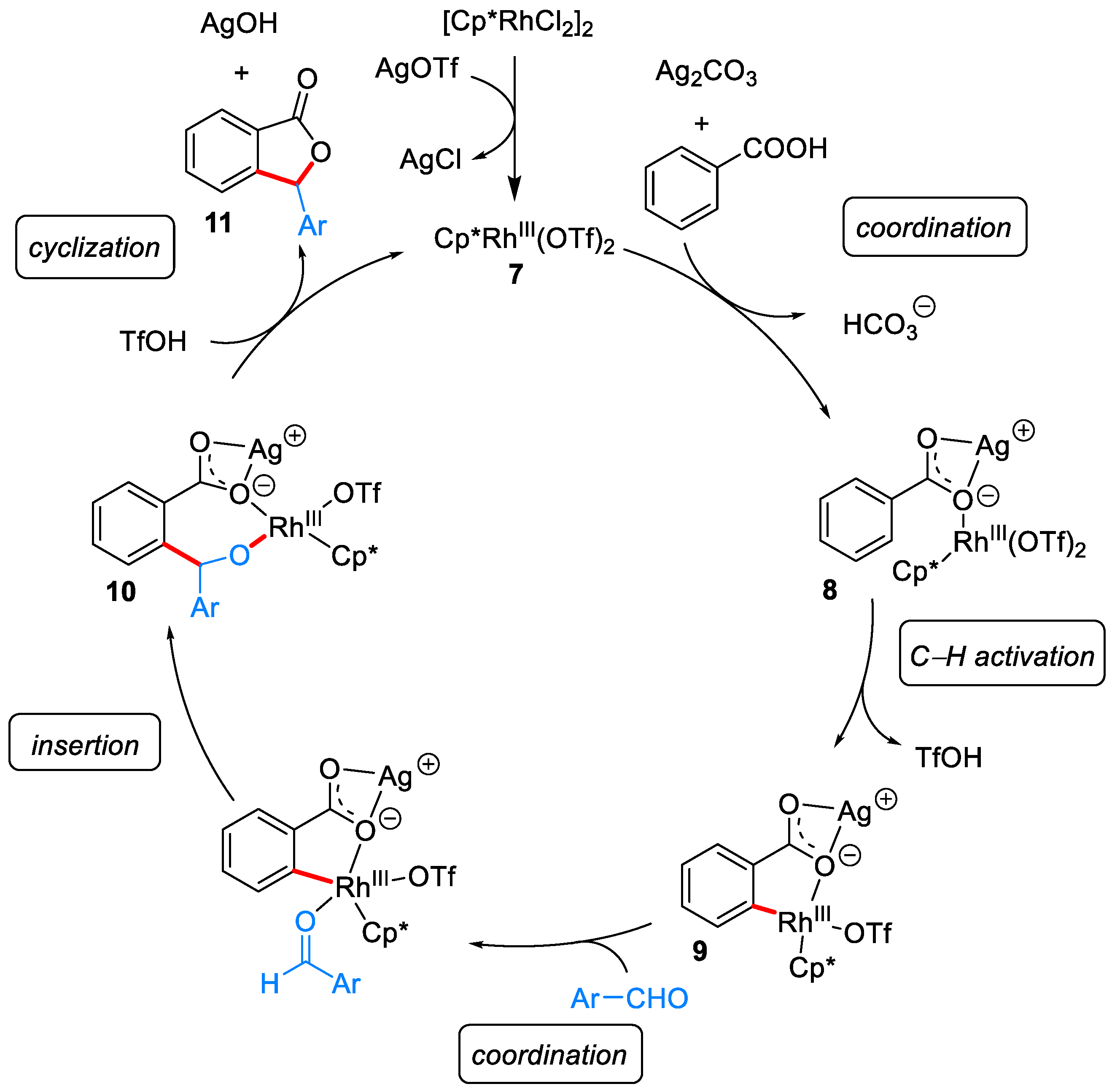
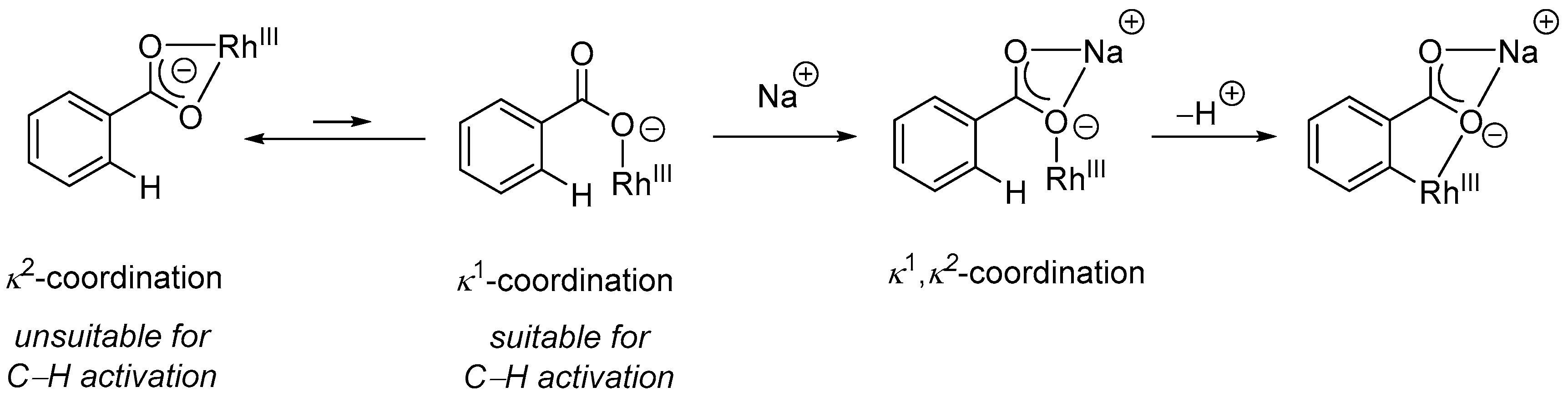
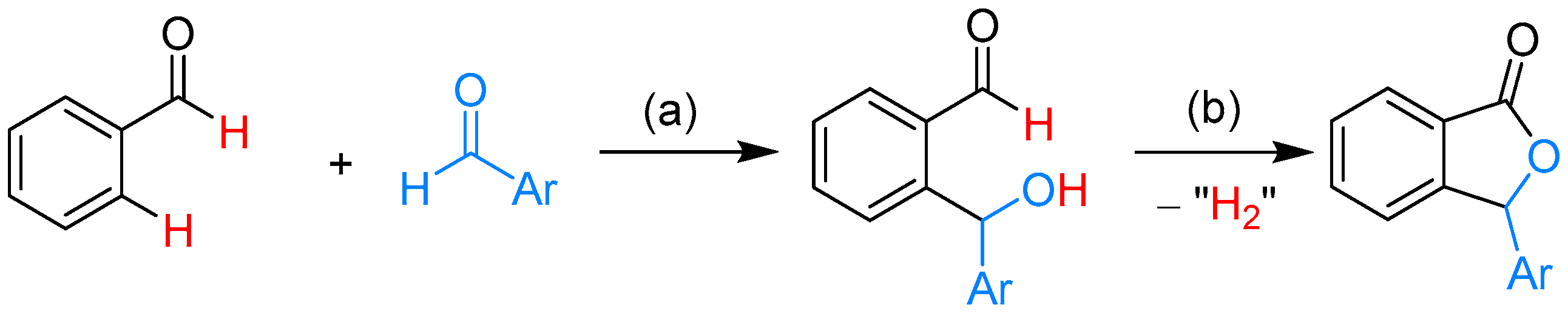
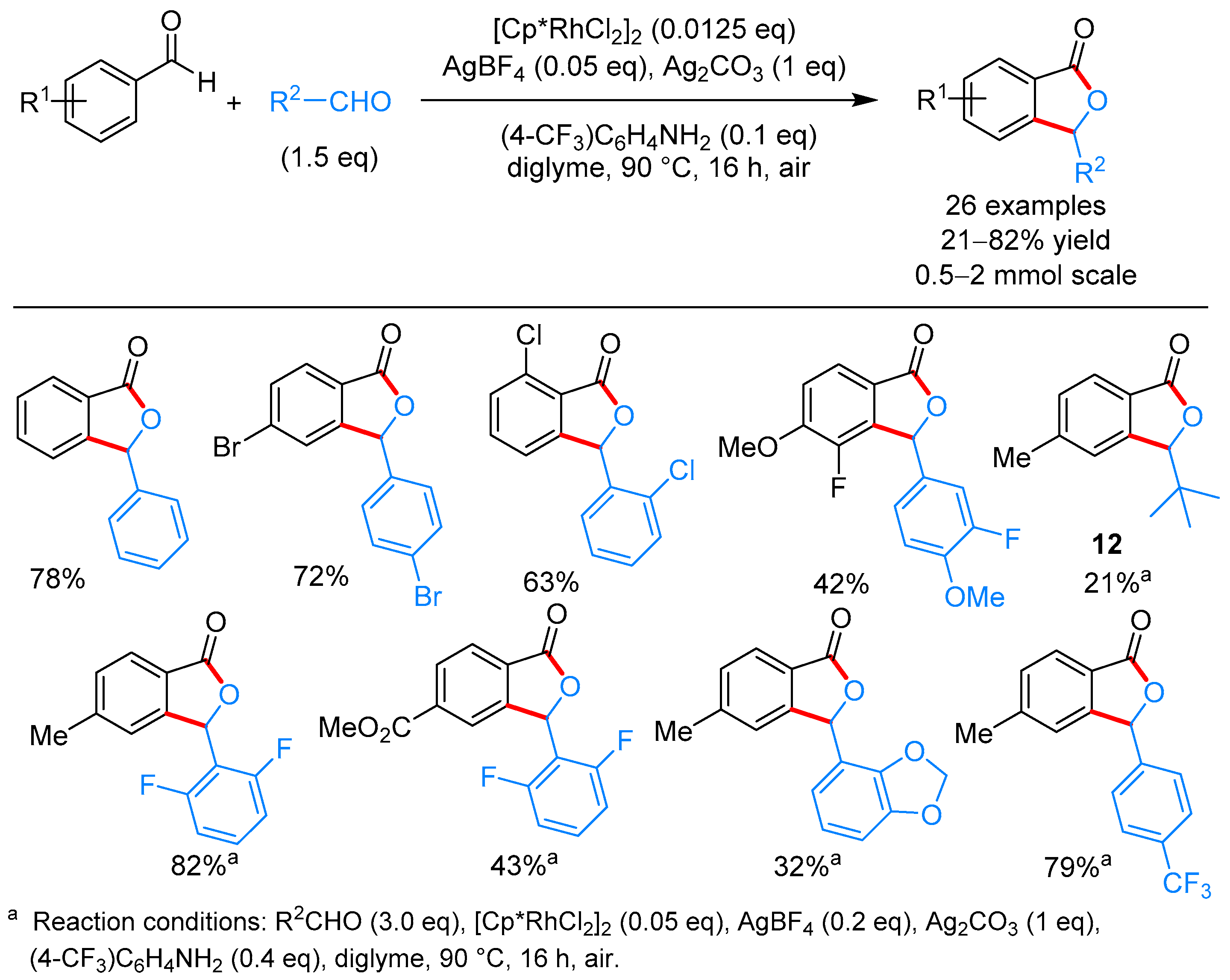

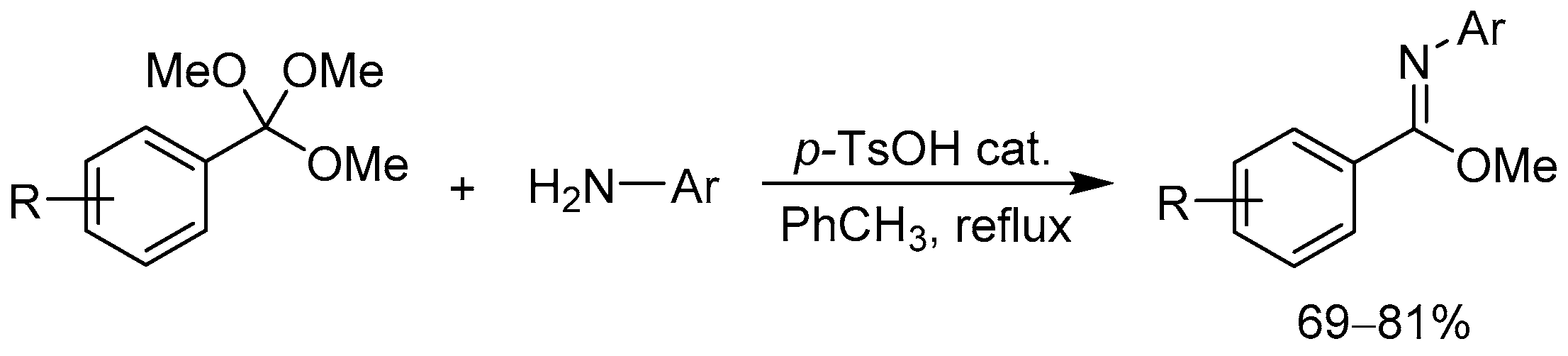















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
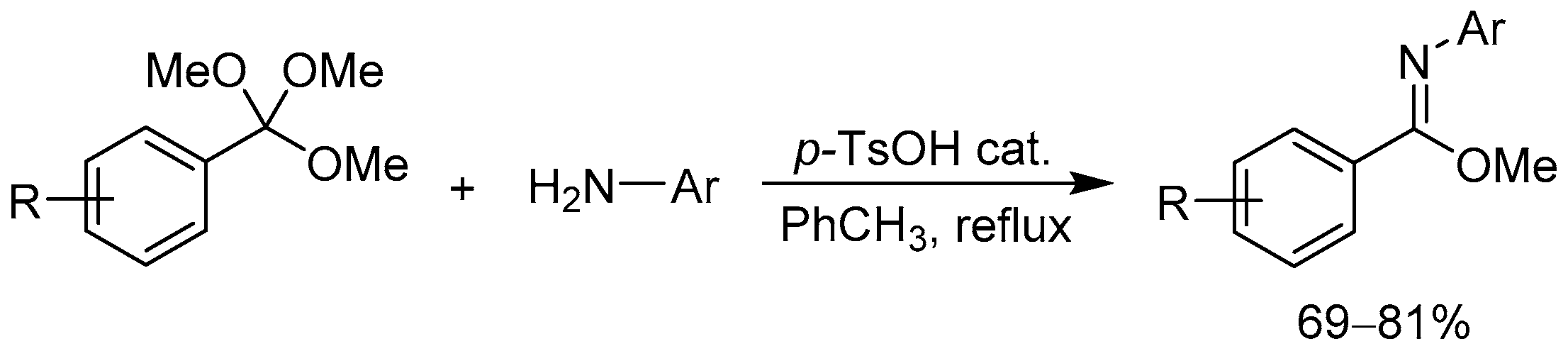{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cat. | eq. AgOTf | Additive | NMR Yield (%) | |
|---|---|---|---|---|
| [Cp*RhCl2]2 (0.08 eq) | 0.40 | - | 23 | 48 |
| [Cp*Rh(MeCN)3](SbF6)2 (0.16 eq) | - | - | 30 | 27 |
| [(COD)RhCl]2 (0.08 eq) | 0.24 | Ph4CpH2 (0.16 eq) | 51 | 22 |
| [(COD)RhCl]2 (0.08 eq) | 0.24 | Me5CpH (0.16 eq) | 21 | 20 |
| Product | Coupling Partners | Conditions | Ref. | |
|---|---|---|---|---|
 | ArCOOH | Ar’CHO | [Cp*RhCl2]2/AgOTf, Ag2CO3, dioxane, 150 °C | [20] |
| ArCHO | Ar’CHO | [Cp*RhCl2]2, AgBF4, (4-CF3)C6H4NH2, Ag2CO3, diglyme, 90 °C | [22] | |
| ArC(=NOMe)OMe | Ar’CHO | [Cp*RhCl2]2, AgSbF6, DCE, 110 °C | [23] | |
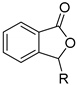 | ArCOOH | alkene | [RuCl2(p-cymene)]2, Cu(OAc)2·H2O, H2O, 80 °C | [24] |
| [RuCl2(p-cymene)]2, Cu(OAc)2·H2O, PEG-400/H2O, 80 °C | [25] | |||
| [RuCl2(p-cymene)]2, Cu(OAc)2·H2O, CH3CN, 100 °C | [26] | |||
| [RuCl2(p-cymene)]2, O2, MOAc/ROH, 60–80 °C | [27] | |||
| [RuCl2(p-cymene)]2, AcOH, O2, GVL, 80 °C | [28] | |||
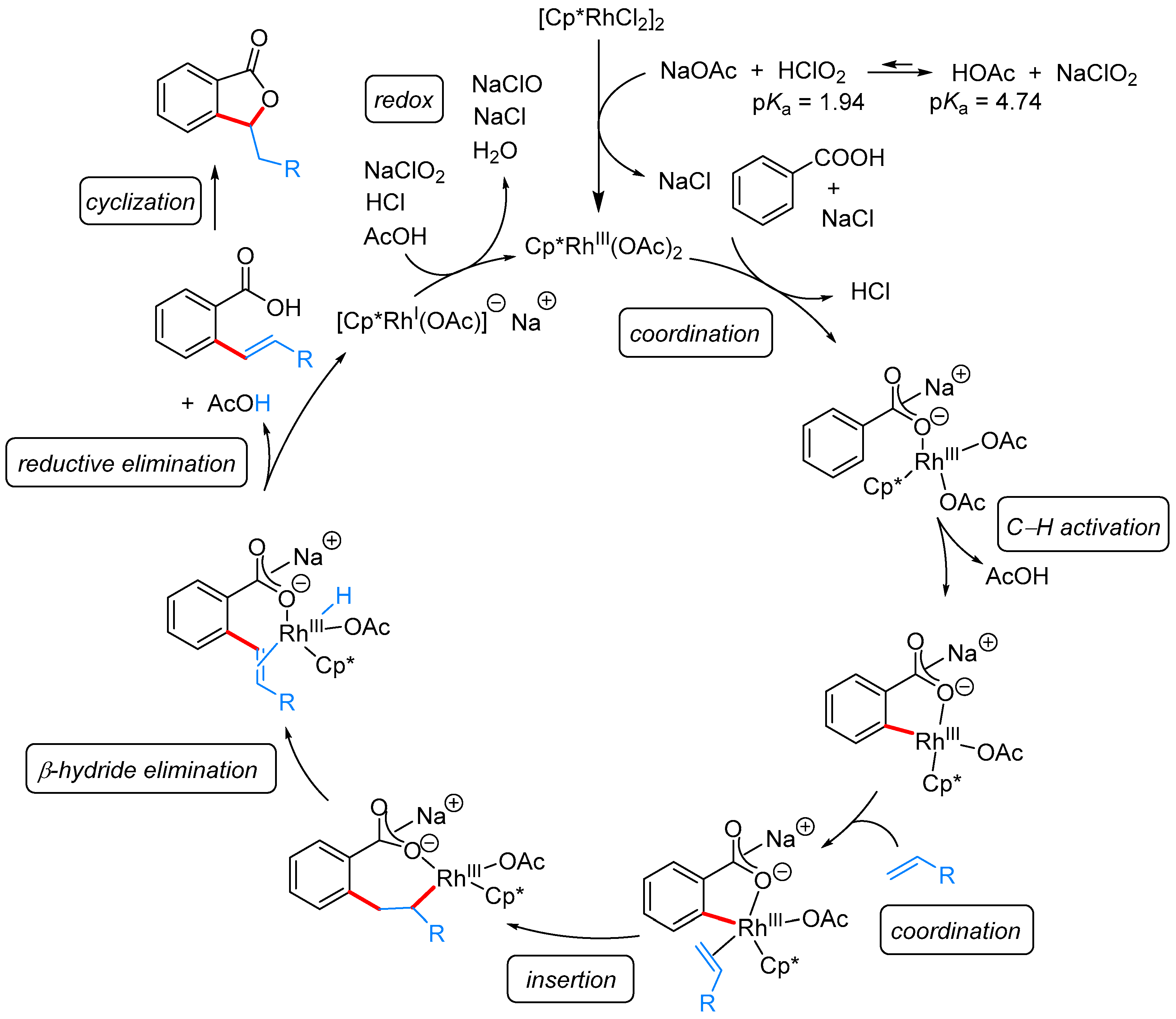
| [Cp*RhCl2]2, NaClO2, AcOH, H2O, 120 °C | [29] | |||
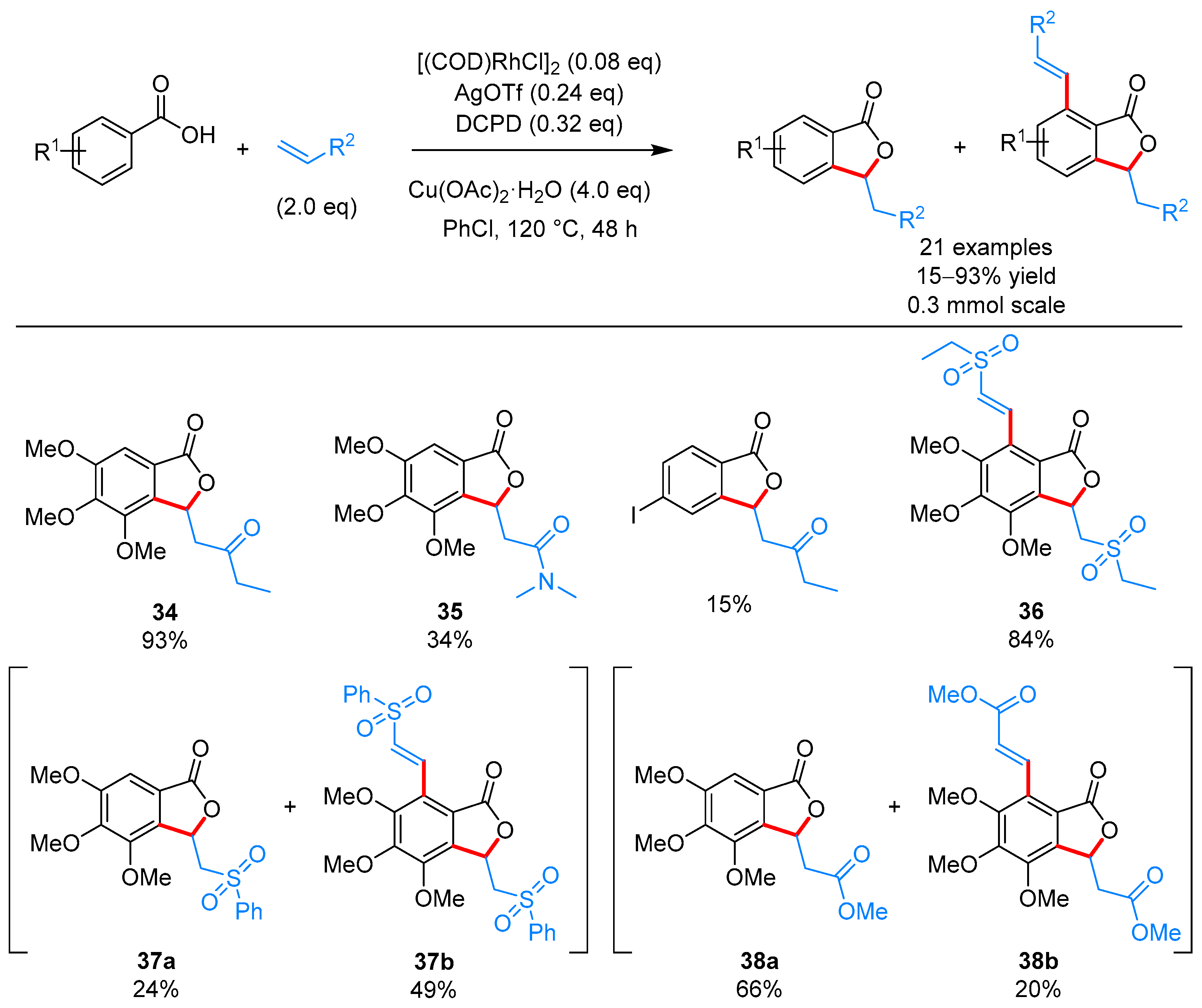
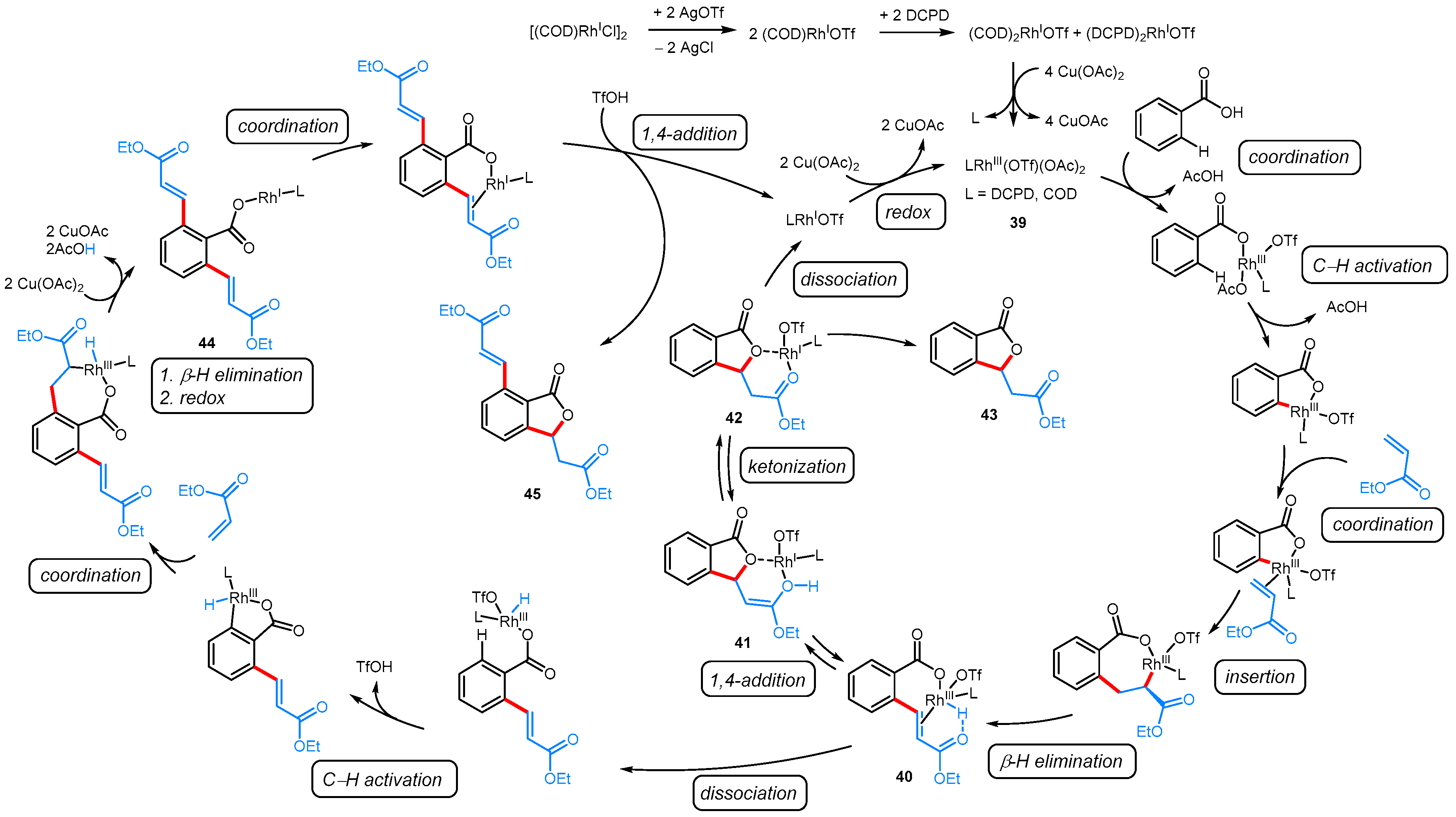
| [(COD)RhCl]2, AgOTf, DCPD, Cu(OAc)2·H2O, PhCl, 120 °C | [30] | |||
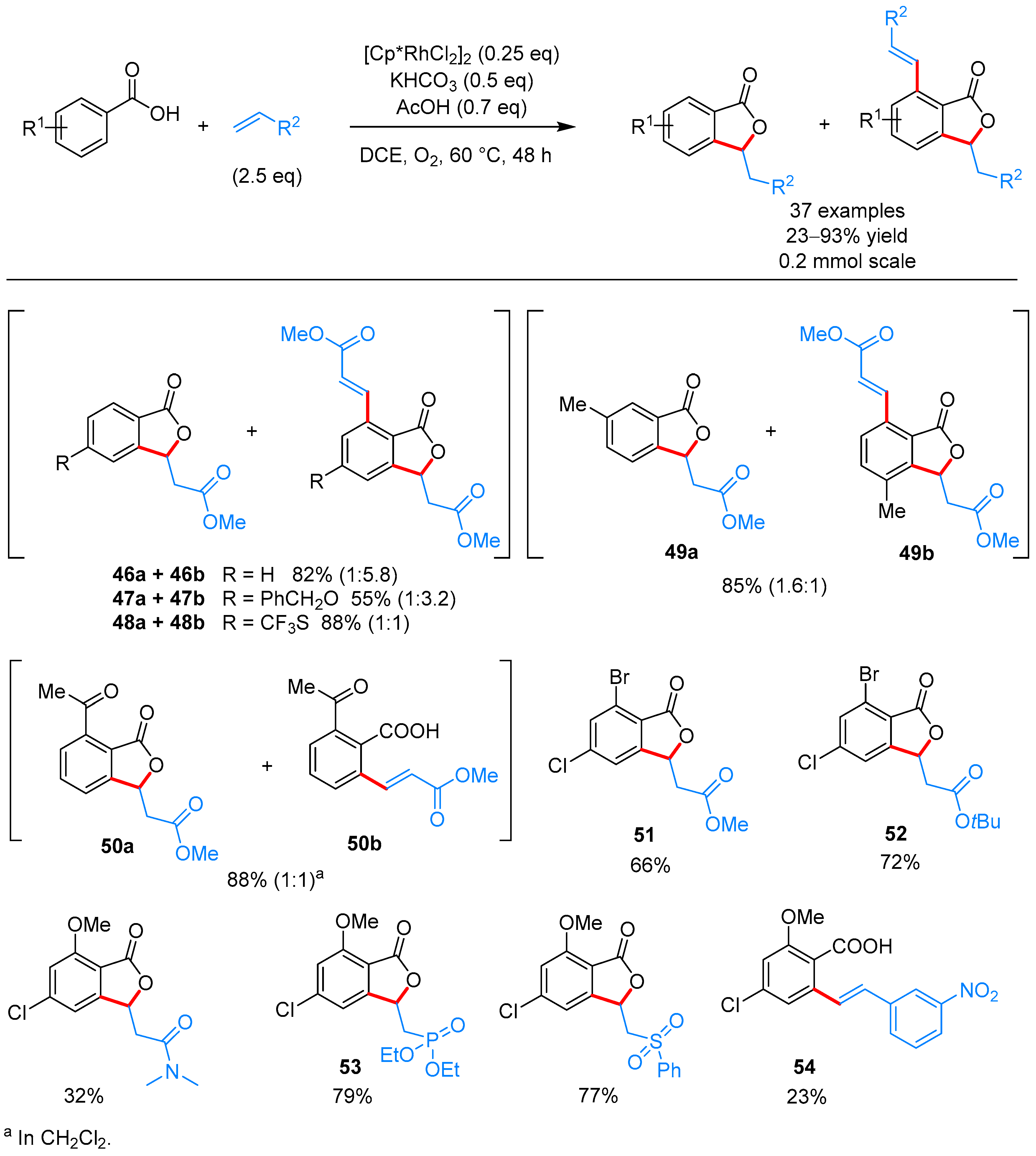
| [Cp*RhCl2]2, O2, KHCO3, AcOH, DCE, 60 °C | [31] | |||
| Pd(OAc)2, Cu(OAc)2·H2O, DMF, 120 °C | [33] | |||
| [Cp*RhCl2]2, Cu(OAc)2·H2O, o-xylene, 120 °C | [34] | |||
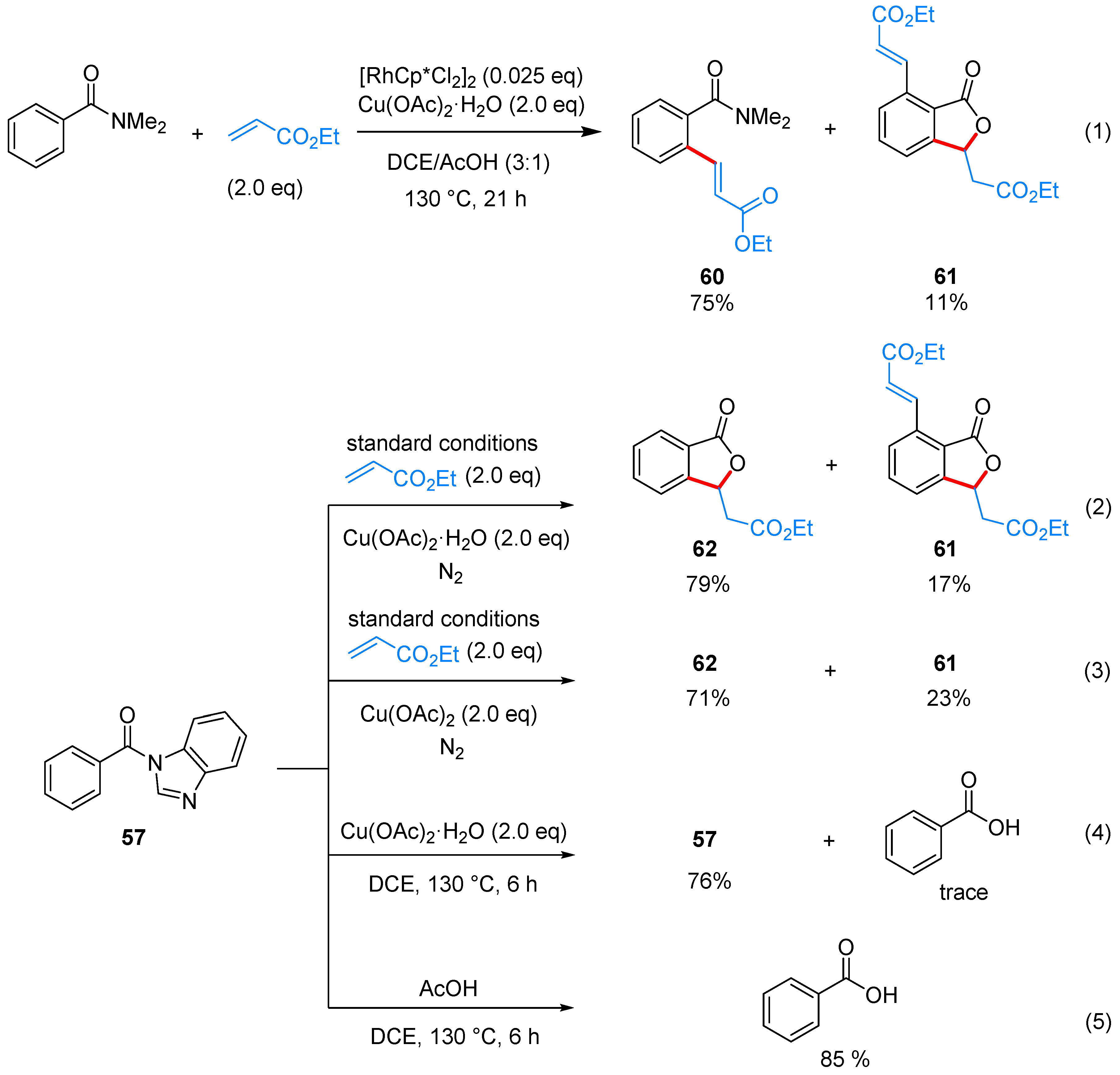
| ArCONR2 | alkene | [Cp*RhCl2]2, Cu(OAc)2·H2O, DCE/AcOH, 130 °C | [35] | |
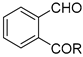 | – | RuHCl(CO)(PPh3)3, PhCH3, 90 °C | [39] | |
| [(COD)RhCl]2, AgNO3, duanphos, PhCH3, 75–100 °C | [40] | |||
| CoCl2, (R,R)-Ph-BPE, In, CH3CN | [41] | |||
 | ArCOOH | alkene | [Cp*RhCl2]2, Cu2O, Ag2CO3, PhCH3, 105 °C | [45] |
| ArCOOH | vinyl arene | Pd(OAc)2, Cu(OAc)2·H2O, DMF, 100 °C | [33] | |
| Pd(OAc)2, Ag2O, DMF, 110 °C | [49] | |||
| ArCOOH | vinyl acetate | [Cp*RhCl2]2, CuO, LiCl, KI, PhCH3/t-AmOH, 120 °C | [50] | |
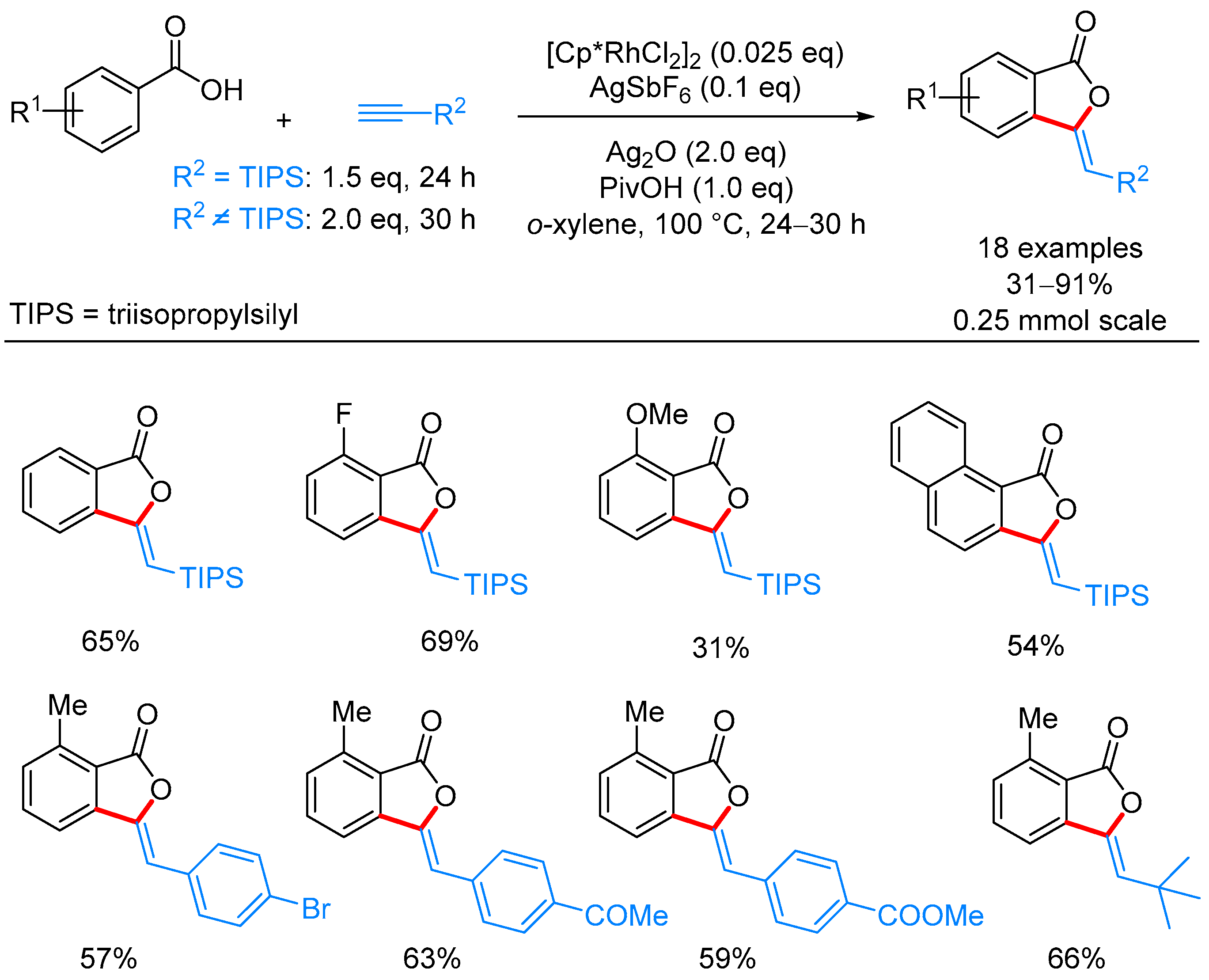
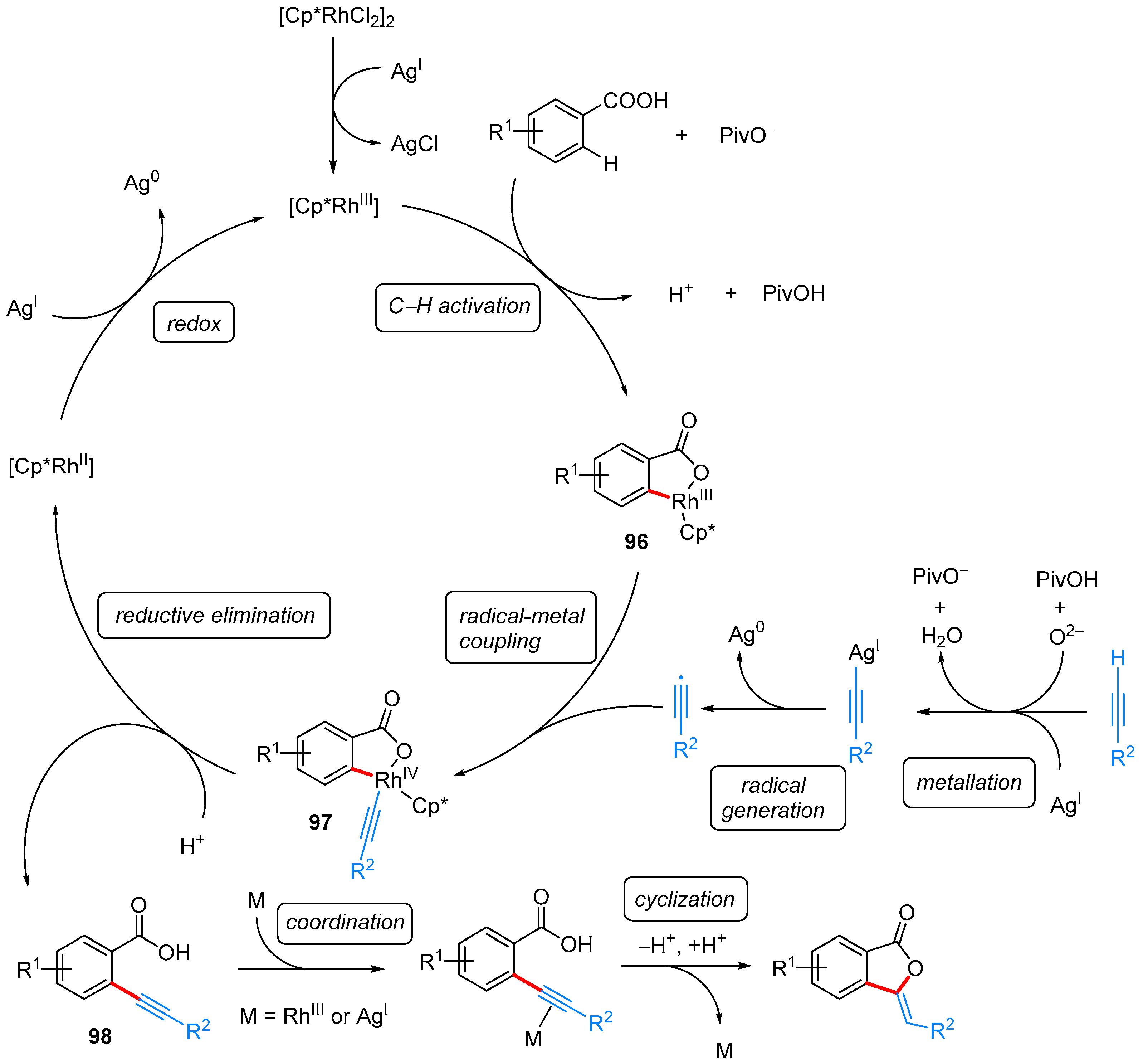
| ArCOOH | alkyne | [Cp*RhCl2]2, AgSbF6, Ag2O, PivOH, o-xylene, 100 °C | [51] | |
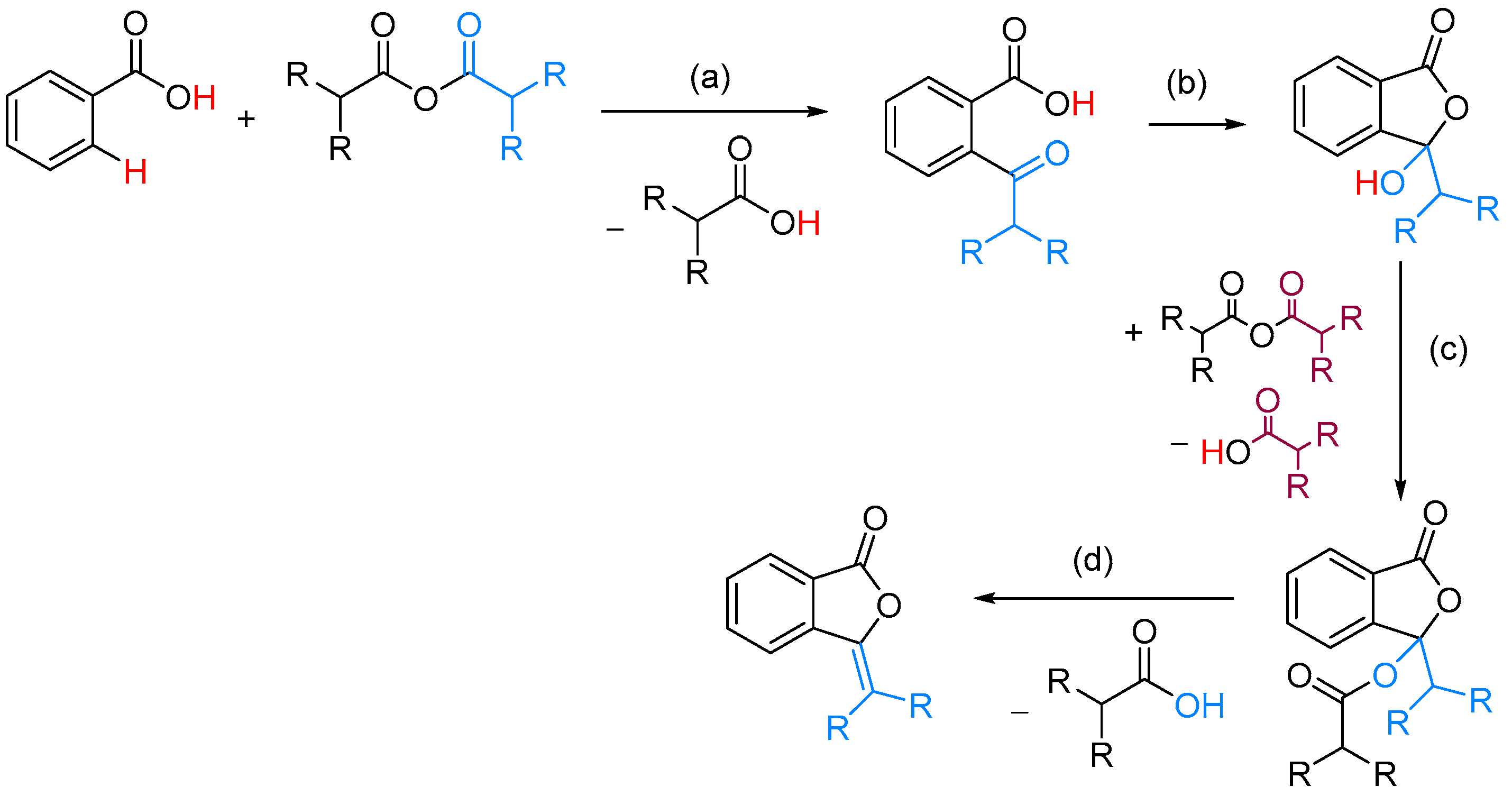
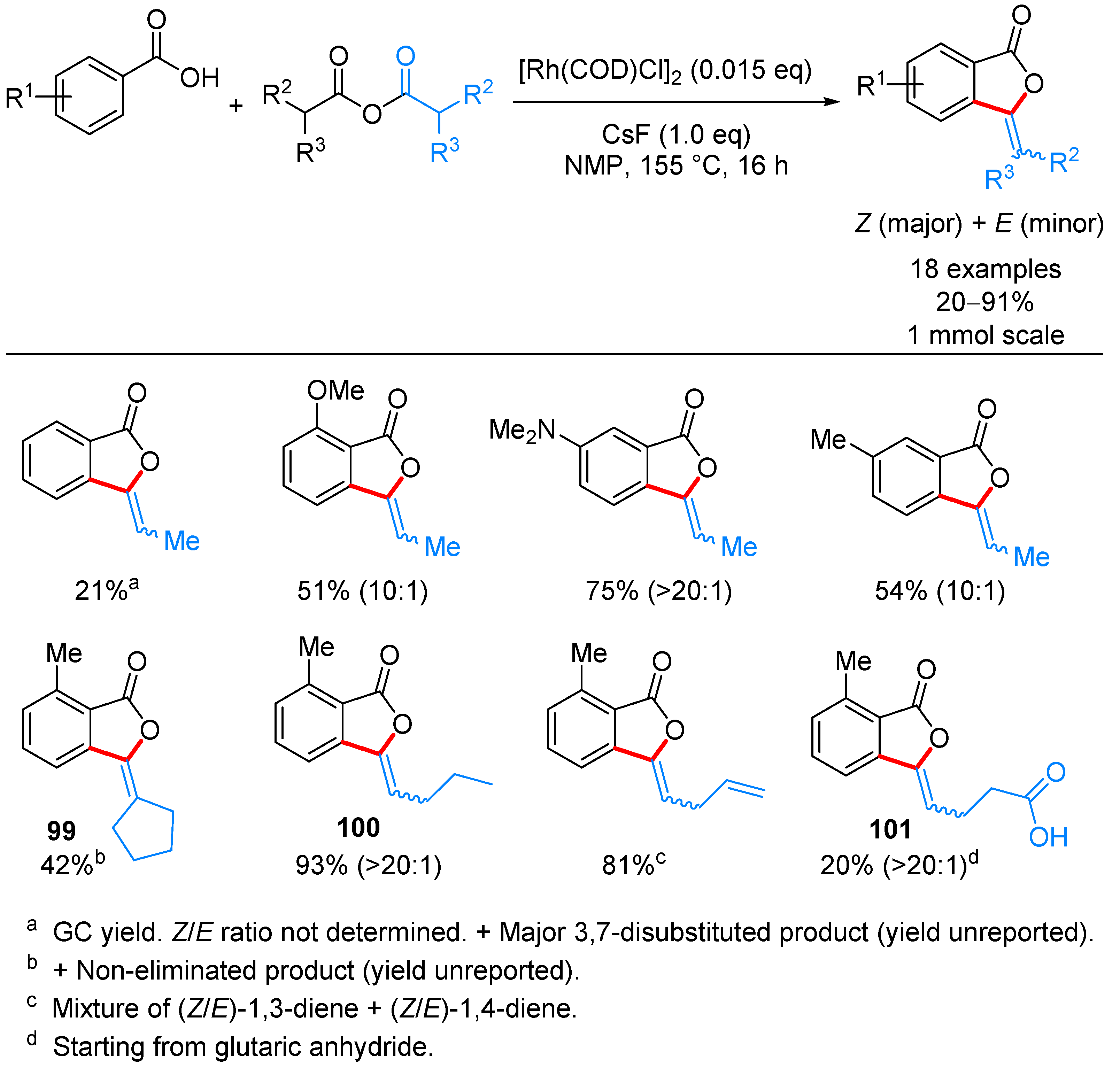
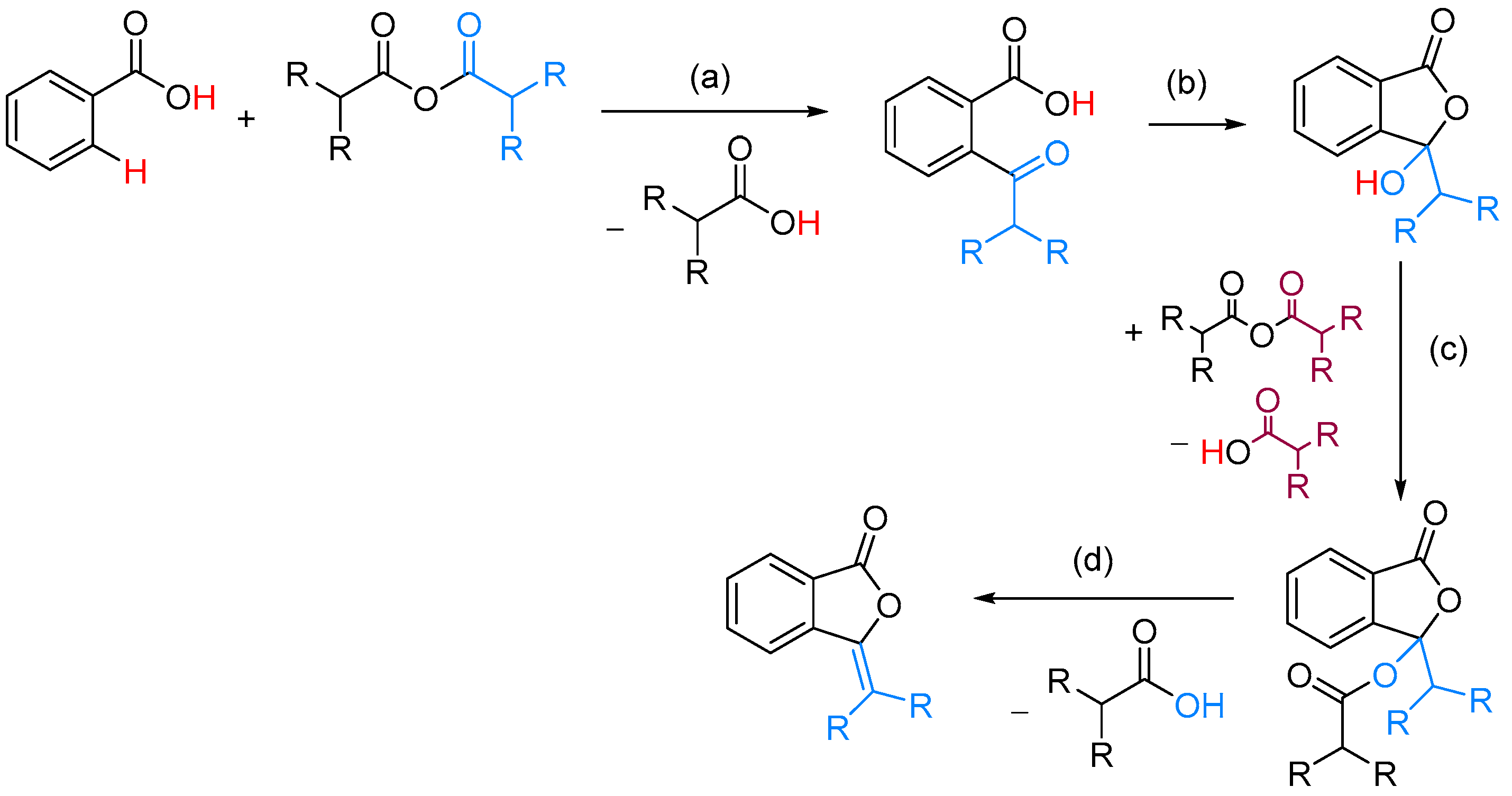
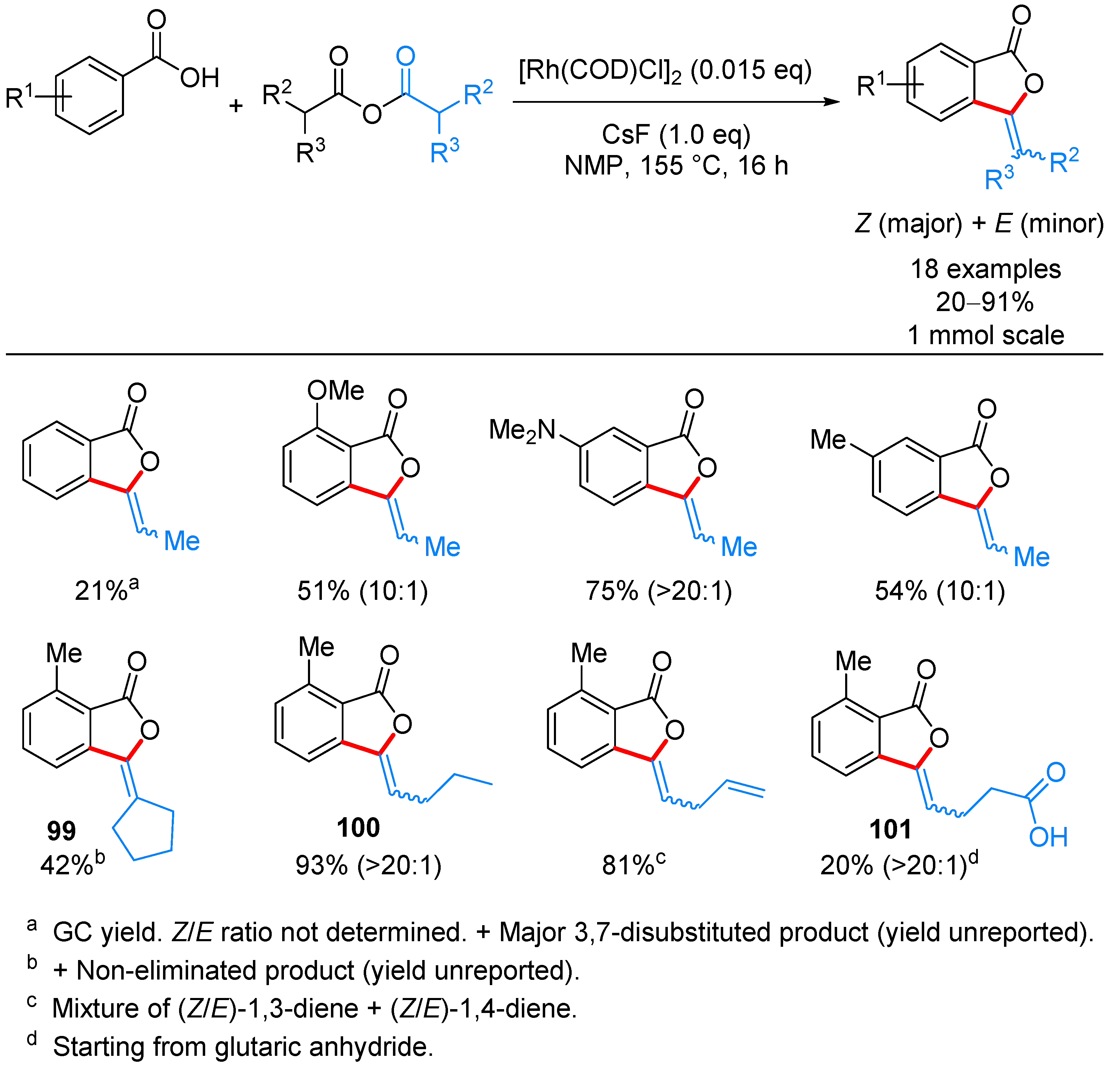
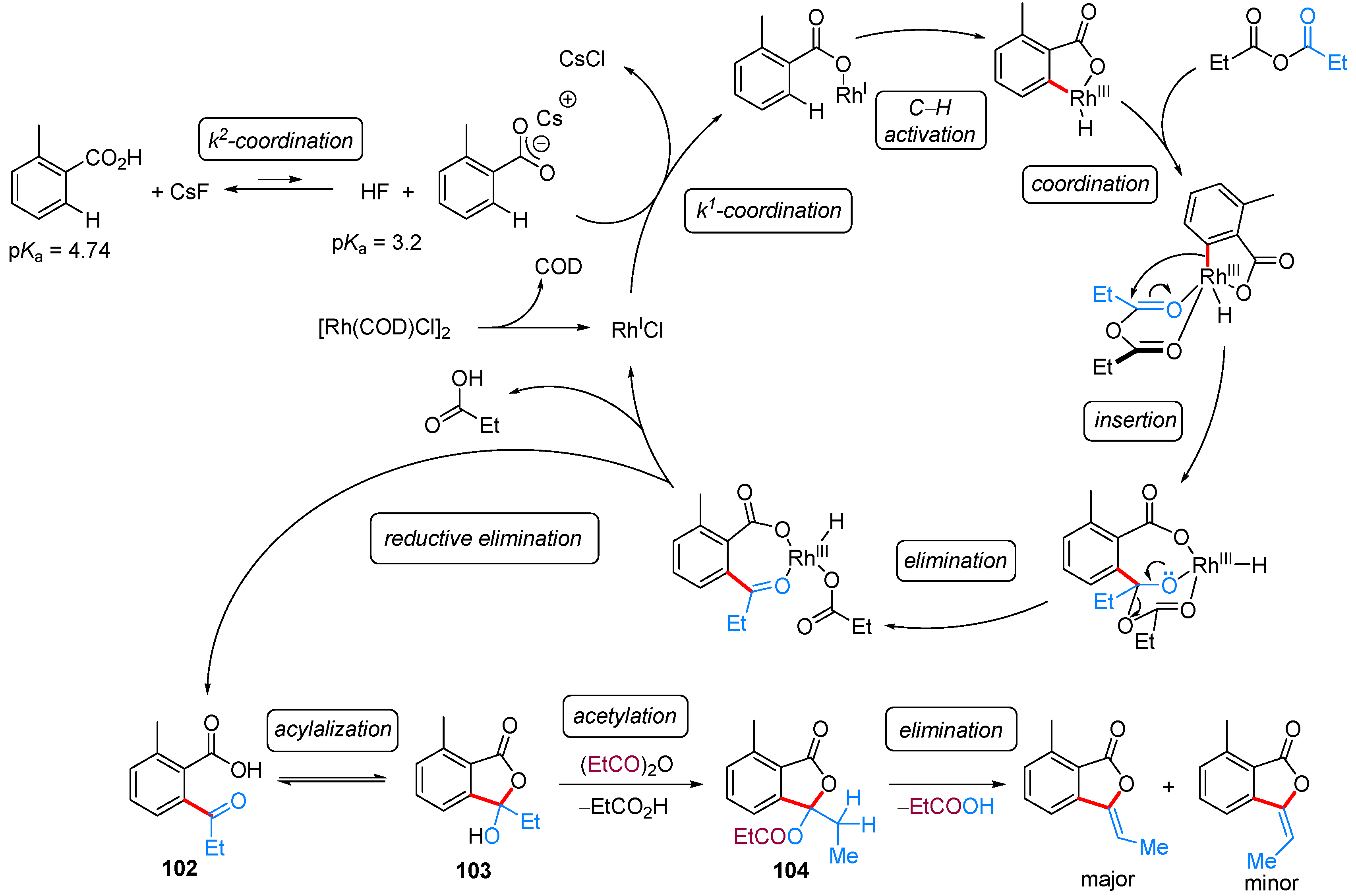
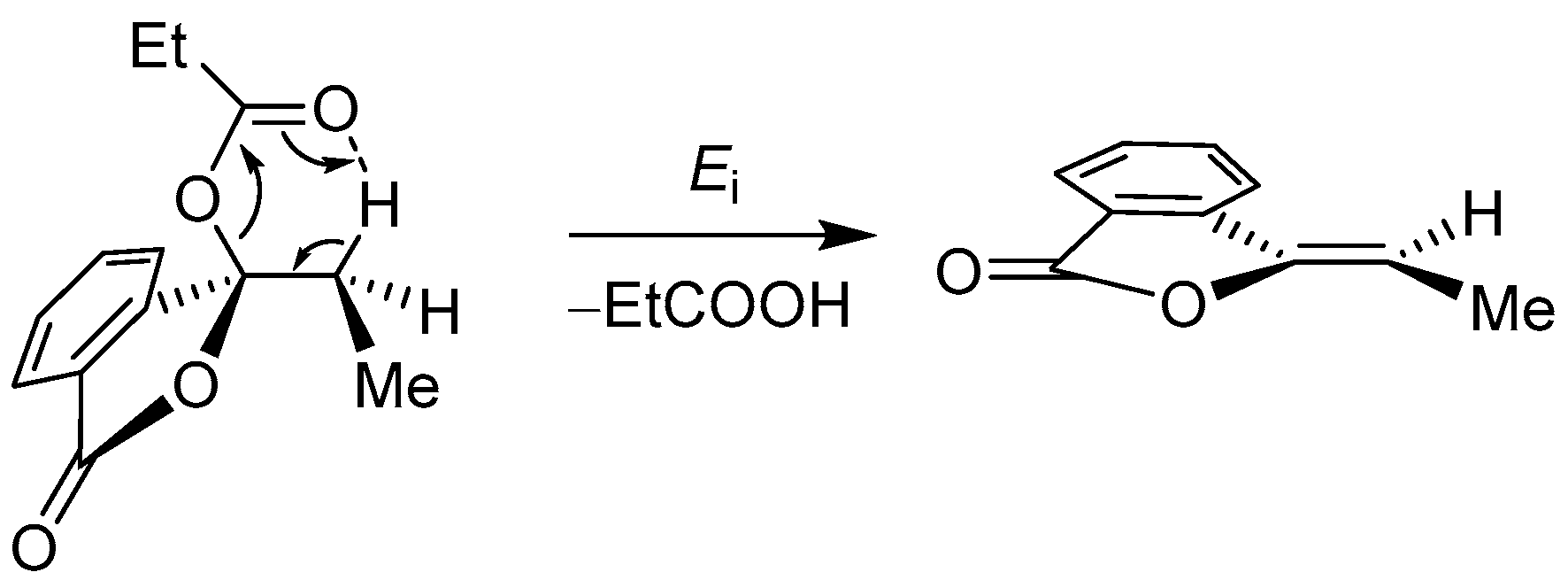
| ArCOOH | anhydride | [(COD)RhCl]2, CsF, NMP, 155 °C | [52] | |
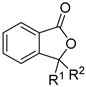 | ArCOOH | allene | [Cp*RhCl2]2, AgOAc, CH3CN, 60 °C | [54] |
| ArCOOH | alkyne | Ru/ZrO2, KOAc, mesitylene, 170 °C | [55] | |
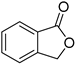 | 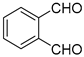 | – | HRh(CO)(PPh3)3, EtOH, 80 °C | [56] |
| RhCl3·3H2O, PPh3, Na2CO3, EtOH, 80 °C | [56] | |||
| RhH(PPh3)3, PhH, 19 °C | [57] | |||
| Et4N+[(μ-H)Cr2(CO)10]− | [58] | |||
| [Rh(dcpe)]2ClO4, acetone, 34 °C | [60] | |||
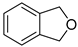 | – | CuCl2, neocuproin, t-BuOOH, H2O, r.t. | [61] | |
| CuI(aas-TPB)]n, t-BuOOH, H2O, r.t. | [63] | |||
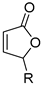 | acrylic acid | acrylate | Ag2CO3, DMF, 120 °C | [65] |
| Cu(OAc)2·H2O, DMF, 100 °C | [65] | |||
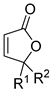 | acrylic acid | allene | [Cp*RhCl2]2, AgOAc, CH3CN, 80 °C | [54] |
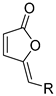 | acrylic acid | acrylate | [Cp*RhCl2]2, Cu(OAc)2·H2O, CH3CN, 120 °C | [66] |
 | alkyne | HCHO | [(COD)RhCl]2, dppp, TPPTS, SDS, H2O, 100 °C | [68] |
| (CH2O)n | [(COD)RhCl]2, dppp, xylene, 100 °C | [68] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renzetti, A.; Fukumoto, K. Synthesis of Phthalides and ?,?-butenolides by Transition Metal-Catalyzed Activation of C—H Bonds. Molecules 2019, 24, 824. https://doi.org/10.3390/molecules24040824
Renzetti A, Fukumoto K. Synthesis of Phthalides and ?,?-butenolides by Transition Metal-Catalyzed Activation of C—H Bonds. Molecules. 2019; 24(4):824. https://doi.org/10.3390/molecules24040824
Chicago/Turabian StyleRenzetti, Andrea, and Kozo Fukumoto. 2019. "Synthesis of Phthalides and ?,?-butenolides by Transition Metal-Catalyzed Activation of C—H Bonds" Molecules 24, no. 4: 824. https://doi.org/10.3390/molecules24040824
APA StyleRenzetti, A., & Fukumoto, K. (2019). Synthesis of Phthalides and ?,?-butenolides by Transition Metal-Catalyzed Activation of C—H Bonds. Molecules, 24(4), 824. https://doi.org/10.3390/molecules24040824