
Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening

 ,
,
Abstract
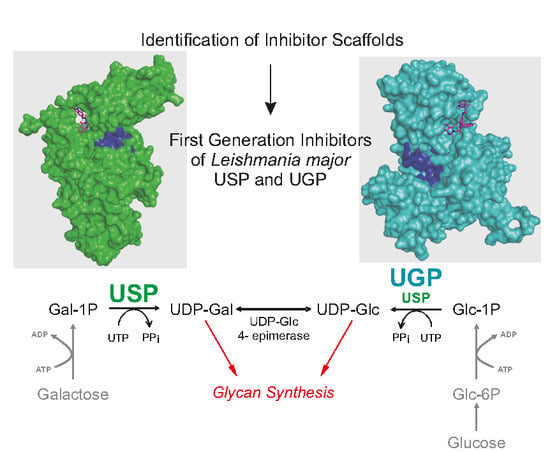
1. Introduction
2. Results
2.1. Identification of Scaffolds for Inhibitor Development by Small-Molecule Fragment Screening
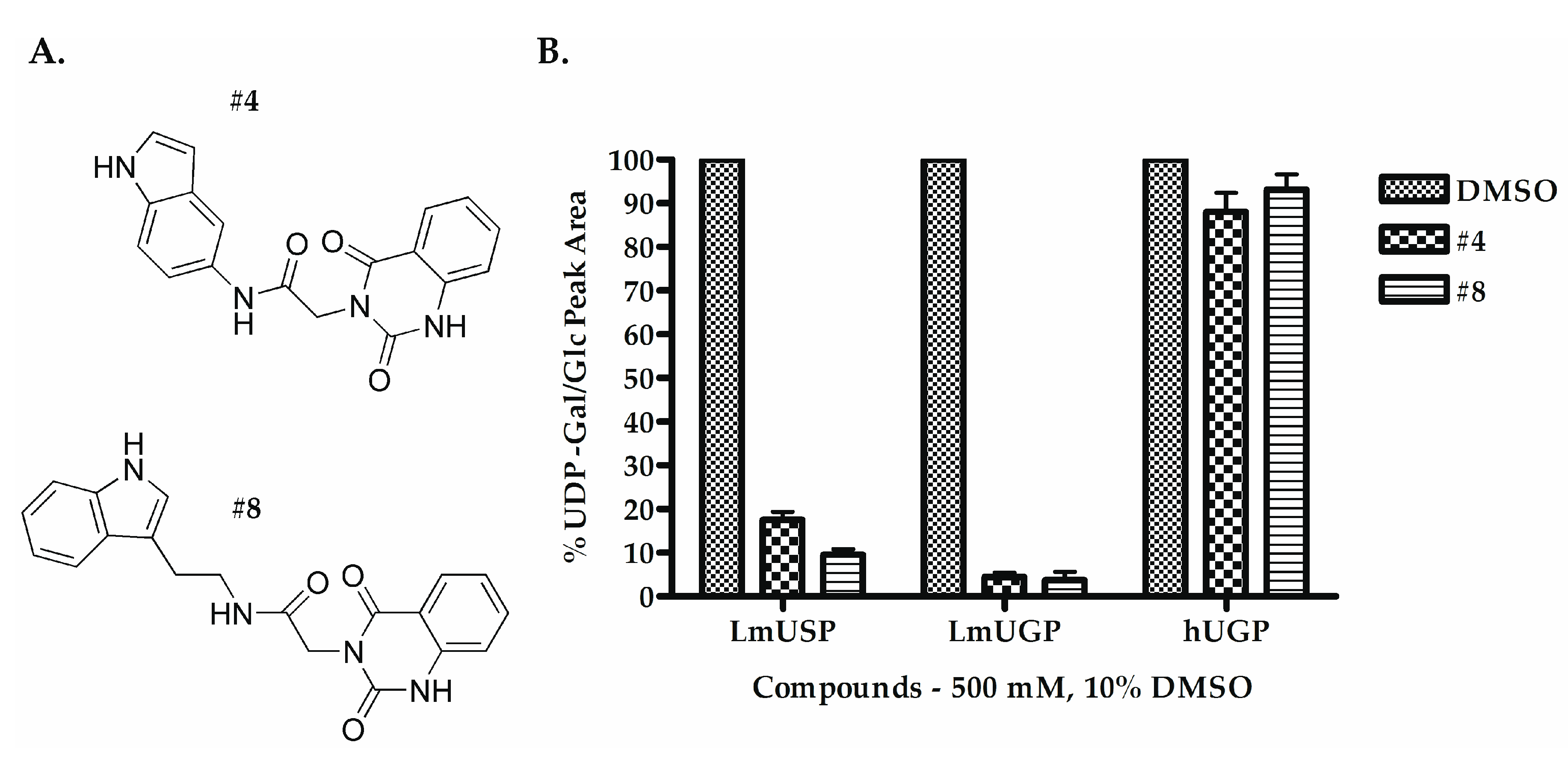
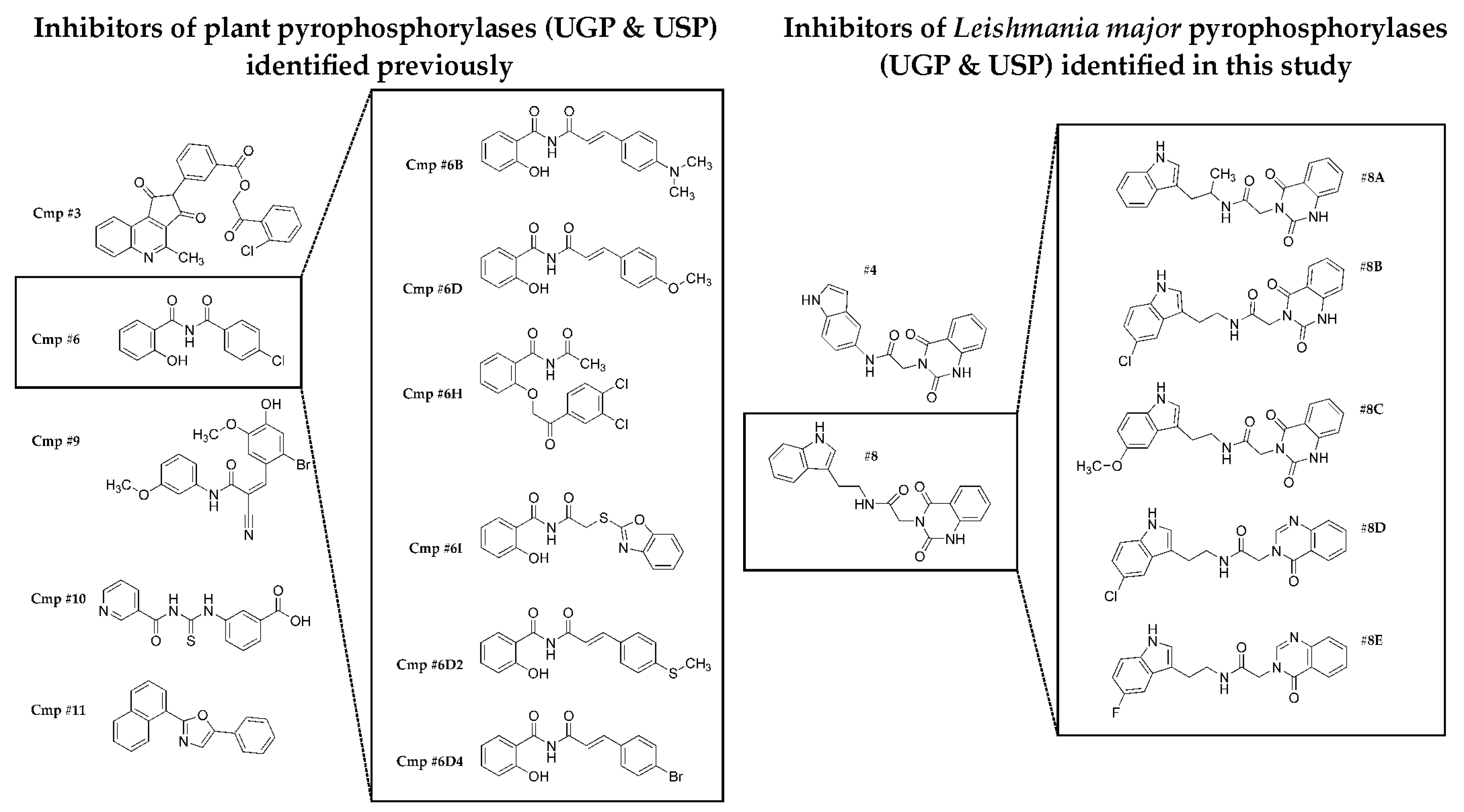
2.2. Selecting the First-Generation Inhibitors
2.3. Predictions of the Compounds’ Binding Sites on LmUGP and LmUSP
2.4. Binding Site Validation through Site-Directed Mutagenesis
3. Discussion
4. Materials and Methods
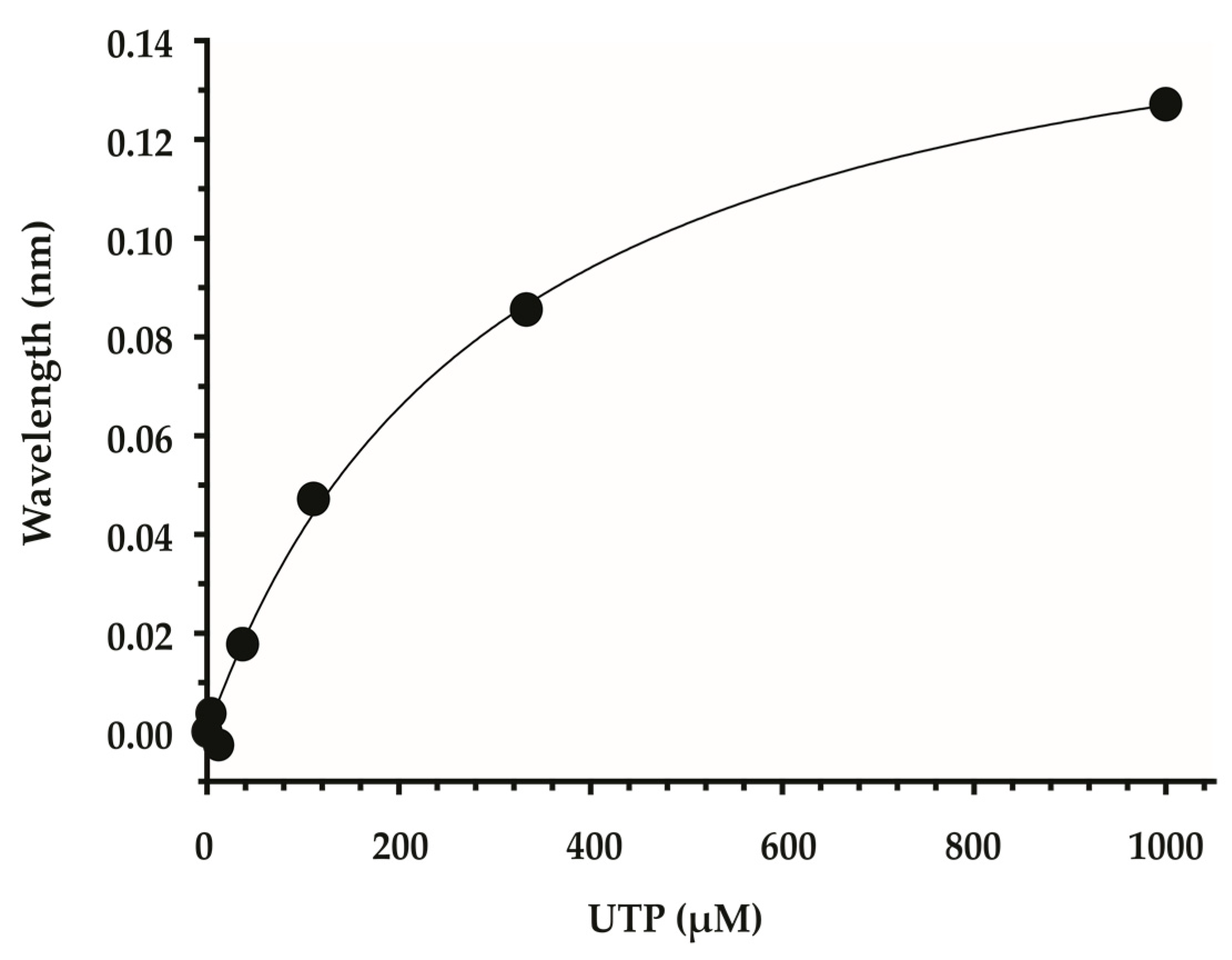
4.1. A BLI Screen Using Recombinant LmUSP
4.2. Fragment Similarity Search
4.3. Protein Purification and Mutant Generation
4.4. Enzyme Activity Assay and Dose Response Study
4.5. Docking Analysis
4.6. Enzyme Kinetics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Pearson, R.D.; Sousa, A.Q. Clinical Spectrum of Leishmaniasis. Clin. Infect. Dis. 1996, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Piscopo, T.V.; Mallia Azzopardi, C. Leishmaniasis. Postgrad. Med. J. 2007, 83, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Kanneganti, T.D. Innate immunity against Leishmania infections. Cell. Microbiol. 2015, 17, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Ozgoren, A.A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Naghavi, M.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Adetokunboh, O.; Afshin, A.; Agrawal, A.; et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Moura, A.P.V.; Santos, L.C.B.; Brito, C.R.N.; Valencia, E.; Junqueira, C.; Filho, A.A.P.; Sant’Anna, M.R.V.; Gontijo, N.F.; Bartholomeu, D.C.; Fujiwara, R.T.; et al. Virus-like Particle Display of the α-Gal Carbohydrate for Vaccination against Leishmania Infection. ACS Cent. Sci. 2017, 3, 1026–1031. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug Resistance in Leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef]
- Dostálová, A.; Volf, P. Leishmania development in sand flies: Parasite-vector interactions overview. Parasit. Vectors 2012, 5, 276:1–276:12. [Google Scholar] [CrossRef]
- Naderer, T.; Vince, J.E.; McConville, M.J. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr. Mol. Med. 2004, 4, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Novozhilova, N.M.; Bovin, N.V. Structure, functions, and biosynthesis of glycoconjugates of Leishmania spp. cell surface. Biochemistry 2010, 75, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Turnock, D.C.; Ferguson, M.A.J. Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot. Cell 2007, 6, 1450–1463. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Sabatini, R. Base J: Discovery, Biosynthesis, and Possible Functions. Annu. Rev. Microbiol. 2008, 62, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Bullard, W.; Lopes Da Rosa-Spiegler, J.; Liu, S.; Wang, Y.; Sabatini, R. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J. Biol. Chem. 2014, 289, 20273–20282. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, L.J.; Kieft, R.; Southern, T.; Birkeland, S.R.; Marshall, M.; Sweeney, K.; Sabatini, R. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 2009, 37, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Van Luenen, H.G.A.M.; Farris, C.; Jan, S.; Genest, P.A.; Tripathi, P.; Velds, A.; Kerkhoven, R.M.; Nieuwland, M.; Haydock, A.; Ramasamy, G.; et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 2012, 150, 909–921. [Google Scholar] [CrossRef]
- Trombetta, S.E.; Bosch, M.; Parodi, A.J. Glucosylation of Glycoproteins by Mammalian, Plant, Fungal, and Trypanosomatid Protozoa Microsomal Membranes. Biochemistry 1989, 28, 8108–8116. [Google Scholar] [CrossRef]
- Izquierdo, L.; Atrih, A.; Rodrigues, J.A.; Jones, D.C.; Ferguson, M.A.J. Trypanosoma brucei UDP-glucose:glycoprotein glucosyltransferase has unusual substrate specificity and protects the parasite from stress. Eukaryot. Cell 2009, 8, 230–240. [Google Scholar] [CrossRef]
- Freeze, H.H.; Hart, G.W.; Schnaar, R.L. Glycosylation Precursors. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 51–63. ISBN 978-1-621821-32-8. [Google Scholar]
- Damerow, S.; Lamerz, A.C.; Haselhorst, T.; Führing, J.; Zarnovican, P.; von Itsztein, M.; Routier, F.H. Leishmania UDP-sugar pyrophosphorylase: The missing link in galactose salvage? J. Biol. Chem. 2010, 285, 878–887. [Google Scholar] [CrossRef]
- Damerow, S.; Hoppe, C.; Bandini, G.; Zarnovican, P.; Buettner, F.F.R.; Ferguson, M.A.J.; Routier, F.H. Leishmania major UDP-sugar pyrophosphorylase salvages galactose for glycoconjugate biosynthesis. Int. J. Parasitol. 2015, 45, 783–790. [Google Scholar] [CrossRef]
- Geserick, C.; Tenhaken, R. UDP-sugar pyrophosphorylase controls the activity of proceeding sugar-1-kinases enzymes. Plant Signal. Behav. 2013, 8, e25478:1–e25478:3. [Google Scholar] [CrossRef]
- Decker, D.; Kleczkowski, L.A. UDP-Sugar Producing Pyrophosphorylases: Distinct and Essential Enzymes With Overlapping Substrate Specificities, Providing de novo Precursors for Glycosylation Reactions. Front. Plant Sci. 2019, 9, 1822:1–1822:19. [Google Scholar] [CrossRef]
- Decker, D.; Oberg, C.; Kleczkowski, L.A. Identification and characterization of inhibitors of UDP-glucose and UDP-sugar pyrophosphorylases for in vivo studies. Plant J. 2017, 90, 1093–1107. [Google Scholar] [CrossRef]
- Lamerz, A.C.; Haselhorst, T.; Bergfeld, A.K.; Von Itzstein, M.; Gerardy-Schahn, R. Molecular cloning of the Leishmania major UDP-glucose pyrophosphorylase, functional characterization, and ligand binding analyses using NMR spectroscopy. J. Biol. Chem. 2006, 281, 16314–16322. [Google Scholar] [CrossRef]
- Ellin, L. Kinetic characterization of UDP-glucose pyrophosphorylase from germinated barley (malt). Phytochemistry 1996, 42, 955–960. [Google Scholar] [CrossRef]
- Dickmanns, A.; Damerow, S.; Neumann, P.; Schulz, E.C.; Lamerz, A.C.; Routier, F.H.; Ficner, R. Structural basis for the broad substrate range of the UDP-sugar pyrophosphorylase from leishmania major. J. Mol. Biol. 2011, 405, 461–478. [Google Scholar] [CrossRef]
- Roeben, A.; Plitzko, J.M.; Körner, R.; Böttcher, U.M.K.; Siegers, K.; Hayer-Hartl, M.; Bracher, A. Structural Basis for Subunit Assembly in UDP-glucose Pyrophosphorylase from Saccharomyces cerevisiae. J. Mol. Biol. 2006, 364, 551–560. [Google Scholar] [CrossRef]
- McCoy, J.G.; Bitto, E.; Bingman, C.A.; Wesenberg, G.E.; Bannen, R.M.; Kondrashov, D.A.; Phillips, G.N. Structure and Dynamics of UDP–Glucose Pyrophosphorylase from Arabidopsis thaliana with Bound UDP–Glucose and UTP. J. Mol. Biol. 2007, 366, 830–841. [Google Scholar] [CrossRef]
- Steiner, T.; Lamerz, A.C.; Hess, P.; Breithaupt, C.; Krapp, S.; Bourenkov, G.; Huber, R.; Gerardy-Schahn, R.; Jacob, U. Open and closed structures of the UDP-glucose pyrophosphorylase from Leishmania major. J. Biol. Chem. 2007, 282, 13003–13010. [Google Scholar] [CrossRef]
- Yu, Q.; Zheng, X. The crystal structure of human UDP-glucose pyrophosphorylase reveals a latch effect that influences enzymatic activity. Biochem. J. 2012, 442, 283–291. [Google Scholar] [CrossRef]
- Mariño, K.; Güther, M.L.S.; Wernimont, A.K.; Amani, M.; Hui, R.; Ferguson, M.A.J. Identification, subcellular localization, biochemical properties, and high-resolution crystal structure of Trypanosoma brucei UDP-glucose pyrophosphorylase. Glycobiology 2010, 20, 1619–1630. [Google Scholar] [CrossRef]
- Führing, J.I.; Cramer, J.T.; Schneider, J.; Baruch, P.; Gerardy-Schahn, R.; Fedorov, R. A Quaternary Mechanism Enables the Complex Biological Functions of Octameric Human UDP-glucose Pyrophosphorylase, a Key Enzyme in Cell Metabolism. Sci. Rep. 2015, 5, 9618:1–9618:11. [Google Scholar] [CrossRef]
- Führing, J.; Damerow, S.; Fedorov, R.; Schneider, J.; Münster-Kühnel, A.K.; Gerardy-Schahn, R. Octamerization is essential for enzymatic function of human UDP-glucose pyrophosphorylase. Glycobiology 2013, 23, 426–437. [Google Scholar] [CrossRef]
- Lamerz, A.C.; Damerow, S.; Kleczka, B.; Wiese, M.; Van Zandbergen, G.; Lamerz, J.; Wenzel, A.; Hsu, F.F.; Turk, J.; Beverley, S.M.; et al. Deletion of UDP-glucose pyrophosphorylase reveals a UDP-glucose independent UDP-galactose salvage pathway in Leishmania major. Glycobiology 2010, 20, 872–882. [Google Scholar] [CrossRef]
- Damerow, S.; Hoppe, C.; Bandini, G.; Zarnovican, P.; Buettner, F.R.; Lüder, C.G.K.; Ferguson, M.A.J.; Routier, F.H. Depletion of UDP-Glucose and UDP-Galactose Using a Degron System Leads to Growth Cessation of Leishmania major. PLoS Negl. Trop. Dis. 2015, 9, e0004205:1–e0004205:15. [Google Scholar] [CrossRef]
- Roper, J.R.; Guther, M.L.S.; Macrae, J.I.; Prescott, A.R.; Hallyburton, I.; Acosta-Serrano, A.; Ferguson, M.A.J. The suppression of galactose metabolism in procylic form Trypanosoma brucei causes cessation of cell growth and alters procyclin glycoprotein structure and copy number. J. Biol. Chem. 2005, 280, 19728–19736. [Google Scholar] [CrossRef]
- Urbaniak, M.D.; Turnock, D.C.; Ferguson, M.A.J. Galactose starvation in a bloodstream form Trypanosoma brucei UDP-glucose 4’-epimerase conditional null mutant. Eukaryot. Cell 2006, 5, 1906–1913. [Google Scholar] [CrossRef]
- MacRae, J.I.; Obado, S.O.; Turnock, D.C.; Roper, J.R.; Kierans, M.; Kelly, J.M.; Ferguson, M.A.J. The suppression of galactose metabolism in Trypanosoma cruzi epimastigotes causes changes in cell surface molecular architecture and cell morphology. Mol. Biochem. Parasitol. 2006, 147, 126–136. [Google Scholar] [CrossRef]
- Tetaud, E.; Barrett, M.P.; Bringaud, F.; Baltz, T. Kinetoplastid glucose transporters. Biochem. J. 1997, 325, 569–580. [Google Scholar] [CrossRef]
- Cramer, J.T.; Führing, J.I.; Baruch, P.; Brütting, C.; Knölker, H.J.; Gerardy-Schahn, R.; Fedorov, R. Decoding Allosteric Networks in Biocatalysts: Rational Approach to Therapies and Biotechnologies. ACS Catal. 2018, 8, 2683–2692. [Google Scholar] [CrossRef]
- Goodey, N.M.; Benkovic, S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008, 4, 474–482. [Google Scholar] [CrossRef]
- Wartchow, C.A.; Podlaski, F.; Li, S.; Rowan, K.; Zhang, X.; Mark, D.; Huang, K.S. Biosensor-based small molecule fragment screening with biolayer interferometry. J. Comput. Aided. Mol. Des. 2011, 25, 669–676. [Google Scholar] [CrossRef]
- Shah, N.B.; Duncan, T.M. Bio-layer Interferometry for Measuring Kinetics of Protein-protein Interactions and Allosteric Ligand Effects. J. Vis. Exp. 2014, e51383:1–e51383:7. [Google Scholar] [CrossRef]
- Erlanson, D.A. Introduction to Fragment-Based Drug Discovery. In Fragment-Based Drug Discovery and X-Ray Crystallography; Davies, T.G., Hyvönen, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–32. ISBN 978-3-642-27540-1. [Google Scholar]
- Führing, J.; Cramer, J.T.; Routier, F.H.; Lamerz, A.C.; Baruch, P.; Gerardy-Schahn, R.; Fedorov, R. Catalytic mechanism and allosteric regulation of UDP-glucose pyrophosphorylase from Leishmania major. ACS Catal. 2013, 3, 2976–2985. [Google Scholar] [CrossRef]
- Lackovic, K.; Parisot, J.P.; Sleebs, N.; Baell, J.B.; Debien, L.; Watson, K.G.; Curtis, J.M.; Handman, E.; Street, I.P. Inhibitors of Leishmania GDP-Mannose Pyrophosphorylase Identified by High-Throughput Screening of Small-Molecule Chemical Library. Antimicrob. Agents Chemother. 2010, 54, 1712–1719. [Google Scholar] [CrossRef]
- Mao, W.; Daligaux, P.; Lazar, N.; Ha-duong, T.; Cavé, C.; Tilbeurgh, V.; Loiseau, P.M.; Pomel, S. Biochemical analysis of leishmanial and human GDP-Mannose Pyrophosphorylases and selection of inhibitors as new leads. Sci. Rep. 2017, 7, e751:1–e751:14. [Google Scholar] [CrossRef]
- Urbaniak, M.D.; Collie, I.T.; Fang, W.; Aristotelous, T.; Eskilsson, S.; Raimi, O.G.; Harrison, J.; Navratilova, I.H.; Frearson, J.A.; Van Aalten, D.M.F.; et al. A novel allosteric inhibitor of the uridine diphosphate N-acetylglucosamine pyrophosphorylase from Trypanosoma brucei. ACS Chem. Biol. 2013, 8, 1981–1987. [Google Scholar] [CrossRef]
- Decker, D.; Öberg, C. The structure-activity relationship of the salicylimide derived inhibitors of UDP-sugar producing pyrophosphorylases. Plant Signal. Behav. 2018, 13, e1507406:1–e1507406:3. [Google Scholar] [CrossRef]
- ChemBridge Chemical Store. Available online: https://www.hit2lead.com/ (accessed on 27 March 2018).
- ExPASy—SIB Bioinformatics Resource Portal—ProtParam. Available online: https://web.expasy.org/protparam/ (accessed on 16 April 2018).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Awale, M.; Kumar, V.; Saravanan, P.; Mohan, C.G. Homology modeling and atomic level binding study of Leishmania MAPK with inhibitors. J. Mol. Model. 2010, 16, 475–488. [Google Scholar] [CrossRef]
- Nagpal, I.; Raj, I.; Subbarao, N.; Gourinath, S. Virtual Screening, Identification and In Vitro Testing of Novel Inhibitors of O-Acetyl-l-Serine Sulfhydrylase of Entamoeba histolytica. PLoS ONE 2012, 7, e30305:1–e30305:7. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment | Structure | Primary Binding Response | Estimated KD (µM) |
|---|---|---|---|
| DDD00095351 |  | 0.127 | 83.3 µM |
| DDD00102262 |  | 0.077 | 72.6 µM |
| DDD00808259 |  | 0.1728 | 67.6 µM |
| DDD01305586 |  | 0.1541 | 60.1 µM |
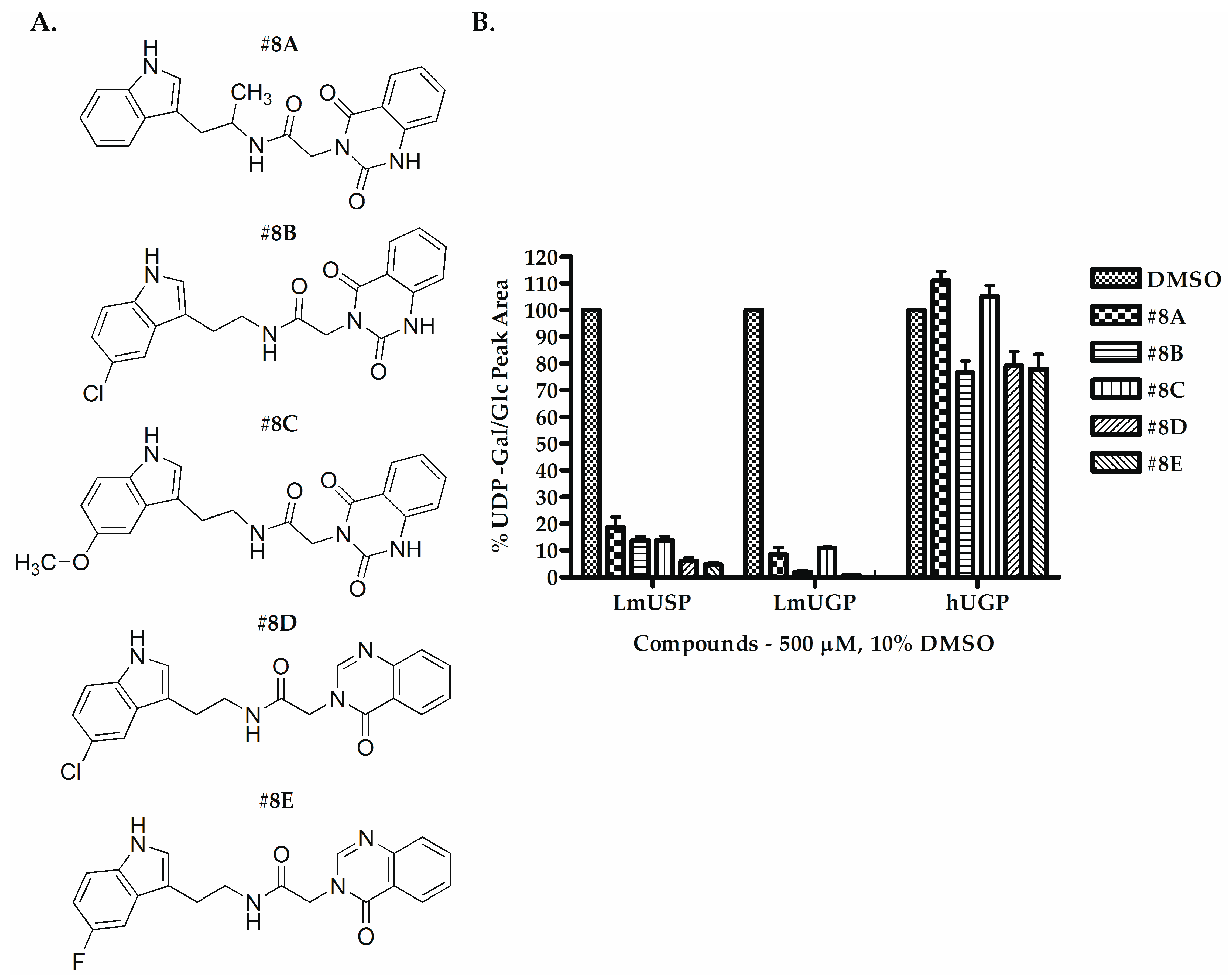
| Compounds | IC50 (µM) | |
|---|---|---|
| LmUSP (UDP-Gal) | LmUGP (UDP-Glc) | |
| #4 | 135.00–175.00 | 126.30–134.00 |
| #8 | 78.06–106.60 | 119.80–151.20 |
| #8A | >250 | >250 |
| #8B | 47.17–66.43 | 55.06–69.13 |
| #8C | >250 | >250 |
| #8D | 169.80–194.80 | 192.70–242.10 |
| #8E | 40.39–46.29 | 37.99–46.76 |
| Ligand Pose | Compound #4 Binding Energy (kcal/mol) | Compound #8 Binding Energy (kcal/mol) | ||
|---|---|---|---|---|
| LmUGP | LmUSP | LmUGP | LmUSP | |
| Pose 1 | −7.61 | −7.83 | −7.92 | −8.22 |
| Pose 2 | −7.61 | −7.78 | −7.71 | −7.80 |
| Pose 3 | - | −7.74 | - | −7.41 |
| Pose 4 | - | −7.70 | - | −7.30 |
| LmUSP | Vmax (µmol min−1 mg−1) | Km (µM) Gal-1P | kcat (s−1) | kcat/Km (µM−1 s−1) | % Catalytic Efficiency |
|---|---|---|---|---|---|
| Wild type | 184.4 ± 6.36 | 529.20 ± 24.60 | 216.45 ± 7.47 | 0.409 ± 0.03 | 100 ± 8.094 |
| V330W | 271.70 ± 19.37 | 1255.00 ± 145.66 | 319.32 ± 22.77 | 0.255 ± 0.01 | 62.25 ± 2.79 |
| F383R | 14.77 ± 0.24 | 364.00 ± 22.20 | 17.33 ± 0.28 | 0.047 ± 0.002 | 11.64 ± 0.52 |
| V330W/F383R | 21.76 ± 1.26 | 276.45 ± 51.26 | 25.58 ± 1.48 | 0.093 ± 0.01 | 22.85 ± 2.92 |
| V199W | 76.79 ±2.94 | 1007.25 ± 18.03 | 90.25 ± 3.46 | 0.089 ± 0.001 | 21.86 ± 0.44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prakash, O.; Führing, J.; Post, J.; Shepherd, S.M.; Eadsforth, T.C.; Gray, D.; Fedorov, R.; Routier, F.H. Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening. Molecules 2019, 24, 996. https://doi.org/10.3390/molecules24050996
Prakash O, Führing J, Post J, Shepherd SM, Eadsforth TC, Gray D, Fedorov R, Routier FH. Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening. Molecules. 2019; 24(5):996. https://doi.org/10.3390/molecules24050996
Chicago/Turabian StylePrakash, Ohm, Jana Führing, John Post, Sharon M. Shepherd, Thomas C. Eadsforth, David Gray, Roman Fedorov, and Françoise H. Routier. 2019. "Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening" Molecules 24, no. 5: 996. https://doi.org/10.3390/molecules24050996
APA StylePrakash, O., Führing, J., Post, J., Shepherd, S. M., Eadsforth, T. C., Gray, D., Fedorov, R., & Routier, F. H. (2019). Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening. Molecules, 24(5), 996. https://doi.org/10.3390/molecules24050996