3.1. Chemistry
All reagents and solvents used are commercially available and were used without further purification. 1H-NMR spectra were recorded in CDCl3 or DMSO-d6 on a Bruker Avance III 400MHz or a WNMR-1 500 MHz spectrometer (Wuhan Zhongke-Niujin, Wuhan, China). The chemical shift was reported in parts per million, relative to tetramethylsilane as the internal standard. High Resolution Mass Spectrum (HRMS) was recorded on a Triple TOF 5600+LC/MS/MS system (CADM-YQ-086) with an Electrospray Ionization (ESI) mass selective detector. The reaction was monitored through TLC Silica gel 60F254 Aluminium plate (Merck, 60F254D, Darmstadt, Germany). Flash chromatography was performed on a CombiflashRf 200 (Teledyne, Lincoln, NE, USA) with gel silica column.
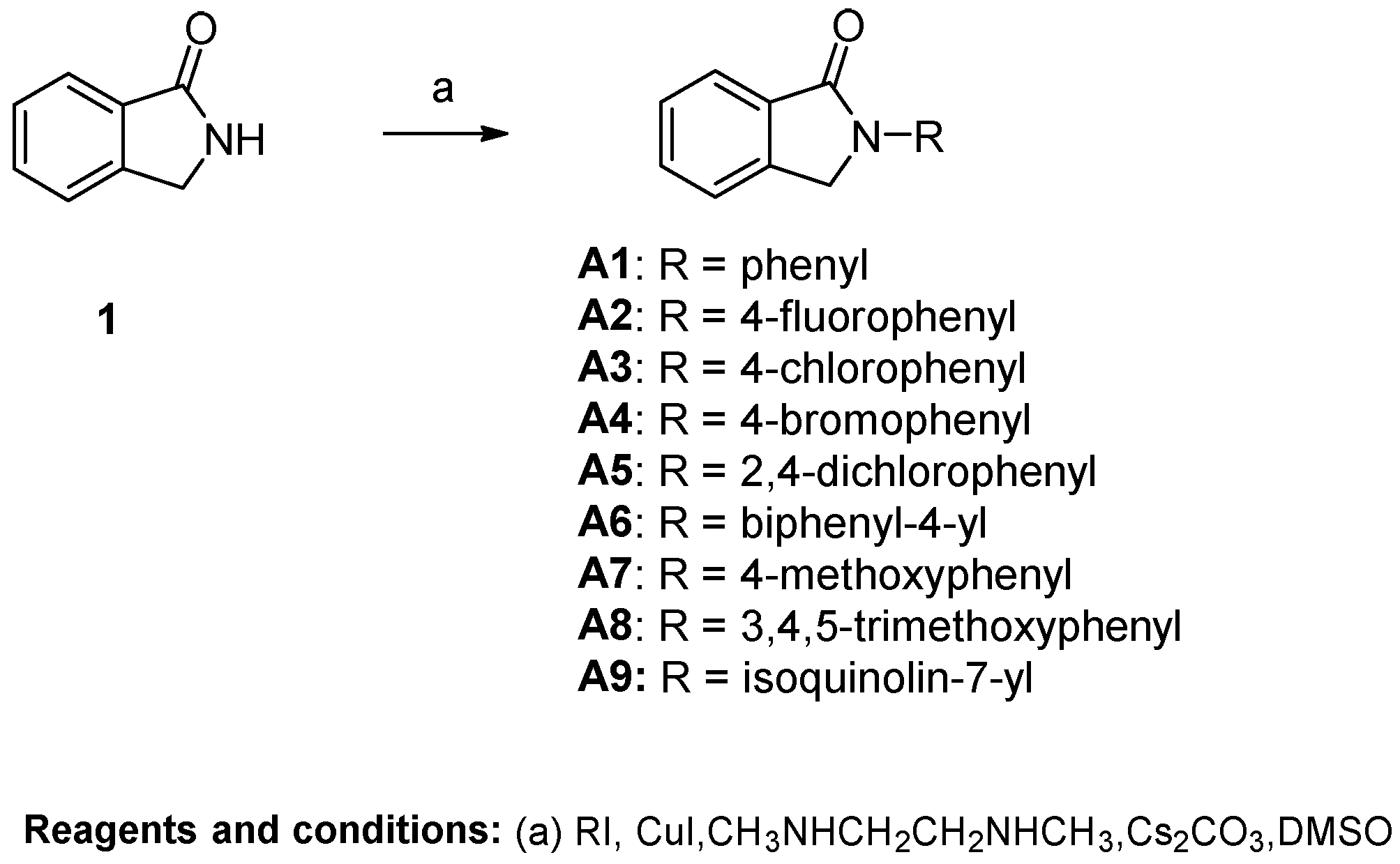
General Procedure A for the Synthesis of Compounds A1~A12. Isoindolin-1-one (200 mg, 1.50 mmol) was dissolved in super-dry DMSO (4 mL), and Cs2CO3 (1215 mg, 3.75 mmol), CuI (58 mg, 0.30 mmol) and N1,N2-dimethylethane-1,2-diamine (27 mg, 33 μL, 0.30 mmol) were added to the solution. The resulting mixture was stirred at room temperature for 10 min, after which iodobenzene (2.25 mmol) was added. Then the mixture was heated to 120 °C. When TLC showed that isoindolin-1-one had been fully converted, the reaction was stopped. The mixture was extracted with ethyl acetate (20 mL) and H2O (10 mL). The water phase was re-extracted with ethyl acetate (20 mL). The organic layer was combined and washed with brine (10 mL). Then the solution was dried over anhydrous MgSO4, filtered and concentrated, and the crude residue was purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient elution to afford the title compounds.
22-phenylisoindolin-1-one (A1). Compound A1 was synthesized from isoindolin-1-one and iodobenzene using general procedure A as a white solid. The yield was 50%. 1H-NMR (500 MHz, Chloroform-d) δ 7.98 (d, J = 7.4 Hz, 1H), 7.92 (d, J = 7.8 Hz, 2H), 7.64 (t, J = 7.5 Hz, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 7.3 Hz, 1H), 4.91 (s, 2H). HRMS(ESI+) m/z calcd for C14H12NO [M + H]+ 210.0919, found 210.0921.
2-(4-fluorophenyl)isoindolin-1-one (A2). Compound A2 was synthesized from isoindolin-1-one and 1-fluoro-4-iodobenzene using general procedure A as a yellow solid. The yield was 32%. 1H-NMR (500 MHz, DMSO-d6) δ 7.98 (dd, J = 8.8, 4.8 Hz, 2H), 7.84 (d, J = 7.5 Hz, 1H), 7.73 (s, 2H), 7.61 (d, J = 7.4 Hz, 1H), 7.35 (t, J = 8.7 Hz, 2H), 5.08 (s, 2H). HRMS(ESI+) m/z calcd for C14H11FNO [M + H]+ 228.0825, found 228.0827.
2-(4-chlorophenyl)isoindolin-1-one (A3). Compound A3 was synthesized from isolindolin-1-one and 1-chloro-4-iodobenzene using general procedure A as a white solid. The yield was 49%. 1H-NMR (500 MHz, Chloroform-d) δ 7.97 (d, J = 7.7 Hz, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.57 (d, J = 7.1 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 4.89 (s, 2H). HRMS(ESI+) m/z calcd for C14H11NOCl [M + H]+ 244.0529, found 244.0522.
2-(4-bromophenyl)isoindolin-1-one (A4). Compound A4 was synthesized from isoindolin-1-one and 1-bromo-4-iodobenzene using general procedure A as a brown solid. The yield was 44%. 1H-NMR (500 MHz, DMSO-d6) δ 7.93 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 7.6 Hz, 1H), 7.75–7.62 (m, 4H), 7.58 (t, J = 7.3 Hz, 1H), 5.06 (s, 2H). HRMS(ESI+) m/z calcd for C14H11BrNO [M + H]+ 288.0024, found 288.0026.
2-(2,4-dichlorophenyl)isoindolin-1-one (A5). Compound A5 was synthesized from isolindolin-1-one and 2,4-dichloro-1-iodobenzene using general procedure A as a white solid. The yield was 48%. 1H-NMR (500 MHz, Chloroform-d) δ 8.01 (d, J = 7.6 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.58 (dd, J = 12.0, 5.9 Hz, 3H), 7.43 (t, J = 6.5 Hz, 2H), 4.85 (s, 2H). HRMS(ESI+) m/z calcd for C14H10NOCl2 [M + H]+ 278.0139, found 278.0132.
2-([1,1’-biphenyl]-4-yl)isoindolin-1-one (A6). Compound A6 was synthesized from isoindolin-1-one and 4-iodo-1,1’-biphenyl using general procedure A as a yellow solid. The yield was 26%. 1H-NMR (500 MHz, DMSO-d6) δ 8.05 (d, J = 8.3 Hz, 2H), 7.84 (d, J = 7.6 Hz, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.73 (t, J = 5.6 Hz, 4H), 7.60 (d, J = 7.1 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 5.12 (s, 2H). HRMS(ESI+) m/z calcd for C20H16NO [M + H]+ 286.1232, found 286.1220.
2-(4-methoxyphenyl)isoindolin-1-one (A7). Compound A7 was synthesized from isolindolin-1-one and 1-iodo-4-methoxybenzene using general procedure A as a white solid. The yield was 53%. 1H-NMR (500 MHz, Chloroform-d) δ 7.96 (d, J = 7.6 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.62 (t, J = 7.4 Hz, 1H), 7.54 (s, 2H), 7.01 (d, J = 8.5 Hz, 2H), 4.85 (s, 2H), 3.87 (s, 3H). HRMS(ESI+) m/z calcd for C15H14NO2 [M + H]+ 240.1025, found 240.1020.
2-(3,4,5-trimethoxyphenyl)isoindolin-1-one (A8). Compound A8 was synthesized from isoindolin-1-one and 3,4,5-trimethoxy-1-iodobenzene using general procedure A as a yellow solid. The yield was 37%. 1H-NMR (500 MHz, DMSO-d6) δ 7.77 (d, J = 7.6 Hz, 1H), 7.69 (dd, J = 6.7, 1.0 Hz, 2H), 7.60–7.51 (m, 1H), 7.28 (s, 2H), 5.05 (s, 2H), 3.83 (s, 6H), 3.67 (s, 3H). HRMS(ESI+) m/z calcd for C17H18NO4 [M + H]+ 300.1236, found 300.1222.
2-(isoquinolin-7-yl)isoindolin-1-one (A9). Compound A9 was synthesized from isoindolin-1-one and 7-iodoisoquinoline using general procedure A as a yellow solid. The yield was 48%. 1H-NMR (500 MHz, Chloroform-d) δ 9.34 (s, 1H), 8.56 (d, J = 9.0 Hz, 2H), 8.32 (s, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.69 (s, 2H), 7.66–7.50 (m, 2H), 5.05 (s, 2H). HRMS(ESI+) m/z calcd for C17H13N2O [M + H]+ 261.1028, found 261.1034.
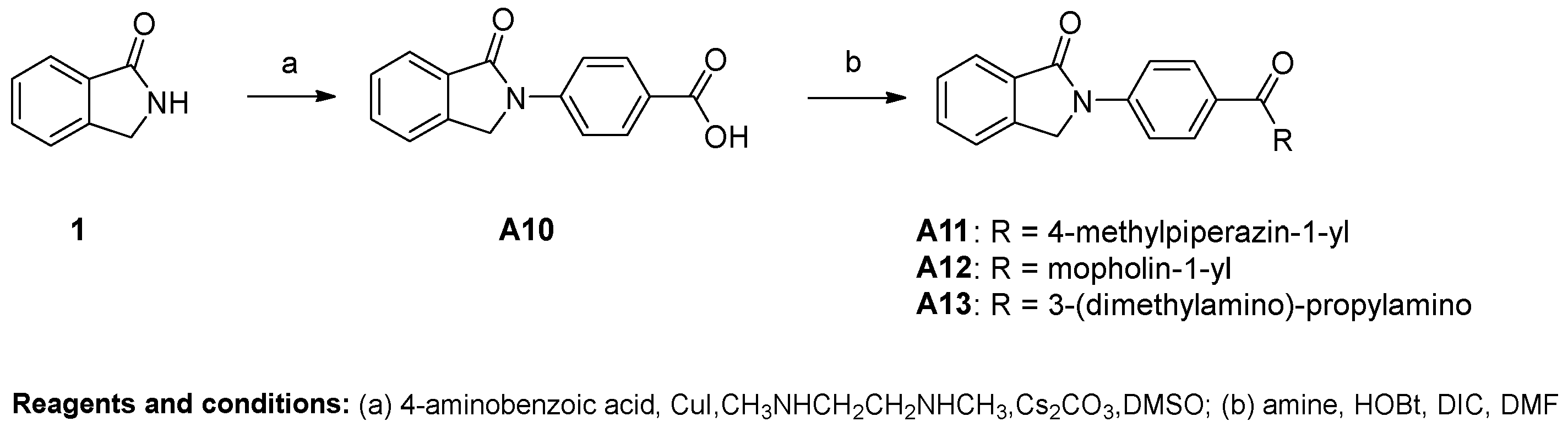
4-(1-oxoisoindolin-2-yl)benzoic acid (A10). Compound A10 was synthesized from isoindolin-1-one and 4-iodobenzoic acid using general procedure A as a white solid. The yield was 31%. 1H-NMR (500 MHz, DMSO-d6) δ 12.87 (s, 1H), 8.11–8.05 (m, 1H), 8.02 (dd, J = 9.1, 2.2 Hz, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.75–7.67 (m, 1H), 7.59–7.54 (m, 0H), 5.09 (s, 1H). HRMS(ESI+) m/z calcd for C15H12NO3 [M + H]+ 254.0817, found 254.0805
General Procedure for the Synthesis of Compounds A11–A13. Compound A10 (100 mg, 0.3 mmol) was dissolved in DMF (3 mL). HOBt (69 mg, 0.4 mmol) and DIC (80 μL, 0.4 mmol) were added to the solution and the resulting solution was stirred for 20 min at room temperature. Amine was added into the resulting mixture and stirred at room temperature overnight. When TLC showed that the reaction was complete, the mixture was extracted with EA and H2O. The water phase was re-extracted with EA. The organic layer was combined and washed with brine (10 mL). Then the solution was dried over anhydrous MgSO4, filtered and concentrated, and the crude residue was purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient elution to afford the title compounds.
2-(4-(4-methylpiperazine-1-carbonyl)phenyl)isoindolin-1-one (A11). Compound A11 was prepared from A10 and 1-methypiperazine using the above procedure as a white solid. The yield was 61%. 1H-NMR (500 MHz, DMSO-d6) δ 8.03–7.98 (m, 2H), 7.82 (d, J = 7.6 Hz, 1H), 7.74–7.67 (m, 2H), 7.59–7.54 (m, 1H), 7.52–7.47 (m, 2H), 5.07 (s, 2H), 3.45 (dd, J = 59.5, 52.5 Hz, 4H), 2.35 (d, J = 16.8 Hz, 4H), 2.20 (s, 3H). HRMS(ESI+) m/z calcd for C20H22N3O2 [M + H]+ 336.1712, found 336.1697.
2-(4-(morpholin-4-carbonyl)phenyl)isoindolin-1-one (A12). Compound A12 was prepared from A10 and morpholine using the above procedure as a white solid. The yield was 65%. 1H-NMR (500 MHz, DMSO-d6) δ 8.01 (d, J = 8.7 Hz, 2H), 7.82 (d, J = 7.6 Hz, 1H), 7.76–7.65 (m, 2H), 7.61–7.47 (m, 3H), 5.08 (s, 2H), 3.62 (s, 8H). HRMS(ESI+) m/z calcd for C19H19N2O3 [M + H]+ 323.1396, found 323.1383.
N-(3-(dimethylamino)propyl)-N-methyl-4-(1-oxoisoindolin-2-yl)benzamide (A13). Compound A13 was prepared from A10 and 3-dimethylamino-propylamine using the above procedure as a shallow yellow solid. The yield was 40%. 1H-NMR (500 MHz, DMSO-d6) δ 8.54 (t, J = 5.5 Hz, 1H), 8.02 (d, J = 8.9 Hz, 2H), 7.93 (d, J = 8.9 Hz, 2H), 7.81 (d, J = 7.6 Hz, 1H), 7.76–7.65 (m, 2H), 7.61–7.52 (m, 1H), 5.08 (s, 2H), 3.30 (dd, J = 12.8, 6.8 Hz, 3H), 2.41 (t, J = 7.1 Hz, 2H), 2.25 (s, 6H), 1.75–1.63 (m, 2H). HRMS(ESI+) m/z calcd for C20H24N3O2 [M + H]+ 338.1869, found 338.1852.
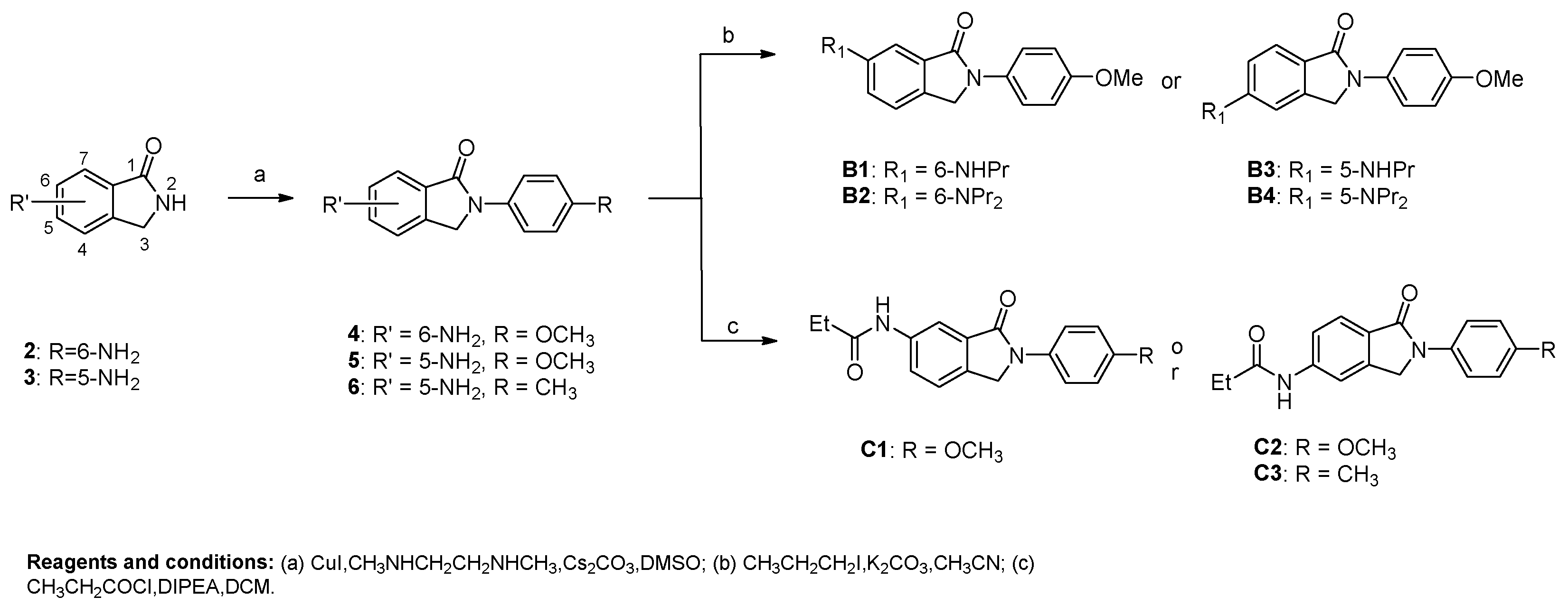
6-amino-2-(4-methoxyphenyl)isoindolin-1-one (4). Compound 4 was synthesized from 6-aminoisoindolin-1-one 2 and 1-iodo-4-methoxybenzene using general procedure A as a white solid. The yield was 56.97%. 1H-NMR (400 MHz, DMSO-d6) δ 7.80–7.73 (m, 2H), 7.26 (d, J = 8.1 Hz, 1H), 7.03–6.95 (m, 2H), 6.93–6.82 (m, 2H), 5.38 (s, 2H), 4.77 (s, 2H), 3.77 (s, 3H), 2.55 (s, 7H). HRMS(ESI+) m/z calcd for C15H15N2O2 [M + H]+ 255.1134, found 255.1140.
5-amino-2-(4-methoxyphenyl)isoindolin-1-one (5). Compound 5 was synthesized from 5-aminoisolindolin-1-one 3 and 1-iodo-4-methoxybenzene using general procedure A as a yellow solid. The yield was 51.54%. 1H-NMR (500 MHz, DMSO-d6) δ 7.77 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 8.1 Hz, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.87–6.50 (m, 2H), 5.92 (s, 2H), 4.78 (s, 2H), 3.78 (s, 3H). HRMS(ESI+) m/z calcd for C15H15N2O2 [M + H]+ 255.1134, found 255.1124.
5-amino-2-(p-tolyl)isoindolin-1-one (6). Compound 6 was synthesized from 5-aminoisoindolin-1-one 3 and 4-iodotoluene using general procedure A as a yellow solid. The yield was 45.69%. 1H-NMR (400 MHz, DMSO-d6) δ 7.74 (dd, J = 8.9, 2.4 Hz, 2H), 7.39 (d, J = 8.1 Hz, 1H), 7.26–7.12 (m, 2H), 6.65 (dd, J = 10.2, 2.1 Hz, 2H), 5.93 (s, 2H), 4.77 (s, 2H), 2.29 (s, 3H). HRMS(ESI+) m/z calcd for C15H15N2O [M + H]+ 239.1184, found 239.1185.
General Procedure B for the Synthesis of Compounds B1–B4. 6-amino-2-(4-methoxyphenyl)isoindolin-1-one 4 (300 mg, 1.18 mmol) was dissolved in superdry CH3CN (5 mL), and K2CO3 (449 mg, 3.54 mmol) was added to the solution. The resulting mixture was stirred at room temperature, after which 1-iodopropane (2.25 mmol) was added. When TLC showed that 6-amino-2-(4-methoxyphenyl)isoindolin-1-one was fully converted, the reaction was stopped. The mixture was filtered and the filtrate was evaporated in vacuo. The residue was dissolved in CH2Cl2 and then washed with H2O. The water phase was re-extracted with CH2Cl2. The organic layer was combined and washed with brine (10 mL). Then the solution was dried over anhydrous MgSO4, filtered and concentrated, and the crude residue was purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient elution to afford 2-(4-methoxyphenyl)-6-(propylamino)isoindolin-1-one (B1) and 6-(dipropylamino)-2-(4-methoxyphenyl)isoindolin-1-one (B2) as a yellow solid with yields of 32% and 26% for B1 and B2, respectively.
Compound B1 1H-NMR (500 MHz, Chloroform-d) δ 7.75 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 7.4 Hz, 2H), 7.22 (s, 1H), 7.00 (d, J = 8.5 Hz, 2H), 4.77 (s, 2H), 3.88 (s, 3H), 3.25 (t, J = 7.7 Hz, 2H), 1.81 (q, J = 7.6 Hz, 2H), 1.07 (t, J = 7.4 Hz, 3H). HRMS(ESI+) m/z calcd for C18H21N2O2 [M + H]+ 297.1603, found 297.1611.
Compound B2 1H-NMR (500 MHz, Chloroform-d) δ 7.77 (d, J = 8.5 Hz, 2H), 7.16 (s, 1H), 7.01 (d, J = 8.6 Hz, 2H), 6.90 (s, 1H), 4.87–4.66 (m, 2H), 3.87 (s, 3H), 3.35 (s, 4H), 1.68 (s, 4H), 0.98 (t, J = 7.4 Hz, 6H). HRMS(ESI+) m/z calcd for C21H27N2O2 [M + H]+ 339.2073, found 339.2055.
2-(4-methoxyphenyl)-5-(propylamino)isoindolin-1-one (B3) and 5-(dipropylamino)-2-(4-methoxyphenyl)isoindolin-1-one (B4) were synthesized from 5-amino-2-(4-methoxyphenyl)isoindolin-1-one (5) and 1-iodopropane using general procedure B, which were both yellow solids. The yields were 34% and 28% for B3 and B4, respectively.
Compound B3 1H-NMR (500 MHz, Chloroform-d) δ 7.74 (dd, J = 14.5, 8.5 Hz, 3H), 6.99 (d, J = 8.6 Hz, 2H), 6.71 (d, J = 24.0 Hz, 2H), 4.75 (s, 2H), 3.87 (s, 3H), 3.21 (t, J = 7.1 Hz, 2H), 1.75 (q, J = 7.1 Hz, 2H), 1.07 (t, J = 7.4 Hz, 3H).
Compound B4 1H-NMR (500 MHz, Chloroform-d) δ 7.75 (dd, J = 13.3, 8.8 Hz, 3H), 6.99 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.6 Hz, 1H), 6.66 (s, 1H), 4.76 (s, 2H), 3.87 (s, 3H), 3.37 (t, J = 7.8 Hz, 4H), 1.71 (h, J = 7.5 Hz, 4H), 1.01 (t, J = 7.4 Hz, 6H). HRMS(ESI+) m/z calcd for C21H27N2O2 [M + H]+ 339.2073, found 339.2076.
General Procedure C for the Synthesis of Compounds C1–C3. 5-amino-2-(p-tolyl)isoindolin-1-one 6 (70 mg, 0.29 mmol) was dissolved in CH2Cl2 (3 mL), and DIPEA (154 μL, 0.87 mmol) was added to the solution. Propionyl chloride was dissolved in CH2Cl2 (1 mL) and transferred to a drip funnel, and dropped into the flask, at a rate of about 1–2 drop(s) per second. After completion of the addition, the mixture was stirred at room temperature. When TLC showed that 6-amino-2-(p-tolyl)isoindolin-1-one had been fully converted, the reaction was stopped. The mixture was extracted with H2O (3 mL). The water phase was re-extracted with CH2Cl2 (10 mL). The organic layer was combined and washed with brine (10 mL). Then the solution was dried over anhydrous MgSO4, filtered and concentrated, and the crude residue was purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient elution to afford compound N-(1-oxo-2-(p-tolyl)isoindolin-5-yl)propionamide (C3) as a yellow solid. The yield was 46%. 1H-NMR (500 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.07 (s, 1H), 7.80 (d, J = 7.9 Hz, 2H), 7.71 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H), 4.97 (s, 2H), 2.41 (q, J = 7.5 Hz, 2H), 2.33 (s, 3H), 1.14 (t, J = 7.5 Hz, 3H). HRMS(ESI+) m/z calcd for C18H19N2O2 [M + H]+ 295.1447, found 295.1433.
N-(2-(4-methoxyphenyl)-3-oxoisoindolin-5-yl)propionamide (C1). Compound C1 was synthesized from 6-amino-2-(4-methoxyphenyl)isoindolin-1-one (4) and propionyl chloride using general procedure C as a yellow solid. The yield was 49%. 1H-NMR (500 MHz, Chloroform-d) δ 8.22 (s, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.6 Hz, 2H), 7.63 (s, 1H), 7.00 (d, J = 8.5 Hz, 2H), 4.83 (s, 2H), 3.87 (s, 3H), 2.77–2.30 (m, 2H), 1.31 (t, J = 7.5 Hz, 3H). HRMS(ESI+) calcd for C18H19N2O3 [M + H]+ 311.1396, found 311.1386.
N-(2-(4-methoxyphenyl)-1-oxoisoindolin-5-yl)propionamide (C2). Compound C2 was synthesized from 5-amino-2-(4-methoxyphenyl)isoindolin-1-one (5) and propionyl chloride using general procedure C as a yellow solid. The yield was 50%. 1H-NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 8.04 (d, J = 1.6 Hz, 1H), 7.82–7.75 (m, 2H), 7.68 (d, J = 8.2 Hz, 2H), 7.61 (dd, J = 8.3, 1.7 Hz, 1H), 7.04–6.97 (m, 2H), 4.94 (s, 2H), 3.77 (s, 3H), 2.39 (q, J = 7.5 Hz, 2H), 1.11 (t, J = 7.5 Hz, 3H). HRMS(ESI+) calcd for C18H19N2O3 [M + H]+ 311.1396, found 311.1382.
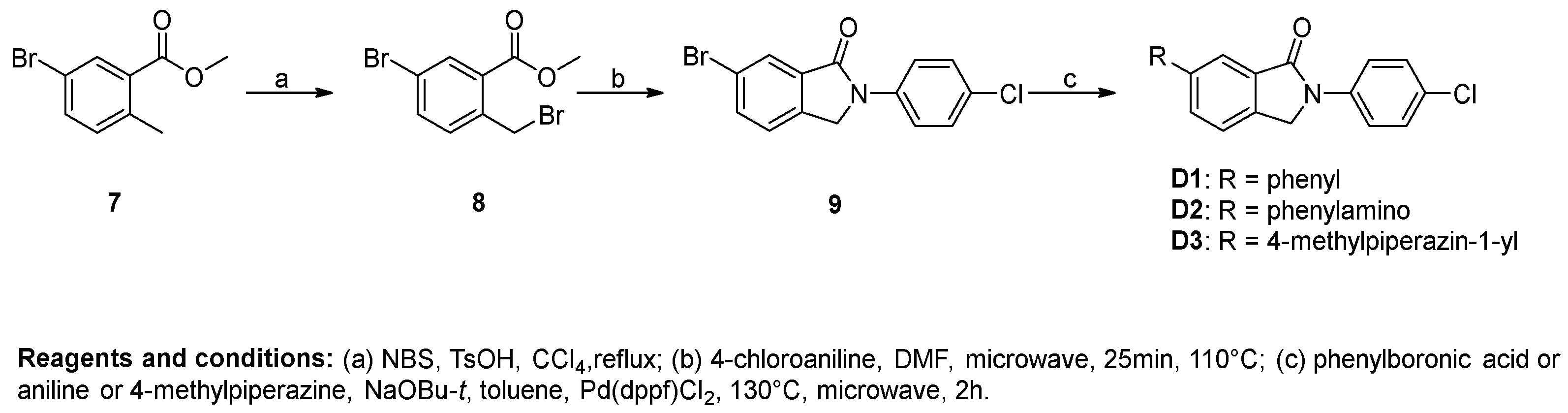
Procedure D for the synthesis of compounds D1–D3. Methyl 5-bromo-2-methylbenzoate (2.29 g, 10 mmol) was added to a flask containing 20 mL CCl4, and then N-bromosuccinimide (1.78 g, 10 mmol) and TsOH (172 mg, 1 mmol) were added. The mixture was refluxed until methyl 5-bromo-2-methylbenzoate was fully converted. The solution was washed with saturated NaHCO3 solution, water and brine successively. The organic phase was evaporated under vacuum to afford the crude product methyl 5-bromo-2-(bromomethyl)benzoate (9).
A 10 mL microwave vial was charged with 4-chloroaniline (638 mg, 5 mmol), methyl 5-bromo-2-(bromomethyl)benzoate (1.57 g, 5.5 mmol), and DMF (4 mL). The vial was sealed and microwaved at 110 °C for 25 min. The reaction mixture was diluted with MeOH (50 mL) and an off-white precipitate was formed, which was filtered and dried under vacuum to give 10 (1.19 g, 73.78%) as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ 7.96 (d, J = 8.9 Hz, 3H), 7.90 (d, J = 8.1 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 5.03 (s, 2H).
A microwave vial was charged with 10 (97 mg, 0.30 mmol), phenylboronic acid (41 mg, 0.33 mmol), NaOBu-t (58 mg, 0.60 mmol), DMSO (2 mL), and H2O (0.3 mL). The solution was degassed with nitrogen for 10 min, and Pd(dppf)Cl2 (12 mg, 0.015 mmol) was added. The vial was sealed and microwaved at 130 °C for 2 h. The reaction mixture was poured into EtOAc/H2O and the organic layer was separated and subsequently passed through a plug of florisil. The obtained organic phase was concentrated into a residue, which was triturated with MeOH, filtered, and dried under vacuum to afford D1 (43 mg, 44.82%) as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ 8.05–7.98 (m, 4H), 7.80 (d, J = 7.3 Hz, 3H), 7.58–7.50 (m, 4H), 7.45 (t, J = 7.3 Hz, 1H), 5.11 (s, 2H).
Compound D2 was synthesized using 10 and aniline using the same method of synthesis as that of D1. D2 was purified by flash chromatography to afford a yellow solid. The yield was 20%. 1H-NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H), 8.00–7.89 (m, 2H), 7.51 (dd, J = 8.5, 4.1 Hz, 3H), 7.45–7.24 (m, 4H), 7.14 (d, J = 7.9 Hz, 2H), 6.91 (t, J = 7.3 Hz, 1H), 4.93 (s, 2H).
Compound D3 was synthesized using 10 and 4-methylpiperazine using the same method of synthesis as that of D1. D3 was purified by flash chromatography to afford a pale yellow solid. The yield was 22%. 1H-NMR (500 MHz, DMSO-d6) δ 7.96 (d, J = 8.9 Hz, 3H), 7.90 (d, J = 8.1 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 5.03 (s, 2H).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}