
Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH)



Abstract
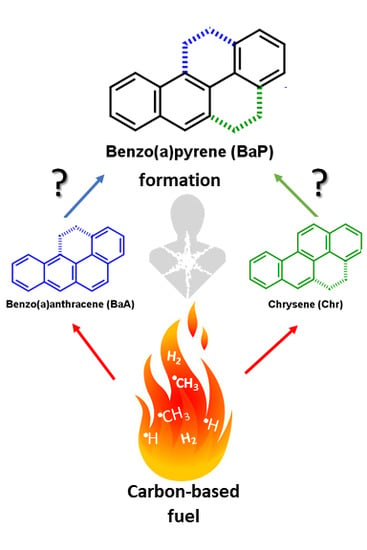
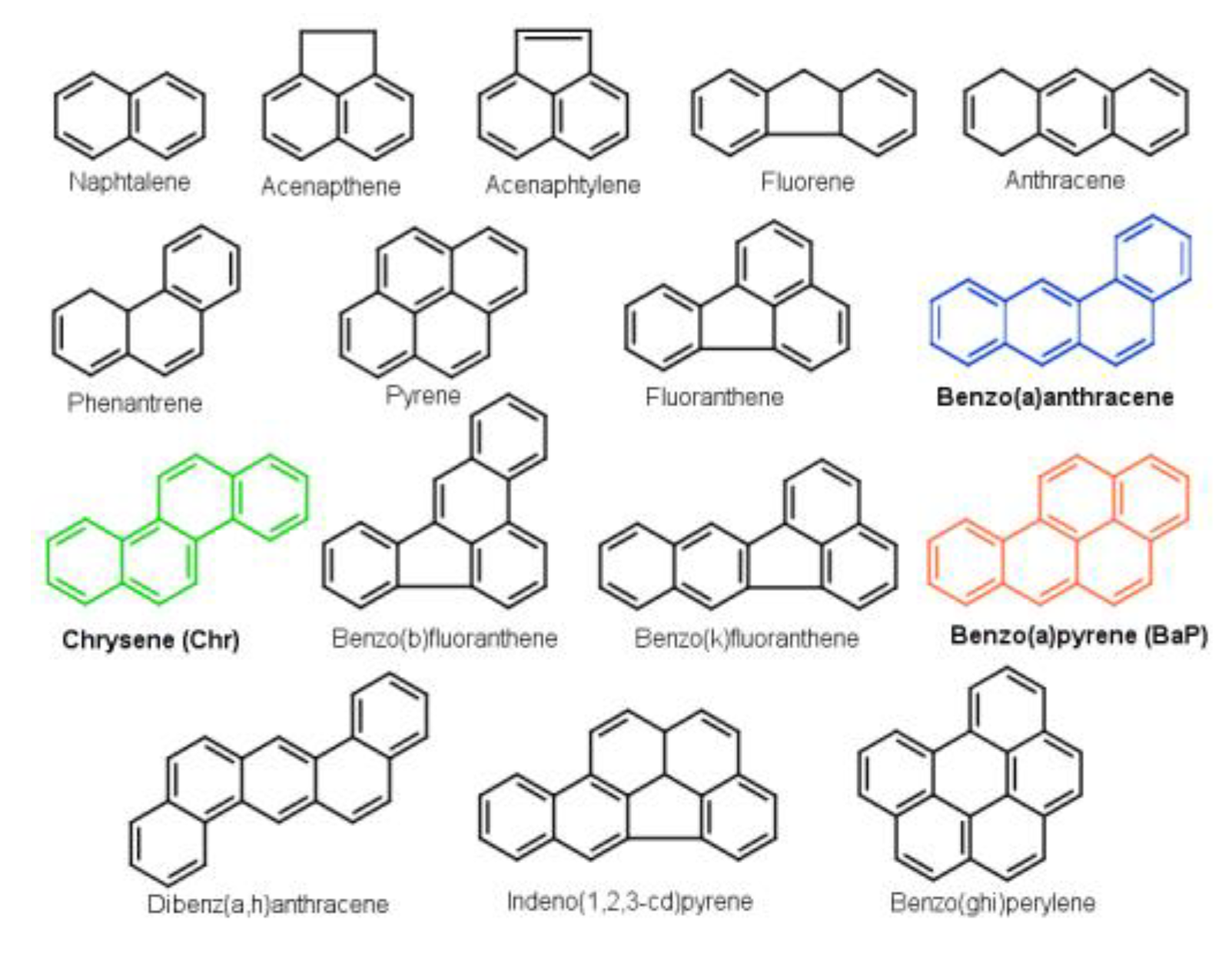
1. Introduction
2. Computational Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tiwari, M.; Sahu, S.K.; Pandit, G.G. Distribution of PAHs in different compartment of creek ecosystem: Ecotoxicological concern and human health risk. Environ. Toxicol. Pharmacol. 2017, 50, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Sahu, S.K.; Pandit, G.G. Inhalation risk assessment of PAH exposure due to combustion aerosols generated from household fuels. Aerosol Air Qual. Res. 2015, 15, 582–590. [Google Scholar] [CrossRef]
- Keith, L.; Telliard, W. ES&T. Special Report Priority pollutants: I-a perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar]
- Keith, L.H. The Source of U.S. EPA’s Sixteen PAH Priority Pollutants. Polycycl. Aromat. Compd. 2015, 35, 147–160. [Google Scholar] [CrossRef]
- Andersson, J.T.; Achten, C. A Critical Look at the 16 EPA PAHs. Polycycl. Aromat. Compd. 2015, 35, 143–146. [Google Scholar] [CrossRef]
- Andersson, J.T.; Achten, C. Time to Say Goodbye to the 16 EPA PAHs? Toward an Up-to-Date Use of PACs for Environmental Purposes. Polycycl. Aromat. Compd. 2015, 35, 330–354. [Google Scholar] [CrossRef] [PubMed]
- Samburova, V.; Zielinska, B.; Khlystov, A. Do 16 Polycyclic Aromatic Hydrocarbons Represent PAH Air Toxicity? Toxics 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- United Nations Environment Programme (UNEP). Guidance for a Global Monitoring Programme for Persistent Organic Pollutants; UNEP: Nairobi, Kenya, 2004. [Google Scholar]
- Stogiannidis, E.; Laane, R. Source characterization of polycyclic aromatic hydrocarbons by using their molecular indices: An overview of possibilities. Rev. Environ. Contam. Toxicol. 2015, 234, 49–133. [Google Scholar] [PubMed]
- Abdel-Shafy, H.; Mansour, M.S. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egyp. J. Pet. 2016, 25, 107–123. [Google Scholar] [CrossRef]
- Shen, H.; Huang, Y.; Wang, R.; Zhu, D.; Li, W.; Shen, G.; Wang, B.; Zhang, Y.; Chen, Y.; Lu, Y.; et al. Global Atmospheric Emissions of Polycyclic Aromatic Hydrocarbons from 1960 to 2008 and Future Predictions. Environ. Sci. Technol. 2013, 47, 6415–6424. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Man, P.L.; Totton, T.S.; Sander, M.; Shirley, R.A.; Kraft, M. New polycyclic aromatic hydrocarbon (PAH) surface processes to improve the model prediction of the composition of combustion-generated PAHs and soot. Carbon 2010, 48, 319–332. [Google Scholar] [CrossRef]
- Frenklach, M. Reaction mechanism of soot formation in flames. Proc. Phys. Chem. Chem. Phys. 2002, 4, 2028–2037. [Google Scholar] [CrossRef]
- Georganta, E.; Rahman, R.; Raj, A. Growth of polycyclic aromatic hydrocarbons (PAHs) by methyl radicals: Pyrene formation from phenanthrene. Combust. Flame 2017, 85, 129–141. [Google Scholar] [CrossRef]
- Hansen, N.; Schenk, M.; Moshammer, K.; Kohse-Höinghaus, K. Investigating repetitive reaction pathways for the formation of polycyclic aromatic hydrocarbons in combustion processes. Combust. Flame 2017, 180, 250–261. [Google Scholar] [CrossRef]
- Hansen, N.; Miller, J.A.; Klippenstein, S.J.; Westmoreland, P.R.; Kohse-Höinghaus, K. Exploring formation pathways of aromatic compounds in laboratory-based model flames of aliphatic fuels. Combust. Explos. Shock Waves 2012, 48, 508–515. [Google Scholar] [CrossRef]
- Hansen, N.; Li, W.; Law, M.E.; Kasper, T.; Westmoreland, P.R.; Yang, B.; Cool, T.A.; Lucassen, A. The importance of fuel dissociation and propargyl + allyl association for the formation of benzene in a fuel-rich 1-hexene flame. Phys. Chem. Chem. Phys. 2010, 12, 12112–12122. [Google Scholar] [CrossRef] [PubMed]
- McEnally, C.; Pfefferle, L.; Atakan, B.; Kohse-Höinghaus, K. Studies of aromatic hydrocarbon formation mechanisms in flames: Progress towards closing the fuel gap. Prog. Energy Combust. Sci. 2006, 32, 247–294. [Google Scholar] [CrossRef]
- Richter, H.; Howard, J.B. Formation of polycyclic aromatic hydrocarbons and their growth to soot—A review of chemical reaction pathways. Prog. Energy Combust. Sci. 2000, 26, 565–608. [Google Scholar] [CrossRef]
- Cavallotti, C.; Polino, D. On the kinetics of the C5H5 + C5H5 reaction. Proc. Combust. Inst. 2013, 34, 557–564. [Google Scholar] [CrossRef]
- Wang, H.; Frenklach, M. A detailed kinetic modeling study of aromatics formation in laminar premixed acetylene and ethylene flames. Combust. Flame 1997, 110, 173–221. [Google Scholar] [CrossRef]
- Raj, A.; Prada, I.D.C.; Amer, A.A.; Chung, S.H. A reaction mechanism for gasoline surrogate fuels for large polycyclic aromatic hydrocarbons. Combust. Flame 2012, 159, 500–515. [Google Scholar] [CrossRef]
- Thomas Mckinnon, J.; Howard, J.B. The roles of pah and acetylene in soot nucleation and growth. Symp. Combust. 1992, 24, 965–971. [Google Scholar] [CrossRef]
- D’Anna, A.; Violi, A. A kinetic model for the formation of aromatic hydrocarbons in premixed laminar flames. Symp. Combust. 1998, 27, 425–433. [Google Scholar] [CrossRef]
- Kislov, V.V.; Sadovnikov, A.I.; Mebel, A.M. Formation Mechanism of Polycyclic Aromatic Hydrocarbons beyond the Second Aromatic Ring. J. Phys. Chem. A 2013, 117, 4794–4816. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Koshi, M. A novel route for PAH growth in HACA based mechanisms. Combust. Flame 2012, 159, 3589–3596. [Google Scholar] [CrossRef]
- Miller, J.A.; Melius, C.F. Kinetic and thermodynamic issues in the formation of aromatic compounds in flames of aliphatic fuels. Combust. Flame 1992, 91, 21–39. [Google Scholar] [CrossRef]
- Lindstedt, P.; Maurice, L.; Meyer, M. Thermodynamic and kinetic issues in the formation and oxidation of aromatic species. Faraday Discuss. 2001, 119, 409–432. [Google Scholar] [CrossRef]
- Raj, A.; Al Rashidi, M.J.; Chung, S.; Sarathy, S. PAH Growth Initiated by Propargyl Addition: Mechanism Development and Computational Kinetics. J. Phys. Chem. A 2014, 118, 2865–2885. [Google Scholar] [CrossRef] [PubMed]
- Melius, C.F.; Colvin, M.E.; Marinov, N.M.; Pit, W.J.; Senkan, S.M. Reaction mechanisms in aromatic hydrocarbon formation involving the C5H5 cyclopentadienyl moiety. Symp. Combust. 1996, 26, 685–692. [Google Scholar] [CrossRef]
- Kim, D.H.; Mulholland, J.A.; Wang, D.; Violi, A. Pyrolytic Hydrocarbon Growth from Cyclopentadiene. J. Phys. Chem. A 2010, 114, 12411–12416. [Google Scholar] [CrossRef] [PubMed]
- Frenklach, M.; Yuan, T.; Ramachandra, M.K. Soot formation in binary hydrocarbon mixtures. Energy & Fuels 1988, 2, 462–480. [Google Scholar]
- Froese, R.; Coxon, J.; West, S.; Morokuma, K. Theoretical Studies of Diels−Alder Reactions of Acetylenic Compounds. J. Org. Chem. 1997, 62, 6991–6996. [Google Scholar] [CrossRef]
- Siegmann, K.; Sattler, K. Formation mechanism for polycyclic aromatic hydrocarbons in methane flames. J. Chem. Phys. 1999, 112, 698. [Google Scholar] [CrossRef]
- Kislov, V.; Islamova, N.; Kolker, A.M.; Lin, S.H.; Mebel, A.M. Hydrogen Abstraction Acetylene Addition and Diels−Alder Mechanisms of PAH Formation: A Detailed Study Using First Principles Calculations. J. Chem. Theory Comput. 2005, 1, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Susa, A.; Miyoshi, A.; Koshi, M. Role of Phenyl Radicals in the Growth of Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 2008, 112, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Koshi, M. A highly efficient growth mechanism of polycyclic aromatic hydrocarbons. Phys. Chem. Chem. Phys. 2010, 12, 2427. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Miyoshi, A.; Koshi, M. Role of Methyl Radicals in the Growth of PAHs. J. Am. Soc. Mass Spectrom. 2010, 21, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Comandini, A.; Abid, S.; Chaumeix, N. Polycyclic Aromatic Hydrocarbon Growth by Diradical Cycloaddition/Fragmentation. J. Phys. Chem. A 2017, 21, 5921–5931. [Google Scholar] [CrossRef] [PubMed]
- Desgroux, P.; Mercier, X.; Thomson, K. Study of the formation of soot and its precursors in flames using optical diagnostics. Proc. Combust. Inst. 2013, 34, 1713–1738. [Google Scholar] [CrossRef]
- Raj, A.; Celnik, M.; Shirley, R.; Sander, M. A statistical approach to develop a detailed soot growth model using PAH characteristics. Combust. Flame 2009, 56, 896–913. [Google Scholar] [CrossRef]
- Shukla, B.; Susa, A.; Miyoshi, A.; Koshi, M. In Situ Direct Sampling Mass Spectrometric Study on Formation of Polycyclic Aromatic Hydrocarbons in Toluene Pyrolysis. J. Phys. Chem. A 2007, 111, 8308–8324. [Google Scholar] [CrossRef] [PubMed]
- Bauschlicher, J.C.; Ricca, A. Mechanisms for polycyclic aromatic hydrocarbon (PAH) growth. Chem. Phys. Lett. 2000, 326, 283–287. [Google Scholar] [CrossRef]
- Xu, F.; Shi, X.; Zhang, Q.; Wang, W. Mechanism for the growth of polycyclic aromatic hydrocarbons from the reactions of naphthalene with cyclopentadienyl and indenyl. Chemosphere 2016, 162, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Shi, X.; Sun, Y.; Zhang, Q.; Wang, W. The growth mechanism of polycyclic aromatic hydrocarbons from the reactions of anthracene and phenanthrene with cyclopentadienyl and indenyl. Chemosphere 2017, 189, 265–276. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Monogrpah on the Evaluation of Carcinogenic Risks to Human; List of Classifications; IARC: Lyon, France, 2018. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.B.; Schlegel, H.; Scuseria, G.E.; Robb, M.A.; Cheeseman, R.; Montgomery, J.J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, G.D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. IV. An improved atomic pair natural orbital method. J. Chem. Phys. 1994, 101, 5900–5909. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row atoms. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Hou, D.; You, X. Reaction kinetics of hydrogen abstraction from polycyclic aromatic hydrocarbons by H atoms. Phys. Chem. Chem. Phys. 2017, 19, 30772–30780. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R. Searching for zwitterionic intermediates in Hetero Diels–Alder reactions between methyl α,p-dinitrocinnamate and vinyl-alkyl ethers. Comput. Theor. Chem. 2014, 1046, 93–98. [Google Scholar] [CrossRef]
- Gonzales, C.; Schlegel, H.B. Reaction Path Following in Mass-Wighted Internal Coordinates. J. Phys. Chem. A 1990, 90, 2154. [Google Scholar]
- Burns, D.M.; Iball, I. The bond lengths in chrysene. Acta Crystallogr. 1956, 9, 314–315. [Google Scholar] [CrossRef]
- Gittins, C.M.; Rohlfing, E.A.; Rohlfing, C.M. Experimental and theoretical characterization of the S1–S0 transition of benzo[a]pyrene. J. Chem. Phys. 1998, 105, 7323. [Google Scholar] [CrossRef]
- Reed, D.R.; Kass, S.R. Experimental Determination of the a and b C-H Bond Dissociation Energies in Naphthalene; Wiley: Hoboken, NJ, USA, 2000; Volume 35. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
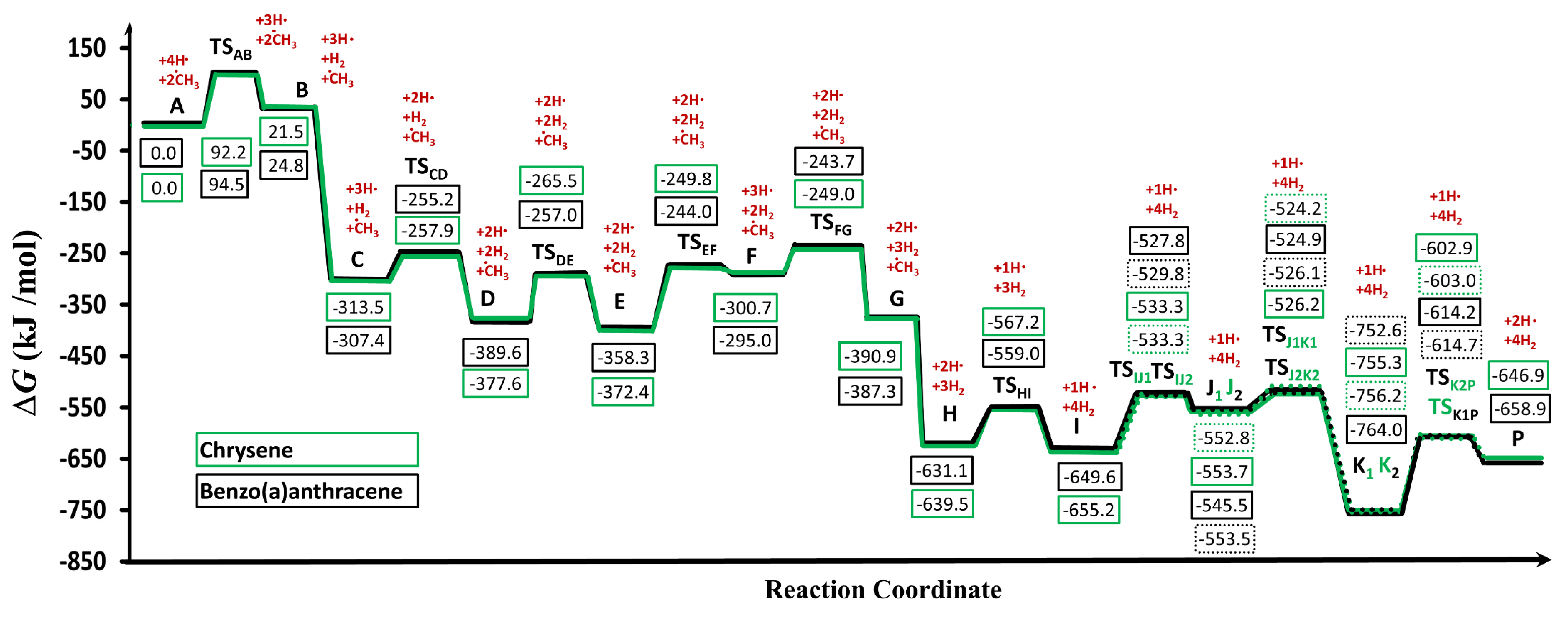
| BaA→BaP | ΔG | ΔH | S | Chr→BaP | ΔG | ΔH | S |
|---|---|---|---|---|---|---|---|
| a | 0.00 | 0.00 | 108.46 | A | 0.00 | 0.00 | 108.91 |
| TSab | 94.50 | 67.86 | 114.51 | TSAB | 92.24 | 64.50 | 114.06 |
| b | 24.76 | 30.63 | 109.44 | B | 21.46 | 26.89 | 109.54 |
| c | −307.39 | −359.74 | 113.13 | C | −313.47 | −364.87 | 114.34 |
| TScd | −255.24 | −336.50 | 117.34 | TSCD | −257.86 | −339.06 | 117.84 |
| d | −389.58 | −435.92 | 114.22 | D | −377.64 | −424.03 | 114.62 |
| TSde | −256.98 | −306.65 | 111.54 | TSDE | −265.48 | −315.72 | 111.53 |
| e | −358.30 | −408.32 | 111.27 | E | −372.38 | −422.91 | 111.31 |
| TSef | −244.05 | −294.14 | 111.21 | TSEF | −249.81 | −300.47 | 111.21 |
| f | −295.00 | −314.47 | 108.36 | F | −300.71 | −320.71 | 108.38 |
| TSfg | −243.72 | −291.55 | 113.02 | TSFG | −248.99 | −297.58 | 112.86 |
| g | −387.31 | −401.11 | 109.19 | G | −390.87 | −405.51 | 108.96 |
| h | −631.14 | −700.50 | 115.01 | H | −639.50 | −708.80 | 115.49 |
| TShi | −558.99 | −656.73 | 119.65 | TSHI | −567.16 | −664.70 | 120.25 |
| i | −649.59 | −710.62 | 117.95 | I | −655.24 | −715.79 | 118.78 |
| TSij1 | −527.83 | −590.35 | 117.05 | TSIJ1 | −533.35 | −595.99 | 117.10 |
| TSij2 | −529.88 | −592.03 | 116.76 | TSIJ2 | −533.31 | −596.02 | 117.05 |
| j1 | −545.54 | −603.31 | 120.90 | J1 | −553.65 | −611.28 | 121.12 |
| j2 | −553.48 | −610.83 | 120.57 | J2 | −552.85 | −610.59 | 121.03 |
| TSj1k1 | −524.89 | −587.64 | 116.58 | TSJ1K1 | −526.23 | −588.62 | 117.31 |
| TSj2k2 | −526.08 | −588.47 | 116.87 | TSJ2K2 | −524.15 | −586.46 | 117.38 |
| k1 | −764.02 | −827.12 | 116.29 | K1 | −755.42 | −818.33 | 116.88 |
| k2 | −752.55 | −818.11 | 114.32 | K2 | −756.29 | −820.14 | 116.13 |
| TSk1P | −614.19 | −678.89 | 115.01 | TSK1P | −602.87 | −668.16 | 114.99 |
| TSk2P | −614.69 | −679.93 | 114.58 | TSK2P | −602.96 | −668.26 | 114.97 |
| P | −658.89 | −694.43 | 110.99 | P | −646.93 | −683.02 | 110.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reizer, E.; Csizmadia, I.G.; Palotás, Á.B.; Viskolcz, B.; Fiser, B. Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH). Molecules 2019, 24, 1040. https://doi.org/10.3390/molecules24061040
Reizer E, Csizmadia IG, Palotás ÁB, Viskolcz B, Fiser B. Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH). Molecules. 2019; 24(6):1040. https://doi.org/10.3390/molecules24061040
Chicago/Turabian StyleReizer, Edina, Imre G. Csizmadia, Árpád B. Palotás, Béla Viskolcz, and Béla Fiser. 2019. "Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH)" Molecules 24, no. 6: 1040. https://doi.org/10.3390/molecules24061040
APA StyleReizer, E., Csizmadia, I. G., Palotás, Á. B., Viskolcz, B., & Fiser, B. (2019). Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH). Molecules, 24(6), 1040. https://doi.org/10.3390/molecules24061040