Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System




Abstract
:1. Introduction
1.1. The Barrier and Beyond: Novel Aspects of Cutaneous (Patho)physiology
1.2. (Endo)cannabinoid Signaling and its most Important Interactions
1.3. Cannabinoids in the Skin: Brief Overview of the “c(ut)annabinoid” Signaling
2. Translational Potential of the Cutaneous Cannabinoid Signaling
2.1. Sebaceous Gland (SG)-Related Disorders: Acne and Skin Dryness
2.2. Hair Growth Disorders: Alopecia, Effluvium, Hirsutism, Hypertrichosis
2.3. Melanocytes & Pigmentation Disorders
2.4. Epidermal Keratinocytes
2.4.1. Proliferation and Differentiation
2.4.2. Barrier Formation
2.4.3. Keratin Disorders
2.5. Cutaneous Inflammation
2.5.1. General Considerations
2.5.2. Role of “Non-Classical” Cannabinoid Targets
2.5.3. Role of “Non-Classical” Cannabinoid Ligands
2.5.4. Putative ECS- Endogenous Opioid System (EOS) Interplay
2.5.5. Selected “Skin-Relevant” Professional Immune Cells: Langerhans Cells and Mast Cells (MC)
2.5.6. Selected Inflammatory Diseases: Psoriasis (PSO)
2.5.7. Selected Inflammatory Diseases: AD
2.5.8. Selected Inflammatory Diseases: Systemic Sclerosis (SSc)
2.6. Wound Healing
2.7. Itch
2.8. Skin Tumors
2.8.1. General Considerations
2.8.2. Melanoma
2.8.3. Non-Melanoma Skin Cancers
3. Challenges, Open Questions, Promising Future Directions
3.1. Potential Side Effects
3.2. Unidentified Players: Intercellular Transport, Cellular (Re-)uptake, Intracellular Trafficking
3.3. Identification of “Disease—Cannabinoid” Pairs
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Jensen, J.M.; Proksch, E. The skin’s barrier. G. Ital. Dermatol. Venereol. 2009, 144, 689–700. [Google Scholar] [PubMed]
- Proksch, E.; Brandner, J.M.; Jensen, J.-M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Szöllősi, A.G.; Bíró, T. The channel physiology of the skin. Rev. Physiol. Biochem. Pharmacol. 2012, 163, 65–131. [Google Scholar] [PubMed]
- Wilson, S.R.; Thé, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Boulais, N.; Misery, L. The epidermis: A sensory tissue. Eur. J. Dermatol. 2008, 18, 119–127. [Google Scholar] [PubMed]
- Chéret, J.; Bertolini, M.; Ponce, L.; Lehmann, J.; Tsai, T.; Alam, M.; Hatt, H.; Paus, R. Olfactory receptor OR2AT4 regulates human hair growth. Nat. Commun. 2018, 9, 3624. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Veitinger, S.; Peek, I.; Busse, D.; Eckardt, J.; Vladimirova, D.; Jovancevic, N.; Wojcik, S.; Gisselmann, G.; Altmüller, J.; et al. Two olfactory receptors-OR2A4/7 and OR51B5-differentially affect epidermal proliferation and differentiation. Exp. Dermatol. 2017, 26, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Toh, P.P.C.; Bigliardi-Qi, M.; Yap, A.M.Y.; Sriram, G.; Stelmashenko, O.; Bigliardi, P. Expression of peropsin in human skin is related to phototransduction of violet light in keratinocytes. Exp. Dermatol. 2016, 25, 1002–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscone, S.; Mardaryev, A.N.; Raafs, B.; Bikker, J.W.; Sticht, C.; Gretz, N.; Farjo, N.; Uzunbajakava, N.E.; Botchkareva, N.V. A new path in defining light parameters for hair growth: Discovery and modulation of photoreceptors in human hair follicle. Lasers Surg. Med. 2017, 49, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanukoglu, I.; Boggula, V.R.; Vaknine, H.; Sharma, S.; Kleyman, T.; Hanukoglu, A. Expression of epithelial sodium channel (ENaC) and CFTR in the human epidermis and epidermal appendages. Histochem. Cell Biol. 2017, 147, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Arantes, E.L.; Dragano, N.; Ramalho, A.; Vitorino, D.; de-Souza, G.F.; Lima, M.H.M.; Velloso, L.A.; Araújo, E.P. Topical Docosahexaenoic Acid (DHA) Accelerates Skin Wound Healing in Rats and Activates GPR120. Biol. Res. Nurs. 2016, 18, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Matsuoka, T.; Honda, T.; Kabashima, K.; Hirata, T.; Narumiya, S. A GPR40 agonist GW9508 suppresses CCL5, CCL17, and CXCL10 induction in keratinocytes and attenuates cutaneous immune inflammation. J. Investig. Dermatol. 2011, 131, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Brettmann, E.A.; de Guzman Strong, C. Recent evolution of the human skin barrier. Exp. Dermatol. 2018, 27, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Ishida-Yamamoto, A.; Igawa, S.; Kishibe, M. Molecular basis of the skin barrier structures revealed by electron microscopy. Exp. Dermatol. 2018, 27, 841–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokouchi, M.; Kubo, A. Maintenance of tight junction barrier integrity in cell turnover and skin diseases. Exp. Dermatol. 2018, 27, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.; Herman, A.P. Antimicrobial peptides activity in the skin. Skin Res. Technol. 2018, 00, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.A.; Sánchez-Borges, M.; Ansotegui, I.J.; Ebisawa, M.; Ortega Martell, J.A. Skin as an immune organ and clinical applications of skin-based immunotherapy. World Allergy Organ. J. 2018, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Denda, M.; Nakatani, M.; Ikeyama, K.; Tsutsumi, M.; Denda, S. Epidermal keratinocytes as the forefront of the sensory system. Exp. Dermatol. 2007, 16, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.; Kabashima, K. Novel insights into the role of immune cells in skin and inducible skin-associated lymphoid tissue (iSALT). Allergo J. Int. 2015, 24, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal control of skin function: The skin as a neuroimmunoendocrine organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Armstrong, C.A.; Bunnett, N.W.; Luger, T.A.; Olerud, J.E.; Ansel, J.C. Neuropeptides in the skin: Interactions between the neuroendocrine and the skin immune systems. Exp. Dermatol. 1998, 7, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A. Neuroendocrine system of the skin. Dermatology 2005, 211, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–115. [Google Scholar]
- Elphick, M.R.; Egertová, M. The phylogenetic distribution and evolutionary origins of endocannabinoid signalling. In Cannabinoids; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 283–297. [Google Scholar]
- McPartland, J.M.; Matias, I.; Di Marzo, V.; Glass, M. Evolutionary origins of the endocannabinoid system. Gene 2006, 370, 64–74. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M.; Norris, R.W.; Kilpatrick, C.W. Coevolution between cannabinoid receptors and endocannabinoid ligands. Gene 2007, 397, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Pacioni, G.; Rapino, C.; Zarivi, O.; Falconi, A.; Leonardi, M.; Battista, N.; Colafarina, S.; Sergi, M.; Bonfigli, A.; Miranda, M.; et al. Truffles contain endocannabinoid metabolic enzymes and anandamide. Phytochemistry 2015, 110, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Gertsch, J. Cannabimimetic phytochemicals in the diet—An evolutionary link to food selection and metabolic stress adaptation? Br. J. Pharmacol. 2017, 174, 1464–1483. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Szekanecz, Z.; Bíró, T. Targeting Cannabinoid Signaling in the Immune System: “High”-ly Exciting Questions, Possibilities, and Challenges. Front. Immunol. 2017, 8, 1487. [Google Scholar] [CrossRef] [PubMed]
- Solymosi, K.; Köfalvi, A. Cannabis: A Treasure Trove or Pandora’s Box? Mini Rev. Med. Chem. 2017, 17, 1223–1291. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Păunescu, H.; Coman, O.A.; Coman, L.; Ghiţă, I.; Georgescu, S.R.; Drăghia, F.; Fulga, I. Cannabinoid system and cyclooxygenases inhibitors. J. Med. Life 2011, 4, 11–20. [Google Scholar] [PubMed]
- Chicca, A.; Marazzi, J.; Nicolussi, S.; Gertsch, J. Evidence for bidirectional endocannabinoid transport across cell membranes. J. Biol. Chem. 2012, 287, 34660–34682. [Google Scholar] [CrossRef] [PubMed]
- Abood, M.E.; Sorensen, R.G.; Stella, N. (Eds.) endoCANNABINOIDS: Actions at Non-CB1/CB2 Cannabinoid Receptors; The Receptors; Springer: New York, NY, USA, 2013; ISBN 978-1-4614-4668-2. [Google Scholar]
- Kaczocha, M.; Rebecchi, M.J.; Ralph, B.P.; Teng, Y.-H.G.; Berger, W.T.; Galbavy, W.; Elmes, M.W.; Glaser, S.T.; Wang, L.; Rizzo, R.C.; et al. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS ONE 2014, 9, e94200. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Piscitelli, F. The Endocannabinoid System and its Modulation by Phytocannabinoids. Neurotherapeutics 2015, 12, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chicca, A.; Nicolussi, S.; Bartholomäus, R.; Blunder, M.; Aparisi Rey, A.; Petrucci, V.; Reynoso-Moreno, I.D.C.; Viveros-Paredes, J.M.; Dalghi Gens, M.; Lutz, B.; et al. Chemical probes to potently and selectively inhibit endocannabinoid cellular reuptake. Proc. Natl. Acad. Sci. USA 2017, 114, E5006–E5015. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Metabolism of the Endocannabinoid Anandamide: Open Questions after 25 Years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, G.; Offertáler, L.; Wagner, J.A.; Kunos, G. Receptors for acylethanolamides-GPR55 and GPR119. Prostaglandins Other Lipid Mediat. 2009, 89, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J. TRP channel cannabinoid receptors in skin sensation, homeostasis, and inflammation. ACS Chem. Neurosci. 2014, 5, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Chicca, A.; Schafroth, M.A.; Reynoso-Moreno, I.; Erni, R.; Petrucci, V.; Carreira, E.M.; Gertsch, J. Uncovering the psychoactivity of a cannabinoid from liverworts associated with a legal high. Sci. Adv. 2018, 4, eaat2166. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.-Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.Q.; Mikkelsen, T.S.; McKinney, M.K.; Lander, E.S.; Cravatt, B.F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006, 281, 36569–36578. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Premoli, M.; Ferrari-Toninelli, G.; Tambaro, S.; Maccarinelli, G.; Memo, M.; Bonini, S.A. Cannabinoids in health and disease: Pharmacological potential in metabolic syndrome and neuroinflammation. Horm. Mol. Biol. Clin. Investig. 2018, 36. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Wang, J. The Endocannabinoidome; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12-420126-2. [Google Scholar]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Pistis, M.; O’Sullivan, S.E. The Role of Nuclear Hormone Receptors in Cannabinoid Function. Adv. Pharmacol. 2017, 80, 291–328. [Google Scholar] [PubMed]
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Orlando, P.; Moriello, A.S.; Aviello, G.; Stott, C.; Izzo, A.A.; Di Marzo, V. Cannabinoid actions at TRPV channels: Effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol. 2012, 204, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: Potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Maldonado, R.; Casas, M.; Henze, T.; Centonze, D. Cannabinoids therapeutic use: What is our current understanding following the introduction of THC, THC:CBD oromucosal spray and others? Expert Rev. Clin. Pharmacol. 2017, 10, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M.; Rapino, C.; Di Francesco, A.; Dainese, E.; D’Addario, C.; Maccarrone, M. Epigenetic control of skin differentiation genes by phytocannabinoids. Br. J. Pharmacol. 2013, 170, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.S.; Kim, Y.-J.; Kim, M.O.; Kang, M.; Oh, S.W.; Nho, Y.H.; Park, S.-H.; Lee, J. Cannabidiol upregulates melanogenesis through CB1 dependent pathway by activating p38 MAPK and p42/44 MAPK. Chem. Biol. Interact. 2017, 273, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirrs, S.; van Karnebeek, C.D.M.; Peng, X.; Shyr, C.; Tarailo-Graovac, M.; Mandal, R.; Testa, D.; Dubin, D.; Carbonetti, G.; Glynn, S.E.; et al. Defects in fatty acid amide hydrolase 2 in a male with neurologic and psychiatric symptoms. Orphanet J. Rare Dis. 2015, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Denovan-Wright, E.M. Cannabinoid receptor ligand bias: Implications in the central nervous system. Curr. Opin. Pharmacol. 2017, 32, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Goya, P.; Jagerovic, N. Emerging strategies targeting CB2 cannabinoid receptor: Biased agonism and allosterism. Biochem. Pharmacol. 2018, 157, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Priestley, R.; Glass, M.; Kendall, D. Functional Selectivity at Cannabinoid Receptors. Adv. Pharmacol. 2017, 80, 207–221. [Google Scholar] [PubMed]
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; de Petrocellis, L.; Ullmer, C.; Rothenhäusler, B.; Perret, C.; van Gils, N.; et al. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. [Google Scholar] [CrossRef] [PubMed]
- Nogueras-Ortiz, C.; Yudowski, G.A. The Multiple Waves of Cannabinoid 1 Receptor Signaling. Mol. Pharmacol. 2016, 90, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Ford, B.M.; Franks, L.N.; Tai, S.; Fantegrossi, W.E.; Stahl, E.L.; Berquist, M.D.; Cabanlong, C.V.; Wilson, C.D.; Penthala, N.R.; Crooks, P.A.; et al. Characterization of structurally novel G protein biased CB1 agonists: Implications for drug development. Pharmacol. Res. 2017, 125, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, S.; Janero, D.R.; Zvonok, N.; Makriyannis, A. Functional selectivity at G-protein coupled receptors: Advancing cannabinoid receptors as drug targets. Biochem. Pharmacol. 2017, 128, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassing, H.A.; Fares, S.; Larsen, O.; Pad, H.; Hauge, M.; Jones, R.M.; Schwartz, T.W.; Hansen, H.S.; Rosenkilde, M.M. Biased signaling of lipids and allosteric actions of synthetic molecules for GPR119. Biochem. Pharmacol. 2016, 119, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, R.; Bushlin, I.; Gomes, I.; Tzavaras, N.; Gupta, A.; Neves, S.; Battini, L.; Gusella, G.L.; Lachmann, A.; Ma’ayan, A.; et al. Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS ONE 2012, 7, e29239. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Natale, M.; Gianchecchi, E.; Capecchi, P.L.; Montilli, C.; Zimbone, S.; Castrichini, M.; Balistreri, E.; Ricci, G.; Selvi, E.; et al. Adenosine A2A receptor activation stimulates collagen production in sclerodermic dermal fibroblasts either directly and through a cross-talk with the cannabinoid system. J. Mol. Med. 2012, 90, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Bagher, A.M.; Laprairie, R.B.; Toguri, J.T.; Kelly, M.E.M.; Denovan-Wright, E.M. Bidirectional allosteric interactions between cannabinoid receptor 1 (CB1) and dopamine receptor 2 long (D2L) heterotetramers. Eur. J. Pharmacol. 2017, 813, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Milligan, G. Heteromultimerization of cannabinoid CB(1) receptor and orexin OX(1) receptor generates a unique complex in which both protomers are regulated by orexin A. J. Biol. Chem. 2011, 286, 37414–37428. [Google Scholar] [CrossRef] [PubMed]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef] [PubMed]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; Rooney, N.; Feeney, M.; Tate, J.; Robertson, D.; Welham, M.; Ward, S. Differential expression of cannabinoid receptors in the human colon: Cannabinoids promote epithelial wound healing. Gastroenterology 2005, 129, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, G.C.; Oprea, T.I.; Zhao, P.; Abood, M.E.; Brailoiu, E. Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J. Biol. Chem. 2011, 286, 29166–29174. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.; Battista, N.; Fezza, F.; Finazzi-Agrò, A.; Maccarrone, M. Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J. Biol. Chem. 2005, 280, 12212–12220. [Google Scholar] [CrossRef] [PubMed]
- Dudok, B.; Barna, L.; Ledri, M.; Szabó, S.I.; Szabadits, E.; Pintér, B.; Woodhams, S.G.; Henstridge, C.M.; Balla, G.Y.; Nyilas, R.; et al. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat. Neurosci. 2015, 18, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Cevikbas, F.; Pasolli, H.A.; Chen, Y.; Kong, W.; Kempkes, C.; Parekh, P.; Lee, S.H.; Kontchou, N.-A.; Yeh, I.; et al. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E3225–E3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Bode, A.M.; Zhao, Q.; Cho, Y.-Y.; Zhu, F.; Ma, W.-Y.; Dong, Z. The cannabinoid receptors are required for ultraviolet-induced inflammation and skin cancer development. Cancer Res. 2008, 68, 3992–3998. [Google Scholar] [CrossRef] [PubMed]
- Inci, R.; Kelekci, K.H.; Oguz, N.; Karaca, S.; Karadas, B.; Bayrakci, A. Dermatological aspects of synthetic cannabinoid addiction. Cutan. Ocul. Toxicol. 2017, 36, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Río, C.D.; Millán, E.; García, V.; Appendino, G.; DeMesa, J.; Muñoz, E. The endocannabinoid system of the skin. A potential approach for the treatment of skin disorders. Biochem. Pharmacol. 2018, 157, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Eagleston, L.R.M.; Kalani, N.K.; Patel, R.R.; Flaten, H.K.; Dunnick, C.A.; Dellavalle, R.P. Cannabinoids in dermatology: A scoping review. Dermatol. Online J. 2018, 24, 1. [Google Scholar]
- Bíró, T.; Tóth, B.I.; Haskó, G.; Paus, R.; Pacher, P. The endocannabinoid system of the skin in health and disease: Novel perspectives and therapeutic opportunities. Trends Pharmacol. Sci. 2009, 30, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Kupczyk, P.; Reich, A.; Szepietowski, J.C. Cannabinoid system in the skin—A possible target for future therapies in dermatology. Exp. Dermatol. 2009, 18, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Bíró, T. Targeting Cutaneous Cannabinoid Signaling in Inflammation—A “High”-way to Heal? EBioMedicine 2017, 16, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Pang, Z. TRP Channels in Skin Biology and Pathophysiology. Pharmaceuticals 2016, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.I.; Oláh, A.; Szöllősi, A.G.; Bíró, T. TRP channels in the skin. Br. J. Pharmacol. 2014, 171, 2568–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Kirchhof, M.G. Dermatology-Related Uses of Medical Cannabis Promoted by Dispensaries in Canada, Europe, and the United States. J. Cutan. Med. Surg. 2018, 1203475418808761. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.H.; Friedman, A. The Therapeutic Potential of Cannabinoids in Dermatology. Skin Ther. Lett. 2018, 23, 1–5. [Google Scholar]
- Liszewski, W.; Farah, R.S. Response to: “The role of cannabinoids in dermatology”. J. Am. Acad. Dermatol. 2017, 77, e87–e88. [Google Scholar] [CrossRef] [PubMed]
- Mounessa, J.S.; Siegel, J.A.; Dunnick, C.A.; Dellavalle, R.P. The role of cannabinoids in dermatology. J. Am. Acad. Dermatol. 2017, 77, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Milando, R.; Friedman, A. Cannabinoids: Potential Role in Inflammatory and Neoplastic Skin Diseases. Am. J. Clin. Dermatol. 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M.; Pirazzi, V.; Pasquariello, N.; Maccarrone, M. Endocannabinoid signaling and epidermal differentiation. Eur. J. Dermatol. 2011, 21 (Suppl. 2), 29–34. [Google Scholar]
- Pappas, A. Epidermal surface lipids. Dermato-Endocrinology 2009, 1, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, V.Y.; Leo, M.; Hassoun, L.; Chahal, D.S.; Maibach, H.I.; Sivamani, R.K. Role of sebaceous glands in inflammatory dermatoses. J. Am. Acad. Dermatol. 2015, 73, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Surber, C.; Abels, C.; Mailbach, H. pH of the Skin: Issues and Challenges|Karger Book; Current Problems in Dermatology; Karger AG: Basel, Switzerland, 2018; ISBN 978-3-318-06384-4. [Google Scholar]
- Zouboulis, C.C.; Katsambas, A.; Kligman, A.M. (Eds.) Pathogenesis and Treatment of Acne and Rosacea; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-540-69374-1. [Google Scholar]
- Zouboulis, C.C.; Picardo, M.; Ju, Q.; Kurokawa, I.; Törőcsik, D.; Bíró, T.; Schneider, M.R. Beyond acne: Current aspects of sebaceous gland biology and function. Rev. Endocr. Metab. Disord. 2016, 17, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouboulis, C.C.; Baron, J.M.; Böhm, M.; Kippenberger, S.; Kurzen, H.; Reichrath, J.; Thielitz, A. Frontiers in sebaceous gland biology and pathology. Exp. Dermatol. 2008, 17, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szöllősi, A.G.; Oláh, A.; Bíró, T.; Tóth, B.I. Recent advances in the endocrinology of the sebaceous gland. Dermatoendocrinology 2017, 9, e1361576. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.I.; Oláh, A.; Szöllosi, A.G.; Czifra, G.; Bíró, T. “Sebocytes” makeup: “Novel mechanisms and concepts in the physiology of the human sebaceous glands”. Pflug. Arch. 2011, 461, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Jourdan, E.; Picardo, M. Acne is an inflammatory disease and alterations of sebum composition initiate acne lesions. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Schmelz, M.; Metze, D.; Luger, T.; Rukwied, R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J. Dermatol. Sci. 2005, 38, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Dobrosi, N.; Tóth, B.I.; Nagy, G.; Dózsa, A.; Géczy, T.; Nagy, L.; Zouboulis, C.C.; Paus, R.; Kovács, L.; Bíró, T. Endocannabinoids enhance lipid synthesis and apoptosis of human sebocytes via cannabinoid receptor-2-mediated signaling. FASEB J. 2008, 22, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Seltmann, H.; Neitzel, H.; Orfanos, C.E. Establishment and characterization of an immortalized human sebaceous gland cell line (SZ95). J. Investig. Dermatol. 1999, 113, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Zákány, N.; Oláh, A.; Markovics, A.; Takács, E.; Aranyász, A.; Nicolussi, S.; Piscitelli, F.; Allarà, M.; Pór, Á.; Kovács, I.; et al. Endocannabinoid Tone Regulates Human Sebocyte Biology. J. Investig. Dermatol. 2018, 138, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Czifra, G.; Szöllősi, A.G.; Tóth, B.I.; Demaude, J.; Bouez, C.; Breton, L.; Bíró, T. Endocannabinoids regulate growth and survival of human eccrine sweat gland-derived epithelial cells. J. Investig. Dermatol. 2012, 132, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.F.; Markovics, A.; Angyal, Á.; Magi, J.; Pór, Á.; Kovács, I.; Zouboulis, C.C.; Bíró, T.; Oláh, A. 1321 Endocannabinoid-like molecule oleoylethanolamide promotes sebaceous lipid synthesis. J. Investig. Dermatol. 2018, 138, S224. [Google Scholar] [CrossRef]
- Yang, J.W.; Kim, H.S.; Choi, Y.-W.; Kim, Y.-M.; Kang, K.W. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes. Metab. 2018, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.I.; Géczy, T.; Griger, Z.; Dózsa, A.; Seltmann, H.; Kovács, L.; Nagy, L.; Zouboulis, C.C.; Paus, R.; Bíró, T. Transient receptor potential vanilloid-1 signaling as a regulator of human sebocyte biology. J. Investig. Dermatol. 2009, 129, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Szántó, M.; Oláh, A.; Szöllősi, A.G.; Tóth, K.F.; Páyer, E.; Czakó, N.; Pór, Á.; Kovács, I.; Zouboulis, C.C.; Kemény, L.; et al. Activation of TRPV3 inhibits lipogenesis and stimulates production of inflammatory mediators in human sebocytes—A putative contributor to dry skin dermatoses. J. Investig. Dermatol. 2019, 139, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Tóth, B.I.; Borbíró, I.; Sugawara, K.; Szöllõsi, A.G.; Czifra, G.; Pál, B.; Ambrus, L.; Kloepper, J.; Camera, E.; et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J. Clin. Investig. 2014, 124, 3713–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szöllősi, A.G.; Vasas, N.; Angyal, Á.; Kistamás, K.; Nánási, P.P.; Mihály, J.; Béke, G.; Herczeg-Lisztes, E.; Szegedi, A.; Kawada, N.; et al. Activation of TRPV3 Regulates Inflammatory Actions of Human Epidermal Keratinocytes. J. Investig. Dermatol. 2018, 138, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, J.D.; Williamson, E.M. Cannabinoids inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism and have a potential therapeutic value in the treatment of psoriasis. J. Dermatol. Sci. 2007, 45, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial cannabinoids from Cannabis sativa: A structure-activity study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Akhtar, N. The safety and efficacy of 3% Cannabis seeds extract cream for reduction of human cheek skin sebum and erythema content. Pak. J. Pharm. Sci. 2015, 28, 1389–1395. [Google Scholar] [PubMed]
- Spleman, L.; Sinclair, R.; Freeman, M.; Davis, M.; Gebauer, K. 1061 The safety of topical cannabidiol (CBD) for the treatment of acne. J. Investig. Dermatol. 2018, 138, S180. [Google Scholar] [CrossRef]
- Oláh, A.; Markovics, A.; Szabó-Papp, J.; Szabó, P.T.; Stott, C.; Zouboulis, C.C.; Bíró, T. Differential effectiveness of selected non-psychotropic phytocannabinoids on human sebocyte functions implicates their introduction in dry/seborrhoeic skin and acne treatment. Exp. Dermatol. 2016, 25, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Bulfone-Paus, S.; Bertolini, M. Hair Follicle Immune Privilege Revisited: The Key to Alopecia Areata Management. J. Investig. Dermatol. Symp. Proc. 2018, 19, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Azzawi, S.; Penzi, L.R.; Senna, M.M. Immune Privilege Collapse and Alopecia Development: Is Stress a Factor. Skin Appendage Disord. 2018, 4, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.W.; Kloepper, J.; Langan, E.A.; Kim, Y.; Yeo, J.; Kim, M.J.; Hsi, T.-C.; Rose, C.; Yoon, G.S.; Lee, S.-J.; et al. A Guide to Studying Human Hair Follicle Cycling In Vivo. J. Investig. Dermatol. 2016, 136, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telek, A.; Bíró, T.; Bodó, E.; Tóth, B.I.; Borbíró, I.; Kunos, G.; Paus, R. Inhibition of human hair follicle growth by endo- and exocannabinoids. FASEB J. 2007, 21, 3534–3541. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-L.; Yu, T.-S.; Li, X.-N.; Fan, Y.-Y.; Ma, W.-X.; Du, Y.; Zhao, R.; Guan, D.-W. Cannabinoid receptor type 2 is time-dependently expressed during skin wound healing in mice. Int. J. Leg. Med. 2012, 126, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Mercati, F.; Dall’Aglio, C.; Pascucci, L.; Boiti, C.; Ceccarelli, P. Identification of cannabinoid type 1 receptor in dog hair follicles. Acta Histochem. 2012, 114, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.L.; Blázquez, C.; Martínez-Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, B.K.; Soni, R.; Patel, J.Z.; Joharapurkar, A.; Sadhwani, N.; Kshirsagar, S.; Mishra, B.; Takale, V.; Gupta, S.; Pandya, P.; et al. Hair growth stimulator property of thienyl substituted pyrazole carboxamide derivatives as a CB1 receptor antagonist with in vivo antiobesity effect. Bioorg. Med. Chem. Lett. 2009, 19, 2546–2550. [Google Scholar] [CrossRef] [PubMed]
- Bíró, T.; Bodó, E.; Telek, A.; Géczy, T.; Tychsen, B.; Kovács, L.; Paus, R. Hair cycle control by vanilloid receptor-1 (TRPV1): Evidence from TRPV1 knockout mice. J. Investig. Dermatol. 2006, 126, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Bodó, E.; Bíró, T.; Telek, A.; Czifra, G.; Griger, Z.; Tóth, B.I.; Mescalchin, A.; Ito, T.; Bettermann, A.; Kovács, L.; et al. A hot new twist to hair biology: Involvement of vanilloid receptor-1 (VR1/TRPV1) signaling in human hair growth control. Am. J. Pathol. 2005, 166, 985–998. [Google Scholar] [CrossRef]
- Borbíró, I.; Lisztes, E.; Tóth, B.I.; Czifra, G.; Oláh, A.; Szöllosi, A.G.; Szentandrássy, N.; Nánási, P.P.; Péter, Z.; Paus, R.; et al. Activation of transient receptor potential vanilloid-3 inhibits human hair growth. J. Investig. Dermatol. 2011, 131, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.L.; Herczeg-Lisztes, E.; Szegedi, A.; Nemes, B.; Paus, R.; Bíró, T.; Szöllősi, A.G. Transient Receptor Potential Vanilloid 4 is Expressed in Human Hair Follicles and Inhibits Hair Growth in Vitro. J. Investig. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.L.; Herczeg-Lisztes, E.; Szollosi, A.G.; Szegedi, A.; Bíró, T.; Oláh, A. 263 (-)-cannabidiol differentially influences hair growth. J. Investig. Dermatol. 2017, 137, S238. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V. Endocannabinoids and Immunity. Cannabis Cannabinoid Res. 2016, 1, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Karsak, M.; Gaffal, E.; Date, R.; Wang-Eckhardt, L.; Rehnelt, J.; Petrosino, S.; Starowicz, K.; Steuder, R.; Schlicker, E.; Cravatt, B.; et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science 2007, 316, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Oláh, A.; Ambrus, L.; Nicolussi, S.; Gertsch, J.; Tubak, V.; Kemény, L.; Soeberdt, M.; Abels, C.; Bíró, T. Inhibition of fatty acid amide hydrolase exerts cutaneous anti-inflammatory effects both in vitro and in vivo. Exp. Dermatol. 2016, 25, 328–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid Delivery Systems for Pain and Inflammation Treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Santander, M.; Sánchez-Domínguez, C.; Cantú-Salinas, C.; Gonzalez-Cárdenas, H.; Cepeda-Nieto, A.C.; Cerda-Flores, R.M.; Ortiz-López, R.; Ocampo-Candiani, J. Association between PTPN22 C1858T polymorphism and alopecia areata risk. Exp. Ther. Med. 2015, 10, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moravvej, H.; Tabatabaei-Panah, P.-S.; Abgoon, R.; Khaksar, L.; Sokhandan, M.; Tarshaei, S.; Ghaderian, S.M.H.; Ludwig, R.J.; Akbarzadeh, R. Genetic variant association of PTPN22, CTLA4, IL2RA, as well as HLA frequencies in susceptibility to alopecia areata. Immunol. Investig. 2018, 47, 666–679. [Google Scholar]
- Bhanusali, D.G.; Sachdev, A.; Olson, M.A.; Gerlach, J.A.; Sinha, A.A. PTPN22 profile indicates a novel risk group in Alopecia areata. Hum. Immunol. 2014, 75, 81–87. [Google Scholar] [CrossRef] [PubMed]
- El-Zawahry, B.M.; Azzam, O.A.; Zaki, N.S.; Abdel-Raheem, H.M.; Bassiouny, D.A.; Khorshied, M.M. PTPN22 gene polymorphism in Egyptian alopecia areata patients and its impact on response to diphencyprone immunotherapy. Gene 2013, 523, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Alzolibani, A.A.; Zari, S.; Ahmed, A.A. Epidemiologic and genetic characteristics of alopecia areata (part 2). Acta Dermatovenerol. APA 2012, 21, 15–19. [Google Scholar]
- Betz, R.C.; König, K.; Flaquer, A.; Redler, S.; Eigelshoven, S.; Kortüm, A.-K.; Hanneken, S.; Hillmer, A.; Tüting, T.; Lambert, J.; et al. The R620W polymorphism in PTPN22 confers general susceptibility for the development of alopecia areata. Br. J. Dermatol. 2008, 158, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kemp, E.H.; McDonagh, A.J.G.; Wengraf, D.A.; Messenger, A.G.; Gawkrodger, D.J.; Cork, M.J.; Tazi-Ahnini, R. The non-synonymous C1858T substitution in the PTPN22 gene is associated with susceptibility to the severe forms of alopecia areata. Hum. Immunol. 2006, 67, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Pike, K.A.; Tremblay, M.L. Protein Tyrosine Phosphatases: Regulators of CD4 T Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2504. [Google Scholar] [CrossRef] [PubMed]
- International Association for Cannabis as Medicine. Available online: https://www.cannabis-med.org/english/bulletin/ww_en_db_cannabis_artikel.php?id=460 (accessed on 4 January 2019).
- Pucci, M.; Pasquariello, N.; Battista, N.; Di Tommaso, M.; Rapino, C.; Fezza, F.; Zuccolo, M.; Jourdain, R.; Finazzi Agrò, A.; Breton, L.; et al. Endocannabinoids stimulate human melanogenesis via type-1 cannabinoid receptor. J. Biol. Chem. 2012, 287, 15466–15478. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.A.; Jacobs, S.E.; Pentland, A.P. sPLA2-X stimulates cutaneous melanocyte dendricity and pigmentation through a lysophosphatidylcholine-dependent mechanism. J. Investig. Dermatol. 2006, 126, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Funakoshi, T.; Mori, M.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-H.; Huang, Y.-C.; Tsai, M.-L.; Cheng, C.-Y.; Liu, L.-L.; Yen, Y.-W.; Chen, W.-L. Inhibition of melanogenesis by β-caryophyllene from lime mint essential oil in mouse B16 melanoma cells. Int. J. Cosmet. Sci. 2015, 37, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magina, S.; Esteves-Pinto, C.; Moura, E.; Serrão, M.P.; Moura, D.; Petrosino, S.; Di Marzo, V.; Vieira-Coelho, M.A. Inhibition of basal and ultraviolet B-induced melanogenesis by cannabinoid CB(1) receptors: A keratinocyte-dependent effect. Arch. Dermatol. Res. 2011, 303, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ren, T.; Li, Y.; Cheng, A.; Xie, W.; Xu, L.; Peng, L.; Lin, J.; Lian, L.; Diao, Y.; et al. Oleoylethanolamide inhibits α-melanocyte stimulating hormone-stimulated melanogenesis via ERK, Akt and CREB signaling pathways in B16 melanoma cells. Oncotarget 2017, 8, 56868–56879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastonini, E.; Bellei, B.; Filoni, A.; Kovacs, D.; Iacovelli, P.; Picardo, M. Involvement of non melanocytic skin cells in vitiligo. Exp. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Delmas, V.; Larue, L. Molecular and cellular basis of depigmentation in vitiligo patients. Exp. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Melendez, M.E.; Salinas-Santander, M.; Sanchez-Dominguez, C.; Gonzalez-Cardenas, H.; Cerda-Flores, R.M.; Ocampo-Candiani, J.; Ortiz-López, R. Protein tyrosine phosphatase PTPN22 +1858C/T polymorphism is associated with active vitiligo. Exp. Ther. Med. 2014, 8, 1433–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBerge, G.S.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. PTPN22 is genetically associated with risk of generalized vitiligo, but CTLA4 is not. J. Investig. Dermatol. 2008, 128, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Laberge, G.S.; Birlea, S.A.; Fain, P.R.; Spritz, R.A. The PTPN22-1858C>T (R620W) functional polymorphism is associated with generalized vitiligo in the Romanian population. Pigment Cell Melanoma Res. 2008, 21, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran, K.S.; Rajappa, M.; Chandrashekar, L.; Thappa, D.M. Association of PTPN22 gene polymorphism with non-segmental vitiligo in South Indian Tamils. Postep. Dermatol. Alergol. 2018, 35, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Akbas, H.; Dertlioglu, S.B.; Dilmec, F.; Atay, A.E. Lack of Association between PTPN22 Gene +1858 C>T Polymorphism and Susceptibility to Generalized Vitiligo in a Turkish Population. Ann. Dermatol. 2014, 26, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, A.; Qarqaz, F.; Al-Sabah, J.; Al Rashaideh, T. Clinical characteristics and PTPN22 1858C/T variant analysis in Jordanian Arab vitiligo patients. Mol. Diagn. Ther. 2010, 14, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Di Rienzo, M.; Battista, N.; Gasperi, V.; Guerrieri, P.; Rossi, A.; Finazzi-Agrò, A. The endocannabinoid system in human keratinocytes. Evidence that anandamide inhibits epidermal differentiation through CB1 receptor-dependent inhibition of protein kinase C, activation protein-1, and transglutaminase. J. Biol. Chem. 2003, 278, 33896–33903. [Google Scholar] [CrossRef] [PubMed]
- Oddi, S.; Bari, M.; Battista, N.; Barsacchi, D.; Cozzani, I.; Maccarrone, M. Confocal microscopy and biochemical analysis reveal spatial and functional separation between anandamide uptake and hydrolysis in human keratinocytes. Cell. Mol. Life Sci. 2005, 62, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, A.; Pasquariello, N.; Barcaroli, D.; Maccarrone, M. Anandamide regulates keratinocyte differentiation by inducing DNA methylation in a CB1 receptor-dependent manner. J. Biol. Chem. 2008, 283, 6005–6012. [Google Scholar] [CrossRef] [PubMed]
- Pasquariello, N.; Oddi, S.; Malaponti, M.; Maccarrone, M. Regulation of gene transcription and keratinocyte differentiation by anandamide. Vitam. Horm. 2009, 81, 441–467. [Google Scholar] [PubMed]
- Tóth, B.I.; Dobrosi, N.; Dajnoki, A.; Czifra, G.; Oláh, A.; Szöllosi, A.G.; Juhász, I.; Sugawara, K.; Paus, R.; Bíró, T. Endocannabinoids modulate human epidermal keratinocyte proliferation and survival via the sequential engagement of cannabinoid receptor-1 and transient receptor potential vanilloid-1. J. Investig. Dermatol. 2011, 131, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Sugawara, K.; Zákány, N.; Tóth, B.I.; Bíró, T.; Paus, R. A novel control of human keratin expression: Cannabinoid receptor 1-mediated signaling down-regulates the expression of keratins K6 and K16 in human keratinocytes in vitro and in situ. PeerJ 2013, 1, e40. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Oláh, A.; Paus, R. Cover Image: Neuroendocrine treatment of inherited keratin disorders by cannabinoids? Br. J. Dermatol. 2018, 178, 1469. [Google Scholar] [CrossRef] [PubMed]
- Roelandt, T.; Heughebaert, C.; Bredif, S.; Giddelo, C.; Baudouin, C.; Msika, P.; Roseeuw, D.; Uchida, Y.; Elias, P.M.; Hachem, J.-P. Cannabinoid receptors 1 and 2 oppositely regulate epidermal permeability barrier status and differentiation. Exp. Dermatol. 2012, 21, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, B.; Park, B.M.; Jeon, J.E.; Lee, S.H.; Mann, S.; Ahn, S.K.; Hong, S.-P.; Jeong, S.K. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int. J. Dermatol. 2015, 54, e401–e408. [Google Scholar] [CrossRef] [PubMed]
- Gaffal, E.; Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T. Cannabinoid 1 receptors in keratinocytes attenuate fluorescein isothiocyanate-induced mouse atopic-like dermatitis. Exp. Dermatol. 2014, 23, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Kabashima, K. Advances in atopic dermatitis in 2015. J. Allergy Clin. Immunol. 2016, 138, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Paus, R. Harnessing neuroendocrine controls of keratin expression: A new therapeutic strategy for skin diseases? Bioessays 2014, 36, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, M.P.; Zinn, Z.; Khuu, P.; Teng, J.M.C. Self-initiated use of topical cannabidiol oil for epidermolysis bullosa. Pediatr. Dermatol. 2018, 35, e224–e227. [Google Scholar] [CrossRef] [PubMed]
- Schräder, N.H.B.; Duipmans, J.C.; Molenbuur, B.; Wolff, A.P.; Jonkman, M.F. Combined tetrahydrocannabinol and cannabidiol to treat pain in epidermolysis bullosa: A report of three cases. Br. J. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Alvarado-Vazquez, P.A.; Moracho-Vilrriales, C.; Virga, K.G.; Gumina, G.; Romero-Sandoval, A.; Asbill, S. Effects of JWH015 in cytokine secretion in primary human keratinocytes and fibroblasts and its suitability for topical/transdermal delivery. Mol. Pain 2017, 13, 1744806916688220. [Google Scholar] [CrossRef] [PubMed]
- Mugnaini, C.; Rabbito, A.; Brizzi, A.; Palombi, N.; Petrosino, S.; Verde, R.; Di Marzo, V.; Ligresti, A.; Corelli, F. Synthesis of novel 2-(1-adamantanylcarboxamido)thiophene derivatives. Selective cannabinoid type 2 (CB2) receptor agonists as potential agents for the treatment of skin inflammatory disease. Eur. J. Med. Chem. 2019, 161, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Wohlman, I.M.; Composto, G.M.; Heck, D.E.; Heindel, N.D.; Lacey, C.J.; Guillon, C.D.; Casillas, R.P.; Croutch, C.R.; Gerecke, D.R.; Laskin, D.L.; et al. Mustard vesicants alter expression of the endocannabinoid system in mouse skin. Toxicol. Appl. Pharmacol. 2016, 303, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gábor, M. Models of acute inflammation in the ear. Methods Mol. Biol. 2003, 225, 129–137. [Google Scholar] [PubMed]
- Tubaro, A.; Giangaspero, A.; Sosa, S.; Negri, R.; Grassi, G.; Casano, S.; Della Loggia, R.; Appendino, G. Comparative topical anti-inflammatory activity of cannabinoids and cannabivarins. Fitoterapia 2010, 81, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Verde, R.; Vaia, M.; Allarà, M.; Iuvone, T.; Di Marzo, V. Anti-inflammatory Properties of Cannabidiol, a Nonpsychotropic Cannabinoid, in Experimental Allergic Contact Dermatitis. J. Pharmacol. Exp. Ther. 2018, 365, 652–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiù, V.; Rapino, C.; Talamonti, E.; Leuti, A.; Lanuti, M.; Gueniche, A.; Jourdain, R.; Breton, L.; Maccarrone, M. Anandamide Suppresses Proinflammatory T Cell Responses In Vitro through Type-1 Cannabinoid Receptor-Mediated mTOR Inhibition in Human Keratinocytes. J. Immunol. 2016, 197, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- Gaffal, E.; Cron, M.; Glodde, N.; Bald, T.; Kuner, R.; Zimmer, A.; Lutz, B.; Tüting, T. Cannabinoid 1 receptors in keratinocytes modulate proinflammatory chemokine secretion and attenuate contact allergic inflammation. J. Immunol. 2013, 190, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Halasz, C.L. Narrowband UVB phototherapy for psoriasis: Results with fixed increments by skin type (as opposed to percentage increments). Photodermatol. Photoimmunol. Photomed. 1999, 15, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Benáková, N. Phototherapy of psoriasis in the era of biologics: Still in. Acta Dermatovenerol. Croat. 2011, 19, 195–205. [Google Scholar] [PubMed]
- Keyal, U.; Bhatta, A.K.; Wang, X.L. UVA1 a promising approach for scleroderma. Am. J. Transl. Res. 2017, 9, 4280–4287. [Google Scholar] [PubMed]
- Gaffal, E.; Cron, M.; Glodde, N.; Tüting, T. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy 2013, 68, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Bíró, T. TRPV3: A “more than skinny” channel. Exp. Dermatol. 2013, 22, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oláh, A.; Szabó-Papp, J.; Soeberdt, M.; Knie, U.; Dähnhardt-Pfeiffer, S.; Abels, C.; Bíró, T. Echinacea purpurea-derived alkylamides exhibit potent anti-inflammatory effects and alleviate clinical symptoms of atopic eczema. J. Dermatol. Sci. 2017, 88, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, S.; Brazis, P.; Della Valle, M.F.; Miolo, A.; Petrosino, S.; Di Marzo, V.; Puigdemont, A. Effects of palmitoylethanolamide on the cutaneous allergic inflammatory response in Ascaris hypersensitive Beagle dogs. Vet. J. 2012, 191, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Cristino, L.; Karsak, M.; Gaffal, E.; Ueda, N.; Tüting, T.; Bisogno, T.; De Filippis, D.; D’Amico, A.; Saturnino, C.; et al. Protective role of palmitoylethanolamide in contact allergic dermatitis. Allergy 2010, 65, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Vaia, M.; Petrosino, S.; De Filippis, D.; Negro, L.; Guarino, A.; Carnuccio, R.; Di Marzo, V.; Iuvone, T. Palmitoylethanolamide reduces inflammation and itch in a mouse model of contact allergic dermatitis. Eur. J. Pharmacol. 2016, 791, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Kendall, A.C.; Pilkington, S.M.; Sassano, G.; Rhodes, L.E.; Nicolaou, A. N-Acyl ethanolamide and eicosanoid involvement in irritant dermatitis. Br. J. Dermatol. 2016, 175, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Biernacki, M.; Ambrożewicz, E.; Surażyński, A.; Wroński, A.; Skrzydlewska, E. The cross-talk between electrophiles, antioxidant defence and the endocannabinoid system in fibroblasts and keratinocytes after UVA and UVB irradiation. J. Dermatol. Sci. 2016, 81, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, L.; Koreck, A.; Kis, K.; Kenderessy-Szabo, A.; Bodai, L.; Cimpean, A.; Paunescu, V.; Raica, M.; Ghyczy, M. Endogenous phospholipid metabolite containing topical product inhibits ultraviolet light-induced inflammation and DNA damage in human skin. Skin Pharmacol. Physiol. 2007, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, L.-H.; Su, T.-F.; Li, L.; Zhou, R.; Peng, M.; Wu, C.-H.; Yuan, X.-C.; Sun, N.; Meng, X.-F.; et al. Signaling Mechanism of Cannabinoid Receptor-2 Activation-Induced β-Endorphin Release. Mol. Neurobiol. 2016, 53, 3616–3625. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Porreca, F.; Lai, J.; Albrecht, P.J.; Rice, F.L.; Khodorova, A.; Davar, G.; Makriyannis, A.; Vanderah, T.W.; Mata, H.P.; et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc. Natl. Acad. Sci. USA 2005, 102, 3093–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuyama, S.; Mizoguchi, H.; Kuwahata, H.; Komatsu, T.; Nagaoka, K.; Nakamura, H.; Bagetta, G.; Sakurada, T.; Sakurada, S. Involvement of peripheral cannabinoid and opioid receptors in β-caryophyllene-induced antinociception. Eur. J. Pain 2013, 17, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.; Su, T.; Cao, F.; Meng, X.; Pei, L.; Shi, J.; Pan, H.-L.; Li, M. Electroacupuncture increases CB2 receptor expression on keratinocytes and infiltrating inflammatory cells in inflamed skin tissues of rats. J. Pain 2010, 11, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Zhang, L.; Peng, M.; Wu, C.; Pan, W.; Tian, B.; Shi, J.; Pan, H.; Li, M. Cannabinoid CB2 receptors contribute to upregulation of β-endorphin in inflamed skin tissues by electroacupuncture. Mol. Pain 2011, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-F.; Zhao, Y.-Q.; Zhang, L.-H.; Peng, M.; Wu, C.-H.; Pei, L.; Tian, B.; Zhang, J.; Shi, J.; Pan, H.-L.; et al. Electroacupuncture reduces the expression of proinflammatory cytokines in inflamed skin tissues through activation of cannabinoid CB2 receptors. Eur. J. Pain 2012, 16, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Cannabinoid Signaling and Neuroinflammatory Diseases: A Melting pot for the Regulation of Brain Immune Responses. J. Neuroimmune Pharmacol. 2015, 10, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; van der Stelt, M.; Centonze, D.; Maccarrone, M. The endocannabinoid system and its therapeutic exploitation in multiple sclerosis: Clues for other neuroinflammatory diseases. Prog. Neurobiol. 2018, 160, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Wakui, J.; Ikeda, S.; Yanagimoto, S.; Kishimoto, S.; Gokoh, M.; Nasui, M.; Sugiura, T. Involvement of the cannabinoid CB2 receptor and its endogenous ligand 2-arachidonoylglycerol in oxazolone-induced contact dermatitis in mice. J. Immunol. 2006, 177, 8796–8805. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Harvima, I.T. Mast cell-neural interactions contribute to pain and itch. Immunol. Rev. 2018, 282, 168–187. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Halova, I.; Rönnberg, E.; Draberova, L.; Vliagoftis, H.; Nilsson, G.P.; Draber, P. Changing the threshold—Signals and mechanisms of mast cell priming. Immunol. Rev. 2018, 282, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Redegeld, F.A.; Yu, Y.; Kumari, S.; Charles, N.; Blank, U. Non-IgE mediated mast cell activation. Immunol. Rev. 2018, 282, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.; Buddenkotte, J.; Lerner, E.A. Role of mast cells and basophils in pruritus. Immunol. Rev. 2018, 282, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Bonnekoh, H.; Scheffel, J.; Kambe, N.; Krause, K. The role of mast cells in autoinflammation. Immunol. Rev. 2018, 282, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Pavón-Martínez, R.; Ruiz, C.; Melguizo-Rodríguez, L. Role of mast cells in autoimmunity. Life Sci. 2018, 209, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Weaver, B.; Choi, H.W.; Abraham, S.N.; Staats, H.F. Mast cell activators as novel immune regulators. Curr. Opin. Pharmacol. 2018, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Hobo, A.; Harada, K.; Maeda, T.; Uchiyama, M.; Irisawa, R.; Yamazaki, M.; Tsuboi, R. IL-17-positive mast cell infiltration in the lesional skin of lichen planopilaris: Possible role of mast cells in inducing inflammation and dermal fibrosis in cicatricial alopecia. Exp. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Grace, S.A.; Sutton, A.M.; Abraham, N.; Armbrecht, E.S.; Vidal, C.I. Presence of Mast Cells and Mast Cell Degranulation in Scalp Biopsies of Telogen Effluvium. Int. J. Trichol. 2017, 9, 25–29. [Google Scholar]
- Guhl, S.; Babina, M.; Neou, A.; Zuberbier, T.; Artuc, M. Mast cell lines HMC-1 and LAD2 in comparison with mature human skin mast cells--drastically reduced levels of tryptase and chymase in mast cell lines. Exp. Dermatol. 2010, 19, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Facci, L.; Dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.D.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. USA 1995, 92, 3376–3380. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, S.; Brazis, P.; della Valle, M.F.; Miolo, A.; Puigdemont, A. Effects of palmitoylethanolamide on immunologically induced histamine, PGD2 and TNFalpha release from canine skin mast cells. Vet. Immunol. Immunopathol. 2010, 133, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Abramo, F.; Campora, L.; Albanese, F.; della Valle, M.F.; Cristino, L.; Petrosino, S.; Di Marzo, V.; Miragliotta, V. Increased levels of palmitoylethanolamide and other bioactive lipid mediators and enhanced local mast cell proliferation in canine atopic dermatitis. BMC Vet. Res. 2014, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Campolo, M.; Impellizzeri, D.; Paterniti, I.; Allarà, M.; Gugliandolo, E.; D’Amico, R.; Siracusa, R.; Cordaro, M.; Esposito, E.; et al. 2-Pentadecyl-2-Oxazoline, the Oxazoline of Pea, Modulates Carrageenan-Induced Acute Inflammation. Front. Pharmacol. 2017, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Abramo, F.; Lazzarini, G.; Pirone, A.; Lenzi, C.; Albertini, S.; Della Valle, M.F.; Schievano, C.; Vannozzi, I.; Miragliotta, V. Ultramicronized palmitoylethanolamide counteracts the effects of compound 48/80 in a canine skin organ culture model. Vet. Dermatol. 2017, 28, 456-e104. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, D.; Negro, L.; Vaia, M.; Cinelli, M.P.; Iuvone, T. New insights in mast cell modulation by palmitoylethanolamide. CNS Neurol. Disord. Drug Targets 2013, 12, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Small-Howard, A.L.; Shimoda, L.M.N.; Adra, C.N.; Turner, H. Anti-inflammatory potential of CB1-mediated cAMP elevation in mast cells. Biochem. J. 2005, 388, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Moore, C.D.; Zhang, J.Y.; Hall, R.P.; MacLeod, A.S.; Liedtke, W. TRPV4 Moves toward Center-Fold in Rosacea Pathogenesis. J. Investig. Dermatol. 2017, 137, 801–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, N.L.; Wang, Z.; Chang, Y.-L.; Di Nardo, A. TRPV4 Mediates Mast Cell Activation in Cathelicidin-Induced Rosacea Inflammation. J. Investig. Dermatol. 2017, 137, 972–975. [Google Scholar] [CrossRef] [PubMed]
- Turner, H.; del Carmen, K.A.; Stokes, A. Link between TRPV channels and mast cell function. In Transient Receptor Potential (TRP) Channels; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 457–471. [Google Scholar]
- Giudice, E.D.; Rinaldi, L.; Passarotto, M.; Facchinetti, F.; D’Arrigo, A.; Guiotto, A.; Carbonare, M.D.; Battistin, L.; Leon, A. Cannabidiol, unlike synthetic cannabinoids, triggers activation of RBL-2H3 mast cells. J. Leukoc. Biol. 2007, 81, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.; Jeong, S.K.; Park, B.M.; Lee, S.H.; Kim, H.J.; Hong, S.-P.; Kim, B.; Kim, B.-W. Selective Cannabinoid Receptor-1 Agonists Regulate Mast Cell Activation in an Oxazolone-Induced Atopic Dermatitis Model. Ann. Dermatol. 2016, 28, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Fiorucci, L.; Erba, F.; Bari, M.; Finazzi-Agrò, A.; Ascoli, F. Human mast cells take up and hydrolyze anandamide under the control of 5-lipoxygenase and do not express cannabinoid receptors. FEBS Lett. 2000, 468, 176–180. [Google Scholar] [CrossRef] [Green Version]
- Cantarella, G.; Scollo, M.; Lempereur, L.; Saccani-Jotti, G.; Basile, F.; Bernardini, R. Endocannabinoids inhibit release of nerve growth factor by inflammation-activated mast cells. Biochem. Pharmacol. 2011, 82, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.I.; Boza, Y.; Yefi, R.; Luza, S.; Andrews, E.; Penissi, A.; Garrido, P.; Rojas, I.G. The influence of mast cell mediators on migration of SW756 cervical carcinoma cells. J. Pharmacol. Sci. 2008, 106, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.L.; Sánchez-Miranda, E.; Castillo-Arellano, J.I.; Cervantes-Villagrana, R.D.; Ibarra-Sánchez, A.; González-Espinosa, C. Anandamide inhibits FcεRI-dependent degranulation and cytokine synthesis in mast cells through CB2 and GPR55 receptor activation. Possible involvement of CB2-GPR55 heteromers. Int. Immunopharmacol. 2018, 64, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, C.; Cantarero, I.; Palomares, B.; Gómez-Cañas, M.; Fernández-Ruiz, J.; Pavicic, C.; García-Martín, A.; Luz Bellido, M.; Ortega-Castro, R.; Pérez-Sánchez, C.; et al. VCE-004.3, a cannabidiol aminoquinone derivative, prevents bleomycin-induced skin fibrosis and inflammation through PPARγ- and CB2 receptor-dependent pathways. Br. J. Pharmacol. 2018, 175, 3813–3831. [Google Scholar] [CrossRef] [PubMed]
- Del Río, C.; Navarrete, C.; Collado, J.A.; Bellido, M.L.; Gómez-Cañas, M.; Pazos, M.R.; Fernández-Ruiz, J.; Pollastro, F.; Appendino, G.; Calzado, M.A.; et al. The cannabinoid quinol VCE-004.8 alleviates bleomycin-induced scleroderma and exerts potent antifibrotic effects through peroxisome proliferator-activated receptor-γ and CB2 pathways. Sci. Rep. 2016, 6, 21703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asakawa, M.; Yoshioka, T.; Matsutani, T.; Hikita, I.; Suzuki, M.; Oshima, I.; Tsukahara, K.; Arimura, A.; Horikawa, T.; Hirasawa, T.; et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J. Investig. Dermatol. 2006, 126, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Bíró, T.; Tsuruta, D.; Tóth, B.I.; Kromminga, A.; Zákány, N.; Zimmer, A.; Funk, W.; Gibbs, B.F.; Zimmer, A.; et al. Endocannabinoids limit excessive mast cell maturation and activation in human skin. J. Allergy Clin. Immunol. 2012, 129, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Zákány, N.; Hundt, T.; Emelianov, V.; Tsuruta, D.; Schäfer, C.; Kloepper, J.E.; Bíró, T.; Paus, R. Cannabinoid receptor 1 controls human mucosal-type mast cell degranulation and maturation in situ. J. Allergy Clin. Immunol. 2013, 132, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pradhan, D.; Puri, P.; Ramesh, V.; Aggarwal, S.; Nayek, A.; Jain, A.K. Genomic alterations driving psoriasis pathogenesis. Gene 2019, 683, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.L.; Orasan, R.I. Understanding psoriasis: Role of miRNAs. Biomed. Rep. 2018, 9, 367–374. [Google Scholar] [PubMed]
- Benhadou, F.; Mintoff, D.; Schnebert, B.; Thio, H.B. Psoriasis and Microbiota: A Systematic Review. Diseases 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Bigliardi, P.L. Role of Skin pH in Psoriasis. Curr. Probl. Dermatol. 2018, 54, 108–114. [Google Scholar] [PubMed]
- Brembilla, N.C.; Senra, L.; Boehncke, W.-H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Benhadou, F.; Mintoff, D.; Del Marmol, V. Psoriasis: Keratinocytes or Immune Cells—Which Is the Trigger? Dermatology 2019, 23, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Staubach, P.; Zimmer, S. Plaque psoriasis—More than a skin disorder. Medizinische Monatsschrift für Pharmazeuten 2017, 40, 231–233. [Google Scholar] [PubMed]
- Katayama, H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, N.; Kazemi, M. Cannabis for Refractory Psoriasis-High Hopes for a Novel Treatment and a Literature Review. Curr. Clin. Pharmacol. 2016, 11, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Pharmacological properties and therapeutic possibilities for drugs acting upon endocannabinoid receptors. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Namazi, M.R. Cannabinoids, loratadine and allopurinol as novel additions to the antipsoriatic ammunition. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Kendall, A.C.; Nicolaou, A. Bioactive lipid mediators in skin inflammation and immunity. Prog. Lipid Res. 2013, 52, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Norooznezhad, A.H.; Norooznezhad, F. Cannabinoids: Possible agents for treatment of psoriasis via suppression of angiogenesis and inflammation. Med. Hypotheses 2017, 99, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Senapati, S.; Roy, S.; Chatterjee, G.; Chatterjee, R. Epigenome-wide DNA methylation regulates cardinal pathological features of psoriasis. Clin. Epigenet. 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Bin Huraib, G.; Al Harthi, F.; Arfin, M.; Rizvi, S.; Al-Asmari, A. The Protein Tyrosine Phosphatase Nonreceptor 22 (PTPN22) R620W Functional Polymorphism in Psoriasis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2018, 11, 1179544117751434. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liao, W.; Chang, M.; Schrodi, S.J.; Bui, N.; Catanese, J.J.; Poon, A.; Matsunami, N.; Callis-Duffin, K.P.; Leppert, M.F.; et al. Further genetic evidence for three psoriasis-risk genes: ADAM33, CDKAL1, and PTPN22. J. Investig. Dermatol. 2009, 129, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Warren, R.B.; Eyre, S.; Ke, X.; Young, H.S.; Allen, M.; Strachan, D.; McArdle, W.; Gittins, M.P.; Barker, J.N.W.N.; et al. Polymorphisms in the PTPN22 region are associated with psoriasis of early onset. Br. J. Dermatol. 2008, 158, 962–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Z.; Rani, P.L.; Fu, X.; Yu, W.; Bao, F.; Yu, G.; Li, J.; Li, L.; Sun, L.; et al. Identification of PTPN22, ST6GAL1 and JAZF1 as psoriasis risk genes demonstrates shared pathogenesis between psoriasis and diabetes. Exp. Dermatol. 2017, 26, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Loehr, S.; Budu-Aggrey, A.; Uebe, S.; Bruce, I.N.; Feletar, M.; Marzo-Ortega, H.; Helliwell, P.; Ryan, A.W.; Kane, D.; et al. PTPN22 is associated with susceptibility to psoriatic arthritis but not psoriasis: Evidence for a further PsA-specific risk locus. Ann. Rheum. Dis. 2015, 74, 1882–1885. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Dang, E.; Yang, C.; Song, H. Progress in genetic research on psoriatic arthritis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2017, 34, 606–610. [Google Scholar] [PubMed]
- Juneblad, K.; Johansson, M.; Rantapää-Dahlqvist, S.; Alenius, G.-M. Association between the PTPN22 +1858 C/T polymorphism and psoriatic arthritis. Arthritis Res. Ther. 2011, 13, R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amur, S.; Parekh, A.; Mummaneni, P. Sex differences and genomics in autoimmune diseases. J. Autoimmun. 2012, 38, J254–J265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Chang, J.S. PTPN22 C1858T and the risk of psoriasis: A meta-analysis. Mol. Biol. Rep. 2012, 39, 7861–7870. [Google Scholar] [CrossRef] [PubMed]
- Hüffmeier, U.; Steffens, M.; Burkhardt, H.; Lascorz, J.; Schürmeier-Horst, F.; Ständer, M.; Kelsch, R.; Baumann, C.; Küster, W.; Mössner, R.; et al. Evidence for susceptibility determinant(s) to psoriasis vulgaris in or near PTPN22 in German patients. J. Med. Genet. 2006, 43, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Ibrahim, S.; Petersen, F.; Yu, X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. 2012, 13, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zervou, M.I.; Castro-Giner, F.; Sidiropoulos, P.; Boumpas, D.T.; Tosca, A.D.; Krueger-Krasagakis, S. The protein tyrosine phosphatase, non-receptor type 22 R620W polymorphism does not confer susceptibility to psoriasis in the genetic homogeneous population of Crete. Genet. Test. Mol. Biomark. 2010, 14, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ambrożewicz, E.; Wójcik, P.; Wroński, A.; Łuczaj, W.; Jastrząb, A.; Žarković, N.; Skrzydlewska, E. Pathophysiological Alterations of Redox Signaling and Endocannabinoid System in Granulocytes and Plasma of Psoriatic Patients. Cells 2018, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Nattkemper, L.A.; Tey, H.L.; Valdes-Rodriguez, R.; Lee, H.; Mollanazar, N.K.; Albornoz, C.; Sanders, K.M.; Yosipovitch, G. The Genetics of Chronic Itch: Gene Expression in the Skin of Patients with Atopic Dermatitis and Psoriasis with Severe Itch. J. Investig. Dermatol. 2018, 138, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Chen, X.; Hu, Y.; Hao, Z.; Osland, J.M.; Chen, X.; Gerber, S.D.; Chen, M.; Gu, H.; Yuan, R. Overexpression and potential roles of NRIP1 in psoriasis. Oncotarget 2016, 7, 74236–74246. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.P.; Zhang, H.H.; Hwang, S.T.; Farber, J.M. IL-23- and Imiquimod-Induced Models of Experimental Psoriasis in Mice. Curr. Protoc. Immunol. 2019, e71. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-J.; Song, T.-J.; Zhang, L.-W.; Su, Y.; Wang, K.-Y.; Sun, Q. TRB3 is elevated in psoriasis vulgaris lesions and mediates HaCaT cells proliferation in vitro. J. Investig. Med. 2017, 65, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rerknimitr, P.; Otsuka, A.; Nakashima, C.; Kabashima, K. The etiopathogenesis of atopic dermatitis: Barrier disruption, immunological derangement, and pruritus. Inflamm. Regen. 2017, 37, 14. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, T. Immune Dysregulation in the Pathogenesis of Atopic Dermatitis. Dermatitis 2018, 29, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Nomura, T.; Common, J.; Kabashima, K. Insights into atopic dermatitis gained from genetically defined mouse models. J. Allergy Clin. Immunol. 2019, 143, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M. Primary role of barrier dysfunction in the pathogenesis of atopic dermatitis. Exp. Dermatol. 2018, 27, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Rangel, S.M.; Paller, A.S. Bacterial colonization, overgrowth, and superinfection in atopic dermatitis. Clin. Dermatol. 2018, 36, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Moriyama, M.; Feld, M.; Buddenkotte, J.; Buhl, T.; Szöllösi, A.; Zhang, J.; Miller, P.; Ghetti, A.; Fischer, M.; et al. New mechanism underlying IL-31-induced atopic dermatitis. J. Allergy Clin. Immunol. 2018, 141, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Seba, A.; Antal, A.S. Immunological and molecular targets of atopic dermatitis treatment. Br. J. Dermatol. 2014, 170 (Suppl. 1), 7–11. [Google Scholar] [CrossRef] [Green Version]
- Trusler, A.R.; Clark, A.K.; Sivamani, R.K.; Shi, V.Y. The Endocannabinoid System and Its Role in Eczematous Dermatoses. Dermatitis 2017, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.; Patel, N.U.; Blake, A.; Prabakaran, S.; Reimer, D.; Feldman, S.R.; Strowd, L.C. Insights into future therapeutics for atopic dermatitis. Expert Opin. Pharmacother. 2018, 19, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Campora, L.; Miragliotta, V.; Ricci, E.; Cristino, L.; Di Marzo, V.; Albanese, F.; Federica Della Valle, M.; Abramo, F. Cannabinoid receptor type 1 and 2 expression in the skin of healthy dogs and dogs with atopic dermatitis. Am. J. Vet. Res. 2012, 73, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Roque, J.B.; O’Leary, C.A.; Kyaw-Tanner, M.; Duffy, D.L.; Gharahkhani, P.; Vogelnest, L.; Mason, K.; Shipstone, M. PTPN22 polymorphisms may indicate a role for this gene in atopic dermatitis in West Highland white terriers. BMC Res. Notes 2011, 4, 571. [Google Scholar] [CrossRef] [PubMed]
- Bonchak, J.G.; Swerlick, R.A. Emerging therapies for atopic dermatitis: TRPV1 antagonists. J. Am. Acad. Dermatol. 2018, 78, S63–S66. [Google Scholar] [CrossRef] [PubMed]
- Haruna, T.; Soga, M.; Morioka, Y.; Imura, K.; Furue, Y.; Yamamoto, M.; Hayakawa, J.; Deguchi, M.; Arimura, A.; Yasui, K. The Inhibitory Effect of S-777469, a Cannabinoid Type 2 Receptor Agonist, on Skin Inflammation in Mice. Pharmacology 2017, 99, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Nojima, H.; Kuraishi, Y.; Aisaka, K. The cannabinoid CB2 receptor inverse agonist JTE-907 suppresses spontaneous itch-associated responses of NC mice, a model of atopic dermatitis. Eur. J. Pharmacol. 2006, 542, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Miyagawa, N.; Wakitani, K. Involvement of cannabinoid CB2 receptors in the IgE-mediated triphasic cutaneous reaction in mice. Life Sci. 2007, 80, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Sasso, O.; Summa, M.; Armirotti, A.; Pontis, S.; De Mei, C.; Piomelli, D. The N-Acylethanolamine Acid Amidase Inhibitor ARN077 Suppresses Inflammation and Pruritus in a Mouse Model of Allergic Dermatitis. J. Investig. Dermatol. 2018, 138, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Noli, C.; Della Valle, M.F.; Miolo, A.; Medori, C.; Schievano, C. Skinalia Clinical Research Group Efficacy of ultra-micronized palmitoylethanolamide in canine atopic dermatitis: An open-label multi-centre study. Vet. Dermatol. 2015, 26, 432-e101. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, B.; Eicke, C.; Reinhardt, H.-W.; Ring, J. Adjuvant treatment of atopic eczema: Assessment of an emollient containing N-palmitoylethanolamine (ATOPA study). J. Eur. Acad. Dermatol. Venereol. 2008, 22, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wang, X.-M.; Guichard, A.; Tan, Y.-M.; Qian, C.-Y.; Yang, L.-J.; Humbert, P. N-palmitoylethanolamine and N-acetylethanolamine are effective in asteatotic eczema: Results of a randomized, double-blind, controlled study in 60 patients. Clin. Interv. Aging 2014, 9, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, R.; Aslani, S.; Dashti, N.; Jamshidi, A.; Gharibdoost, F.; Mahmoudi, M. Genetic implications in the pathogenesis of systemic sclerosis. Int. J. Rheum. Dis. 2018, 21, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Singhvi, G.; Manchanda, P.; Krishna Rapalli, V.; Kumar Dubey, S.; Gupta, G.; Dua, K. MicroRNAs as biological regulators in skin disorders. Biomed. Pharmacother. 2018, 108, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Aslani, S.; Sobhani, S.; Gharibdoost, F.; Jamshidi, A.; Mahmoudi, M. Epigenetics and pathogenesis of systemic sclerosis; the ins and outs. Hum. Immunol. 2018, 79, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Bellocchi, C.; Volkmann, E.R. Update on the Gastrointestinal Microbiome in Systemic Sclerosis. Curr. Rheumatol. Rep. 2018, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Giovannetti, A.; Gambardella, L.; Malorni, W.; Pietraforte, D.; Straface, E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J. Cell. Mol. Med. 2018, 22, 3308–3314. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y. Systemic sclerosis. J. Dermatol. 2018, 45, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Dieudé, P.; Boileau, C.; Allanore, Y. Immunogenetics of systemic sclerosis. Autoimmun. Rev. 2011, 10, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Dieudé, P.; Guedj, M.; Wipff, J.; Avouac, J.; Hachulla, E.; Diot, E.; Granel, B.; Sibilia, J.; Cabane, J.; Meyer, O.; et al. The PTPN22 620W allele confers susceptibility to systemic sclerosis: Findings of a large case-control study of European Caucasians and a meta-analysis. Arthritis Rheum. 2008, 58, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Gourh, P.; Tan, F.K.; Assassi, S.; Ahn, C.W.; McNearney, T.A.; Fischbach, M.; Arnett, F.C.; Mayes, M.D. Association of the PTPN22 R620W polymorphism with anti-topoisomerase I- and anticentromere antibody-positive systemic sclerosis. Arthritis Rheum. 2006, 54, 3945–3953. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. The association between the PTPN22 C1858T polymorphism and systemic sclerosis: A meta-analysis. Mol. Biol. Rep. 2012, 39, 3103–3108. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Dieude, P.; Boileau, C. Genetic background of systemic sclerosis: Autoimmune genes take centre stage. Rheumatology 2010, 49, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gallo, L.M.; Gourh, P.; Broen, J.; Simeon, C.; Fonollosa, V.; Ortego-Centeno, N.; Agarwal, S.; Vonk, M.C.; Coenen, M.; Riemekasten, G.; et al. Analysis of the influence of PTPN22 gene polymorphisms in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Balada, E.; Simeón-Aznar, C.P.; Serrano-Acedo, S.; Martínez-Lostao, L.; Selva-O’Callaghan, A.; Fonollosa-Pla, V.; Vilardell-Tarrés, M. Lack of association of the PTPN22 gene polymorphism R620W with systemic sclerosis. Clin. Exp. Rheumatol. 2006, 24, 321–324. [Google Scholar] [PubMed]
- Ramirez, M.; Quintana, G.; Diaz-Gallo, L.M.; Caminos, J.; Garces, M.; Cepeda, L.; Rondon, F.; Restrepo, J.F.; Egea, E.; Garavito, G.; et al. The PTPN22 C1858T variant as a risk factor for rheumatoid arthritis and systemic lupus erythematosus but not for systemic sclerosis in the Colombian population. Clin. Exp. Rheumatol. 2012, 30, 520–524. [Google Scholar] [PubMed]
- Wipff, J.; Allanore, Y.; Kahan, A.; Meyer, O.; Mouthon, L.; Guillevin, L.; Pierlot, C.; Glikmans, E.; Bardin, T.; Boileau, C.; et al. Lack of association between the protein tyrosine phosphatase non-receptor 22 (PTPN22)*620W allele and systemic sclerosis in the French Caucasian population. Ann. Rheum. Dis. 2006, 65, 1230–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.-C.; Lee, Y.H. Association between the functional PTPN22 G788A (R263Q) polymorphism and susceptibility to autoimmune diseases: A meta-analysis. Cell. Mol. Biol. 2018, 64, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ochoa, Á.; Quirantes-Piné, R.; Borrás-Linares, I.; Gemperline, D.; PRECISESADS Clinical Consortium; Alarcón Riquelme, M.E.; Beretta, L.; Segura-Carretero, A. Urinary and plasma metabolite differences detected by HPLC-ESI-QTOF-MS in systemic sclerosis patients. J. Pharm. Biomed. Anal. 2019, 162, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S.H. Ajulemic acid: Potential treatment for chronic inflammation. Pharmacol. Res. Perspect. 2018, 6, e00394. [Google Scholar] [CrossRef] [PubMed]
- Zurier, R.B.; Burstein, S.H. Cannabinoids, inflammation, and fibrosis. FASEB J. 2016, 30, 3682–3689. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Rybałtowska-Kawałko, P.; Skrzydlewska, E. Rutin as a Mediator of Lipid Metabolism and Cellular Signaling Pathways Interactions in Fibroblasts Altered by UVA and UVB Radiation. Oxid. Med. Cell. Longev. 2017, 2017, 4721352. [Google Scholar] [CrossRef] [PubMed]
- Palumbo-Zerr, K.; Horn, A.; Distler, A.; Zerr, P.; Dees, C.; Beyer, C.; Selvi, E.; Cravatt, B.F.; Distler, O.; Schett, G.; et al. Inactivation of fatty acid amide hydrolase exacerbates experimental fibrosis by enhanced endocannabinoid-mediated activation of CB1. Ann. Rheum. Dis. 2012, 71, 2051–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, E.; Selvi, E.; Balistreri, E.; Lorenzini, S.; Maggio, R.; Natale, M.-R.; Capecchi, P.-L.; Lazzerini, P.-E.; Bardelli, M.; Laghi-Pasini, F.; et al. Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology 2009, 48, 1050–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, R.; Cohen, J.; Sharma, S.; Zhang, D.X.; Lafyatis, R.; Bhawan, J.; Rahaman, S.O. TRPV4 ION Channel Is Associated with Scleroderma. J. Investig. Dermatol. 2017, 137, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Marquart, S.; Zerr, P.; Akhmetshina, A.; Palumbo, K.; Reich, N.; Tomcik, M.; Horn, A.; Dees, C.; Engel, M.; Zwerina, J.; et al. Inactivation of the cannabinoid receptor CB1 prevents leukocyte infiltration and experimental fibrosis. Arthritis Rheum. 2010, 62, 3467–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.C.; Sweet, H.O.; Bunker, L.E. Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am. J. Pathol. 1976, 82, 493–512. [Google Scholar] [PubMed]
- Bolognini, D.; Cascio, M.G.; Parolaro, D.; Pertwee, R.G. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br. J. Pharmacol. 2012, 165, 2561–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-S.; Wang, L.-L.; Liu, M.; Jiang, S.-K.; Zhang, M.; Tian, Z.-L.; Wang, M.; Li, J.-Y.; Zhao, R.; Guan, D.-W. Cannabinoid CB2 receptors are involved in the regulation of fibrogenesis during skin wound repair in mice. Mol. Med. Rep. 2016, 13, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Servettaz, A.; Kavian, N.; Nicco, C.; Deveaux, V.; Chéreau, C.; Wang, A.; Zimmer, A.; Lotersztajn, S.; Weill, B.; Batteux, F. Targeting the cannabinoid pathway limits the development of fibrosis and autoimmunity in a mouse model of systemic sclerosis. Am. J. Pathol. 2010, 177, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Dees, C.; Busch, N.; Beer, J.; Sarter, K.; Zwerina, J.; Zimmer, A.; Distler, O.; Schett, G.; Distler, J.H.W. The cannabinoid receptor CB2 exerts antifibrotic effects in experimental dermal fibrosis. Arthritis Rheum. 2009, 60, 1129–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, H.; Burstein, S.H.; Zurier, R.B.; Chen, J.D. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol. Pharmacol. 2003, 63, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.G.; Selvi, E.; Balistreri, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Lazzerini, P.E.; Montilli, C.; Capecchi, P.L.; Lucattelli, M.; et al. Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1545–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucattelli, M.; Fineschi, S.; Selvi, E.; Garcia Gonzalez, E.; Bartalesi, B.; De Cunto, G.; Lorenzini, S.; Galeazzi, M.; Lungarella, G. Ajulemic acid exerts potent anti-fibrotic effect during the fibrogenic phase of bleomycin lung. Respir. Res. 2016, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, A.; Garrido-Rodríguez, M.; Navarrete, C.; Del Río, C.; Bellido, M.L.; Appendino, G.; Calzado, M.A.; Muñoz, E. EHP-101, an oral formulation of the cannabidiol aminoquinone VCE-004.8, alleviates bleomycin-induced skin and lung fibrosis. Biochem. Pharmacol. 2018, 157, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, E.; Garcia-Gonzalez, E.; Selvi, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Maggio, R.; Lucattelli, M.; Galeazzi, M.; Distler, J.W.H. The cannabinoid WIN55, 212–2 abrogates dermal fibrosis in scleroderma bleomycin model. Ann. Rheum. Dis. 2011, 70, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Tomcik, M.; Palumbo-Zerr, K.; Zerr, P.; Sumova, B.; Avouac, J.; Dees, C.; Distler, A.; Becvar, R.; Distler, O.; Schett, G.; et al. Tribbles homologue 3 stimulates canonical TGF-β signalling to regulate fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X.; Osland, J.; Gerber, S.J.; Luan, C.; Delfino, K.; Goodwin, L.; Yuan, R. Deletion of Nrip1 Extends Female Mice Longevity, Increases Autophagy, and Delays Cell Senescence. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, V.; Chicca, A.; Glasmacher, S.; Paloczi, J.; Cao, Z.; Pacher, P.; Gertsch, J. Pepcan-12 (RVD-hemopressin) is a CB2 receptor positive allosteric modulator constitutively secreted by adrenals and in liver upon tissue damage. Sci. Rep. 2017, 7, 9560. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Goswami, R.; Merth, M.; Cohen, J.; Lei, K.Y.; Zhang, D.X.; Rahaman, S.O. TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 2017, 312, C562–C572. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Czirják, L.; Sándor, Z.; Helyes, Z.; László, T.; Elekes, K.; Czömpöly, T.; Starr, A.; Brain, S.; Szolcsányi, J.; et al. Investigation of sensory neurogenic components in a bleomycin-induced scleroderma model using transient receptor potential vanilloid 1 receptor- and calcitonin gene-related peptide-knockout mice. Arthritis Rheum. 2008, 58, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Guo, X. A review: Therapeutic potential of adipose-derived stem cells in cutaneous wound healing and regeneration. Stem Cell Res. Ther. 2018, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; Gómez, B.I.; McIntyre, M.K.; Dubick, M.A.; Christy, R.J.; Nicholson, S.E.; Burmeister, D.M. The Cutaneous Microbiome and Wounds: New Molecular Targets to Promote Wound Healing. Int. J. Mol. Sci. 2018, 19, 2699. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.; Lin, E.J.; Tartar, D. Immunology of Wound Healing. Curr. Dermatol. Rep. 2018, 7, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Guan, D.; Liu, W.; Wang, T.; Fan, Y.; Cheng, Z.; Zheng, J.; Hu, G. Expression of cannabinoid receptor I during mice skin incised wound healing course. Fa Yi Xue Za Zhi 2010, 26, 241–245. [Google Scholar] [PubMed]
- Wang, L.-L.; Zhao, R.; Li, J.-Y.; Li, S.-S.; Liu, M.; Wang, M.; Zhang, M.-Z.; Dong, W.-W.; Jiang, S.-K.; Zhang, M.; et al. Pharmacological activation of cannabinoid 2 receptor attenuates inflammation, fibrogenesis, and promotes re-epithelialization during skin wound healing. Eur. J. Pharmacol. 2016, 786, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Sasso, O.; Pontis, S.; Armirotti, A.; Cardinali, G.; Kovacs, D.; Migliore, M.; Summa, M.; Moreno-Sanz, G.; Picardo, M.; Piomelli, D. Endogenous N-acyl taurines regulate skin wound healing. Proc. Natl. Acad. Sci. USA 2016, 113, E4397–E4406. [Google Scholar] [CrossRef] [PubMed]
- Alser, O.H.; Goutos, I. The evidence behind the use of platelet-rich plasma (PRP) in scar management: A literature review. Scars Burn. Heal. 2018, 4, 2059513118808773. [Google Scholar] [CrossRef] [PubMed]
- Descalzi, F.; Ulivi, V.; Cancedda, R.; Piscitelli, F.; Luongo, L.; Guida, F.; Gatta, L.; Maione, S.; Di Marzo, V. Platelet-rich plasma exerts antinociceptive activity by a peripheral endocannabinoid-related mechanism. Tissue Eng. Part A 2013, 19, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Sulk, M.; Steinhoff, M. Chapter 17—Role of TRP Channels in Skin Diseases. In TRP Channels as Therapeutic Targets; Szallasi, A., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 293–323. ISBN 978-0-12-420024-1. [Google Scholar]
- Yang, P.; Feng, J.; Luo, J.; Madison, M.; Hu, H. A Critical Role for TRP Channels in the Skin. In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017; ISBN 978-1-315-15283-7. [Google Scholar]
- Valdes-Rodriguez, R.; Kaushik, S.B.; Yosipovitch, G. Transient receptor potential channels and dermatological disorders. Curr. Top. Med. Chem. 2013, 13, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. TRPV3 regulates nitric oxide synthase-independent nitric oxide synthesis in the skin. Nat. Commun. 2011, 2, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Uchida, K.; Hata, S.; Hatta, M.; Kita, T.; Miyake, Y.; Okamura, K.; Tamaoki, S.; Ishikawa, H.; Yamazaki, J. TRPV2 channel inhibitors attenuate fibroblast differentiation and contraction mediated by keratinocyte-derived TGF-β1 in an in vitro wound healing model of rats. J. Dermatol. Sci. 2018, 90, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, H.; Kikuchi, S.; Arai, N.; Koda, A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Jpn. J. Pharmacol. 1992, 60, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Styrczewska, M.; Kulma, A.; Ratajczak, K.; Amarowicz, R.; Szopa, J. Cannabinoid-like anti-inflammatory compounds from flax fiber. Cell. Mol. Biol. Lett. 2012, 17, 479–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styrczewska, M.; Kostyn, A.; Kulma, A.; Majkowska-Skrobek, G.; Augustyniak, D.; Prescha, A.; Czuj, T.; Szopa, J. Flax Fiber Hydrophobic Extract Inhibits Human Skin Cells Inflammation and Causes Remodeling of Extracellular Matrix and Wound Closure Activation. Biomed Res. Int. 2015, 2015, 862391. [Google Scholar] [CrossRef] [PubMed]
- Yosipovitch, G.; Rosen, J.D.; Hashimoto, T. Itch: From mechanism to (novel) therapeutic approaches. J. Allergy Clin. Immunol. 2018, 142, 1375–1390. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Steinhoff, M. Molecular mechanisms of pruritus. Curr. Res. Transl. Med. 2016, 64, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.I.; Szallasi, A.; Bíró, T. Transient receptor potential channels and itch: How deep should we scratch? In Pharmacology of Itch; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 226, pp. 89–133. [Google Scholar]
- Moore, C.; Gupta, R.; Jordt, S.-E.; Chen, Y.; Liedtke, W.B. Regulation of Pain and Itch by TRP Channels. Neurosci. Bull. 2018, 34, 120–142. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Hu, H. TRP Channels as Drug Targets to Relieve Itch. Pharmaceuticals 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Heisig, M.; Łaczmański, Ł.; Reich, A.; Lwow, F.; Szepietowski, J.C. Uremic Pruritus Is Not Associated with Endocannabinoid Receptor 1 Gene Polymorphisms. Biomed Res. Int. 2016, 2016, 3567527. [Google Scholar] [CrossRef] [PubMed]
- Neff, G.W.; O’Brien, C.B.; Reddy, K.R.; Bergasa, N.V.; Regev, A.; Molina, E.; Amaro, R.; Rodriguez, M.J.; Chase, V.; Jeffers, L.; et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am. J. Gastroenterol. 2002, 97, 2117–2119. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.J.; Lefever, T.W.; Rawls, S.M.; Whiteside, G.T.; Walker, E.A. Age-dependent effects of the cannabinoid CB1 antagonist SR141716A on food intake, body weight change, and pruritus in rats. Psychopharmacology 2009, 206, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Pavon, F.J.; Bilbao, A.; Hernández-Folgado, L.; Cippitelli, A.; Jagerovic, N.; Abellán, G.; Rodríguez-Franco, M.A.I.; Serrano, A.; Macias, M.; Gómez, R.; et al. Antiobesity effects of the novel in vivo neutral cannabinoid receptor antagonist 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1H-1,2,4-triazole--LH 21. Neuropharmacology 2006, 51, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Bilir, K.A.; Anli, G.; Ozkan, E.; Gunduz, O.; Ulugol, A. Involvement of spinal cannabinoid receptors in the antipruritic effects of WIN 55,212-2, a cannabinoid receptor agonist. Clin. Exp. Dermatol. 2018, 43, 553–558. [Google Scholar] [CrossRef] [PubMed]
- HU-210|Ligand Page|IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=731 (accessed on 21 January 2019).
- Dvorak, M.; Watkinson, A.; McGlone, F.; Rukwied, R. Histamine induced responses are attenuated by a cannabinoid receptor agonist in human skin. Inflamm. Res. 2003, 52, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Haruna, T.; Soga, M.; Morioka, Y.; Hikita, I.; Imura, K.; Furue, Y.; Yamamoto, M.; Imura, C.; Ikeda, M.; Yamauchi, A.; et al. S-777469, a novel cannabinoid type 2 receptor agonist, suppresses itch-associated scratching behavior in rodents through inhibition of itch signal transmission. Pharmacology 2015, 95, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Yesilyurt, O.; Cayirli, M.; Sakin, Y.S.; Seyrek, M.; Akar, A.; Dogrul, A. Systemic and spinal administration of FAAH, MAGL inhibitors and dual FAAH/MAGL inhibitors produce antipruritic effect in mice. Arch. Dermatol. Res. 2016, 308, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Tosun, N.C.; Gunduz, O.; Ulugol, A. Attenuation of serotonin-induced itch responses by inhibition of endocannabinoid degradative enzymes, fatty acid amide hydrolase and monoacylglycerol lipase. J. Neural Transm. 2015, 122, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Reynoso-Moreno, I.; Chicca, A.; Flores-Soto, M.E.; Viveros-Paredes, J.M.; Gertsch, J. The Endocannabinoid Reuptake Inhibitor WOBE437 Is Orally Bioavailable and Exerts Indirect Polypharmacological Effects via Different Endocannabinoid Receptors. Front. Mol. Neurosci. 2018, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Schlosburg, J.E.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. Endocannabinoid modulation of scratching response in an acute allergenic model: A new prospective neural therapeutic target for pruritus. J. Pharmacol. Exp. Ther. 2009, 329, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Spradley, J.M.; Davoodi, A.; Gee, L.B.; Carstens, M.I.; Carstens, E. Differences in peripheral endocannabinoid modulation of scratching behavior in facial vs. spinally-innervated skin. Neuropharmacology 2012, 63, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ständer, S.; Reinhardt, H.W.; Luger, T.A. Topical cannabinoid agonists. An effective new possibility for treating chronic pruritus. Hautarzt 2006, 57, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Visse, K.; Blome, C.; Phan, N.Q.; Augustin, M.; Ständer, S. Efficacy of Body Lotion Containing N-palmitoylethanolamine in Subjects with Chronic Pruritus due to Dry Skin: A Dermatocosmetic Study. Acta Derm. Venereol. 2017, 97, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birdsall, S.M.; Birdsall, T.C.; Tims, L.A. The Use of Medical Marijuana in Cancer. Curr. Oncol. Rep. 2016, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.P. Cannabinoids for Symptom Management and Cancer Therapy: The Evidence. J. Natl. Compr. Cancer Netw. 2016, 14, 915–922. [Google Scholar] [CrossRef]
- Blake, A.; Wan, B.A.; Malek, L.; DeAngelis, C.; Diaz, P.; Lao, N.; Chow, E.; O’Hearn, S. A selective review of medical cannabis in cancer pain management. Ann. Palliat. Med. 2017, 6, S215–S222. [Google Scholar] [CrossRef] [PubMed]
- Carpi, S.; Fogli, S.; Polini, B.; Montagnani, V.; Podestà, A.; Breschi, M.C.; Romanini, A.; Stecca, B.; Nieri, P. Tumor-promoting effects of cannabinoid receptor type 1 in human melanoma cells. Toxicol. In Vitro 2017, 40, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Ladin, D.A.; Soliman, E.; Griffin, L.; Van Dross, R. Preclinical and Clinical Assessment of Cannabinoids as Anti-Cancer Agents. Front. Pharmacol. 2016, 7, 361. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, M.; Góralska, J.; Solnica, B. Cannabinoids—A new weapon against cancer? Postepy Higieny i Medycyny Doswiadczalnej 2016, 70, 1309–1320. [Google Scholar] [PubMed]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Tímár, J.; Hegedüs, B.; Rásó, E. The role of lipid signaling in the progression of malignant melanoma. Cancer Metastasis Rev. 2018, 37, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, J.; Zhao, H.; Fang, X.; Li, H. Cannabinoid receptor 2 is upregulated in melanoma. J. Cancer Res. Ther. 2012, 8, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Sailler, S.; Schmitz, K.; Jäger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, M.R.; Cantarella, G.; Scollo, M.; Lempereur, L.; Palumbo, M.; Saccani-Jotti, G.; Bernardini, R. The antimitogenic effect of the cannabinoid receptor agonist WIN55212-2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett. 2011, 310, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, I.; Bánki, B.; Márk, A.; Varga, N.; Tóvári, J.; Ladányi, A.; Rásó, E.; Tímár, J. Revisiting CB1 receptor as drug target in human melanoma. Pathol. Oncol. Res. 2012, 18, 857–866. [Google Scholar] [CrossRef] [PubMed]
- AM251|Ligand Page|IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=3317 (accessed on 23 January 2019).
- Carpi, S.; Fogli, S.; Romanini, A.; Pellegrino, M.; Adinolfi, B.; Podestà, A.; Costa, B.; Da Pozzo, E.; Martini, C.; Breschi, M.C.; et al. AM251 induces apoptosis and G2/M cell cycle arrest in A375 human melanoma cells. Anticancer Drugs 2015, 26, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Verdegaal, E.M.E.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Haskó, J.; Fazakas, C.; Molnár, J.; Nyúl-Tóth, Á.; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A. TRP Channels as Therapeutic Targets: From Basic Science to Clinical Use; Academic Press: Cambridge, MA, USA, 2015; ISBN 978-0-12-420079-1. [Google Scholar]
- Bernardini, M.; Fiorio Pla, A.; Prevarskaya, N.; Gkika, D. Human transient receptor potential (TRP) channel expression profiling in carcinogenesis. Int. J. Dev. Biol. 2015, 59, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wu, H.; Wei, Z.; Wang, X.; Shen, P.; Wang, S.; Wang, A.; Chen, W.; Lu, Y. TRPM8: A potential target for cancer treatment. J. Cancer Res. Clin. Oncol. 2016, 142, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Prevarskaya, N. Oncogenic TRP channels. Adv. Exp. Med. Biol. 2011, 704, 929–945. [Google Scholar] [PubMed]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP channels in cancer. Biochim. Biophys. Acta 2007, 1772, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivan-Viguera, A.; Garcia-Otin, A.L.; Lozano-Gerona, J.; Abarca-Lachen, E.; Garcia-Malinis, A.J.; Hamilton, K.L.; Gilaberte, Y.; Pueyo, E.; Köhler, R. Pharmacological activation of TRPV4 produces immediate cell damage and induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS ONE 2018, 13, e0190307. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Tundis, R.; Menichini, F.; Saab, A.M.; Statti, G.A.; Menichini, F. Antiproliferative effects of essential oils and their major constituents in human renal adenocarcinoma and amelanotic melanoma cells. Cell Prolif. 2008, 41, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.I.; Kim, E.J.; Kwon, G.T.; Jung, Y.J.; Park, T.; Kim, Y.; Yu, R.; Choi, M.-S.; Chun, H.S.; Kwon, S.-H.; et al. β-Caryophyllene potently inhibits solid tumor growth and lymph node metastasis of B16F10 melanoma cells in high-fat diet-induced obese C57BL/6N mice. Carcinogenesis 2015, 36, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, T.-T.; Hua, F.; Hu, Z.-W. Metformin reduces TRIB3 expression and restores autophagy flux: An alternative antitumor action. Autophagy 2018, 14, 1278–1279. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, T.-T.; Wang, F.; Cui, B.; Zhao, C.-X.; Yu, J.-J.; Lv, X.-X.; Zhang, X.-W.; Yang, Z.-N.; Huang, B.; et al. Metformin suppresses melanoma progression by inhibiting KAT5-mediated SMAD3 acetylation, transcriptional activity and TRIB3 expression. Oncogene 2018, 37, 2967–2981. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Yang, J.; Li, H.; Zhao, H. Expression of cannabinoid receptor 2 in squamous cell carcinoma. Nan Fang Yi Ke Da Xue Xue Bao 2010, 30, 593–595. [Google Scholar] [PubMed]
- Nakajima, J.; Nakae, D.; Yasukawa, K. Structure-dependent inhibitory effects of synthetic cannabinoids against 12-O-tetradecanoylphorbol-13-acetate-induced inflammation and skin tumour promotion in mice. J. Pharm. Pharmacol. 2013, 65, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez, E.; Andradas, C.; Flores, J.M.; Quintanilla, M.; Paramio, J.M.; Guzmán, M.; Sánchez, C. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene 2013, 32, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2016, 55, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Soliman, E.; Henderson, K.L.; Danell, A.S.; Van Dross, R. Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: Role of cyclooxygenase-2 and novel J-series prostamides. Mol. Carcinog. 2016, 55, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.T. Metabolism of anandamide by COX-2 is necessary for endocannabinoid-induced cell death in tumorigenic keratinocytes. Mol. Carcinog. 2009, 48, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A.; Albayram, O.; Draffehn, A.; Michel, K.; Piyanova, A.; Oppenheimer, H.; Dvir-Ginzberg, M.; Rácz, I.; Ulas, T.; Imbeault, S.; et al. A chronic low dose of Δ9-tetrahydrocannabinol (THC) restores cognitive function in old mice. Nat. Med. 2017, 23, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A.; Drews, E.; Albayram, Ö.; Piyanova, A.; Gaffal, E.; Tueting, T.; Michel, K.; Mauer, D.; Maier, W.; Zimmer, A. Early onset of aging-like changes is restricted to cognitive abilities and skin structure in Cnr1−/− mice. Neurobiol. Aging 2012, 33, 200.e11–200.e22. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J.; Bousser, M.-G.; Fox, K.A.A.; Creager, M.A.; Despres, J.-P.; Easton, J.D.; Hamm, C.W.; Montalescot, G.; Steg, P.G.; Pearson, T.A.; et al. Rimonabant for prevention of cardiovascular events (CRESCENDO): A randomised, multicentre, placebo-controlled trial. Lancet 2010, 376, 517–523. [Google Scholar] [CrossRef]
- Lazzari, P.; Sanna, A.; Mastinu, A.; Cabasino, S.; Manca, I.; Pani, L. Weight loss induced by rimonabant is associated with an altered leptin expression and hypothalamic leptin signaling in diet-induced obese mice. Behav. Brain Res. 2011, 217, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Pira, M.; Pinna, G.A.; Pisu, C.; Casu, M.A.; Reali, R.; Marcello, S.; Murineddu, G.; Lazzari, P. NESS06SM reduces body weight with an improved profile relative to SR141716A. Pharmacol. Res. 2013, 74, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Manca, I.; Mastinu, A.; Olimpieri, F.; Falzoi, M.; Sani, M.; Ruiu, S.; Loriga, G.; Volonterio, A.; Tambaro, S.; Bottazzi, M.E.H.; et al. Novel pyrazole derivatives as neutral CB1 antagonists with significant activity towards food intake. Eur. J. Med. Chem. 2013, 62, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Pira, M.; Pani, L.; Pinna, G.A.; Lazzari, P. NESS038C6, a novel selective CB1 antagonist agent with anti-obesity activity and improved molecular profile. Behav. Brain Res. 2012, 234, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Stinchcomb, A.L.; Valiveti, S.; Hammell, D.C.; Ramsey, D.R. Human skin permeation of Delta8-tetrahydrocannabinol, cannabidiol and cannabinol. J. Pharm. Pharmacol. 2004, 56, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Von Schaper, E. Bial incident raises FAAH suspicions. Nat. Biotechnol. 2016, 34, 223. [Google Scholar] [CrossRef] [PubMed]
- Van Esbroeck, A.C.M.; Janssen, A.P.A.; Cognetta, A.B.; Ogasawara, D.; Shpak, G.; van der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; de Vrij, F.M.S.; Deng, H.; et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Brodie, J.S.; Di Marzo, V.; Guy, G.W. Polypharmacology Shakes Hands with Complex Aetiopathology. Trends Pharmacol. Sci. 2015, 36, 802–821. [Google Scholar] [CrossRef] [PubMed]


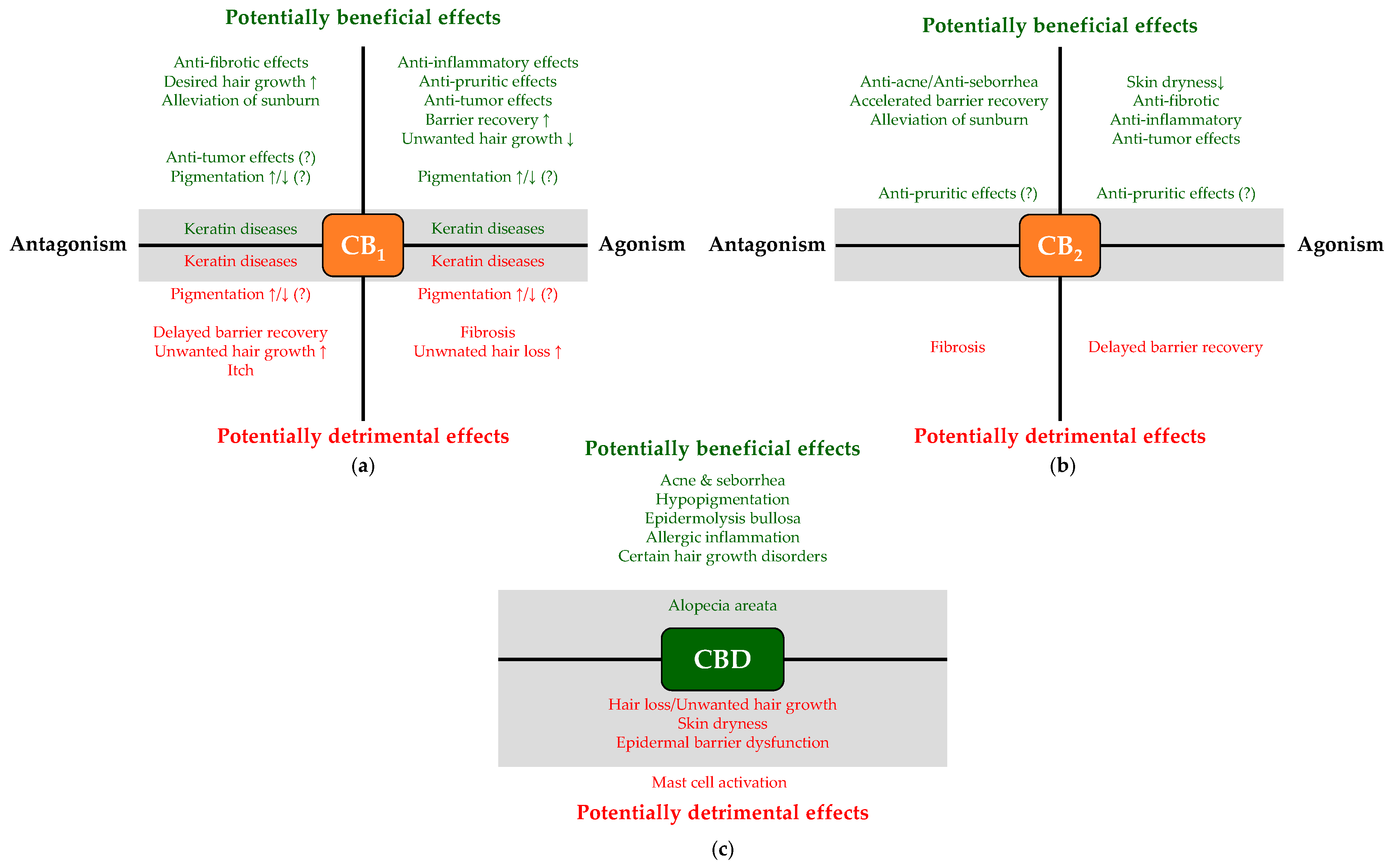

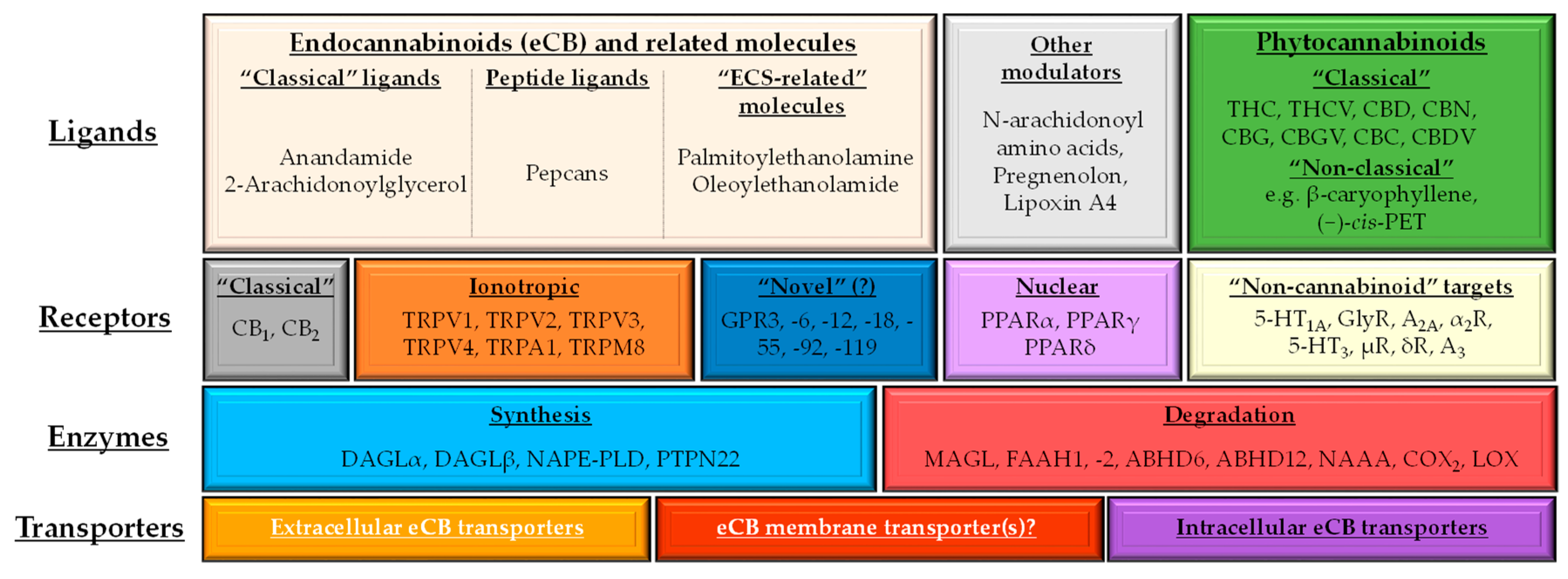{kind=link}
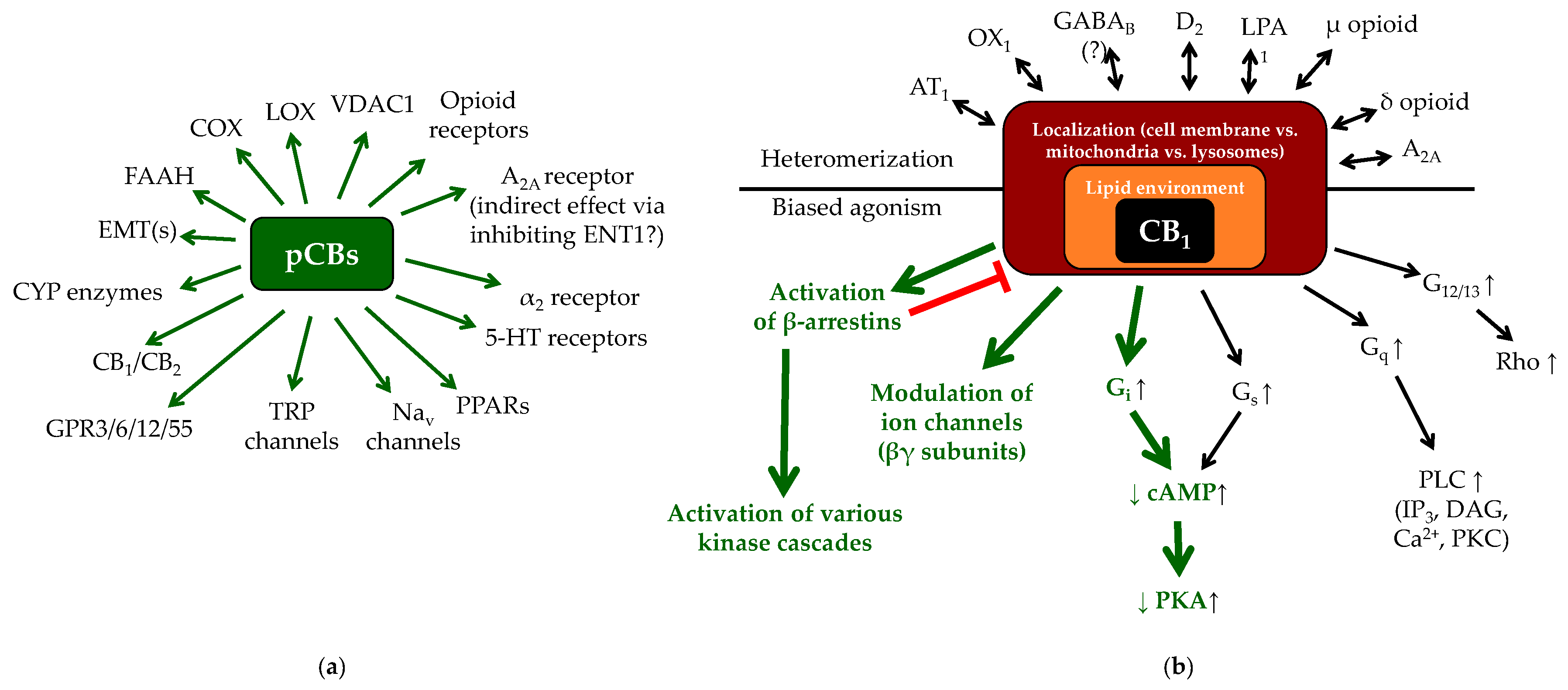{kind=link}
{kind=link}
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Dry skin | EMT-inhibition (elevation of the eCB-tone) | In vitro (cell culture) data | [112,114] |
| CBG, CBGV | In vitro (cell culture) data | [126] | |
| Acne & Seborrhea | CBD (via activating TRPV4 and A2A receptors) | In vitro (cell culture) and ex vivo (organ culture) data | [120] |
| BTX 1503 (synthetic CBD containing cream) | Successful phase Ib and ongoing phase II clinical trials | [125] ClinicalTrials.gov ID: NCT03573518 | |
| THCV, CBC, CBDV | In vitro (cell culture) data | [126] | |
| 3% Cannabis seeds extract cream | single-blind, split-face study | [124] | |
| Reduction of the eCB-tone | In vitro (cell culture) data | [112,114] | |
| GPR119-antagonism 1 | Hypothesis based on preliminary in vitro (cell culture) data | [114,116] |
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Unwanted hair growth (hirsutism, hypertrichosis) | Certain CB1 agonists | Ex vivo (organ culture) data | [130] |
| TRPV1, TRPV3 and TRPV4 activators | Ex vivo (organ culture) data | [135,136,137,138] | |
| Unwanted hair loss (different non-immune alopecia forms) | Certain CB1 antagonists/inverse agonists | Ex vivo (organ culture) and in vivo (mouse) data | [130,134] |
| TRPV1, TRPV3 and TRPV4 antagonists | Ex vivo (organ culture) data | [135,136,137,138] | |
| Alopecia areata | Elevation of the eCB-tone; certain CB1 agonists, low doses of CBD 1 | Hypothesis based on the available data | [31,130,140,146,147,148,149,150,151,152,154] |
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Hypopigmentation | Elevation of the eCB-tone/activation of CB1 (?) | In vitro (monoculture of primary human epidermal melanocytes) | [155] |
| Administration of CBD (via activating CB1) | In vitro (monoculture of primary human epidermal melanocytes) | [60] | |
| Hyperpigmentation | Elevation of the eCB-tone/activation of CB1 (?) | In vitro (co-culture of SK-mel-1 and HaCaT keratinocytes) | [160] |
| β-caryophyllene | In vitro (mono-culture of B16 melanoma cells) | [158] | |
| Vitiligo | Elevation of the eCB-tone 1 | Hypothesis based on literature data | [164,165,166,167] |
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Epidermolysis bullosa | Topical CBD | Case report of 3 patients | [183] |
| Sublingual THC and CBD containing CBM oil | Case report of 3 patients | [184] | |
| Pachyonychia congenita | ACEA (and maybe other CB1 agonists) | Ex vivo (hSOC) | [175] |
| Epidermolytic ichthyosis | ACEA (and maybe other CB1 agonists) | Pilot ex vivo (hSOC) | [176] |
| Barrier disruption | CB1 activation and/or CB2 blockade | In vivo (CB1−/− and CB2−/− mice) | [177] |
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Sunburn | CB1&CB2 antagonism (?) 1 | Cell culture, as well as KO-validated animal data | [87] |
| TRPV4 antagonism | Cell culture, as well as KO-validated animal data | [86] | |
| Allergic inflammation, atopic dermatitis (AD) | CB1 and/or CB2 agonism; FAAH-inhibition | Cell culture, as well as KO-validated animal data | [143,144,186,187,191,192] |
| Topical CBC, CBCV, CBD, CBDV, Δ8-THCV, Δ8-THC, Δ9-THC | In vivo mouse data | [189] | |
| TRPV3 blockade or desensitization | Cell culture data | [119,121] | |
| Echinacea purpurea-derived alkylamides | Cell culture data and clinical trials | [199] | |
| PEA | Cell culture data, animal data and human clinical trials | [200,201,202,205] | |
| CB2 blockade (?) 1 | Animal data | [214] | |
| Excessive MC activity | PEA | Cell culture data | [226] |
| Ex vivo dog skin organ culture data | [227] | ||
| PEA-OXA (NAAA-inhibititor) | Animal data | [229] | |
| Activation of CB1 | Cell culture data | [232,236,237] | |
| Ex vivo human HF and nasal polyp organ culture data | [245,246] | ||
| Activation of CB2 | Cell culture data | [232,236,242,243] | |
| Activation of PPARγ | Cell culture data | [242,243] | |
| TRPV3 blockade or desensitization 2 | Hypothesis predicted based on animal data | [244] | |
| PSO | CB1 activators (e.g., ACEA) via suppressing hyper-proliferation and K6 & K16 expression | Cell culture as well as ex vivo hSOC data | [174,175,182] |
| NRIP1↓ | Cell culture as well as NRIP1−/− mice data | [277] | |
| TRIB3↓ | Cell culture data | [279] | |
| AD | TRPV1 antagonism | Ongoing phase II and III clinical trials | [293] |
| TRPV3 antagonism or desensitization (candidate: CBGV) 2 | Cell culture data | [119,121] | |
| FAAH-inhibition | Animal data | [144] | |
| CB1 activators | Animal data | [178] | |
| CB2 activators | Clinical study | [199] | |
| Animal data | [294] | ||
| CB2 antagonists (?) 1 | Animal data | [295] | |
| NAAA-inhibitors or PPARα agonists | Animal data | [297,298,299] | |
| PEA | Human clinical studies | [300,301] | |
| EMT-inhibition 2 | Hypothesis based on cell culture data | [114] | |
| CBG, CBGV 2 | Hypothesis based on cell culture data | [126] | |
| SSc | TRPV4 blockade | Animal data | [324] |
| CB1 antagonism | Animal data | [325] | |
| Cell culture data | [75] | ||
| A2A antagonism | Cell culture data | [75] | |
| CB2 activators | Cell culture data | [75] | |
| Animal data | [328] | ||
| KO-validated animal data | [329,330] | ||
| AJA (CB2 and PPARγ activator) | Cell culture data | [332] | |
| Animal data | [333] | ||
| Completed phase II clinical trial, ongoing phase III trial | NCT02465437 NCT03398837 [319] | ||
| VCE-004.3 (CB2 and PPARγ activator; CB1 antagonist) | Cell culture and animal data | [242] | |
| VCE-004.8/EHP-101 (CB2 and PPARγ activator) | Cell culture and animal data | [243,334] | |
| TRIB3↓ | Animal data | [336] | |
| Pepcan-12 or THCV 2 | Hypothesis based on the available data | [57,338] |
| Condition | Intervention | Level of Evidence | References |
|---|---|---|---|
| Excisional wound | FAAH-inhibition and the subsequent elevation of N-acyl taurines | Animal data | [348] |
| Full-thickness wound | TRPV3 activation | Animal data | [354] |
| In vitro wound models | TRPV2 antagonism | Cell culture data | [355,356] |
| EB | Topical CBD | Case report of 3 patients | [183] |
| Condition | Intervention | Level of Evidence | References |
|---|---|---|---|
| Intractable cholestasis related itch | Dronabinol (5 mg at bedtime) | Pilot clinical data of 3 patients | [365] |
| Various itch models | CB1 activation | Animal data | [366,368] |
| Human study | [370] | ||
| CB2 activation | Animal data | [371] | |
| CB2 blockade (?) 1 | Animal data | [295] | |
| FAAH- and/or MAGL-inhibition | Animal data | [372,375] | |
| NAAA-inhibition | Animal data | [297] | |
| PEA | Animal data | [202,299] | |
| Human clinical data | [300,377] |
| Disease | Intervention | Level of Evidence | References |
|---|---|---|---|
| Melanoma | CB1 activation | Cell culture data | [390] |
| Cell culture data | [392] | ||
| Cell culture data | [394] | ||
| Animal data | [394] | ||
| CB1 antagonism (?) 1 | Cell culture data | [382] | |
| CB2 activation | Cell culture data | [392] | |
| Animal data | [392] | ||
| Cell culture data | [398] | ||
| GPR18 blockade | Cell culture data | [397] | |
| GPR55 activation | Cell culture data | [390] | |
| PEA | Cell culture data | [391] | |
| Animal data | [391] | ||
| β-caryophyllene | Cell culture data | [406] | |
| Animal data | [407] | ||
| THC | Cell culture data | [392] | |
| Cell culture data and animal data | [409] | ||
| Cell culture data | [410] | ||
| Animal data | [410] | ||
| THC+CBD | Cell culture data | [410] | |
| Animal data | [410] | ||
| Non-melanoma tumors | CB1 activation | Cell culture data | [133] |
| Animal data | [133] | ||
| CB2 activation | Cell culture data | [133] | |
| Animal data | [133] | ||
| GPR55 blockade | Animal data | [415] | |
| CB1/CB2 blockade (?) 1 | Animal data | [87] | |
| AEA administration | Cell culture data | [416,417,418] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, K.F.; Ádám, D.; Bíró, T.; Oláh, A. Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System. Molecules 2019, 24, 918. https://doi.org/10.3390/molecules24050918
Tóth KF, Ádám D, Bíró T, Oláh A. Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System. Molecules. 2019; 24(5):918. https://doi.org/10.3390/molecules24050918
Chicago/Turabian StyleTóth, Kinga Fanni, Dorottya Ádám, Tamás Bíró, and Attila Oláh. 2019. "Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System" Molecules 24, no. 5: 918. https://doi.org/10.3390/molecules24050918
APA StyleTóth, K. F., Ádám, D., Bíró, T., & Oláh, A. (2019). Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System. Molecules, 24(5), 918. https://doi.org/10.3390/molecules24050918