A Non-Enzymatic and Label-Free Fluorescence Bioassay for Ultrasensitive Detection of PSA

Abstract
:1. Introduction
2. Results and Discussion
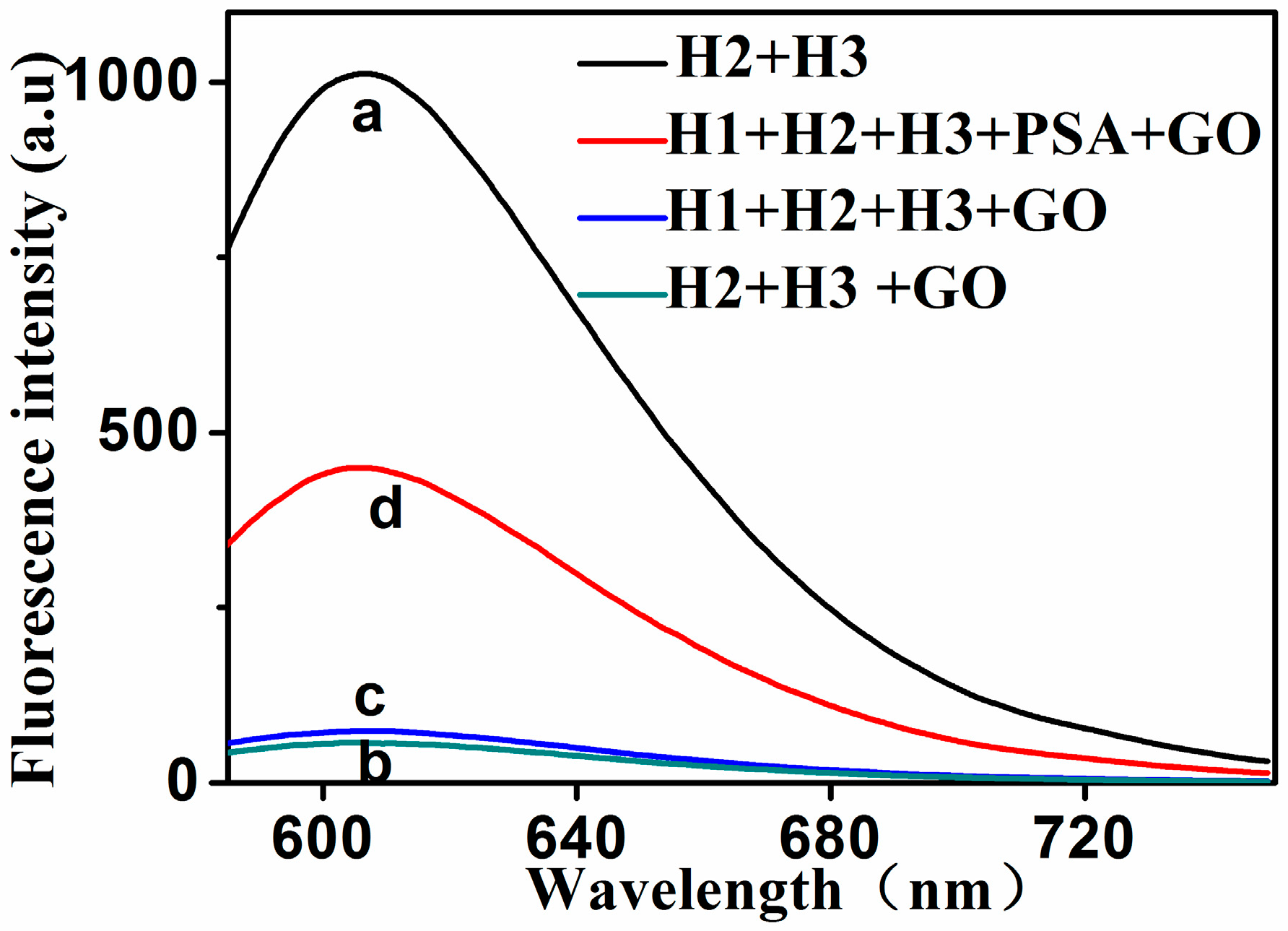
2.1. Principle and Feasibility of the Assay
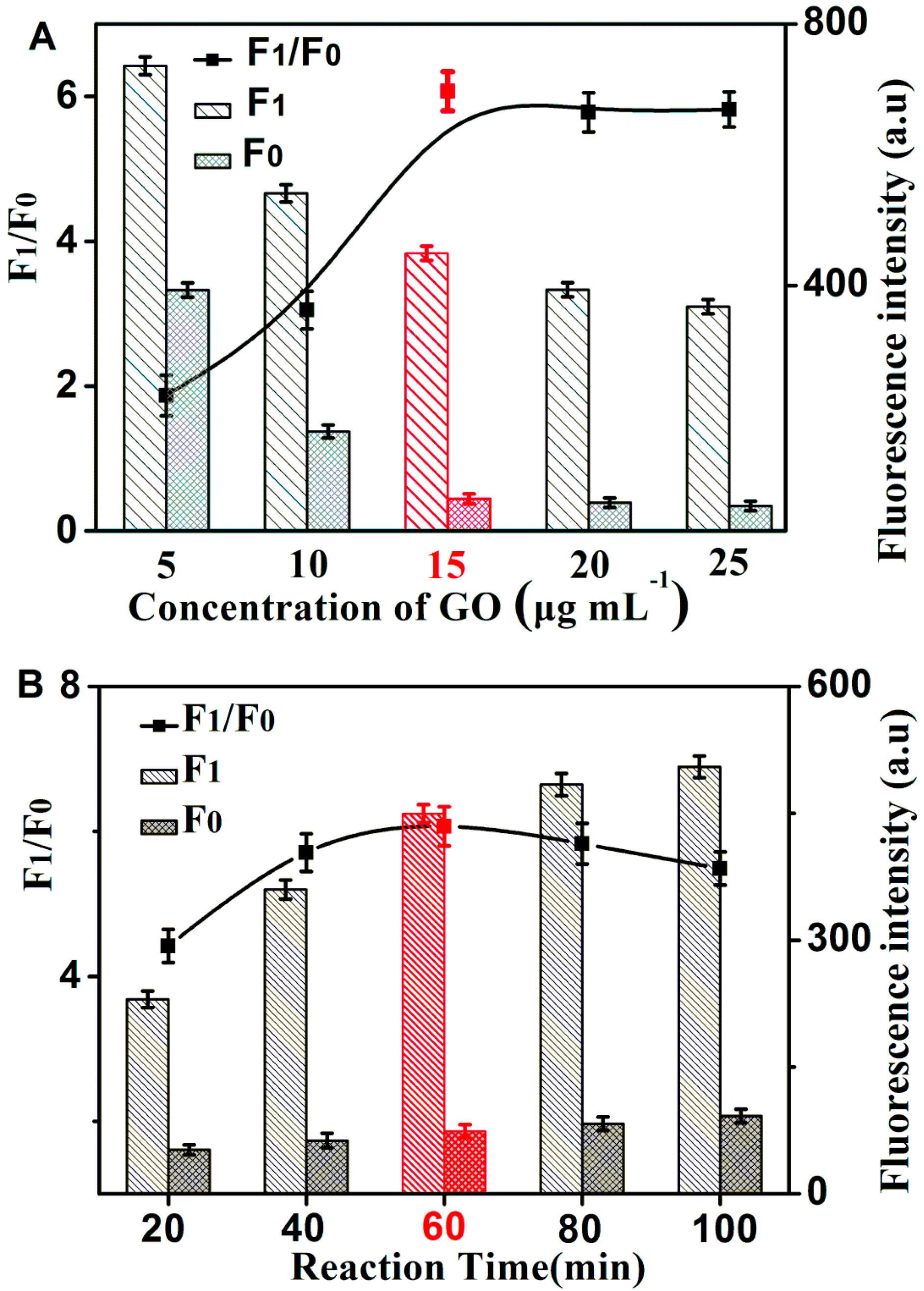
2.2. Optimization of Reaction Conditions
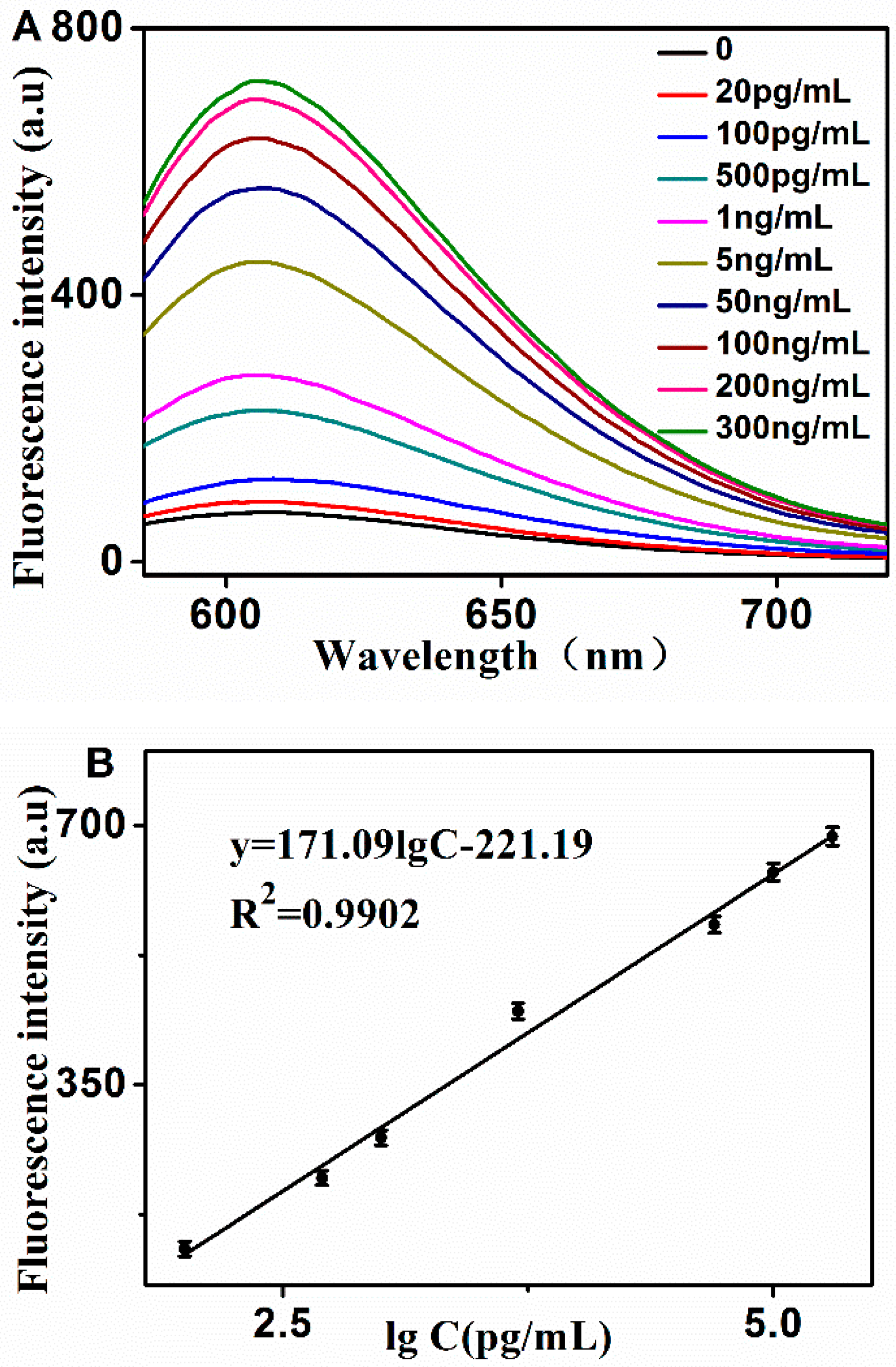
2.3. Sensitivity and Specificity for PSA Detection
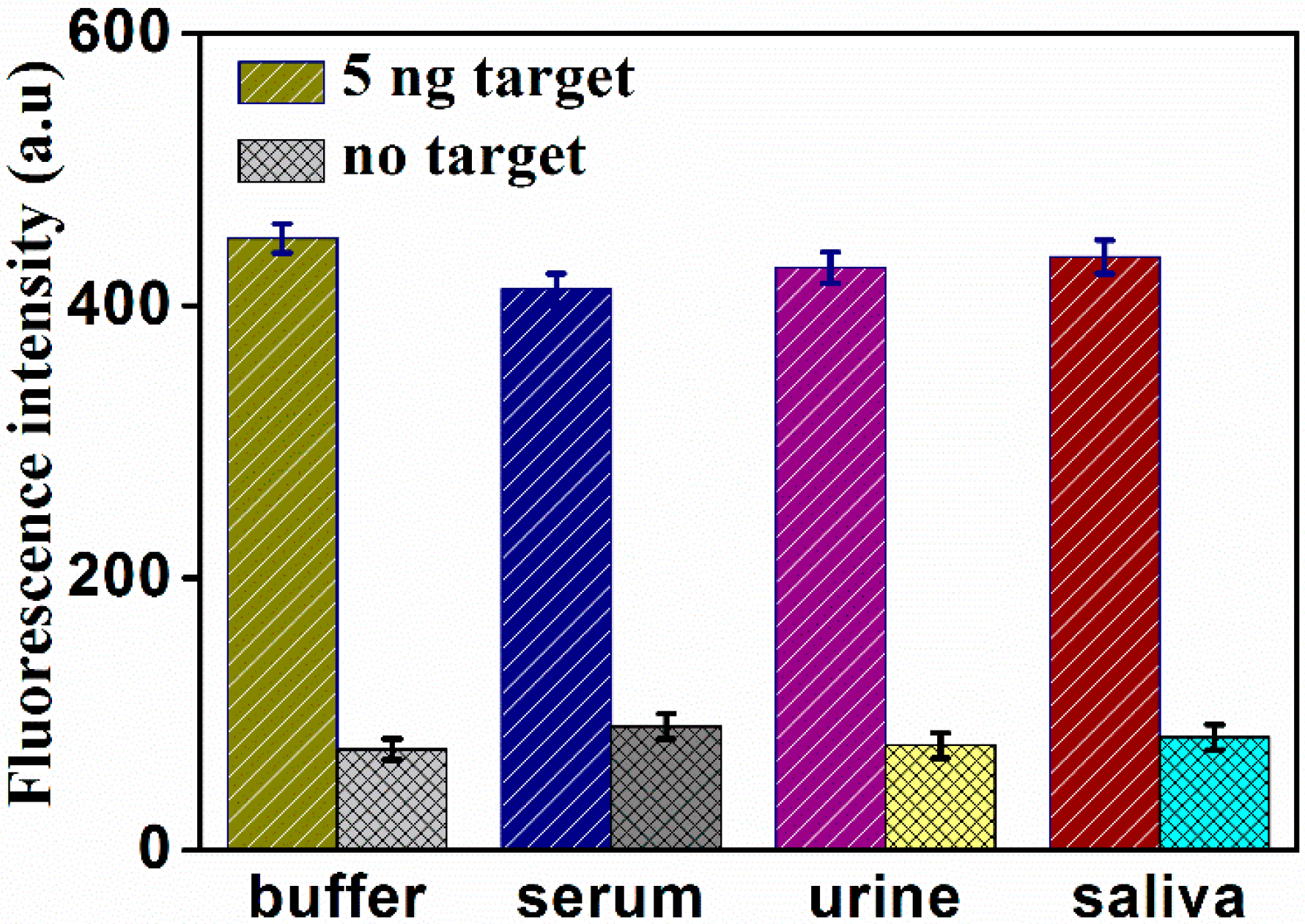
2.4. Determination of PSA in Real Samples
3. Experimental Section
3.1. Reagents and Materials
3.2. Apparatus and Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Saini, S. PSA and beyond: Alternative prostate cancer biomarkers. Cell. Oncol. 2016, 39, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Cochetti, G.; Poli, G.; Guelfi, G.; Boni, A.; Egidi, M.G.; Mearini, E. Different levels of serum microRNAs in prostate cancer and benign prostatic hyperplasia: Evaluation of potential diagnostic and prognostic role. Oncotargets Ther. 2016, 9, 7545–7553. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Wu, Q.; Williams, V.; Clark, G.; Guruli, G.; Zehner, Z. A Panel of MicroRNAs as Diagnostic Biomarkers for the Identification of Prostate Cancer. Int. J. Mol. Sci. 2017, 18, 1281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lei, Y.; Chen, X.; Wang, Z.; Liu, J. An aptamer based resonance light scattering assay of prostate specific antigen. Biosens. Bioelectron. 2012, 36, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.I.; Wichers, J.H.; van Amerongen, A.; Reis, N.M. Towards One-Step Quantitation of Prostate-Specific Antigen (PSA) in Microfluidic Devices: Feasibility of Optical Detection with Nanoparticle Labels. Bionanoscience 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Compton, O.C.; Nguyen, S.T. Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials. Small 2010, 6, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.; Hu, D.; Lin, C.T.; Li, J.; Lin, Y. Aptamer/graphene oxide nanocomplex for in situ molecular probing in living cells. J. Am. Chem. Soc. 2010, 132, 9274–9276. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Rastonm, N.H.A.; Gu, M.B. Aptamer-based nanobiosensors. Biosens. Bioelectron. 2016, 76, 2–19. [Google Scholar] [PubMed]
- Meng, H.M.; Liu, H.; Kuai, H.; Peng, R.; Mo, L.; Zhang, X.B. Aptamer-integrated DNA nanostructures for biosensing, bioimaging and cancer therapy. Chem. Soc. Rev. 2016, 45, 2583–2602. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.M.; Ding, L.; Wang, Z.; You, J.; Qu, F. A novel aptamer-functionalized MoS2 nanosheet fluorescent biosensor for sensitive detection of prostate specific antigen. Anal. Bioanal. Chem. 2015, 407, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Han, D.; Ocsoy, I.; Tan, W. Aptamers selected by cell-SELEX for application in cancer studies. Bioanalysis 2015, 2, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Khoshfetrat, S.M.; Mehrgardi, M.A. Amplified detection of leukemia cancer cells using an aptamer-conjugated gold-coated magnetic nanoparticles on a nitrogen-doped graphene modified electrode. Bioelectrochemistry 2017, 114, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Lee, S.J.; Moskovits, M. Aptamer-mediated surface-enhanced Raman spectroscopy intensity amplification. Nano Lett. 2010, 10, 4181–4185. [Google Scholar] [CrossRef] [PubMed]
- Heydari-Bafrooei, E.; Shamszadeh, N.S. Electrochemical bioassay development for ultrasensitive aptasensing of prostate specific antigen. Biosens. Bioelectron. 2017, 91, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.M.; Zhang, X.; Ding, L.; Yang, D.; Qu, F. Label-free fluorescence turn-on aptasensor for prostate-specific antigen sensing based on aggregation-induced emission-silica nanospheres. Anal. Bioanal. Chem. 2017, 409, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xu, B.; Liu, L.; Tian, W. A Label-free Fluorescent Aptasensor for Turn-on Monitoring Ochratoxin A Based on AIE-active Probe and Graphene Oxide. Chem. Res. Chin. Univ. 2018, 1–6. [Google Scholar] [CrossRef]
- Li, Y.; Dai, W.; Lv, X.; Deng, Y. Aptamer-Based Rolling Circle Amplification coupling with Graphene Oxide-Based Fluorescence Resonance Energy Transfer for Sensitive Detection of Cardiac Troponin I. Anal. Methods 2018, 10, 1767–1773. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Tang, D.; Niessner, R.; Chen, G.; Knopp, D. DNA-based hybridization chain reaction for amplified bioelectronic signal and ultrasensitive detection of proteins. Anal. Chem. 2012, 84, 5392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chu, Z.; Jin, X.; Zhao, S. A fluorescence polarization assay for nucleic acid based on the amplification of hybridization chain reaction and nanoparticles. Sen. Actuators B Chem. 2015, 209, 116–121. [Google Scholar] [CrossRef]
- Zhou, Q.; Yan, H.; Ran, F.; Cao, J.; Chen, L.; Shang, B.; Chen, H.; Wei, J.; Chen, Q. Ultrasensitive enzyme-free fluorescent detection of VEGF 165 based on target-triggered hybridization chain reaction amplification. RSC Adv. 2018, 8, 25955–25960. [Google Scholar] [CrossRef]
- Song, C.; Yang, X.; Wang, K.; Wang, Q.; Huang, J.; Liu, J.; Liu, W.; Liu, P. Label-free and non-enzymatic detection of DNA based on hybridization chain reaction amplification and dsDNA-templated copper nanoparticles. Anal. Chim. Acta 2014, 827, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Z.; Tao, Y.; Wang, Z.; Ren, J.; Qu, X. Hybridization chain reaction engineered dsDNA for Cu metallization: An enzyme-free platform for amplified detection of cancer cells and microRNAs. Chem. Commun. 2015, 51, 11496–11499. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Baum, L.; Fu, W.L. Simple and practical staining of DNA with GelRed in agarose gel electrophoresis. Clin. Lab. 2010, 56, 149–152. [Google Scholar] [PubMed]
- Maurye, P.; Basu, A.; Biswas, J.K.; Bandyopadhyay, T.K. Electrophoresis-staining apparatus for DNA agarose gels with solution exchange and image acquisition. Instrum. Sci. Technol. 2017, 45, 49–61. [Google Scholar] [CrossRef]
- De, M.; Chou, S.S.; Dravid, V.P. Graphene Oxide as an Enzyme Inhibitor: Modulation of Activity of α-Chymotrypsin. J. Am. Chem. Soc. 2016, 133, 17524–17527. [Google Scholar] [CrossRef] [PubMed]
- Moralesnarváez, E.; Merkoçi, A. Graphene oxide as an optical biosensing platform. Adv. Mater. 2012, 24, 3298–3308. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, Y.; Chen, Y.; Zhu, Z.; Yang, X.; Yang, C.J.; Wang, K.; Tan, W. Pyrene-excimer probes based on the hybridization chain reaction for the detection of nucleic acids in complex biological fluids. Angew. Chem. 2012, 50, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhou, J.; Zheng, Y.; Gamson, A.S.; Roembke, B.T.; Nakayama, S.; Sintim, H.O. Isothermal amplified detection of DNA and RNA. Mol. Biosyst. 2014, 10, 970–1003. [Google Scholar] [CrossRef] [PubMed]
- Kalluru, P.; Vankayala, R.; Chiang, C.S.; Hwang, K.C. Nano-graphene oxide-mediated In vivo fluorescence imaging and bimodal photodynamic and photothermal destruction of tumors. Biomaterials 2016, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhao, Y.; Xu, C.; Wu, D.; Cai, Y.; He, J.; Li, H.; Du, B.; Yang, M. Nanoporous gold film based immunosensor for label-free detection of cancer biomarker. Biosens. Bioelectron. 2011, 26, 3714–3718. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, Y.; Wang, T.; Li, J. Positive potential operation of a cathodic electrogenerated chemiluminescence immunosensor based on luminol and graphene for cancer biomarker detection. Anal. Chem. 2011, 83, 3817–3823. [Google Scholar] [CrossRef] [PubMed]
- Uludağ, Y.; Tothill, I.E. Development of a sensitive detection method of cancer biomarkers in human serum (75%) using a quartz crystal microbalance sensor and nanoparticles amplification system. Talanta 2010, 82, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.H.; Kuo, S.H.; Lin, T.Y.; Lin, C.W.; Fang, P.Y.; Yang, H.W. An electrochemical biosensor to simultaneously detect VEGF and PSA for early prostate cancer diagnosis based on graphene oxide/ssDNA/PLLA nanoparticles. Biosens. Bioelectron. 2017, 89, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Presnova, G.; Presnov, D.; Krupenin, V.; Grigorenko, V.; Trifonov, A.; Andreeva, I.; Ignatenko, O.; Egorov, O.; Rubtsova, M. Biosensor based on a silicon nanowire fieldeffect transistor functionalized by gold nanoparticles for the highly sensitive determination of prostate specific antigen. Biosens. Bioelectron. 2017, 88, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Chen, L.B.; Chen, Y.Q.; Bai, H.; Li, L. Three-dimensional porous graphene-based composite materials: Electrochemical synthesis and application. J. Mater. Chem. 2012, 22, 20968–20976. [Google Scholar] [CrossRef]
- Du, C.; Yao, Z.; Chen, Y.; Bai, H.; Li, L. Synthesis of metal nanoparticle@graphene hydrogel composites by substrate-enhanced electroless deposition and their application in electrochemical sensors. RSC Adv. 2014, 4, 9133–9138. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Linear Range | Detection Limit | Reference |
|---|---|---|---|
| Microfluidic Devices | 10–100 ng/mL | 10 ng/mL | [5] |
| Rresonance light scattering | 0.13–110 ng/mL | 0.13 ng/mL | [4] |
| Immunosensor | 0.05–26 ng/mL | 0.05 ng/mL | [30] |
| electrogenerated chemiluminescence immunosensor | 8–10 pg/mL | 8 pg/mL | [31] |
| quartz crystal microbalance sensor | 0.29–150 ng/mL | 0.29 ng/mL | [32] |
| Electrochemistry | 0.05–100 ng/mL | 1000 pg/mL | [33] |
| Field effect transistor | 0.023–500 ng/mL | 23 pg/mL | [34] |
| Fluoroimmunoassay | 0.1–200 ng/mL | 10 pg/mL | This Work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Wang, C.; Zhang, H.; Zhang, Y.; Zhang, G. A Non-Enzymatic and Label-Free Fluorescence Bioassay for Ultrasensitive Detection of PSA. Molecules 2019, 24, 831. https://doi.org/10.3390/molecules24050831
Sun Y, Wang C, Zhang H, Zhang Y, Zhang G. A Non-Enzymatic and Label-Free Fluorescence Bioassay for Ultrasensitive Detection of PSA. Molecules. 2019; 24(5):831. https://doi.org/10.3390/molecules24050831
Chicago/Turabian StyleSun, Yujie, Chenyun Wang, Hong Zhang, Yulin Zhang, and Guojun Zhang. 2019. "A Non-Enzymatic and Label-Free Fluorescence Bioassay for Ultrasensitive Detection of PSA" Molecules 24, no. 5: 831. https://doi.org/10.3390/molecules24050831