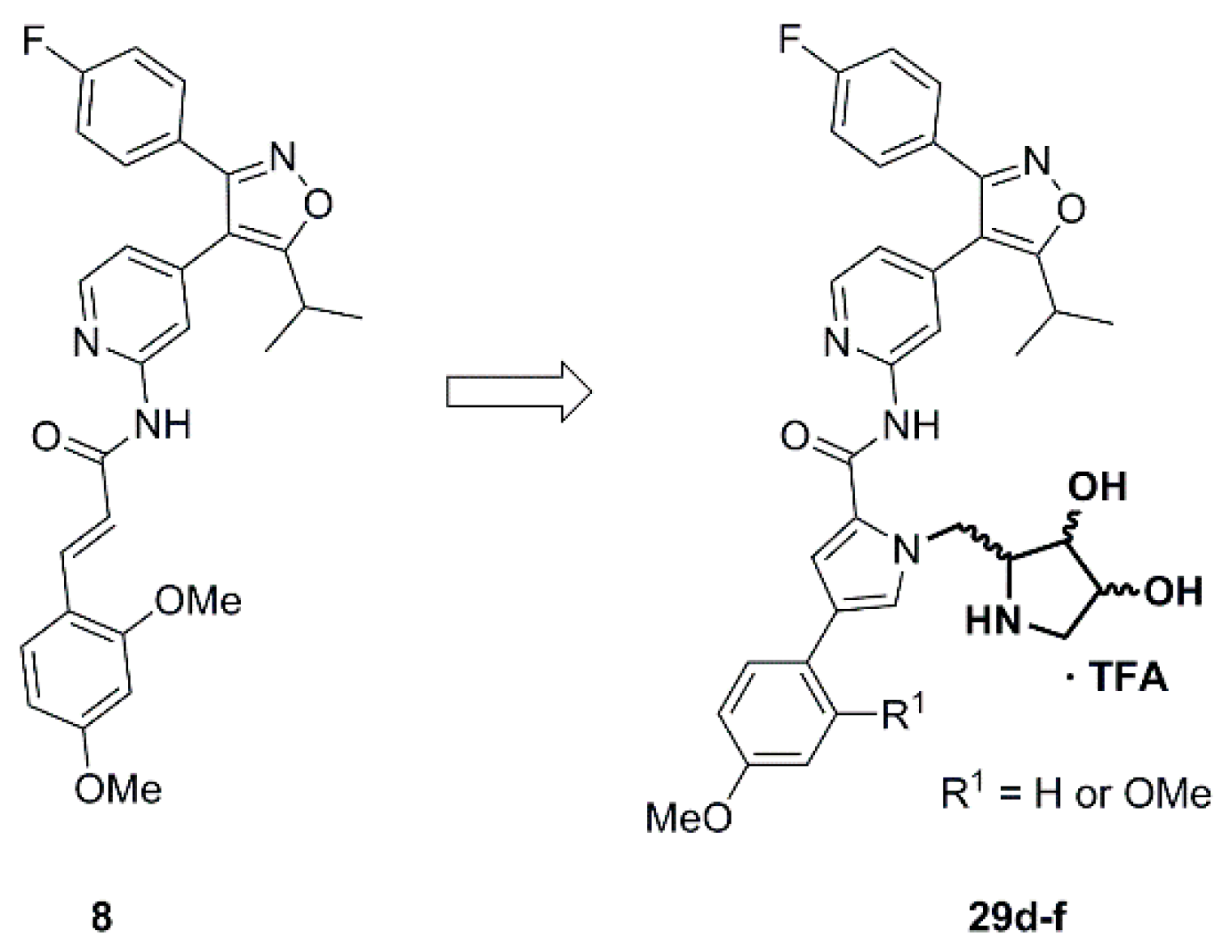
3.2.2. Syntheses
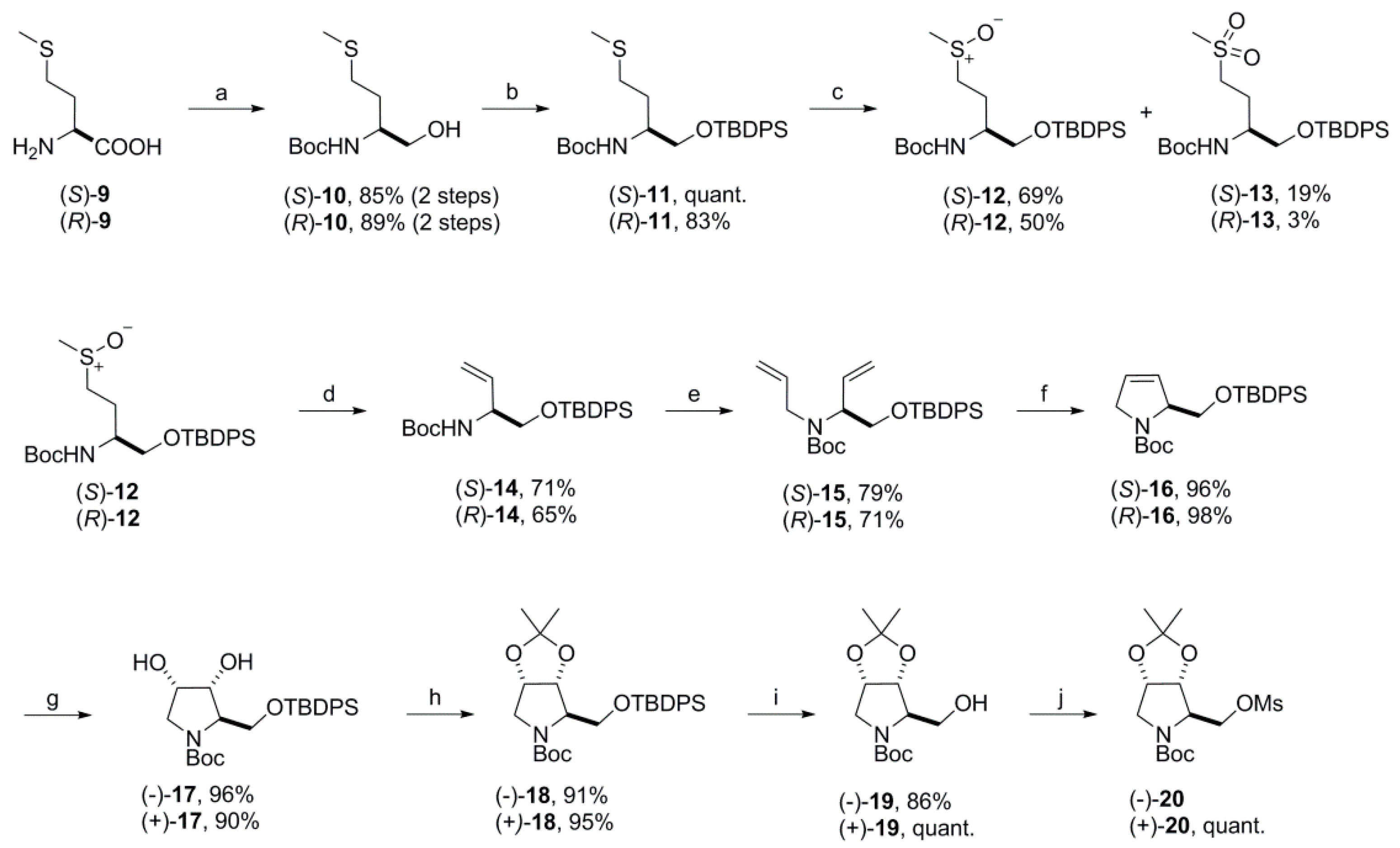
(S)-tert-Butyl 1-(tert-butyldiphenylsilyloxy)-4-(methylthio)-butan-2-ylcarbamate [(S)-11]. To a solution of alcohol (S)-10 (17.4 g, 73.9 mmol) in dry DMF (110 mL) was added imidazole (12.6 g, 185 mmol) and TBDPSCl (21.2 mL, 81.5 mmol), and the resulting reaction mixture was stirred at room temperature overnight. Water (500 mL) was added and the mixture extracted with ethyl acetate (3×). The combined organic layers were washed with water (2×) and brine, dried over MgSO4 and concentrated. The crude product was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether 1:19, 1:9 and 1:6) to yield the desired product (S)-11 quantitatively as a colorless oil. = −11.7 (c 1.20, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.66–7.61 (m, 4H), 7.45–7.34 (m, 6H), 4.76–4.64 (m, 1H), 3.81–3.65 (m, 2H), 3.61 (dd, 10.0, 3.0 Hz, 1H), 2.53–2.42 (m, 2H), 2.07 (s, 3H), 1.90–1.72 (m, 2H), 1.44 (s, 9H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.53, 135.58, 135.56, 133.26, 133.20, 129.82, 129.79, 127.77, 79.22, 65.62, 51.38, 31.75, 30.77, 28.41, 26.91, 19.32, 15.52. HRMS (ES+) m/z calcd for C26H39NO3SSiNa+ 496.2312, found 496.2307.
tert-Butyl {(2S)-1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfinyl)butan-2-yl} carbamate [(S)-12]. To a solution of (S)-11 (1.03 g, 2.17 mmol) in dichloromethane (46 mL) was added a solution of m-CPBA (50–55%; 750 mg, 2.17 mmol) in dichloromethane (4.5 mL) dropwise at −20 °C. After being stirred for 1 h at this temperature the reaction mixture was allowed to warm to 0 °C and was quenched with saturated sodium carbonate solution. The organic layer was separated, washed another two times with saturated sodium carbonate solution, dried over MgSO4 and concentrated. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether 3:7 and 1:0) to give 737 mg (69%) of the sulfoxide (S)-12 as a light-yellow heavy oil that solidified on standing. In addition, 211 mg (19%) of the sulfone (S)-13 were isolated as a colorless solid.
tert-Butyl {(2S)-1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfinyl)butan-2-yl}carbamate [(S)-12]. = −9.3 (c 0.55, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.65–7.61 (m, 4H), 7.46–7.36 (m, 6H), 4.86–4.71 (m, 1H, NH) 3.85–3.61 (m, 3H), 2.78–2.64 (m, 2H), 2.54 (s, 1.5H), 2.53 (s, 1.5H), 2.07–1.90 (m, 2H), 1.44 (s, 9H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.72, 155.66, 135.55, 135.53, 133.00, 132.93, 129.91, 129.89, 127.83, 79.52, 65.85, 65.81, 51.70, 51.40, 51.01, 38.70, 38.65, 28.36, 26.90, 25.73, 25.22, 19.28; HRMS (ES+) m/z calcd for C26H39NO4SSiNa+ 512.2261, found 512.2261.
tert-Butyl (S)-{1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfonyl)butan-2-yl}carbamate [(S)-13]. m.p.: 95 °C; = −6.3 (c 1.115, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.65–7.60 (m, 4H), 7.47–7.37 (m, 6H), 4.75 (d, 7.4 Hz, 1H), 3.80–3.67 overlapping signals (m, 1H and 3.71, dd, 10.4, 4.1 Hz, 1H), 3.63 (dd, 10.3; 3.8 Hz, 1H), 3.10–2.99 (m, 2H), 2.88 (s, 3H), 2.11–1.97 (m, 2H), 1.44 (s, 9H), 1.08 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.64, 135.57, 135.53, 132.87, 132.78, 130.00, 129.96, 127.90, 127.87, 79.78, 65.71, 51.99, 50.87, 40.69, 28.34, 26.90, 25.19, 19.27; HRMS (ES+) m/z calcd for C26H39NO5SSiNa+ 528.2210, found 528.2202.
tert-Butyl (S)-{1-[(tert-butyldiphenylsilyl)oxy]but-3-en-2-yl}carbamate [(
S)-
14]. A mixture of sulfoxide (
S)-
12 (29.9 g, 61.1 mmol) and calcium carbonate (14.7 g, 147 mmol) in 1,2-dichlorobenzene (165 mL) was heated under reflux for 7 h (TLC analysis). After being cooled to room temperature the mixture was filtered through a pad of celite and the celite was additionally washed with ethyl acetate. The filtrate was concentrated to small volume and the residue was purified by flash column chromatography to afford 18.4 g (71%) of vinylglycinol (
S)-
14 as a light-yellow oil.
= −29.5 (
c 1.18, CHCl
3), Ref. [
56]:
= −27.7 (
c 1.02, CHCl
3);
1H NMR (300 MHz, CHCl
3) δ 7.67–7.61 (m, 4H), 7.46–7.33 (m, 6H), 5.84 (ddd, 17.2, 10.4, 5.4 Hz, 1H), 5.22 (dt, 17.3, 1.4 Hz, 1H), 5.16 (dt, 10.5, 1.4 Hz, 1H), 4.86–4.73 (m, 1H), 4.30–4.16 (m, 1H), 3.74 (dd, 10.1, 4.4 Hz, 1H), 3.64 (dd, 10.1, 4.6 Hz, 1H), 1.45 (s, 9H), 1.06 (s, 9H);
13C NMR (75 MHz, CHCl
3) δ 155.42, 136.48, 135.62, 135.56, 133.30, 133.19, 129.77, 127.72, 115.63, 79.35, 66.05, 54.41, 28.41, 26.84, 19.31; HRMS (ESI)
m/
z calcd for C
25H
35NO
3SiNa
+ 448.2278, found 488.2589.
tert-Butyl (S)-allyl{1-[(tert-butyldiphenylsilyl)oxy]but-3-en-2-yl}carbamate [(S)-15]. To a solution of vinylglycinol (S)-14 (18.3 g, 43.0 mmol) and allyl bromide (6.78 mL, 78.4 mmol) in dry DMF (140 mL) was added sodium hydride (60%, in mineral oil; 2.35 g, 58.8 mmol) portionwise at room temperature followed by a catalytic amount of tetra-n-butyl ammonium iodide. The ice bath was removed, and the reaction was stirred at room temperature overnight. Another portion of allyl bromide (6.78 mL, 78.4 mmol) and sodium hydride (60%, in mineral oil; 2.35 g, 58.8 mmol) was added at room temperature and the mixture was again stirred overnight. After a third addition of allyl bromide (3.00 mL, 34.7 mmol) and sodium hydride (60%, in mineral oil; 1.00 g, 25.0 mmol) and stirring at room temperature overnight water was added carefully and the mixture was extracted with ethyl acetate (3×). The organic phases were combined, washed with water (2×) and brine, dried over MgSO4 and concentrated. The obtained crude residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether 1:30) to yield 15.8 g (79%) of N-allylvinylglycinol (S)-15 as a colorless oil. = +0.82 (c 0.90, CHCl3). 1H NMR (500 MHz, d6-DMSO, rotamers) δ 7.64–7.59 (m, 4H), 7.50–7.41 (m, 6H), 5.87–5.74 (m, 2H), 5.15 (dt, 10.6, 1.4 Hz, 1H), 5.13–5.06 (overlapping signals: m, 1H and 5.10, dt, 17.4, 1.4 Hz, 1H), 5.05–5.01 (m, 1H), 4.70–4.55 (m, 0.5H), 4.36–4.20 (m, 0.4H), 3.87–3.64 (overlapping signals: 3.81, dd, 10.2, 8.3 Hz, 1H and m, 3H), 1.37 (sbr, 9H), 0.99 (s, 9H); 13C NMR (125 MHz, d6-DMSO, rotamers) δ 154.48, 135.98, 135.52, 134.93, 134.46, 132.75, 129.79, 127.74, 117.28, 115.69, 115.27, 78.69, 63.81, 63.32, 60.69, 59.29, 47.71, 46.84, 27.90, 26.42, 18.63; 1H NMR (500 MHz, d6-DMSO, 100 °C) δ 7.65–7.61 (m, 4H), 7.48–7.39 (m, 6H), 5.87 (ddd, 17.3, 10.8, 6.4 Hz, 1H), 5.80 (ddt, 17.2, 10.5, 5.7 Hz, 1H), 5.17–5.07 (overlapping signals: 5.15, dt, 10.6, 1.5 Hz, 1H and, m, 2H), 5.02 (ddd, 10.3, 3.0, 1.5 Hz, 1H), 4.47–4.39 (m, 1H), 3.88 (dd, 10.3, 7.6 Hz, 1H), 3.85–3.77 (overlapping signals: 3.82, ddt, 16.1, 5.6, 1.4 Hz, 1H and 3.79, dd, 10.3, 6.1 Hz, 1H), 3.74 (ddt, 16.1, 5.8, 1.4 Hz, 1H), 1.38 (s, 9H), 1.03 (s, 9H), 13C NMR (125 MHz, d6-DMSO, 100 °C) δ 154.04, 135.34, 134.52, 134.47, 132.71, 129.15, 127.12, 116.41, 114.87, 78.38, 63.62, 59.94, 47.19, 27.52, 26.12, 18.20; HRMS (ESI) m/z calcd for C28H39NO3SiNa+ 488.2591, found 488.2589; Anal. calcd for C28H39NO3Si: C, 72.21; H, 8.44; N, 3.01. Found C, 72.43; H, 8.69; N, 3.03.
tert-Butyl (S)-2-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,5-dihydro-1H-pyrrole-1-carboxylate [(
S)-
16]. A solution of
N-allylvinylglycinol (
S)-
15 (4.32 g, 9.28 mmol) in dry DCM (40 mL) was treated with Grubbs 1st-generation catalyst (40.0 mg, 0.049 mmol) employing the procedure described by Brackmann et al. [
55] to afford 3.91 g (96%) of 3,4-dehydroprolinol (
S)-
16 as a colorless oil.
= −116 (
c 0.95, CHCl
3), Ref. [
58]:
= −90.1 (
c 1.06, CHCl
3), Ref. [
57]
= −24.6 (
c 1.00, CHCl
3);
1H NMR (500 MHz, CHCl
3; mixture of rotamers) δ 7.66–7.61 (m, 4H), 7.44–7.32 (m, 6H), 5.93–5.78 (m, 2H), 4.66–4.61 (m, 0.4H), 4.55–4.49 (m, 0.6H), 4.25 (dd, 15.4, 1.5 Hz, 0.6H), 4.17 (dd, 15.4, 1.2 Hz, 0.4H), 4.09 (d, 5.3 Hz, 0.6H), 4.06 (d, 5.3 Hz, 0.4H), 3.99 (dd, 9.8, 5.0 Hz, 0.4H), 3.88 (dd, 9.5, 3.1 Hz, 0.6H), 3.82 (dd, 9.8, 2.1 Hz, 0.4H), 3.67 (dd, 9.4, 6.5 Hz, 0.6H), 1.48 (s, 3.4H), 1.35 (s, 5.2H), 1.03 (s, 5H), 1.02 (s, 4H);
13C NMR (125 MHz, CHCl
3; mixture of rotamers) δ 154.13, 154.08, 135.55, 133.90, 133.80, 133.68, 133.57, 129.64, 129.52, 128.85, 128.75, 127.67, 127.59, 126.29, 79.46, 79.17, 65.57, 65.42, 65.14, 63.74, 54.27, 53.96, 28.57, 28.45, 26.78, 19.35, 19.28; HRMS (ESI)
m/
z calcd for C
26H
35NO
3SiNa
+ 460.2278, found 460.2289.
tert-Butyl (2R,3R,4S)-2-{[(tert-butyldiphenylsilyl)oxy]methyl}-3,4-dihydroxypyrrolidine-1-carboxylate [(-)-
17]. Following the procedure described by Murruzzu and Riera [
57] 3,4-dehydroprolinol (
S)-
16 (461 mg, 1.05 mmol) was dissolved in a 3:1 mixture of acetone (14 mL) and water (4.7 mL) and reacted with osmium tetroxide (7 mg, 0.028 mmol) in the presence of
N-methylmorpholine
N-oxide (325 mg, 2.40 mmol) at room temperature overnight. The crude reaction product was purified by automated column chromatography (silica gel, ethyl acetate/petroleum ether 2–50%) to afford 477 mg (96%) of (−)-
17 as a colorless oil.
= −27.3 (
c 0.59, MeOH), Ref. [
58]:
= −30.5 (
c 1.01, MeOH);
1H NMR (500 MHz, CD
3OD; mixture of rotamers) δ 7.68–7.61 (m, 4H), 7.47–7.36 (m, 6H), 4.41–4.34 (m, 1.6H), 4.30 (t, 3.8 Hz, 0.4H), 4.07 (dd, 10.5, 3.7, Hz, 0.4H), 3.88 (dd, 10.5, 4.3 Hz, 0.6H), 3.77 (dd, 10.5, 2.0 Hz, 0.6H), 3.73 (dd, 10.5, 1.4 Hz, 0.4H), 3.70–3.63 (m, 1H), 3.55 (dd, 11.1, 6.1 Hz, 0.4H), 3.51 (dd, 11.2, 6.2 Hz, 0.6H), 3.44–3.38 (m, 1H), 1.48 (s, 4H), 1.29 (s, 5H), 1.05 (s, 5H), 1.04 (s, 4H);
13C NMR (125 MHz, CD
3OD; mixture of rotamers) δ 156.53, 156.49, 136.71, 136.66, 134.57, 134.46, 134.36, 131.05, 130.98, 130.95, 128.99, 128.95, 128.92, 128.86, 81.25, 80.97, 74.99, 74.50, 71.33, 70.84, 66.35, 66.08, 63.81, 62.94, 52.81, 52.17, 28.89, 28.71, 27.39, 20.14, 20.10; HRMS (ESI)
m/
z calcd for C
26H
37NO
5SiNa
+ 494.2333, found 494.2345.
tert-Butyl(3aR,4R,6aS)-4-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate [(−)-
18]. To a solution of compound (−)-
17 (334 mg, 0.708 mmol) and 2,2-dimethoxypropane (0.20 mL, 1.63 mmol) in acetone (17 mL) was added a catalytic amount of
p-toluenesulfonic acid monohydrate (26.6 mg, 0.140 mmol). After being stirred at room temperature overnight saturated bicarbonate solution (10 mL) and water are added, and the mixture was extracted with ethyl acetate (3×). The combined organic phases washed with brine, dried over MgSO
4 and concentrated. The residue was purified by automated column chromatography (silica gel, ethyl acetate/petroleum ether 2–5%) to yield 328 mg (91%) of (−)-
18 as a colorless oil that solidified on standing.
= −47.9 (
c 0.85, CHCl
3), Ref. [
57]:
= −36.1 (
c 1.05, CHCl
3);
1H NMR (500 MHz, CHCl
3; mixture of rotamers) δ 7.66–7.56 (m, 4H), 7.46–7.34 (6H), 4.84–4.76 (overlapping signals: m, 1H and 4.78, d, 6.0 Hz, 0.6H), 4.74 (d, 6.1 Hz, 0.4H), 4.14–4.11 (m, 0.4H), 4.03–3.98 (m, 1H), 3.84 (dd, 12.4, 0.9 Hz, 0.6H), 3.77 (dd, 10.5, 3.7 Hz, 0.6H), 3.73 (dd, 12.3, 0.9 Hz, 0.4H), 3.70–3.63 (m, 2H), 1.49 (s, 4H), 1.47 (s, 1.6H), 1.46 (s, 1.4H), 1.37 (s, 5H), 1.35 (s, 1.6H), 1.33 (s, 1.4H), 1.05 (s, 4H), 1.04 (s, 5H);
13C NMR (125 MHz, CHCl
3; mixture of rotamers) δ 154.25, 154.06, 135.56, 135.45, 132.99, 132.94, 132.80, 132.70, 129.94, 129.89, 129.87, 129.80, 127.89, 127.84, 127.78, 111.47, 83.31, 82.66, 79.99, 79.67, 79.61, 79.20, 65.00, 64.68, 64.62, 64.42, 54.23, 53.54, 28.52, 28.42, 27.10, 26.90, 26.83, 25.14, 19.16, 19.08; HRMS (ESI)
m/
z calcd for C
29H
41NO
5SiNa
+ 534.2646, found 534.2654.
tert-Butyl(3aR,4R,6aS)-4-(hydroxymethyl)-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate [(−)-
19]. To a solution of (−)-
18 (218 mg, 0.426 mmol) in THF (3 mL) was added a 1 M solution of
tetra-
n-butylammonium fluoride in THF (0.7 mL, 0.70 mmol) at room temperature. After being stirred overnight water was added and the resulting mixture extracted with ethyl acetate (3×). The organic phases were combined, washed with brine, dried over MgSO
4 and concentrated. The crude product was purified by automated column chromatography (silica gel, ethyl acetate/petroleum ether 2–20%) to give 100 mg (86%) of the desired alcohol (−)-
19 as a colorless oil that solidified on standing.
= −47.6 (
c 0.555, CHCl
3), Ref. [
57]:
= −30.3 (
c 0.3, CHCl
3);
1H NMR (500 MHz, CH
3OD; mixture of rotamers) δ 4.78–4.73 (m, 1H), 4.72–4.68 (m, 1H), 3.98 (t, 3.7 Hz, 0.5H), 3.93 (t, 4.0 Hz, 0.5H), 3.69 (d, 11.8 Hz, 0.5H), 3.67 (d, 11.8 Hz, 0.5H), 3.64–3.59 (m, 2H), 3.50 (dd, 12.5 Hz, 5.1 Hz, 0.5H), 3.46 (dd, 12.5 Hz, 5.2 Hz, 0.5H), 1.46 (s, 9H), 1.40 (s, 3H), 1.30 (s, 3H);
13C NMR (125 MHz, CD
3OD; mixture of rotamers) δ 156.41, 156.26, 112.57, 84.29, 83.57, 81.36, 81.29, 81.00, 80.23, 66.93, 66.44, 62.77, 62.34, 54.52, 53.89, 28.75, 27.35, 25.10; HRMS (ESI)
m/
z calcd for C
13H
23NO
5Na
+ 296.1468, found 296.1480.
tert-Butyl(3aR,4R,6aS)-2,2-dimethyl-4-{[(methylsulfonyl)oxy]methyl}tetrahydro-5H-[1,3]dioxo-lo[4,5-c]pyrrole-5-carboxylate [(−)-
20]. To a solution of (−)-
19 (6.98 g, 25.5 mmol) in dry DCM (160 mL) and triethylamine (10.7 mL, 76.7 mmol) was added methanesulfonyl chloride (3.0 mL, 38.8 mmol) dropwise at 0 °C. After being stirred for 30 min the reaction was poured into half concentrated bicarbonate solution. The organic phase was separated, and the aqueous layer extracted with ethyl acetate another three times. The organic phases were combined, washed with brine, dried over MgSO
4 and concentrated. The recovered crude product was used in the next reaction step without further purification. An analytical sample was purified by automated column chromatography (silica gel, ethyl acetate/petroleum ether 2–50%) to yield (−)-
20 as a colorless oil.
= −50.8 (
c 0.62, MeOH);
1H NMR (500 MHz, CD
3OD; mixture of rotamers) δ 4.81–4.77 (m, 1H), 4.72 (ap d, 6.1 Hz, 1H), 4.41 (dd, 10.4, 4.5 Hz, 0.5H), 4.35 (dd, 10.4, 5.0 Hz, 0.5H), 4.31 (ap dd, 10.4, 3.6 Hz, 1H), 4.20–4.14 (m, 1H), 3.74 (d, 12.6 Hz, 0.5H), 3.70 (d, 12.7 Hz, 0.5H), 3.51 (dd, 12.7, 5.1 Hz, 0.5H), 3.47 (dd, 12.7, 5.1 Hz, 0.5H), 3.10 (s, 1.5H), 3.09 (s, 1.5H), 1.48 (s, 4.5H), 1.47 (s, 4.5H), 1.42 (s, 3H), 1.31 (s, 3H);
13C NMR (125 MHz, CD
3OD; mixture of rotamers) δ 156.19, 155.81, 113.06, 83.86, 83.08, 82.08, 81.83, 80.76, 80.00, 70.00, 69.81, 64.45, 64.03, 54.24, 53.46, 37.37, 37.27, 28.67, 27.30, 25.05;
1H and
13C NMR data recorded in CDCl
3 was consistent with that reported in reference [
48,
49]. HRMS (ESI)
m/
z calcd for C
14H
25NO
7SNa
+ 374.1244, found 374.1244.
tert-Butyl (R)-{1-[(tert-butyldiphenylsilyl)oxy]-4-(methylthio)butan-2-yl}carbamate [(R)-11]. Using the same procedure as for the preparation of compound (S)-11 alcohol (R)-10 (35.0 g, 149 mmol) was dissolved in dry DMF (250 mL) and reacted with TBDPSCl (46.4 mL, 179 mmol) in the presence of imidazole (25.3 g, 372 mmol) to yield 58.2 g (83%) of (R)-11 as a colorless oil. = +6.8 (c 1.075, CHCl3); All spectroscopic data was consistent with its enantiomer (S)-11. HRMS (ES+) m/z calcd for C26H39NO3SSiNa+ 496.2318, found 496.2312.
tert-Butyl {(2R)-1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfinyl)butan-2-yl}carbamate [(R)-12]. According to the protocol outlined for the preparation of (S)-12 sulfide (R)-11 (58.0 g, 122 mmol) dissolved in dichloromethane (1700 mL) was oxidized by adding dropwise a solution of m-CPBA (50–55%; 20 g, 61.2 mmol) in dichloromethane (160 mL) (dried over MgSO4) at −40 °C to give 29.8 g (50%) of the desired sulfoxide (R)-12 as a colorless oil that solidified on standing. In addition, 1.77 g (3%) of the sulfone (R)-13 were isolated from the reaction as a colorless solid that also solidified on standing along with 26.1 g (45%) of starting material (R)-11.
tert-Butyl {(2R)-1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfinyl)butan-2-yl}carbamate [(R)-12]. = +9.4 (c 1.075, CHCl3); All spectroscopic data was consistent with its enantiomer (S)-12. HRMS (ES+) m/z calcd for C26H39NO4SSiNa+ 512.2261, found 512.2274.
tert-Butyl (R)-{1-[(tert-butyldiphenylsilyl)oxy]-4-(methylsulfonyl)butan-2-yl}carbamate [(R)-13]. = +6.0 (c 1.14, CHCl3); All spectroscopic data was consistent with its enantiomer (S)-13. HRMS (ES+) m/z calcd for C26H39NO5SSiNa+ 528.2210, obsd 528.2209; Anal. calcd for C26H39NO5SSi: C, 61.75; H, 7.77; N, 2.77. Found C, 62.02; H, 7.96; N, 2.68.
tert-Butyl (R)-{1-[(tert-butyldiphenylsilyl)oxy]but-3-en-2-yl}carbamate [(
R)-
14]. Employing the same procedure as for the preparation of (
S)-
14 a mixture of sulfoxide (
R)-
12 (29.6 g, 60.4 mmol) and calcium carbonate (14.5 g, 145 mmol) in 1,2-dichlorobenzene (165 mL) was heated at 200 °C for 7 h to yield 16.8 g (65%) of vinylglycinol (
R)-
14 as a light-yellow oil.
= +27.0 (
c 1.03, CHCl
3), Ref. [
84]:
= +25.4 (
c 1.7, CHCl
3); The spectroscopic data was consistent with that for its enantiomer (
S)-
14. HRMS (ESI)
m/
z calcd for C
25H
35NO
3SiNa
+ 448.2278, found 448.2275.
tert-Butyl (R)-allyl{1-[(tert-butyldiphenylsilyl)oxy]but-3-en-2-yl}carbamate [(R)-15]. Using the same procedure as for the preparation of N-allylvinylglycinol (S)-15 vinylglycinol (R)-14 (16.4 g, 38.5 mmol) dissolved in dry DMF (125 mL) was reacted with allyl bromide (6.67 mL, 77.1 mmol; second addition after ca. 20 h: 7.0 mL, 80.9 mmol; third addition after another ca. 20 h: 7.0 mL, 80.9 mmol) and sodium hydride (60% in mineral oil, 2.31 g, 57.8 mmol; second addition after ca. 20 h: 2.38 g, 59.5 mmol; third addition after another 20 h: 1.19 g, 29.8 mmol) in the presence of a catalytic amount of n-Bu4NI to afford 12.7 g (71%) of N-allylvinylglycinol (R)-15 as a colorless oil. In addition, 3.86 g of mixed starting material and product were = −0.76 (c 1.07, CHCl3). The spectroscopic data was consistent with that reported for its enantiomer (S)-15. HRMS (ESI) m/z calcd for C28H39NO3SiNa+ 488.2591, found 488.2596; Anal. calcd for C28H39NO3Si: C, 72.21; H, 8.44; N, 3.01. Found C, 72.30; H, 8.52; N, 3.07.
tert-Butyl (R)-2-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,5-dihydro-1H-pyrrole-1-carboxylate [(
R)-
16]. Following the method described by Brackmann et al. [
55]
N-allylvinylglycinol (
R)-
15 (9.80 g, 21.0 mmol) was dissolved in dry DCM (94 mL) and treated with Grubbs 1
st-generation catalyst (250 mg, 0.304 mmol) for 24 h at room temperature to yield 9.04 g (98%) of 3,4-dehydroprolinol (
R)-
16 as a colorless oil.
= +115 (
c 1.38, CHCl
3). The spectroscopic data was consistent with those reported for its enantiomer (
S)-
16. HRMS (ES+)
m/
z calcd for C
26H
35NO
3SiNa
+ 460.2274, found 460.2281.
tert-Butyl (2S,3S,4R)-2-{[(tert-butyldiphenylsilyl)oxy]methyl}-3,4-dihydroxypyrrolidine-1-carboxylate [(+)-
17]. Deploying the procedure described by Murruzzu and Riera [
57] compound (
R)-
16 (8.85 g, 20.2 mmol) was dissolved in a 10:1 mixture of acetone (360 mL) and water (36 mL) and treated with osmium tetroxide (59.9 mg, 0.236 mmol) in the presence of
N-methylmorpholine
N-oxide (6.01 g, 44.5 mmol) at room temperature overnight to yield 8.61 g (90%) of (+)-
17 as a colorless oil.
= +28.6 (
c 0.545, MeOH); All spectroscopic data was consistent with the data described for its enantiomer (−)-
17. HRMS (ESI)
m/
z calcd for C
26H
37NO
5SiNa
+ 494.2333, found 494.2344.
tert-Butyl(3aS,4S,6aR)-4-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate [(+)-18]. Using the same procedure as for the preparation of its enantiomer (−)-18 compound (+)-17 (8.50 g, 18.0 mmol) and 2,2-dimethoxypropane (4.43 mL, 36.0 mmol) in acetone (430 mL) was treated with a catalytic amount of p-toluenesulfonic acid monohydrate (189 mg, 0.994 mmol) to give 8.76 g (95%) of (+)-18 as a colorless oil that solidified on standing. = +46.1 (c 0.92, CHCl3); All spectroscopic data was consistent with that of its enantiomer (−)-18. HRMS (ESI) m/z calcd for C29H41NO5SiNa+ 534.2646, found 534.2652; Anal. calcd for C29H41NO5: C, 68.07; H, 8.08; N, 2.74. Found C, 68.33; H, 8.20; N, 2.78.
tert-Butyl(3aS,4S,6aR)-4-(hydroxymethyl)-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate [(+)-
19]. Following the same protocol as outlined for the preparation of its enantiomer (−)-
19 the fully protected pyrrolidine (+)-
18 (8.67 g, 16.9 mmol) was dissolved in THF (85 mL) and treated with a 1 M solution of
tetra-
n-butylammonium fluoride in THF (25 mL, 25 mmol) at room temperature overnight to yield 4.65 g (quant.) of (+)-
19 as a colorless oil that solidified on standing.
= +46.7 (
c 0.505, CHCl
3), Ref. [
85]:
= +29 (
c 1.0, CHCl
3), Ref. [
86]:
= +29.4 (
c 1.04, CHCl
3); All spectroscopic data was consistent with its enantiomer (−)-
19. HRMS (ESI)
m/
z calcd for C
13H
23NO
5Na
+ 296.1468, found 296.1468; Anal. calcd for C
13H
23NO
5; C, 57.13; H, 8.48; N, 5.12. Found C, 57.16; H, 8.53; N, 5.11.
tert-Butyl(3aS,4S,6aR)-2,2-dimethyl-4-{[(methylsulfonyl)oxy]methyl}tetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate [(+)-20]. Deploying the same protocol as described for the preparation of compound (−)-20 alcohol (+)-19 (2.51 g, 9.18 mmol) was dissolved in dry DCM (40 mL) and reacted with methanesulfonyl chloride (1.1 mL, 14.2 mmol) in the presence of triethylamine (3.9 mL, 28.0 mmol) to yield 3.21 g (quant.) of (+)-20 as a colorless oil upon purification by automated column chromatography (silica gel, ethyl acetate/petroleum ether 2–50%). = +51.5 (c 0.565, MeOH); All spectroscopic data was consistent with that reported for its enantiomer (−)-20. HRMS (ESI) m/z calcd for C14H25NO7SNa+ 374.1244, found 374.1242; Anal. calcd for C14H25NO7S: C, 47.85; H, 7.17; N, 3.99; S, 9.12. Found C, 47.55; H, 7.21; N, 3.98; S, 8.95.
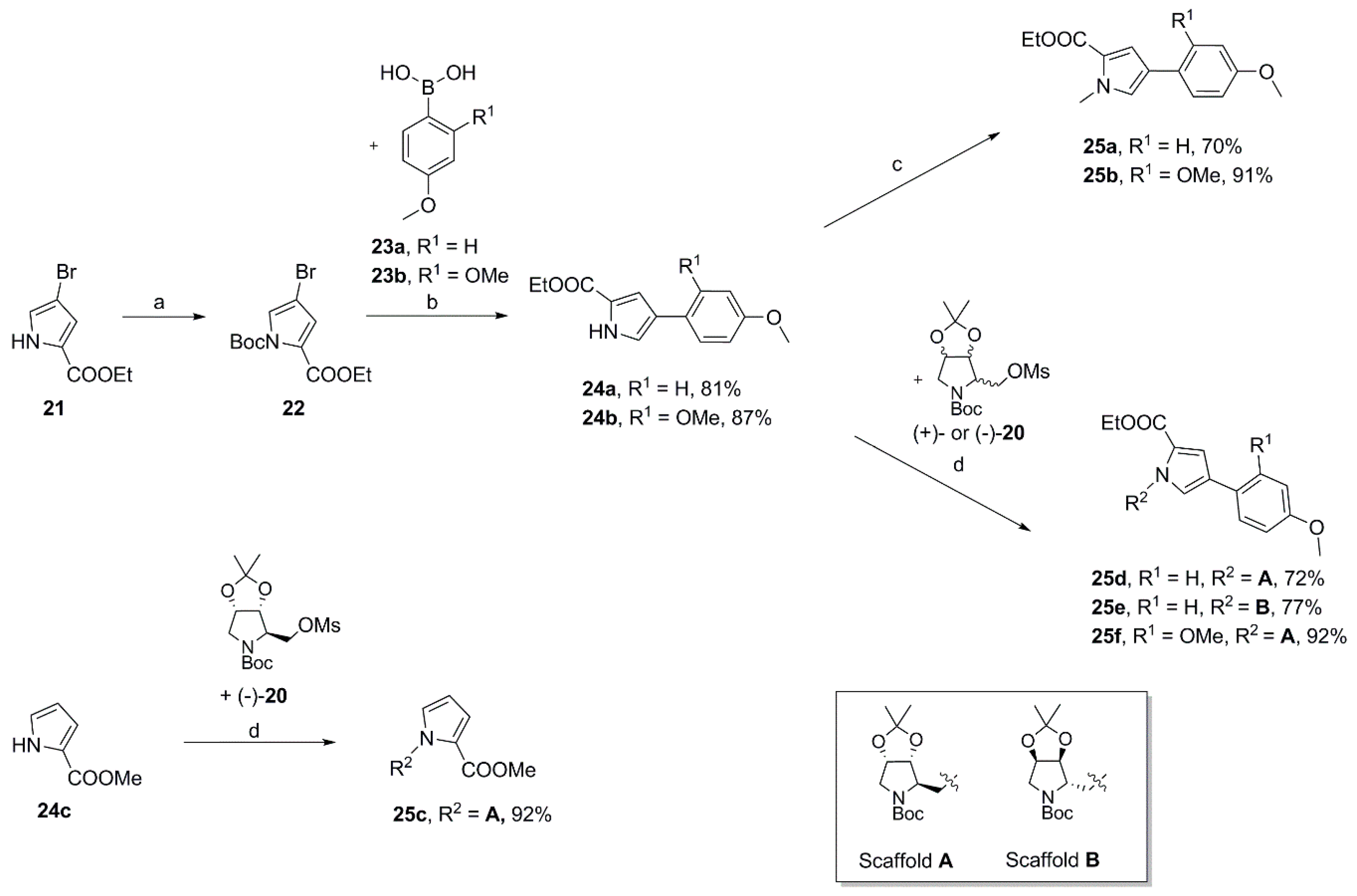
Ethyl 4-(4-methoxyphenyl)-1H-pyrrole-2-carboxylate (
24a). To a solution of 1-(
tert-butyl) 2-ethyl 4-bromo-1
H-pyrrole-1,2-dicarboxylate
22 [
59] (141 mg, 0.443 mmol) and 4-methoxyphenylboronic acid
23a (95%; 213 mg, 1.33 mmol) in DMF (7.5 mL) was added palladium-
tetrakis(triphenylphosphine) (29 mg, 0.025 mmol) and a 2 M aqueous solution of sodium carbonate (2.25 mL) and the resulting reaction mixture was heated at 110 °C overnight. After being cooled to room temperature water was added and the mixture was extracted with ethyl acetate (3×). The combined organic phases were washed with water (2×) and brine, dried over MgSO
4 and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–20%) to afford 88 mg (81%) of
24a as a colorless solid. m.p. 136–138 °C (DSC);
1H NMR (500 MHz, CDCl
3) δ 9.22 (s
br, 1H), 7.44 (AA’XX’,
JAX = 8.8 Hz, 2H), 7.15–7.12 (m, 2H), 6.90 (AA’XX’,
JAX = 8.8 Hz, 2H), 4.34 (q, 7.2 Hz, 2H), 3.82 (s, 3H), 1.38 (t, 7.2 Hz, 3H);
13C NMR (125 MHz, CDCl
3) δ 161.18, 158.31, 127.35, 126.64, 126.45, 123.60, 118.73, 114.22, 112.18, 60.43, 55.32, 14.45; HRMS (ESI)
m/
z calcd for C
14H
15NO
3Na
+ 268.0944, found 268.0947.
Ethyl 4-(4-methoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (25a). To a solution of 24a (727 mg, 2.96 mmol) in dry DMF (10 mL) was added sodium hydride (60% in mineral oil; 150 mg, 3.75 mmol) at 0 °C. After 20 min iodomethane (0.25 mL, 4.02 mmol) was introduced dropwise, the ice bath was removed after a further 15 min and the reaction was stirred for another 2.5 h. Subsequently, water was added, and the aqueous mixture was extracted with ethyl acetate (3×). The combined organic layers were washed with water (2×) and brine, dried over MgSO4 and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–20%) to yield 540 mg (70%) of 25a as a colorless oil that solidified on standing. 1H NMR, (500 MHz, CDCl3) δ 7.41 (AA’XX’, JAX = 8.9 Hz, 2H), 7.15 (d, 2.1 Hz, 1H), 6.98 (d, 2.1 Hz, 1H), 6.89 (AA’XX’, JAX = 8.9 Hz, 2H), 4.30 (q, 7.1 Hz, 2H), 3.95 (s, 3H), 3.81 (s, 3H), 1.37 (t, 7.2 Hz, 3H); 13C NMR, (125 MHz, CDCl3) δ 161.34, 158.18, 127.38, 126.25, 125.57, 123.82, 123.31, 114.50, 114.21, 59.89, 55.33, 36.89, 14.48; HRMS (ESI) m/z calcd for C15H17NO3Na+ 282.1101, found 282.1105; Anal. calcd for C15H17NO3: C, 69.48; H, 6.61; N, 5.40. Found C, 69.45; H, 6.73; N, 5.43.
4-(4-Methoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (26a). To solution of 25a (479 mg, 1.85 mmol) in a 1:1 mixture of THF (10 mL) and methanol (10 mL) was added an 8 M aqueous solution of potassium hydroxide (3.5 mL) and the resulting reaction mixture was heated at 50 °C overnight. After having evaporated the solvents water was added and the resulting mixture was acidified to pH 1 by slowly adding a 2 M aqueous solution of hydrochloric acid (to pH 3-4 by slowly adding a 1 M aqueous solution of hydrochloric acid in the case of O-isopropylidene-protected compounds: 26c–f). The precipitate was extracted with ethyl acetate (3×) and the combined organic phases were washed with brine, dried over MgSO4 and concentrated to small volume which was then treated with a small amount of petroleum ether. The ochre crystalline solid was filtered off and washed with a 3:7 mixture of ethyl acetate and petroleum ether. The collected solid was dried under high vacuum to give 344 mg (81%) of 26a. m.p. 203–204 °C (DSC); 1H NMR, (500 MHz, d6-DMSO) δ 12.22 (sbr, 1H), 7.47 (AA’XX’, JAX = 8.8 Hz, 2H), 7.42 (d, 2.0 Hz, 1H), 7.09 (d, 2.1 Hz, 1H), 6.90 (AA’XX’, JAX = 8.8 Hz, 2H), 3.86 (s, 3H), 3.75 (s, 3H); 13C NMR, (125 MHz, d6-DMSO) δ 161.86, 157.46, 126.89, 126.06, 125.60, 123.14, 122.37, 114.08, 113.59, 54.95, 36.33; HRMS (ESI) m/z calcd for C13H13NO3Na+ 254.0788, found 254.0794; Anal. calcd for C13H13NO3: C, 67.52; H, 5.67; N, 6.06. Found C, 67.51; H, 5.59; N, 6.21.
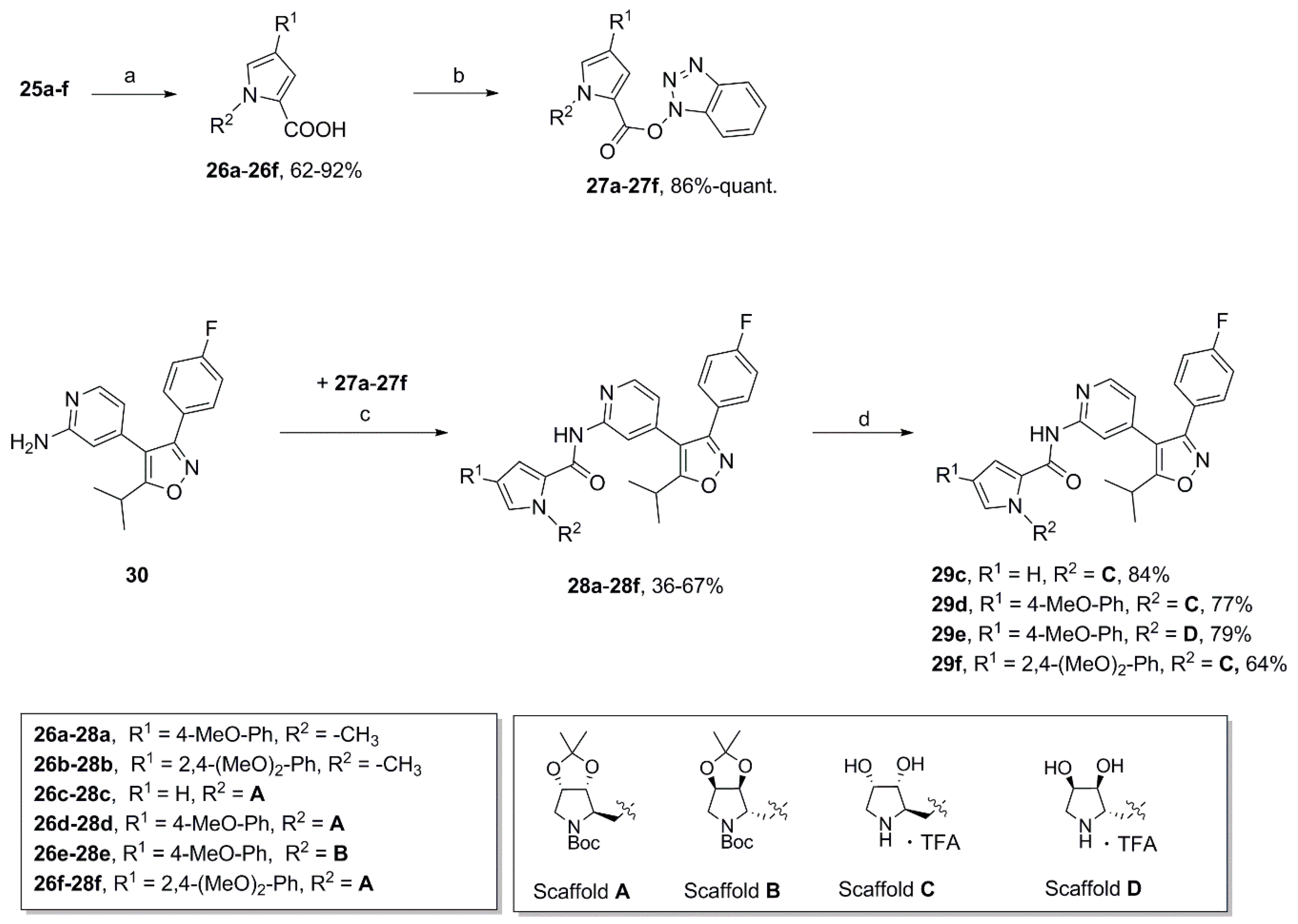
1H-Benzo[d][1,2,3]triazol-1-yl 4-(4-methoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (27a). To a solution of 26a (278 mg, 1.20 mmol) and triethylamine (0.51 mL, 3.66 mmol) in dry DMF (10 mL) was added HBTU (559 mg, 1.47 mmol) in one portion at room temperature. After being stirred overnight water was added and the resulting mixture was extracted with ethyl acetate (3×). The combined organic phases were washed with water (2×) and brine, dried over MgSO4 and concentrated. The crude reaction product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–50%) to yield 403 mg of 27a (96%) as a colorless foam. 1H NMR (500 MHz, CDCl3) δ 8.09 (dt, 8.5 Hz, 0.8 Hz, 1H), 7.63 (d, 2.0 Hz, 1H), 7.55 (ddd, 8.3, 6.6, 0.9 Hz, 1H), 7.51 (dt, 8.3, 1.0 Hz, 1H), 7.47 (AA’XX’, JAX = 8.8 Hz, 2H), 7.43 (ddd, 8.3, 6.7, 1.4 Hz, 1H), 7.28 (d, 1.9 Hz, 1H), 6.95 (AA’XX’, JAX = 8.8 Hz, 2H), 3.98 (s, 3H), 3.84, (s, 3H); 13C NMR (125 MHz, CDCl3) δ 158.79, 156.79, 143.57, 129.54, 129.15, 128.62, 126.55, 126.09, 125.77, 124.72, 120.54, 117.82, 116.83, 114.43, 108.49, 55.38, 36.97; HRMS (ESI) m/z calcd for C19H16N4O3Na+ 371.1115, found 371.1114. Anal. calcd for C19H16N4O3: C, 65.51; H, 4.63; N, 16.08. Found C, 65.54; H, 4.50; N, 16.02.
N-{4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}-4-(4-methoxyphenyl)-1-methyl-1H-pyrrole-2-carboxamide (28a). To a solution of isoxazole 30 (313 mg, 1.05 mmol) in dry DMF (10 mL) was added sodium hydride (60% in mineral oil; 39.7 mg, 0.993 mmol) in one portion at room temperature. After 30 min activated carboxylic acid 27a (195 mg, 0.560 mmol) was introduced in one portion and the resulting reaction was stirred for 48 h. Water was added and the mixture was extracted with ethyl acetate (3×). The combined organic phases were then washed with water (2×) and brine, dried over MgSO4 and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–40%) to afford 191 mg (67%) of 28a. Additional recrystallization from ethyl acetate/petroleum ether yielded 100 mg of pure 28a as a colorless solid. m.p. 157–158 °C (DSC); 1H NMR, (500 MHz, CDCl3) δ 8.42 (sbr, 1H), 8.27–8.23 (m, 2H), 7.43 (AA’BB’X, JAB = 8.9, JAF = 5.4 Hz, 2H), 7.40 (AA’XX’, JAX = 8.8, 2H), 7.06–7.01 (overlapping signals: 7.03, AA’BB’X, JAB = JBF = 8.7 Hz, 2H and 7.02, d, 1.9 Hz, 1H), 7.00 (d, 1.8 Hz, 1H), 6.91 (AA’XX’, JAX = 8.8 Hz, 2H), 6.74 (dd, 5.2, 1.5 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 3H), 3.25 (sp, 7.0 Hz, 1H), 1.39 (d, 7.0 Hz, 6H); 19F NMR (470 MHz, CDCl3) δ −111.12; 13C NMR, (125 MHz, CDCl3) δ 175.49, 164.58/162.60 (d, 248.2 Hz), 160.04, 159.51, 158.39, 152.41, 148.03, 141.33, 130.50/130.43 (d, 8.2 Hz), 126.96, 126.27, 125.94, 125.55, 124.78, 123.98, 120.46, 115.88/115.71 (d, 21.7 Hz), 114.43, 114.35, 112.14, 110.45, 55.36, 37.05, 26.66, 21.00; HRMS (ESI) m/z calcd for C30H27FN4O3H+ 511.2140, found 511.2150; Anal. calcd for C30H27FN4O3: C, 70.57; H, 5.33; N, 10.97. Found C, 70.60; H, 5.41; N, 11.03.
Ethyl 4-(2,4-dimethoxyphenyl)-1H-pyrrole-2-carboxylate (24b). Applying the same method as for the preparation of 24a a mixture of 1-(tert-butyl) 2-ethyl 4-bromo-1H-pyrrole-1,2-dicarboxylate 22 (1.19 g, 3.74 mmol), 2,4-dimethoxyphenylboronic acid 23b (95%; 2.18 g, 11.4 mmol), palladium-tetrakis(triphenylphosphine) (225 mg, 0.195 mmol), and a 2 M aqueous solution of sodium carbonate (19 mL) in DMF (63 mL) was heated at 110 °C overnight to yield 892 mg (87%) of 24b as a colorless solid. M.p. 124–125 °C (DSC); 1H NMR (500 MHz, CDCl3) δ 9.13 (sbr, 1H), 7.44–7.41 (m, 1H), 7.38 (dd, 2.9, 1.6 Hz, 1H), 7.23 (dd, 2.7, 1.7 Hz, 1H), 6.54–6.51 (m, 2H), 4.34 (q, 7.1 Hz, 2H), 3.87 (s, 3H), 3.83 (s, 3H), 1.37 (t, 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 161.25, 159.32, 157.18, 128.48, 122.49, 122.41, 121.87, 116.44, 113.84, 104.74, 99.02, 60.28, 55.39, 14.47; HRMS (ESI) m/z calcd for C15H17NO4Na+ 298.1050, found 298.1049.
Ethyl 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (25b). Employing the same procedure as described for the preparation of 25a pyrrole 24b (790 mg, 2.87 mmol) dissolved in dry DMF (10 mL) was treated with sodium hydride (60% in mineral oil; 140 mg, 3.50 mmol) and iodomethane (0.23 mL, 3.70 mmol) to yield 758 mg (91%) of 25b as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.42–7.38 (m, 1H), 7.24–7.21 (m, 2H), 6.54–6.49 (m, 2H), 4.30 (q, 7.1 Hz, 2H), 3.94 (s, 3H), 3.87 (s, 3H), 3.82 (s, 3H), 1.36 (t, 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 161.47, 159.19, 157.11, 128.77, 128.32, 122.25, 119.63, 116.48, 116.20, 104.75, 98.99, 59.76, 55.41, 36.82, 14.51; HRMS (ESI) m/z calcd for C16H19NO4Na+ 312.1206, found 312.1205; Anal. calcd for C16H19NO4: C, 66.42; H, 6.62; N, 4.84. Found C, 66.59; H, 6.53; N, 4.90.
4-(2,4-Dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (26b). Following the procedure described for the preparation of 26a ethyl ester 25b (522 mg, 1.80 mmol) dissolved in a 1:1 mixture of THF and methanol (10 mL/10 mL) was treated with an 8 M aqueous potassium hydroxide solution (2.5 mL) to afford 294 mg (62%) of 26b as a crystalline, ochre solid. m.p. 175–176 °C (DSC); 1H NMR (500 MHz, d6-DMSO) δ 7.42 (d, 8.5 Hz, 1H), 7.41 (d, 2.0 Hz, 1H), 7.13 (d, 2.0 Hz, 1H), 6.60 (d, 2.5 Hz, 1H), 6.53 (dd, 8.5, 2.5 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.77 (s, 3H); 13C NMR (125 MHz, d6-DMSO) δ 161.96, 158.57, 156.60, 128.46, 127.55, 121.99, 118.68, 115.59, 105.18, 98.77, 55.28, 55.08, 36.22; HRMS (ESI) m/z calcd for C14H15NO4Na+ 284.0893, found 284.0898; Anal. calcd for C14H15NO4: C, 64.36; H, 5.79; N, 5.36. Found C, 64.46; H, 5.68; N, 5.38.
1H-Benzo[d][1,2,3]triazol-1-yl 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (27b). Carboxylic acid 26b (225 mg, 0.861 mmol) in dry DMF (8 mL) was reacted with HBTU (398 mg, 1.05 mmol) in the presence of triethylamine (0.40 mL, 2.87 mmol) according to the procedure outlined for the preparation of compound 27a to give 293 mg (90%) of 27b as a colorless foam. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, 8.5 Hz, 1H), 7.73 (d, 1.8 Hz, 1H), 7.57–7.49 (m, 3H), 7.47–7.40 (m, 2H), 6.58–6.55 (m, 2H), 3.97 (s, 3H), 3.91 (s, 3H), 3.85 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.86, 157.26, 156.87, 143.57, 132.64, 129.21, 128.57, 128.52, 124.68, 121.69, 120.49, 119.67, 115.76, 115.22, 108.56, 104.96, 99.08, 55.48, 36.86; HRMS (ESI) m/z calcd for C20H18N4O4Na+ 401.1220, found 401.1229.
4-(2,4-Dimethoxyphenyl)-N-{4-[3-(4-fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}-1-methyl-1H-pyrrole-2-carboxamide (28b). Deploying the same procedure as for the preparation of compound 28a isoxazole 30 (304 mg, 1.02 mmol) was dissolved in dry DMF (5 mL), deprotonated with sodium hydride (60% in mineral oil; 36 mg, 0.900 mmol) and subsequently reacted with a solution of 27b (212 mg, 0.560 mmol) in dry DMF (6 mL) to yield 172 mg (57%) of 28b as a colorless foam. The removal of a minor impurity required additional automated flash column chromatography on reversed phase C-18 silica gel (water/acetonitrile 10–90%). 1H NMR (500 MHz, CDCl3) δ 8.51 (sbr, 1H), 8.28 (s, 1H), 8.24 (d, 5.1 Hz, 1H), 7.43 (AA’BB’X, JAB = 8.5, JAF = 5.4 Hz, 2H), 7.38–7.34 (m, 1H), 7.23 (s, 1H), 7.14 (s, 1H), 7.03 (AA’BB’X, JAB = JBF = 8.6 Hz, 2H), 6.73 (d, 5.1, Hz, 1H), 6.56–6.51 (m, 2H), 3.99 (s, 3H), 3.89 (s, 3H), 3.83 (s, 3H), 3.26 (sp, 7.0 Hz, 1H), 1.39 (d, 7.0 Hz, 6H); 19F NMR (470 MHz, CDCl3) δ −111.12; 13C NMR (125 MHz, CDCl3) δ 175.46, 164.57/162.59 (d, 248 Hz), 160.03, 159.68, 159.42, 157.16, 152.58, 148.07, 141.23, 130.50/130.43 (d, 8.2 Hz), 128.89, 128.26, 124.80, 124.52, 120.36, 119.84, 116.05, 115.87/115.69 (d, 21.7 Hz), 114.46, 112.43, 112.18, 104.83, 99.08, 55.44, 36.99, 26.65, 20.99; HRMS (ESI) m/z calcd for C31H29FN4O4H+ 541.2246, found 541.2251. Anal. calcd for C31H29FN4O4·0.2(CHCl3): C, 66.39; H, 5.21; N, 9.93. Found C, 66.35; H, 5.22; N, 9.88.
tert-Butyl(3aR,4R,6aS)-4-{[2-(methoxycarbonyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (
25c) To a solution of methyl 1H-pyrrole-2-carboxylate
24c [
63] (325 mg, 2.60 mmol) and crude mesylate (−)-
20 (1.14 g, 3.24 mmol) in dry DMF (28 mL) was added cesium carbonate (2.22 g, 6.81 mmol) and a catalytic amount of tetra n-butylammonium iodide, and the resulting mixture was heated at 80 °C overnight. After being cooled to room temperature a 10:1 mixture of water and brine was added, and the aqueous mixture was extracted with ethyl acetate (3×). The combined organic phases were washed with a 10:1 mixture of water and brine (2×), and brine, dried over MgSO
4 and concentrated. Finally, the crude reaction product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–40%) to yield 909 mg (92%) of
25c as a colorless oil that solidified on standing.
= +4.42 (
c 0.77, CHCl
3);
1H NMR (500 MHz, CDCl
3, mixture of rotamers) δ 6.95 (dd, 3.9, 1.5 Hz, 1H); 6.88 (s, 0.4H), 6.75-6.72 (m, 0.6H), 6.19–6.16 (m, 0.4H), 6.15 (t, 3.0 Hz, 0.6H), 4.66–4.51 (m, 3H), 4.49 (dd, 13.9, 5.2 Hz, 0.4H), 4.45–4.38 (m, 1H), 4.13 (dd, 13.7, 8.2 Hz, 0.6H), 3.96 (d, 13.1 Hz, 0.6H), 3.80 (s, 3H), 3.68 (d, 12.9 Hz, 0.4H), 3.26 (dd, 13.1, 5.0 Hz, 0.6H), 3.17 (dd, 12.8, 5.0 Hz, 0.4H), 1.46 (s, 3.4H), 1.41 (s, 3H), 1.34 (s, 5.6H), 1.27 (s, 2H), 1.25 (s, 1H);
13C NMR (125 MHz, CDCl
3, mixture of rotamers) δ 161.99, 161.80, 154.27, 129.33, 129.05, 122.00, 121.90, 118.61, 118.48, 111.83, 111.71, 109.22, 108.92, 82.39, 81.51, 80.05, 79.97, 79.32, 78.90, 64.56, 64.02, 52.19, 51.13, 51.06, 47.49, 46.32, 28.41, 28.11, 27.02, 26.93, 25.10, 25.06; HRMS (ESI)
m/
z calcd for C
19H
28N
2O
6Na
+ 403.1840, found 403.1844. Anal. calcd for C
19H
28N
2O
6: C, 59.99; H, 7.42; N, 7.36. Found C, 60.07; H, 7.52; N, 7.39.
1-{[(3aR,4R,6aS)-5-(tert-Butoxycarbonyl)-2,2-dimethyltetrahydro-4H-[1,3]dioxolo[4,5-c]pyrrol-4-yl]methyl}-1H-pyrrole-2-carboxylic acid (26c). According to the procedure described for the preparation of carboxylic acid 26a methyl ester 25c (752 mg, 1.98 mmol) dissolved in a 2:1 mixture of THF (12 mL) and MeOH (6 mL) was saponified with an aqueous 8 M potassium hydroxide solution 82.5 mL). The crude product was subsequently purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether 2–70%) to give 606 mg (84%) of the desired carboxylic acid 26c as a colorless oil that solidified on standing. = +13.2 (c 0.50, CHCl3); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.13–7.10 (m, 1H), 6.95 (s, 0.4H), 6.80–6.77 (s, 0.6H), 6.24–6.21 (m, 0.4H), 6.21–6.18 (m, 0.6H), 4.68–4.59 (m, 2H), 4.56–4.48 (m, 1.4H), 4.48–4.40 (m, 1H), 4.11 (dd, 13.7, 8.6 Hz, 0.6H), 3.99 (d, 13.2 Hz, 0.6H), 3.70 (d, 12.9 Hz, 0.4H), 3.27 (dd, 13.2, 4.9 Hz, 0.6H), 3.16 (dd, 12.8, 4.9 Hz, 0.4H), 1.46 (s, 3.4H), 1.41 (s, 3H), 1.34 (s, 5.6H), 1.29 (s, 2H), 1.26 (s, 1H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 165.39, 165.23, 154.33, 130.71, 130.42, 121.13, 120.98, 120.79, 120.72, 111.92, 111.80, 109.71, 109.41, 82.35, 81.51, 80.20, 80.14, 79.31, 78.90, 64.48, 63.99, 52.23, 51.03, 47.67, 46.49, 28.39, 28.12, 27.01, 26.92, 25.06; HRMS (ESI) m/z calcd for C18H26N2O6Na+ 389.1683, found 389.1687. Anal. calcd for C18H26N2O6: C, 59.00; H, 7.15; N, 7.65. Found C, 59.22; H, 7.19; N, 7.60.
tert-Butyl(3aR,4R,6aS)-4-{[2-{[(1H-benzo[d][1,2,3]triazol-1-yl)oxy]carbonyl}-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (27c). Employing the same protocol as for the preparation of 27a carboxylic acid 26c (492 mg, 1.34 mmol) was dissolved in dry DMF (15 mL) and subsequently activated with HBTU (625 mg, 1.65 mmol) in the presence of triethylamine (0.57 mL, 4.09 mmol) to afford 648 mg (quant.) of 27c as a colorless foam. = +31.4 (c 0.805, CHCl3); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.11–8.06 (m, 1H), 7.59–7.47 (m, 3H), 7.46–7.41 (m, 1H), 7.21 (s, 0.4H), 7.06–7.03 (s, 0.6H), 6.42–6.37 (m, 1H), 4.62 (t, 5.0 Hz, 0.6H), 4.54 (t, 4.8 Hz, 0.4H), 4.51–4.37 (m, 3.4H), 4.18 (dd, 13.5, 9.3 Hz, 0.6H), 4.03 (d, 13.4 Hz, 0.6H), 3.74 (d, 13.1 Hz, 0.4H), 3.22–3.13 (overlapping signals: 3.19, dd, 13.1, 4.5 Hz, 0.6H and m, 0.4H), 1.45 (s, 4H), 1.40 (s, 1H), 1.39 (s, 2H), 1.34 (s, 5H), 1.26 (s, 3H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 157.23, 157.05, 154.32, 143.54, 133.20, 132.73, 129.21, 128.72, 128.59, 124.75, 122.37, 122.18, 120.56, 120.43, 116.27, 116.08, 112.07, 111.00, 110.67, 108.71, 108.49, 82.35, 81.53, 80.41, 80.28, 79.39, 78.98, 64.90, 64.02, 51.93, 50.84, 48.05, 47.09, 28.33, 28.14, 26.91, 26.83, 24.99, 24.92; HRMS (ESI) m/z calcd for C24H29N5O6Na+ 506.2010, found 506.2012.
tert-Butyl(3aR,4R,6aS)-4-{[2-({4-[3-(4-fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (28c). Using the same procedure as for the preparation of compound 28a isoxazole 30 (376 mg, 1.26 mmol) dissolved in dry DMF (5 mL) was firstly deprotonated with sodium hydride (60% in mineral oil; 45.0 mg, 1.13 mmol) and subsequently reacted with a solution of compound 27c (339 mg, 0.701 mmol) in dry DMF (5 mL) at room temperature over a period of 48 h to afford 228 mg (50%) of the desired coupling product 28c as a colorless foam. = +46.8 (c 0.66, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.37 (s, 0.4H), 8.34 (s, 0.6H), 8.25–8.21 (m, 2H), 7.44 (AA’BB’X, JAB = 8.6, JAF = 5.4 Hz, 2H), 7.04 (AA’BB’X, JAB = JBF = 8.6 Hz, 2H), 6.92 (s, 0.4H), 6.84–6.78 (m, 1.6H), 6.76–6.72 (m, 1H), 6.24–6.20 (m, 1H), 4.70–4.54 (m, 3H), 4.47–4.41 (m, 1.4H), 4.26 (dd, 13.6, 8.1 Hz, 0.6H), 3.94 (d, 13.1 Hz, 0.6H), 3.71 (d, 12.9 Hz, 0.4H), 3.31–3.19 (overlapping signals: m, 1H and 3.26, sp, 7.0 Hz, 1H), 1.44 (s, 4H), 1.40 (s, 3H), 1.40 (d, 6.9 Hz, 6H), 1.34 (s, 5H), 1.26 (s, 2H), 1.22 (s, 1H); 19F NMR (470 MHz, CDCl3, mixture of rotamers) δ −110.96, −111.01; 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 175.48, 164.59/162.61 (d, 248.3 Hz), 160.06, 159.78, 159.52, 154.34, 154.23, 152.34, 152.28, 148.20, 141.09, 130.55/130.48 (d, 8.2 Hz), 129.27, 129.11, 124.79/124.76 (d, 3.4 Hz), 124.71, 120.43, 115.89/115.72 (d, 21.6 Hz), 114.46, 114.41, 114.03, 113.95, 112.18, 111.79, 111.69, 109.19, 109.01, 82.35, 81.53, 80.06, 79.94, 79.48, 78.94, 64.75, 64.01, 52.10, 51.09, 47.42, 46.41, 28.37, 28.19, 26.97, 26.92, 26.66, 25.04, 21.00; HRMS (ESI) m/z calcd for C35H40FN5O6H+ 646.3035, found 646.3044.
(2R,3R,4S)-2-{[2-({4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-1H-pyrrol-1-yl]methyl}-3,4-dihydroxypyrrolidin-1-ium trifluoroacetate (29c). Trifluoroacetic acid (4.75 mL) and water (0.50 mL) were added to compound 28c (163 mg, 0.252 mmol) and the resulting mixture was stirred for 3 h at 0 °C. After complete deprotection as indicated by TLC analysis the reaction was concentrated and the residue purified by automated column chromatography (silica gel, methanol/methylene chloride 0–10%) to yield 131 mg (84%) of 29c as a colorless foam. = −4.5 (c 0.29, MeOH); 1H NMR (500 MHz, CD3OD) δ 8.35 (dd, 5.2, 0.7 Hz, 1H), 8.08 (dd, ~1.3, ~0.6 Hz, 1H), 7.45 (AA’BB’X, JAB = 8.9, JAF = 5.3 Hz, 2H), 7.16 (dd, 2.6, 1.7 Hz, 1H), 7.13 (AA’BB’X, JAB = JBF = 8.9 Hz, 2H), 7.10 (dd, 4.1, 1.6 Hz, 1H), 6.95 (dd, 5.2, 1.5, Hz, 1H), 6.30 (dd, 4.1, 2.6 Hz, 1H), 4.64 (d, 6.4 Hz, 2H), 4.20 (td, 4.1, 2.1 Hz, 1H), 4.02 (dd, 8.4, 4.1 Hz, 1H), 3.77 (dt, ~6.5, ~8.2 Hz, 1H), 3.40 (dd, 12.4, 4.2 Hz, 1H), 3.28 (sp, 7.0 Hz, 1H), 3.14 (dd, 12.4, 2.1 Hz, 1H), 1.38 (d, 7.0 Hz, 6H); 19F NMR (470 MHz, CD3OD) δ −77.10, −112.81; 13C NMR (125 MHz, CD3OD) δ 176.92, 166.15/164.17 (d, 247.1 Hz), 162.97, 161.49, 153.76, 149.81, 142.26, 131.83/131.76 (d, 8.3 Hz), 131.06, 126.52, 126.19, 122.22 117.21, 117.08, 116.96/116.79 (d, 22.1 Hz), 113.62, 110.55, 75.09, 71.13, 63.96, 51.24, 48.77, 27.96, 21.25; HRMS (ESI) m/z calcd for C27H28FN5O4H+ 506.2198, found 506.2203; Anal. calcd for C27H28FN5O4·1.3(CF3COOH): C, 54.38; H, 4.52; N, 10.71. Found C, 54.28; H, 4.77; N, 10.85.
tert-Butyl(3aR,4R,6aS)-4-{[2-(ethoxycarbonyl)-4-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (25d). Using the same procedure as for the preparation of compound 25c mesylate (−)-20 (2.65 g, 7.54 mmol) was dissolved in dry DMF (40 mL) and reacted with pyrrole 24a (1.5 g, 6.12 mmol) in the presence of cesium carbonate (4.38 g, 13.4 mmol) and a catalytic amount of tetrabutylammonium iodide at 80 °C overnight to give 2.19 g (72%) of 25d as a colorless foam. A crystalline sample (rosettes of fine transparent needles) for X-ray analysis was obtained by crystallization from a mixture of ethyl acetate and 2-propanol. m.p. 136–137 °C (DSC); = +14.5 (c 0.725, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.40 (AA’XX’, JAX = 8.7 Hz, 0.8H), 7.38 (AA’XX’, JAX = 8.7 Hz, 1.2H), 7.18–7.15 (m, 1H), 7.10 (d, 1.7 Hz, 0.4H), 6.92 (d, 1.7 Hz, 0.6H), 6.91–6.86 (overlapping AA’XX’ spin systems, JAX = 8.7 Hz, 2H), 4.72–4.60 (m, 2H), 4.60–4.54 (m, 1H), 4.51–4.41 (m, 1.4H), 4.30 (q, 7.1 Hz, 2H), 4.14 (dd, 13.6, 8.5 Hz, 0.6H), 3.99 (d, 13.2 Hz, 0.6H), 3.82 (s, 3H), 3.69 (d, 13.0 Hz, 0.4H), 3.33 (dd, 13.1, 4.9 Hz, 0.6H), 3.21 (dd, 12.8, 5.1 Hz, 0.4H), 1.47 (s, 4H), 1.41 (s, 3H), 1.38/1.36 (overlapping tripletts, 7.0 Hz, 3H), 1.30 (s, 5H), 1.28 (s, 2H), 1.25 (s, 1H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 161.59, 161.35, 158.32, 154.37, 154.27, 127.03, 126.42, 126.32, 125.37, 124.98, 124.87, 124.81, 122.90, 122.73, 115.33, 115.28, 114.21, 111.87, 111.71, 82.45, 81.48, 80.16, 80.00, 79.31, 78.93, 64.69, 64.11, 60.07, 55.33, 52.29, 51.04, 47.68, 46.27, 28.43, 28.09, 27.01, 26.92, 25.04, 14.47; HRMS (ESI) m/z calcd for C27H36N2O7Na+ 523.2415, found 523.2411; Anal. calcd for C27H36N2O7: C, 64.78; H, 7.25; N, 5.60. Found C, 64.78; H, 7.33; N, 5.54.
1-{[(3aR,4R,6aS)-5-(tert-Butoxycarbonyl)-2,2-dimethyltetrahydro-4H-[1,3]dioxolo[4,5-c]pyrrol-4-yl]methyl}-4-(4-methoxyphenyl)-1H-pyrrole-2-carboxylic acid (26d). Deploying the same protocol as described for the preparation of compound 26a ethyl ester 25d (1.96 g, 3.92 mmol) dissolved in 2:1 mixture of THF (20 mL) and methanol (10 mL) was treated with a 4 M aqueous solution of sodium hydroxide (7.5 mL) at 50 °C overnight to yield 1.73 g (94%) of crude 26d. An analytical sample of 26d was obtained by recrystallization from methanol as a colorless and crystalline solid. m.p. 197–199 °C (DSC); = +17.8 (c 0.695, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.43–7.37 (overlapping AA’XX’ spin systems, JAX = 8.7 Hz, 2H), 7.32 (d, 2.0 Hz, 1H), 7.18 (s, 0.4H), 6.99 (d, 1.3 Hz, 0.6H), 6.90 (AA’XX’, JAX = 8.6 Hz, 2H), 4.73–4.55 (m, 3H), 4.51–4.45 (m, 1.4H), 4.13 (dd, 13.7, 8.8 Hz, 0.6H), 4.04 (d, 13.2 Hz, 0.6H), 3.82 (s, 3H), 3.72 (d, 12.8 Hz, 0.4H), 3.35 (dd, 13.2, 4.8 Hz, 0.6H), 3.22 (dd, 12.8, 5.0 Hz, 0.4H), 1.47 (s, 4H), 1.42 (s, 3H), 1.31/1.30 (overlapping singlets, 7H), 1.26 (s, 1H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 165.17, 164.96, 158.46, 154.35, 126.84, 126.65, 126.47, 126.36, 125.51, 125.34, 121.65, 121.49, 117.34, 114.26, 111.95, 111.84, 82.40, 81.48, 80.30, 80.19, 79.31, 78.93, 64.63, 64.03, 55.32, 52.27, 51.00, 47.83, 46.47, 28.39, 28.07, 26.99/26.89, 25.04; HRMS (ESI) m/z calcd for C25H32N2O7H+ 473.2282, found 473.2281; Anal. calcd for C25H32N2O7: C, 63.55; H, 6.83; N, 5.93. Found C, 63.58; H, 6.88; N, 5.89.
tert-Butyl(3aR,4R,6aS)-4-[(2-{[(1H-benzo[d][1,2,3]triazol-1-yl)oxy]carbonyl}-4-(4-methoxyphen-yl)-1H-pyrrol-1-yl)methyl]-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (27d). Applying the same procedure as for the preparation of compound 27a carboxylic acid 26d (1.05 g, 2.22 mmol) dissolved in dry DMF (30 mL) was reacted with HBTU (1.01 g, 2.66 mmol) in the presence of triethylamine (0.93 mL, 6.67 mmol) to yield 1.28 g (98%) of 27d as a colorless foam; = +41.6 (c 0.700, CHCl3); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.12–8.07 (m, 1H), 7.67 (d, 1.6 Hz, 1H), 7.64–7.51 (m, 2H), 7.49–7.41 (m, 3.4H), 7.24 (m, 0.6H), 6.94 (AA’XX’, JAX = 8.7 Hz, 2H), 4.67–4.62 (m, 0.6H), 4.61–4.51 (m, 1.2H), 4.51–4.39 (m, 2.6H), 4.22 (dd, 14.8, 11.5 Hz, 0.6H), 4.07 (d, 13.4 Hz, 0.6H), 3.85 (s, 3H), 3.75 (d, 13.7 Hz, 0.4H), 3.27–3.17 (m, 1H), 1.45 (s, 3H), 1.41 (s, 1H), 1.39 (s, 2H), 1.30 (s, 6H), 1.26 (s, 3H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 158.91, 157.05, 154.43, 154.36, 143.55, 129.49, 129.23, 129.15, 128.78, 128.68, 126.92, 126.67, 125.78, 124.79, 120.59, 120.43, 118.64, 118.52, 116.77, 116.53, 114.47, 112.12, 108.77, 108.47, 82.41, 81.50, 80.50, 80.34, 79.36, 79.03, 65.04, 64.03, 55.37, 51.99, 50.83, 48.22, 47.03, 28.35, 28.09, 26.91, 26.84, 24.94; HRMS (ESI) m/z calcd for C31H35N5O7Na+ 612.2429, found 612.2429.
tert-Butyl(3aR,4R,6aS)-4-{[2-({4-[3-(4-fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carba-moyl)-4-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (28d). Following the procedure as described for the preparation of compound 28a isoxazole 30 (266 mg, 0.895 mmol) dissolved in dry DMF (5 mL) was deprotonated with sodium hydride (60% in mineral oil; 32 mg, 0.80 mmol). Subsequently, a solution of compound 27d (309 mg, 0.524 mmol) in dry DMF (5 mL) was added dropwise via cannula and the reaction was stirred at room temperature for 48 h to obtain 220 mg (56%) of 28d as a colorless foam. = +36.8 (c 0.595, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.49 (s, 0.4H), 8.45 (s, 0.6H), 8.27-8.22 (m, 2H), 7.45 (AA’BB’X, JAB = 8.5, JAF = 5.4 Hz, 2H), 7.42–7.36 (m, 2H), 7.14 (s, 0.4H), 7.08–7.02 (m, 3H), 6.99 (s, 0.6H), 6.92 (AA’XX’, JAX = 8.7 Hz, 2H), 6.78–6.73 (m, 1H), 4.77–4.66 (m, 1.4H), 4.64–4.56 (m, 1.6H), 4.52–4.44 (m, 1H), 4.39 (dd, 13.6, 5.6 Hz, 0.4H), 4.29 (dd, 13.6, 8.4 Hz, 0.6H), 3.98 (d, 13.1 Hz, 0.6H), 3.83 (s, 3H), 3.73 (d, 12.8 Hz, 0.4H), 3.32 (dd, 13.1, 4.8 Hz, 0.6H), 3.30–3.22 (overlapping signals: m, 0.4H; 3.27, sp, 6.9 Hz, 1H), 1.44 (s, 4H), 1.43–1.39 (overlapping signals: 1.41, s, 3H; 1.41, d, 6.9 Hz, 6H), 1.31 (s, 5H), 1.27 (s, 2H), 1.23 (s, 1H); 19F NMR (470 MHz, CDCl3) δ −110.91, −110.96; 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 175.54, 164.61/162.62 (d, 248.4 Hz), 160.07, 159.66, 159.41, 158.53, 154.39, 154.25, 152.29, 152.22, 148.06, 141.25, 130.56/130.50 (d, 7.8 Hz), 126.66, 126.62, 126.41, 126.36, 125.43, 125.31, 125.25, 125.11, 125.04, 124.77, 124.74, 120.46, 115.91/115.74 (d, 21.6 Hz), 114.49, 114.46, 114.37, 114.35, 112.15, 111.85, 111.73, 111.03, 82.41, 81.50, 80.18, 80.00, 79.48, 79.00, 64.87, 64.04, 55.36, 52.17, 51.06, 47.56, 46.41, 28.39, 28.15, 26.98, 26.92, 26.67, 25.05, 21.01; HRMS (ESI) m/z calcd for C42H46FN5O7H+ 752.3454, found 752.3461. Anal. calcd for C42H46FN5O7·0.1(CHCl3): C, 66.20; H, 6.08; N, 9.17. Found C, 66.18; H, 6.09; N, 9.13.
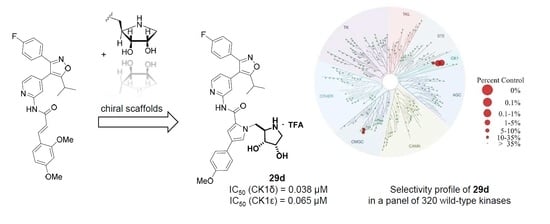
(2R,3R,4S)-2-{[2-({4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-3-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-3,4-dihydroxypyrrolidin-1-ium trifluoroacetate (29d). Employing the same method as for the preparation of 29c compound 28d (161 mg, 0.214 mmol) was treated with a mixture of trifluoroacetic acid (4.75 mL) and water (0.5 mL) at 0 °C for 2.5 h to yield 119 mg (77%) of 29d as a yellow semisolid. Pure 29d was obtained by subsequent recrystallization from ethyl acetate and petroleum ether. m.p. 191–193 °C (DSC); = −2.4 (c 0.655, MeOH); 1H NMR (500 MHz, CD3OD) δ 8.38 (d, 5.3 Hz, 1H), 8.09–8.07 (m, 1H), 7.52 (AA’XX’, JAX = 8.8 Hz, 2H), 7.49–7.44 (overlapping signals: 7.48, d, 1.7 Hz, 1H and 7.46, AA’BB’X, JAB = 8.9, JAF = 5.3 Hz, 2H), 7.42 (d, 1.9 Hz, 1H), 7.14 (AA’BB’X, JAB = JBF = 8.9 Hz, 2H), 6.99 (dd, 5.1, 1.5 Hz, 1H), 6.94 (AA’XX’, JAX = 8.8 Hz, 2H), 4.73 (dd, 15.2, 8.8 Hz, 1H), 4.68 (dd, 15.2, 3.8 Hz, 1H), 4.25 (dt, 3.8, 1.6 Hz, 1H), 4.12 (dd, 8.8, 3.9 Hz, 1H), 3.91 (td, 8.8, 3.8 Hz, 1H), 3.81 (s, 3H), 3.48 (dd, 12.5, 3.9 Hz, 1H), 3.28 (sp, 7.0 Hz, 1H), 3.24 (dd, 12.5, 1.7 Hz, 1H), 1.38 (d, 7.0 Hz, 6H); 19F NMR (470 MHz, CD3OD) δ −76.90, −112.83; 13C NMR (125 MHz, CD3OD) δ 176.93, 166.15/164.18 (d, 247.3 Hz), 162.99, 161.47, 160.20, 153.67, 149.90, 142.32, 131.83/131.76 (d, 8.5 Hz), 127.90, 127.40, 127.11, 126.21/126.18 (d, 3.0 Hz), 122.40, 117.20, 116.98/116.80 (d, 22.1 Hz), 115.37, 114.13, 113.61, 74.82, 70.83, 63.72, 55.81, 51.13, 48.70, 27.98, 21.25; HRMS (ESI) m/z calcd for C34H34FN5O5H+ 612.2617, found 612.2616. Anal. calcd for C34H34FN5O5+·CF3COOH: C, 59.58; H, 4.86; N, 9.65. Found C, 59.49; H, 4.88; N, 9.50.
tert-Butyl(3aS,4S,6aR)-4-{[2-(ethoxycarbonyl)-4-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (25e). Using the same procedure as for the preparation of compound 25c crude mesylate (+)-20 (1.22 g, 3.47 mmol) dissolved in dry DMF (20 mL) was reacted with pyrrole 24a (712 mg, 2.90 mmol), cesium carbonate (2.08 g, 6.38 mmol) and a catalytic amount of tetra-n-butylammonium iodide to afford 1.12 g (77%) of 25e as a colorless foam. = −15.4 (c 0.695, acetone). All spectroscopic data was consistent with that reported for its enantiomer 25d. HRMS (ESI) m/z calcd for C27H36N2O7Na+ 523.2415, found 523.2411; Anal. calcd for C27H36N2O7: C, 64.78; H, 7.25; N, 5.60. Found C, 64.61; H, 7.37; N, 5.59.
1-{[(3aS,4S,6aR)-5-(tert-Butoxycarbonyl)-2,2-dimethyltetrahydro-4H-[1,3]dioxolo[4,5-c]pyrrol-4-yl]methyl}-4-(4-methoxyphenyl)-1H-pyrrole-2-carboxylic acid (26e). Applying the same protocol as for the preparation of compound 26a ethyl ester 25e (1.01 g, 2.02 mmol) was dissolved in a 2:1 mixture of THF and methanol (12 mL/6 mL) and treated with an aqueous solution of potassium hydroxide (8 M, 3.75 mL) at 50 °C overnight to afford 983 mg (quant.) of crude 26e as a light-yellow foam which was subsequently deployed in the next reaction step without further purification. An analytical sample was obtained by recrystallization from methanol. m.p. 196–199 °C (DSC). = −17.7 (c 0.515, acetone). All spectroscopic data was consistent with the data reported for its enantiomer 26d. HRMS (ESI) m/z calcd for C25H32N2O7Na+ 495.2102, found 495.2102; Anal. calcd for C25H32N2O7: C, 63.55; H, 6.83; N, 5.93. Found C, 63.47; H, 6.89; N, 5.91.
tert-Butyl(3aS,4S,6aR)-4-{[2-{[(1H-benzo[d][1,2,3]triazol-1-yl)oxy]carbonyl}-4-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (27e). Using the same method as described for the preparation of compound 27a crude carboxylic acid 26e (916 mg, 1.94 mmol) dissolved in dry DMF (20 mL) was reacted with HBTU (894 mg, 2.36 mmol) in the presence of triethylamine (0.83 mL, 5.96 mmol) to yield 1.05 g (92%) of 27e as a colorless foam. = −39.7 (c 0.635, CHCl3); All spectroscopic data was consistent with the data described for its enantiomer 27d. HRMS (ESI) m/z calcd for C31H35N5O7Na+ 612.2429, found 612.2437.
tert-Butyl(3aS,4S,6aR)-4-{[2-({4-[3-(4-fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-4-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]di-oxolo[4,5-c]pyrrole-5-carboxylate (28e). Following the procedure outlined for the preparation of compound 28a isoxazole 30 (474 mg, 1.59 mmol) dissolved in dry DMF (14 mL) was deprotonated with sodium hydride (60% in mineral oil; 75 mg, 1.88 mmol) and compound 27e (500 mg, 0.848 mmol) was subsequently introduced in one portion at room temperature to yield 335 mg (53%) of 28e as a light-yellow resin. = −32.5 (c 0.76, acetone). All spectroscopic data of 28e was consistent with that reported for its enantiomer 28d. HRMS (ESI) m/z calcd for C42H46FN5O7Na+ 774.3273, found 774.3269. Anal. calcd for C42H46FN5O7: C, 67.10; H, 6.17; N, 9.31. Found C, 67.45; H, 6.53; N, 8.92.
(2S,3S,4R)-2-{[2-({4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-3-(4-methoxyphenyl)-1H-pyrrol-1-yl]methyl}-3,4-dihydroxypyrrolidin-1-ium trifluoroacetate (29e). Employing the same procedure as outlined for the preparation of 29c compound 28e (234 mg, 0.311 mmol) was treated with a mixture of TFA (6.8 mL) and water (0.76 mL) at 0 °C for 3 h and the crude product was purified by automated flash column chromatography (silica gel, methanol/dichloromethane 0–10%) to give 179 mg (79%) of 29e as a light-yellow resin. Subsequent recrystallization from ethyl acetate/petroleum ether provided 145 mg of pure 29e as a colorless solid. m.p. 188–191 °C (DSC); = +2.6 (c 0.53, MeOH). All spectroscopic data was consistent with the data reported for its enantiomer 29d. HRMS (ESI) m/z calcd for C34H34FN5O5H+ 612.2617, found 612.2620. Anal. calcd for C34H34FN5O5+·CF3COOH: C, 59.58; H, 4.86; N, 9.65. Found C, 59.71; H, 4.98; N, 9.68.
tert-Butyl(3aR,4R,6aS)-4-{[4-(2,4-dimethoxyphenyl)-2-(ethoxycarbonyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (25f). Employing the same procedure as for the preparation of compound 25c crude mesylate (−)-20 (2.58 g, 7.34 mmol) dissolved in dry DMF (50 mL) was reacted with pyrrole 24b (1.68 g, 6.10 mmol) and cesium carbonate (4.48 g, 13.8 mmol) to afford 1.86 g (57%) of 25f as a colorless oil that solidified on standing. = +13.2 (c 1.105, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.42–7.37 (m, 1H), 7.32 (d, 1.6 Hz, 0.4H), 7.25 (d, 1.7 Hz, 0.4H), 7.24 (d, 1.7 Hz, 0.6H), 7.21 (d, 1.7 Hz, 0.6H), 6.53–6.49 (m, 2H), 4.67 (dd, 13.9, 7.8 Hz, 0.4H), 4.64–4.57 (m, 2.2H), 4.55 (dd, 14.0, 4.7 Hz, 0.4H), 4.51 (t, 5.2 Hz, 0.4H), 4.47–4.41 (m, 1H), 4.32–4.24 (overlapping signals: 4.29, q, 7.0 Hz, 2H and m, 0.6H), 3.91 (d, 13.0 Hz, 0.6H), 3.86 (s, 1H), 3.85 (s, 2H), 3.82 (s, 3H), 3.66 (d, 13.2 Hz, 0.4H), 3.33 (dd, 13.1, 4.2 Hz, 0.6H), 3.25 (dd, 12.7, 5.3 Hz, 0.4H), 1.47 (s, 3H), 1.41 (s, 3H), 1.39–1.33 (overlapping signals: m, 3H and 1.34, s, 6H), 1.27 (s, 2H), 1.24 (s, 1H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 161.70, 161.52, 159.31, 157.18, 154.39, 154.14, 128.62, 128.35, 128.26, 121.67, 120.85, 120.60, 117.08, 116.84, 116.13, 111.75, 111.58, 104.73, 98.93, 82.42, 81.40, 80.03, 79.91, 79.30, 78.78, 64.61, 63.92, 59.94, 55.41, 55.37, 55.29, 52.39, 51.27, 47.47, 46.21, 28.44, 28.12, 27.06, 26.98, 25.08, 14.50; HRMS (ESI) m/z calcd for C28H38N2O8Na+ 553.2520, found 553.2529.
1-{[(3aR,4R,6aS)-5-(tert-Butoxycarbonyl)-2,2-dimethyltetrahydro-4H-[1,3]dioxolo[4,5-c]pyrrol-4-yl]methyl}-4-(2,4-dimethoxyphenyl)-1H-pyrrole-2-carboxylic acid (26f). Applying the same procedure as reported for the preparation of carboxylic acid 25a ethyl ester 25f (1.21 g, 2.28 mmol) dissolved in a 2:1 mixture of THF (12.5 mL) and methanol (6.5 mL) was treated with a 4 M aqueous sodium hydroxide solution (4.4 mL) at 50 °C overnight to obtain 1.05 g (92%) of 26f as a light-yellow solid. An analytical sample of 26f was obtained as colorless crystals by re-crystallization from ethyl acetate/petroleum ether. m.p. 180–181 °C (DSC); = +12.6 (c 0.840, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.43–7.37 (m, 2H), 7.27–7.25 (m, 1H), 6.54–6.50 (m, 2H), 4.71–4.51 (m, 3.4H), 4.51–4.44 (m, 1H), 4.25 (dd, 13.7, 7.8 Hz, 0.6H), 3.96 (d, 13.0 Hz, 0.6H), 3.87 (s, 1H), 3.86 (s, 2H), 3.83 (s, 3H), 3.70 (d, 13.1 Hz, 0.4H), 3.34 (dd, 13.0, 4.9 Hz, 0.6H), 3.25 (dd, 12.7, 5.2 Hz, 0.4H), 1.47 (s, 3H), 1.42 (s, 3H), 1.35 (s, 6H), 1.29 (s, 2H), 1.26 (s, 1H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 165.50, 165.32, 159.49, 157.21, 154.40, 154.20, 130.07, 129.81, 128.38, 128.32, 121.48, 121.22, 120.43, 119.26, 119.10, 115.78, 111.87, 111.71, 104.78, 98.97, 82.40, 81.41, 80.18, 80.09, 79.33, 78.82, 64.55, 63.89, 55.43, 55.38, 55.31, 52.40, 51.25, 47.67, 46.40, 28.43, 28.13, 27.05, 26.98, 25.11; HRMS (ESI) m/z calcd for C26H34N2O8Na+ 525.2207, found 525.2220. Anal. calcd for C26H34N2O8: C, 62.14; H, 6.82; N, 5.57. Found C, 62.13; H, 6.96; N, 5.48.
tert-Butyl(3aR,4R,6aS)-4-{[2-{[(1H-benzo[d][1,2,3]triazol-1-yl)oxy]carbonyl}-4-(2,4-dimethoxyphenyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo[4,5-c]pyrrole-5-carboxylate (27f). Deploying the same procedure as for the preparation of compound 27a carboxylic acid 26f (317 mg, 0.631 mmol) dissolved in dry DMF (6 mL) was reacted with HBTU (304 mg, 0.802 mmol) in the presence of triethylamine (0.3 mL, 2.15 mmol) to give 335 mg (86%) of 27f as a colorless resin. = +34.6 (c 0.735, CHCl3); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.11–8.06 (m, 1H), 7.78–7.75 (m, 1H), 7.65–7.49 (m, 3H), 7.47–7.41 (m, 2H), 6.58–6.54 (m, 2H), 4.61 (t, 5.0 Hz, 0.6H), 4.57 (dd, 12.3, 6.1 Hz, 0.4H), 4.54–4.39 (m, 3.4H), 4.27 (dd, 15.41, 10.4 Hz, 0.6H), 4.01 (d, 13.3 Hz, 0.6H), 3.91 (s, 1H), 3.90 (s, 2H), 3.85 (s, 3H), 3.72 (d, 12.8 Hz, 0.4H), 3.27–3.19 (overlapping signals: m, 0.4H and 3.24, dd, 13.2, 4.7 Hz, 0.6H), 1.45 (s, 3H), 1.41 (s, 1H), 1.40 (s, 2H), 1.31 (s, 6H), 1.26 (s, 3H); 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 159.91, 157.27, 157.10, 154.35, 154.22, 143.49, 132.53, 132.07, 129.23, 129.16, 128.67, 128.56, 128.52, 128.40, 124.70, 122.87, 122.60, 120.54, 120.49, 120.33, 120.26, 115.47, 115.40, 114.90, 114.86, 111.99, 111.92, 108.79, 108.48, 104.91, 98.99, 82.36, 81.36, 80.32, 80.19, 79.28, 78.90, 64.91, 63.78, 55.41, 55.33, 52.05, 50.92, 47.98, 46.84, 28.31, 28.01, 26.91, 26.83, 24.93; HRMS (ESI) m/z calcd for C32H37N5O8Na+ 642.2534, found 642.2530.
tert-Butyl(3aR,4R,6aS)-4-{[4-(2,4-dimethoxyphenyl)-2-({4-[3-(4-fluorophenyl)-5-isopropylisoxa-zol-4-yl]pyridin-2-yl}carbamoyl)-1H-pyrrol-1-yl]methyl}-2,2-dimethyltetrahydro-5H-[1,3]dioxolo [4,5-c]pyrrole-5-carboxylate (28f). Following the same method as described for the preparation of compound 28a isoxazole 30 (196 mg, 0.659 mmol) dissolved in dry DMF (5 mL) was initially deprotonated with sodium hydride (60% in mineral oil; 24.7 mg, 0.618 mmol) which was then followed by introducing dropwise a solution of compound 27f (230 mg, 0.371 mmol) in dry DMF (5 mL) via cannula to give 103 mg (36%) of 28f as a colorless foam. = +26.2 (c 0.775, acetone); 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 8.49 (sbr, 0.4H), 8.46 (sbr, 0.6H), 8.28-8.23 (m, 2H), 7.45 (AA’BB’X, JAB = 8.4, JAF = 5.5 Hz, 2H), 7.39–7.35 (m, 1H), 7.34 (s, 0.4H), 7.23 (s, 0.6H), 7.18–7.14 (m, 1H), 7.05 (AA’BB’X, JAB = JBF = 8.6 Hz, 2H), 6.74 (t, 4.5 Hz, 1H), 6.56–6.52 (m, 2H), 4.74–4.55 (m, 3H), 4.53–4.43 (m, 1.4H), 4.37 (dd, 13.4, 7.6 Hz, 0.6H), 3.92 (d, 13.1 Hz, 0.6H), 3.89 (s, 1H), 3.88 (s, 2H), 3.84 (s, 3H), 3.71 (d, 12.9 Hz, 0.4H), 3.36–3.22 (overlapping signals: 3.33, dd, 13.1, 4.9 Hz, 0.6H; m, 0.4H and 3.27, sp, 7.0 Hz, 1H), 1.45 (s, 3.5H), 1.43-1.39 (overlapping signals, 9H), 1.34 (s, 5.5H), 1.27 (s, 2H), 1.22 (s, 1H); 19F NMR (470 MHz, CDCl3, mixture of rotamers) δ −110.94, −111.00; 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 175.50, 164.60/162.62 (d, 247.8 Hz), 160.08, 159.81, 159.56, 157.25, 154.42, 154.17, 152.43, 152.38, 148.17, 141.11, 130.56/130.49 (d, 8.2 Hz), 128.40, 128.25, 128.20, 124.79, 124.78, 124.17, 124.13, 121.08, 120.91, 120.39, 115.90/115.73 (d, 21.7 Hz), 114.47, 112.98, 112.80, 112.20, 111.76, 111.60, 104.83, 99.05, 82.40, 81.44, 80.09, 79.92, 79.49, 78.87, 64.78, 63.90, 55.45, 55.34, 52.27, 51.26, 47.42, 46.26, 28.41, 28.15, 27.02, 26.98, 26.66, 25.09, 21.01; HRMS (ESI) m/z calcd for C43H48FN5O8Na+ 804.3379, found 804.3383.
(2R,3R,4S)-2-{[3-(2,4-Dimethoxyphenyl)-2-({4-[3-(4-fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}carbamoyl)-1H-pyrrol-1-yl]methyl}-3,4-dihydroxypyrrolidin-1-ium trifluoroacetate (29f). Compound 28f (110 mg, 0.141 mmol) was treated with a 9:1 mixture of trifluoroacetate (3.6 mL) and water (0.4 mL) at 0 °C for 2.5 h applying the same protocol as for the preparation of compound 29c to yield 68.4 mg (64%) of 29f as a light-yellow foam. = −1.14 (c 0.875, MeOH); 1H NMR (500 MHz, CD3OD) δ 8.37 (d, 3.3 Hz, 1H), 8.07 (s, 1H), 7.63 (d, 1.7 Hz, 1H), 7.52 (d, 1.8 Hz, 1H), 7.49 (d, 8.6 Hz, 1H), 7.46 (AA’BB’X, JAB = 8.9, JAF = 5.3 Hz, 2H), 7.14 (AA’BB’X, JAB = JBF = 8.8 Hz, 2H), 7.00 (d, 4.6 Hz, 1H), 6.61 (d, 2.4 Hz, 1H), 6.57 (dd, 8.5, 2.4 Hz, 1H), 4.72 (d, 6.3 Hz, 2H), 4.26 (td, 3.8, 1.6 Hz, 1H), 4.13 (dd, 8.8, 3.9 Hz, 1H), 3.96–3.88 (overlapping signals: m, 1H and 3.90, s, 3H), 3.82 (s, 3H), 3.49 (dd, 12.5, 3.9 Hz, 1H), 3.33–3.22 (overlapping signals: 3.29, sp, 7.0 Hz, 1H and 3.24, dd, 12.6, 1.7 Hz, 1H), 1.39 (d, 7.0 Hz, 6H); 19F NMR (470 MHz, CD3OD) δ −77.01, −112.77; 13C NMR (125 MHz, CD3OD) δ 177.05, 166.17/164.19 (d, 257.6 Hz), 163.13, 161.47, 161.33, 158.83, 153.53, 149.15, 142.83, 131.86/131.79 (d, 8.3 Hz), 130.25, 129.25, 126.14, 125.69, 123.30, 122.30, 117.22, 117.00, 116.83, 116.60, 116.15, 113.51, 106.38, 99.87, 74.81, 70.80, 63.74, 55.91, 51.11, 48.69, 27.98, 21.25; HRMS (ESI) m/z calcd for C35H36FN5O6H+ 642.2722, found 642.2735. Anal. calcd for C35H36FN5O6·2.3(CF3COOH): C, 52.62; H, 4.27; N, 7.75. Found C, 52.61; H, 4.44; N, 7.73.
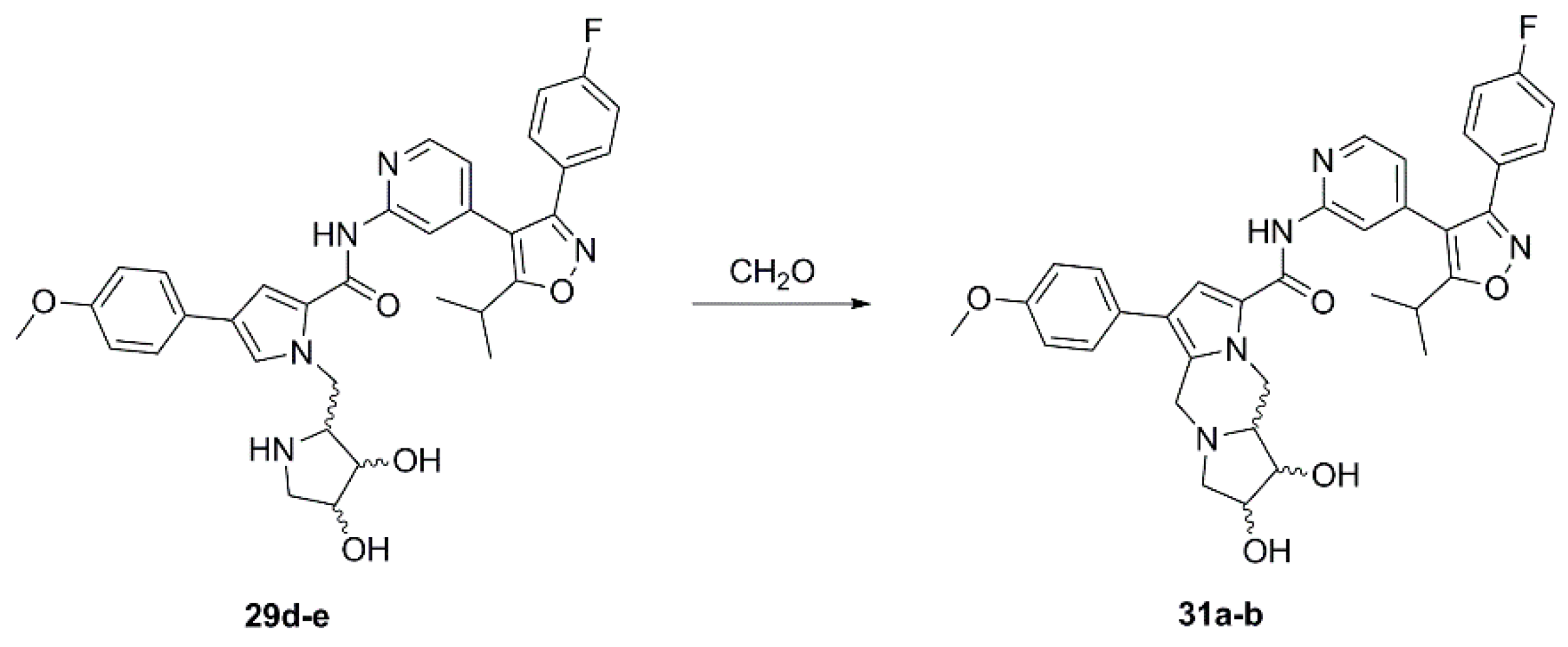
(1R,2S,10aR)-N-{4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}-1,2-dihydroxy-6-(4-methoxyphenyl)-2,3,10,10a-tetrahydro-1H,5H-dipyrrolo[1,2-a:1’,2’-d]pyrazine-8-carboxamide (31a). To a solution of 29d (20 mg, 0.043 mmol) in methanol (2 mL) were added 2 drops of formalin (formaldehyde solution, 37% in water). After being stirred at room temperature for 3 h the reaction was concentrated, and the crude product purified by automated column chromatography (silica gel, methanol/dichloromethane 0–10%) to afford 9 mg of 31a as a colorless solid. 1H NMR (400 MHz, d6-DMSO) δ 10.39 (s, 1H, NH), 8.36 (d, 4.6 Hz, 1H), 8.10 (s, 1H), 7.57 (s, 1H), 7.46 (AA’BB’X, JAB = 8.9, JAF = 5.5 Hz, 2H), 7.30 (AA’XX’, JAX = 8.7 Hz, 2H), 7.27 (AA’BB’X, JAB = JBF = 8.9 Hz, 2H), 6.98 (AA’XX’, JAX = 8.8 Hz, 2H), 6.89 (dd, 5.1, 1.5 Hz, 1H), 4.95–4.84 (overlapping signals: 4.93, dd, 13.0, 3.8 Hz, 1H; 4.93, sbr, 1H, OH), 4.84 (sbr, 1H, OH), 4.10 (d, 14.7 Hz, 1H), 4.09–4.03 (m, 1H), 3.80–3.74 (m, 1H; 3.77, s, 3H), 3.66 (m, 1H), 3.58 (d, 14.5 Hz, 1H), 3.47 (dd, 9.7, 6.4 Hz, 1H), 3.22 (sp, 7.0 Hz, 1H), 2.60–2.54 (m, 1H), 2.29–2.25 (m, 1H), 1.32 (d, 6.9 Hz, 6H); 19F NMR (376 MHz, d6-DMSO) δ −111.04; 13C NMR (125 MHz, d6-DMSO) δ 174.80, 164.0/161.6 (d, 247.3 Hz), 159.80, 159.58, 157.36, 152.97, 148.30, 139.42, 130.45/130.36 (d, 8.6 Hz), 130.1, 127.74, 127.23, 124.56/124.54 (d, 3.2 Hz), 122.83, 119.87, 118.34, 116.03/115.81 (d, 21.9 Hz), 114.49, 114.20, 114.08, 112.13, 73.99, 67.99, 63.13, 61.21, 55.00, 50.64 (br), 49.79 (br), 26.00, 20.60; HRMS (ESI) m/z calcd for C35H34FN5O5H+ 624.2617, found 624.2613.
(1S,2R,10aS)-N-{4-[3-(4-Fluorophenyl)-5-isopropylisoxazol-4-yl]pyridin-2-yl}-1,2-dihydroxy-6-(4-methoxyphenyl)-2,3,10,10a-tetrahydro-1H,5H-dipyrrolo[1,2-a:1’,2’-d]pyrazine-8-carboxamide (31b). Employing the same procedure as for the preparation of 31a compound 29e (31 mg, 0.043 mmol) was dissolved in methanol (2 mL) and treated with 2 drops of formalin (formaldehyde solution, 37% in water) at room temperature overnight to afford 30 mg of 31b as a light-yellow solid. All spectroscopic data of 31b was consistent with that recorded for 31a. HRMS (ESI) m/z calcd for C35H34FN5O5H+ 624.2617, found 624.2615.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












