Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives


Abstract
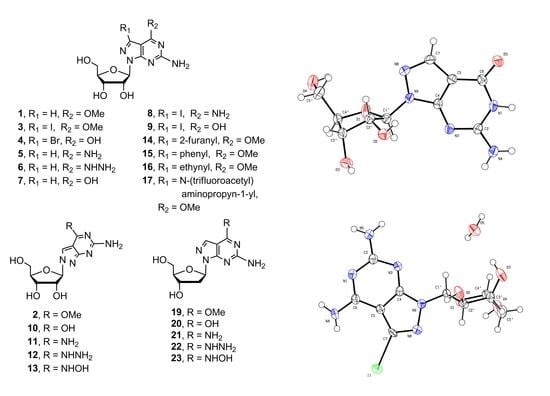
:
1. Introduction
2. Results and Discussion
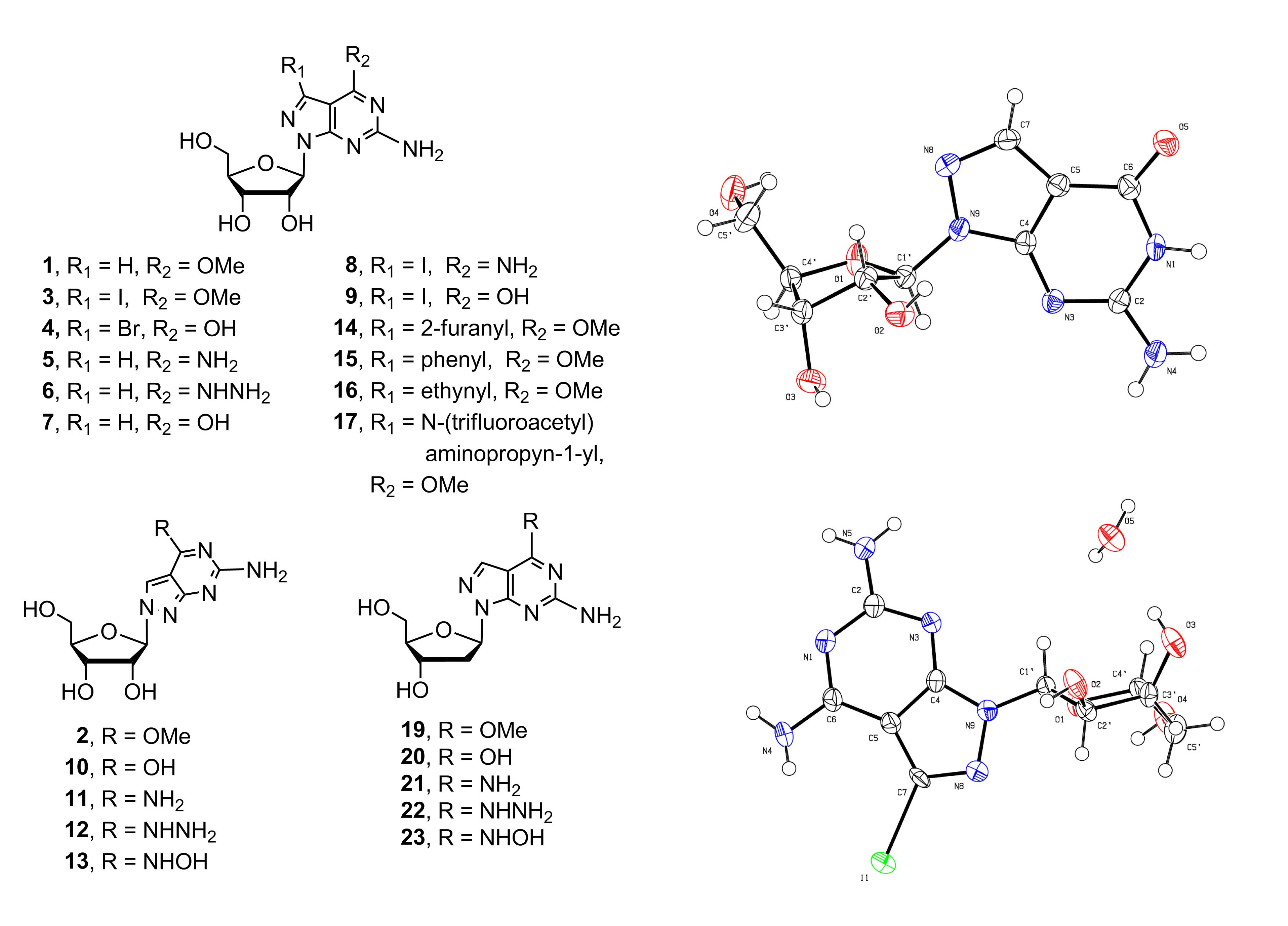
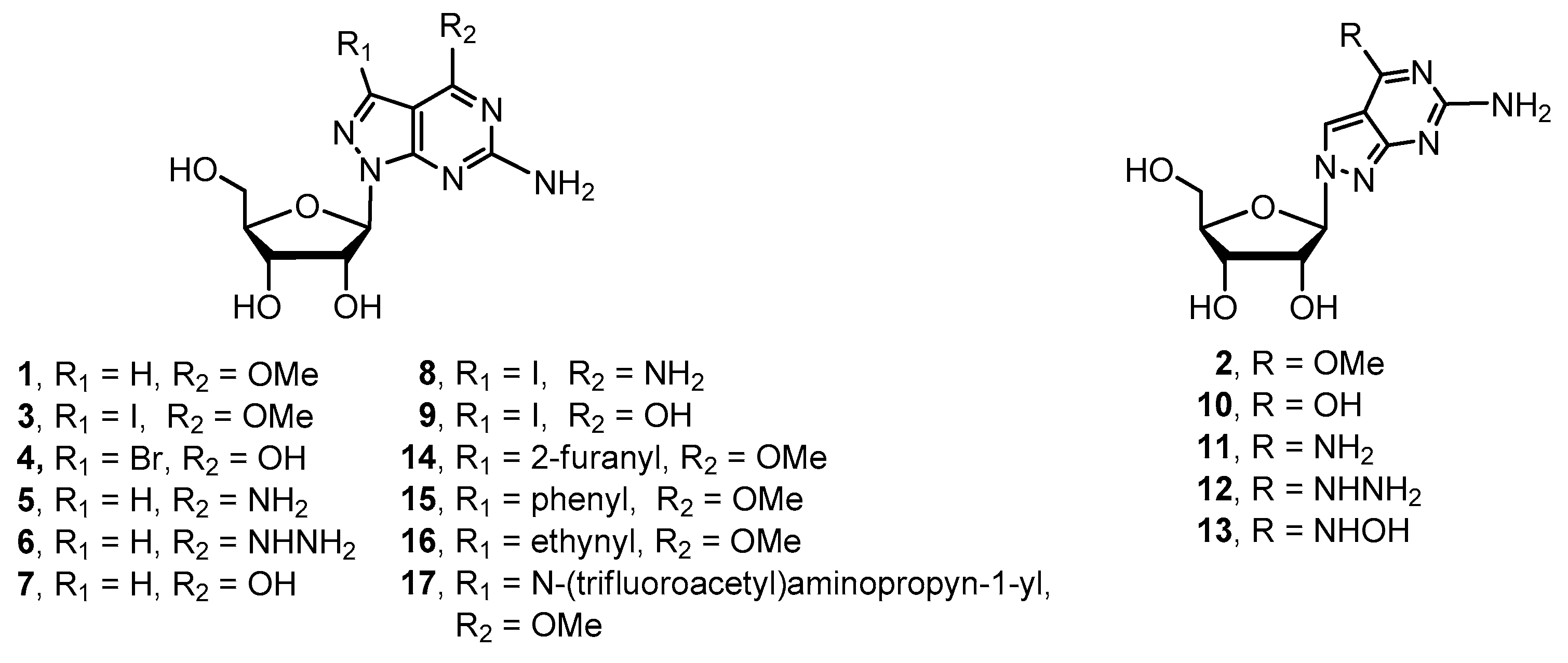
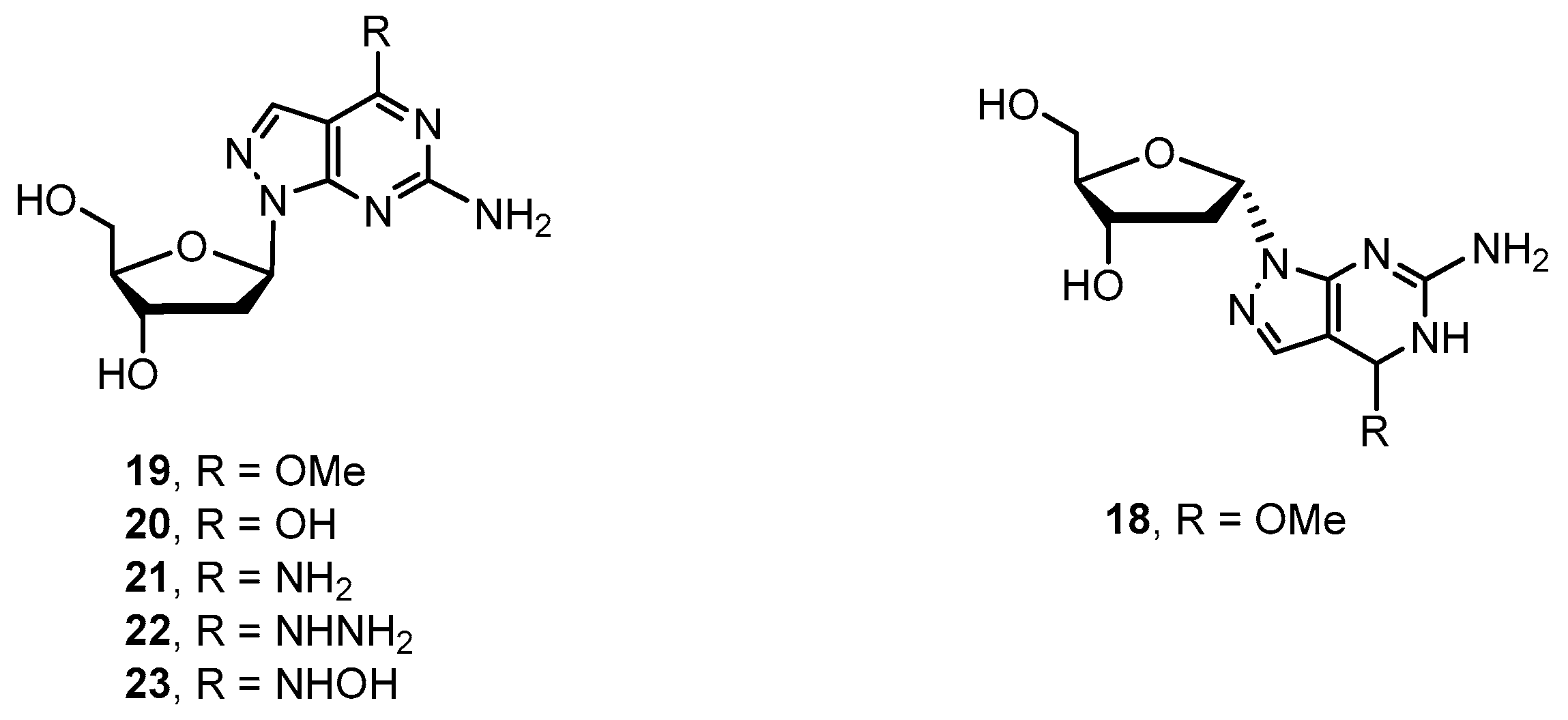
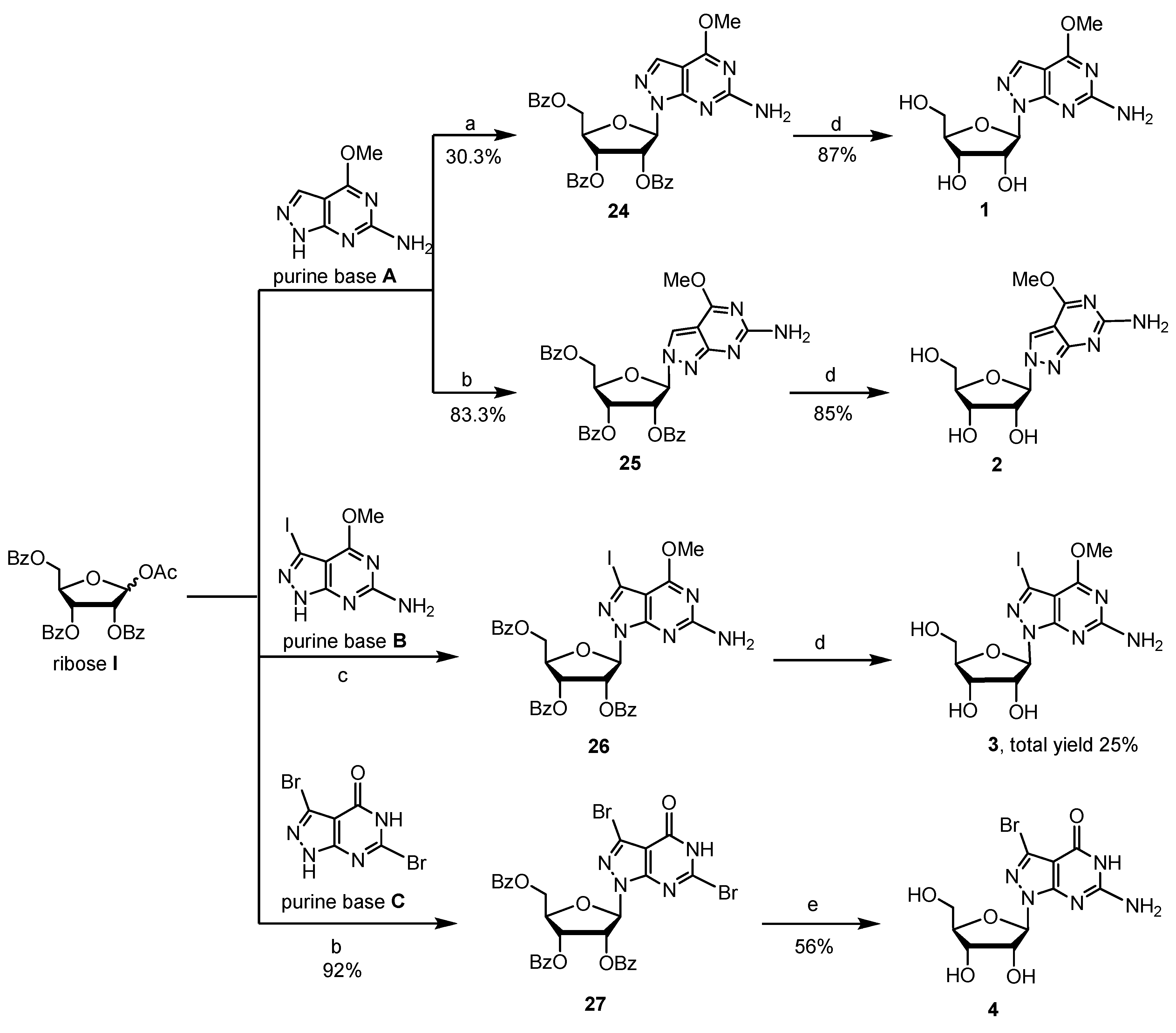
2.1. Synthesis of 8-Aza-7-deazapurine Ribo-Nucleosides
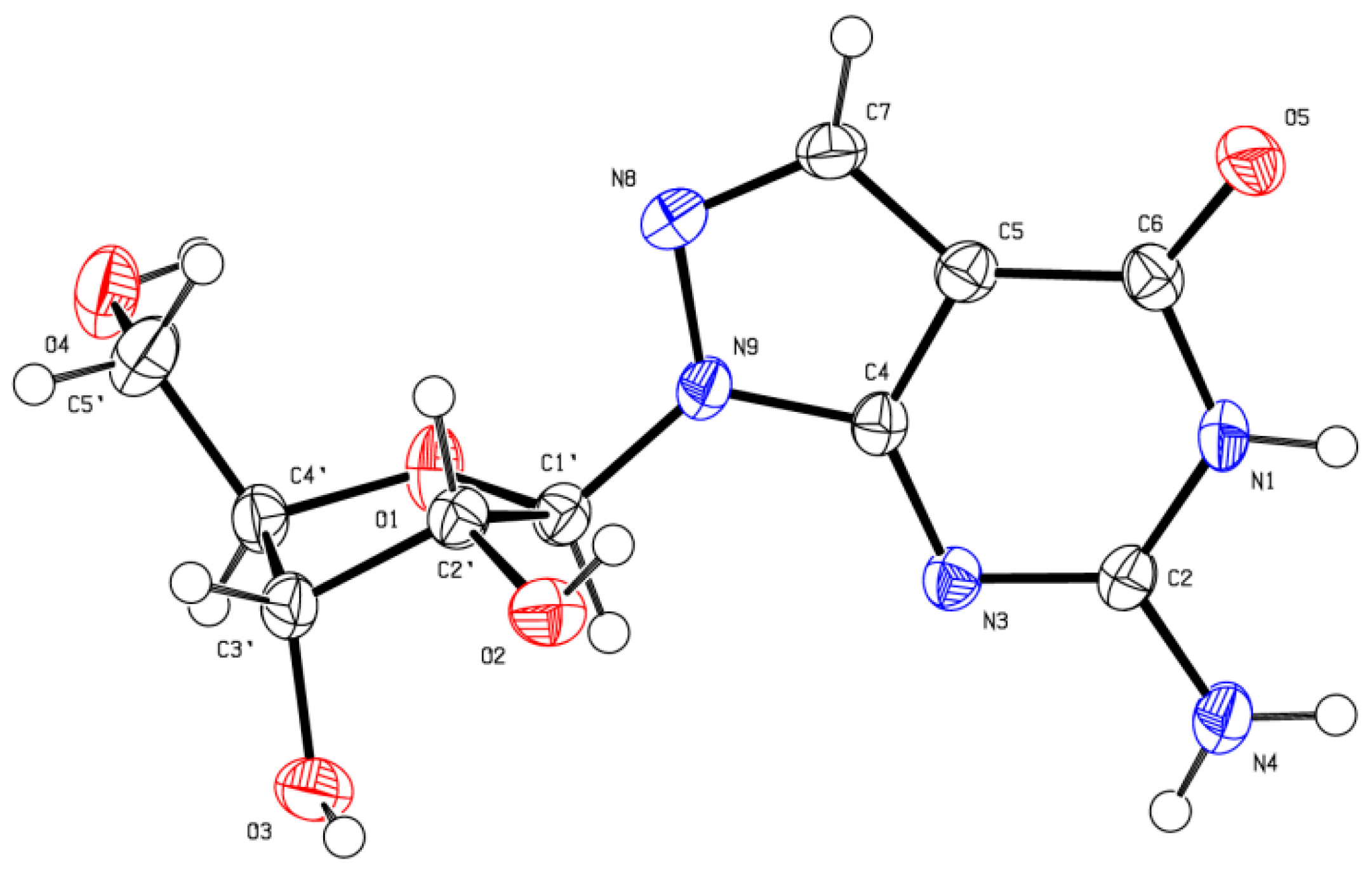
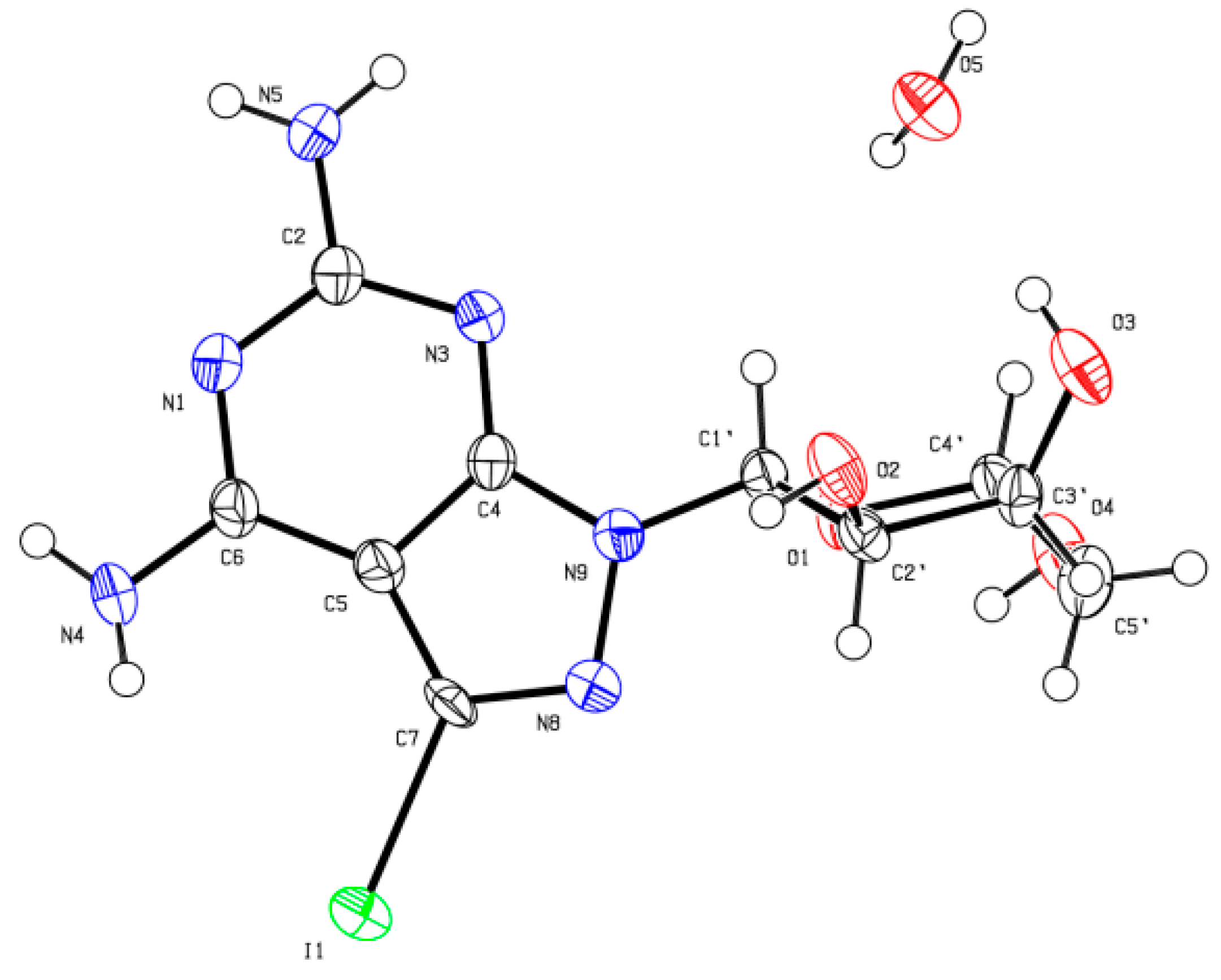
X-ray Crystallographic Study
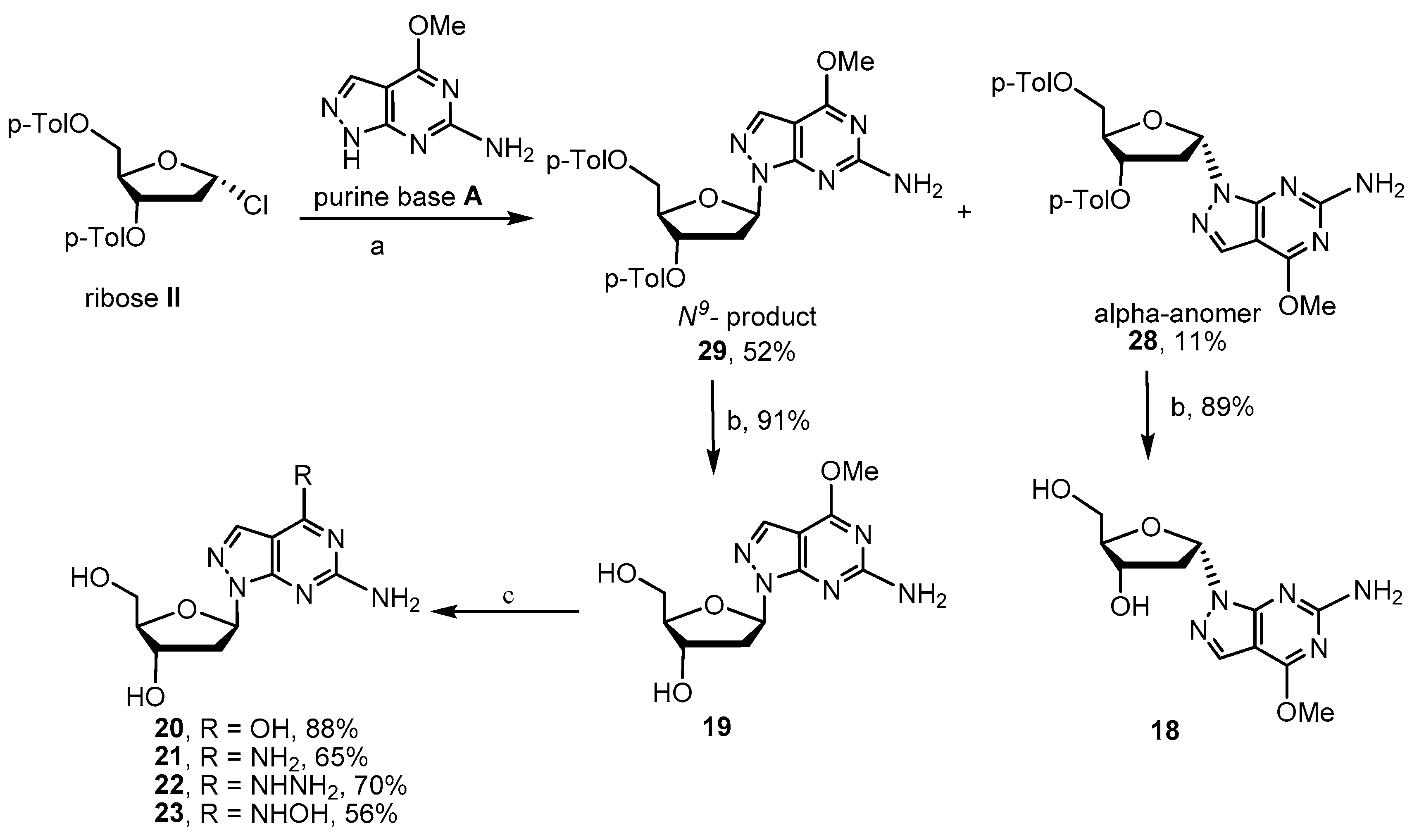
2.2. Synthesis of 8-Aza-7-deaza-2′-deoxy Purine Nucleosides
3. Biological Evaluation
4. Materials and Methods
4.1. Materials and Reagents
4.2. Chemical Synthesis
4.2.1. Procedure for Preparation of Glycosylated Products 24–26 and Key Intermediate 3
4.2.2. Procedure for Preparation of Glycosylated Product 27 and Compound 4
4.2.3. Procedure for Preparation of Key Intermediates 1 and 2
4.2.4. Procedure for Preparation of Target Compounds 5–17
4.2.5. Procedure for Preparation of Glycosylated Products 28–29
4.2.6. Procedure for Preparation of Key Intermediates 18 and 19
4.2.7. Procedure for Preparation of Target Compounds 20–23
4.3. Proliferation Inhibitory Effect Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cottam, H.B.; Wasson, D.B.; Shih, H.C.; Raychaudhuri, A.; Di Pasquale, G.; Carson, D.A. New adenosine kinase inhibitors with oral anti-inflammatory activity: Synthesis and biological evaluation. J. Med. Chem. 1993, 36, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.P., III; Howard, B.C.; Patricia, A.M.; Roland, K.R.; Ganapathi, R.R. Synthesis and biological activity of 6-azacadeguomycin and certain 3,4,6-trisubstituted pyrazolo[3,4-d]pyrimidine ribonucleosides. J. Med. Chem. 1985, 28, 1010–1016. [Google Scholar] [CrossRef]
- Michael, A.P.; Marr, J.J. Antileishmanial effect of allopurinol. Antimicrob. Agents Chemother. 1974, 5, 469–472. [Google Scholar] [CrossRef]
- Garaeva, L.D.; Korbukh, I.A.; Dobrynin, Y.V.; Nikolaeva, T.G.; Preobrazhenskaya, M.N. Synthesis and cytotoxic activity of 4,6-diaminopyrazolo[3,4-d] pyrimidine riboside and its 3-carbamoyl derivative. Pharm. Chem. J. 1988, 22, 523–526. [Google Scholar] [CrossRef]
- Junlin, H.; Mikhailopulo, I.; Seela, F. 3-Bromopyrazolo[3,4-d]pyrimidine 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides: Modified DNA constituents with an unusually rigid sugar N-conformation. J. Org. Chem. 2003, 68, 5519–5524. [Google Scholar] [CrossRef]
- Brian, C.K.; Lisa, L.E.; Justin, D.B.; David, O.M.; Kevan, M.S. Inhibitor scaffolds as new allele specific kinase substrates. J. Am. Chem. Soc. 2002, 124, 12118–12128. [Google Scholar] [CrossRef]
- Ilja, V.F.; Maria, I.K.; Konstantin, V.A.; Igor, A.M. Recognition of artificial nucleobases by E. coli purine nucleoside phosphorylase versus its Ser90Ala mutant in the Synthesis of base-modified nucleosides. Chem. Eur. J. 2015, 21, 13401–13419. [Google Scholar] [CrossRef]
- Wenqing, L.; Kuiying, X.; Seela, F. 7-Substituted 8-aza-7-deazapurines and 2,8-diaza-7-deaza-purines: Synthesis of nucleosides and oligonucleotides. Nucleosides Nucleotides Nucleic Acids 2005, 24, 869–873. [Google Scholar] [CrossRef]
- Junlin, H.; Seela, F. Oligonucleotides incorporating 8-aza-7-deazapurines: synthesis and base pairing of nucleosides with nitrogen-8 as a glycosylation position. Org. Biomol. Chem. 2003, 1, 1873–1883. [Google Scholar] [CrossRef]
- Seela, F.; Kuiying, X. Pyrazolo[3,4-d]pyrimidine ribonucleosides related to 2-aminoadenosine and isoguanosine: synthesis, deamination and tautomerism. Org. Biomol. Chem. 2007, 5, 3034–3045. [Google Scholar] [CrossRef] [PubMed]
- Tiannan, H.; Sutera, S.R.; Mumbleaua, M.M.; Beal, P.A. TLR8 activation and inhibition by guanosine analogs in RNA: Importance of functional groups and chain length. Bioorg. Med. Chem. 2018, 26, 77–83. [Google Scholar] [CrossRef]
- Seela, F.; Suresh, S.P. Hydrogelation and spontaneous fiber formation of 8-aza-7-deazaadenine nucleoside ‘click’ conjugates. Tetrahedron 2011, 67, 7418–7425. [Google Scholar] [CrossRef]
- Seela, F.; Pujari, S.S. Azide-alkyne “Click” conjugation of 8-aza-7-deazaadenine-DNA: Synthesis, duplex stability, and fluorogenic dye labeling. Bioconjugate Chem. 2010, 21, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Yuxuan, Z.; Beal, P.A. Synthesis and evaluation of an alkyne-modified ATP analog for enzymatic incorporation into RNA. Bioorg. Med. Chem. Lett. 2016, 26, 1799–1802. [Google Scholar] [CrossRef]
- Yang, L.; Zhiwen, L.; Gaofeng, L.; Keliang, L.; Junlin, H. Breaking the conservation of guanine residues in the catalytic loop of 10–23 DNAzyme by position-specific nucleobase modifications for rate enhancement. Chem. Commun. 2013, 49, 5037–5039. [Google Scholar] [CrossRef]
- Seela, F.; Steker, H. Synthesis of 2′-deoxyribofuranosides of 8-aza-7-deazaguanine and related pyrazolo[3,4-d]pyrimidines. Helv. Chim. Acta 1986, 69, 1602–1613. [Google Scholar] [CrossRef]
- Seela, F.; Becher, G. Synthesis of 7-halogenated 8-aza-7-deaza-2′-deoxyguanosines and related pyrazolo[3,4-d]pyrimidine 2′-deoxyribonucleosides. Synthesis 1998, 207–214. [Google Scholar] [CrossRef]
- Bontems, R.J.; Anderson, J.D.; Cottam, H.B. Guanosine analogues. Synthesis ofnucleosides of certain 3-substituted 6-aminopyrazolo[3,4-d]pyrimidin-4(5H)-ones as potential immunotherapeutic agents. J. Med. Chem 1990, 33, 2174–2178. [Google Scholar] [CrossRef] [PubMed]
- Seela, F.; Xiaohua, P. 7-Functionalized 7-deazapurine ribonucleosides related to 2-aminoadenosine, guanosine, and xanthosine: Glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-D-ribofuranose. J. Org. Chem. 2006, 71, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Haixin, D.; Yanhui, D.; Ruchun, Y.; Qi, S.; Qiang, X.; Yong, J. Efficient and practical synthesis of 5′-deoxytubercidin and its analogues via vorbrüggen glycosylation. Synthesis 2011, 9, 1442–1446. [Google Scholar] [CrossRef]
- Seela, F.; Becher, G.; Rosemeyer, H.; Reuter, H.; Kastner, G.; Igor, A.M. The high-anti conformation of 7-halogenated 8-aza-7-deaza-2′-deoxyguanosines: A study of the influence of modified bases on the sugar structure of nucleosides. Helv. Chim. Acta 1999, 82, 105–124. [Google Scholar] [CrossRef]
- Becher, G.; He, J.; Seela, F. Major-Groove-Halogenated DNA: The effects of bromo and iodo substituents replacing H-C(7) of 8-aza-7-deazapurine-2,6-diamine or H-C(5) of uracil residues. Helv. Chim. Acta 2001, 84, 1048–1065. [Google Scholar] [CrossRef]
- Yoshinaga, Y.; Kenzo, F. Ultrafast reversible photo-cross-linking reaction: Toward in situ DNA manipulation. Org. Lett. 2008, 10, 3227–3230. [Google Scholar] [CrossRef]
- Hall, L.M.; Gerowska, M.; Brown, T. A highly fluorescent DNA toolkit: Synthesis and properties of oligonucleotides containing new Cy3, Cy5 and Cy3B monomers. Nucleic Acids Res. 2012, 40, e108. [Google Scholar] [CrossRef] [PubMed]
- Ilirian, D.; John, S.J. Efficient preparation of 2-deoxy-3,5-di-O-p-toluoyl-α-D-ribofuranosyl chloride. Synlett 2004, 335–337. [Google Scholar] [CrossRef]
- He, Z.; Yuqi, C.; Yafen, W.; Yuhao, D.; Xiang, Z. A rapidly photo-activatable light-up fluorescent nucleoside and its application in DNA base variation sensing. Chem. Commun. 2016, 52, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–23 are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax/nm (Methanol) |
|---|---|
| Purine base A | 223, 250, 280 |
| Compound 1 | 222, 252, 276 |
| Purine base B | 231, 278 |
| Compound 3 | 232, 278 |
| Compound | A549 | MDA-MB-231 | ||
|---|---|---|---|---|
| Cytotoxicity IC50 (μM) | 100 μM IC% a | Cytotoxicity IC50 (μM) | 100 μM IC% a | |
| 2 | >100 | 41.42 | >100 | 2.69 |
| 6 | >100 | 17.56 | >100 | 17.81 |
| 8 | 7.68 | 61.44 | >100 | 14.04 |
| 9 | >100 | 43.78 | >100 | 14.14 |
| 10 | >100 | 26.76 | >100 | 10.32 |
| 11 | >100 | 39.18 | >100 | 10.69 |
| 12 | >100 | 21.98 | >100 | 5.55 |
| 13 | >100 | 27.25 | >100 | 11.59 |
| 14 | 100 | 56.18 | >100 | 6.41 |
| 16 | >100 | 39.50 | 81.72 | 54.70 |
| 18 | >100 | 37.18 | >100 | 25.67 |
| 19 | >100 | 27.31 | >100 | 3.04 |
| 22 | >100 | 18.64 | >100 | 7.64 |
| 23 | >100 | 19.63 | >100 | 10.37 |
| DOX | 0.019 | 84.32 | 0.001 | 95.94 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, H.; An, H.; Tao, J. Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives. Molecules 2019, 24, 983. https://doi.org/10.3390/molecules24050983
Ren H, An H, Tao J. Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives. Molecules. 2019; 24(5):983. https://doi.org/10.3390/molecules24050983
Chicago/Turabian StyleRen, Hang, Haoyun An, and Jingchao Tao. 2019. "Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives" Molecules 24, no. 5: 983. https://doi.org/10.3390/molecules24050983
APA StyleRen, H., An, H., & Tao, J. (2019). Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives. Molecules, 24(5), 983. https://doi.org/10.3390/molecules24050983