
Enzymatic Synthesis of Trideuterated Sialosides


Abstract
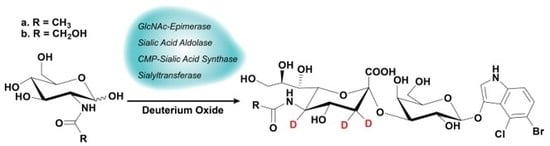
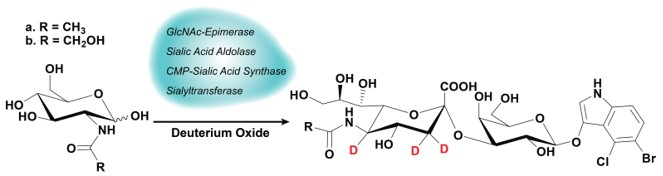
1. Introduction
2. Results
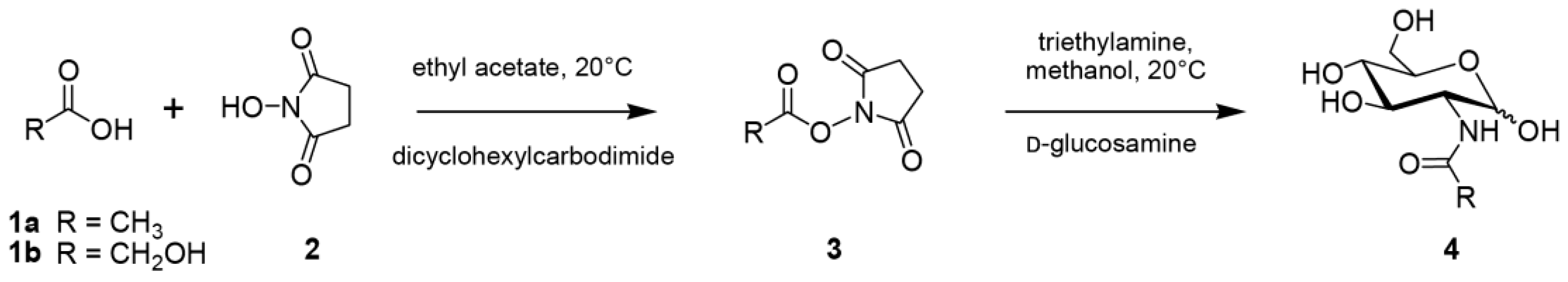
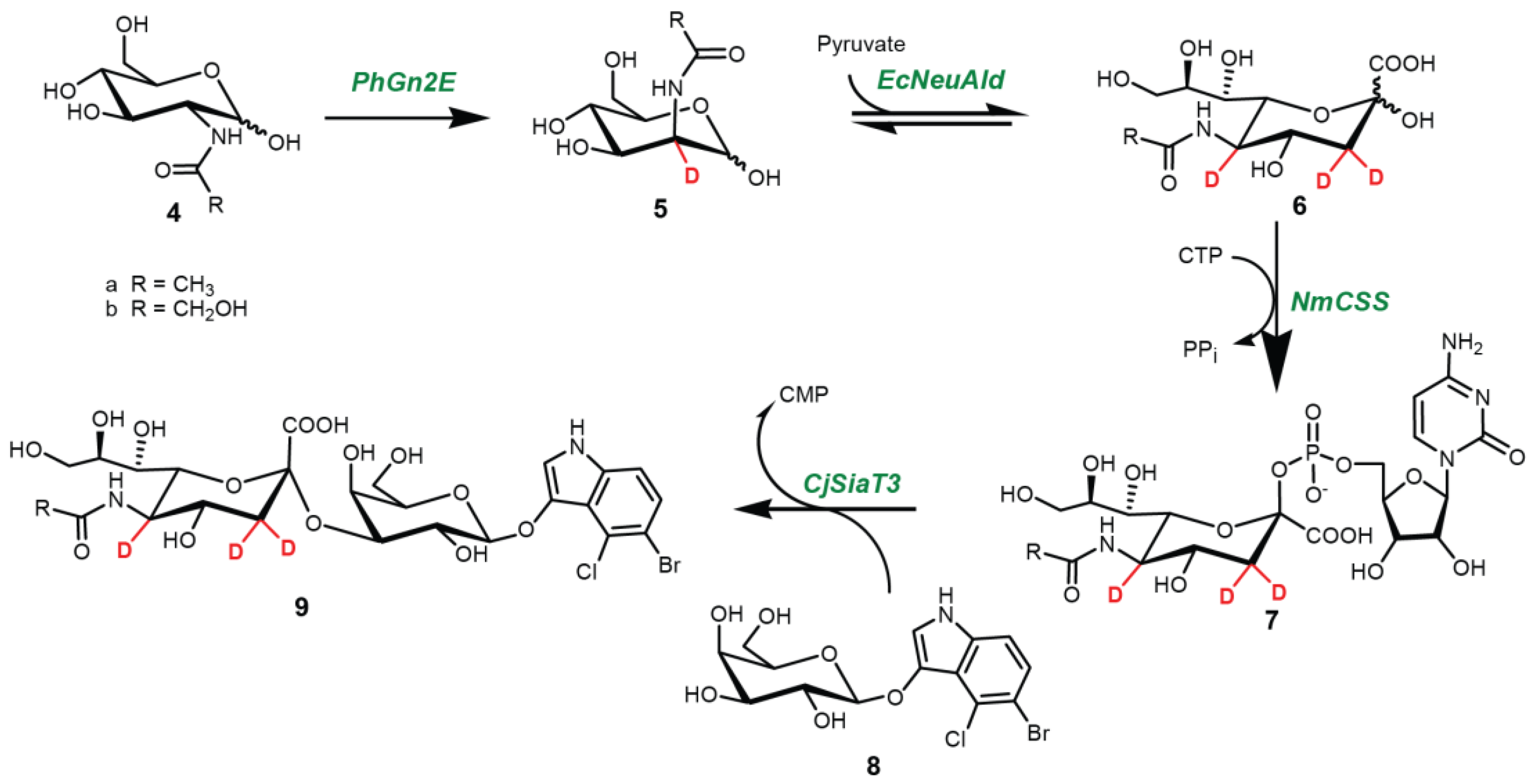
2.1. Synthesis
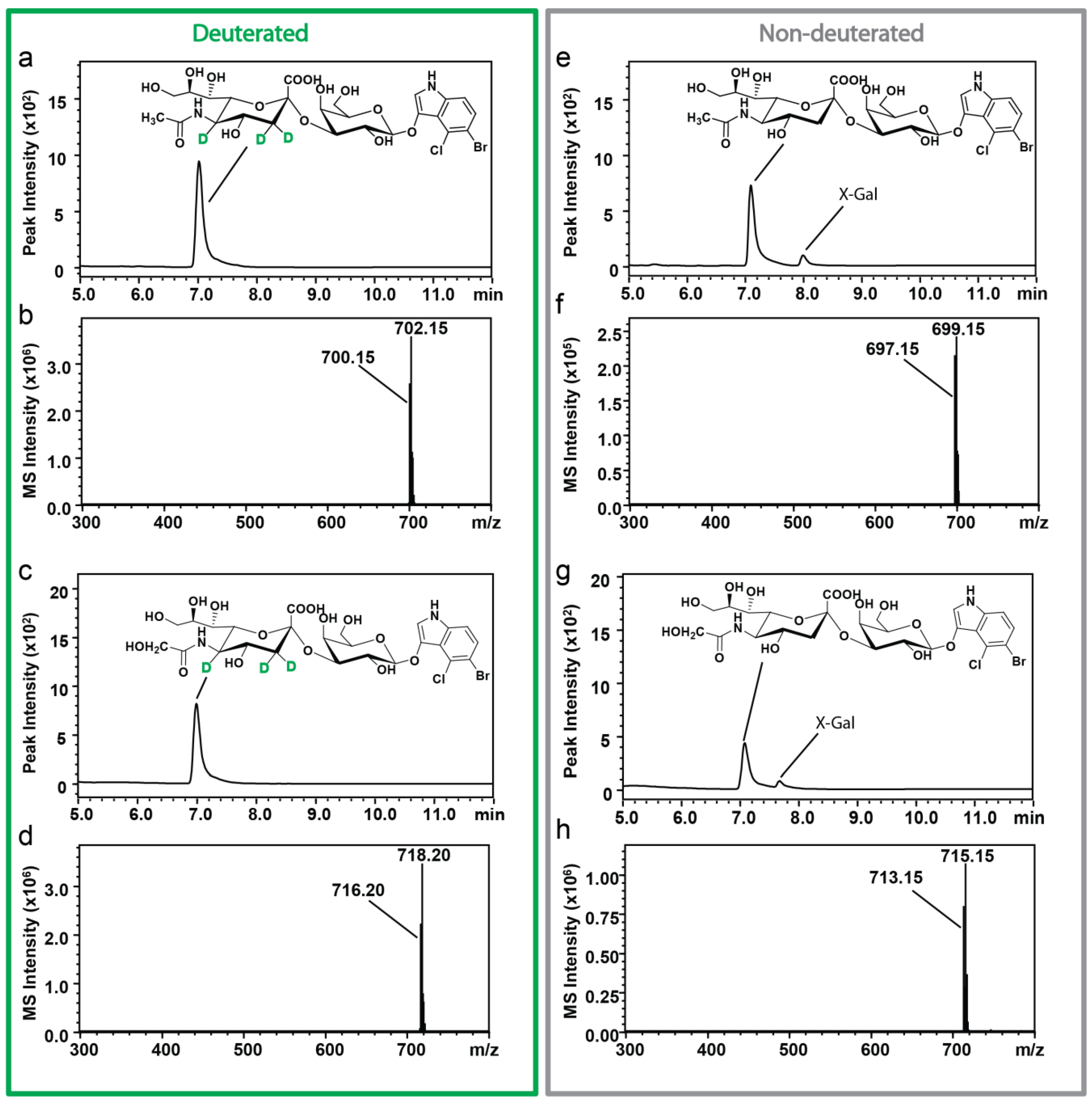
2.2. Characterization of Sialosides by HPLC-ESI-MS
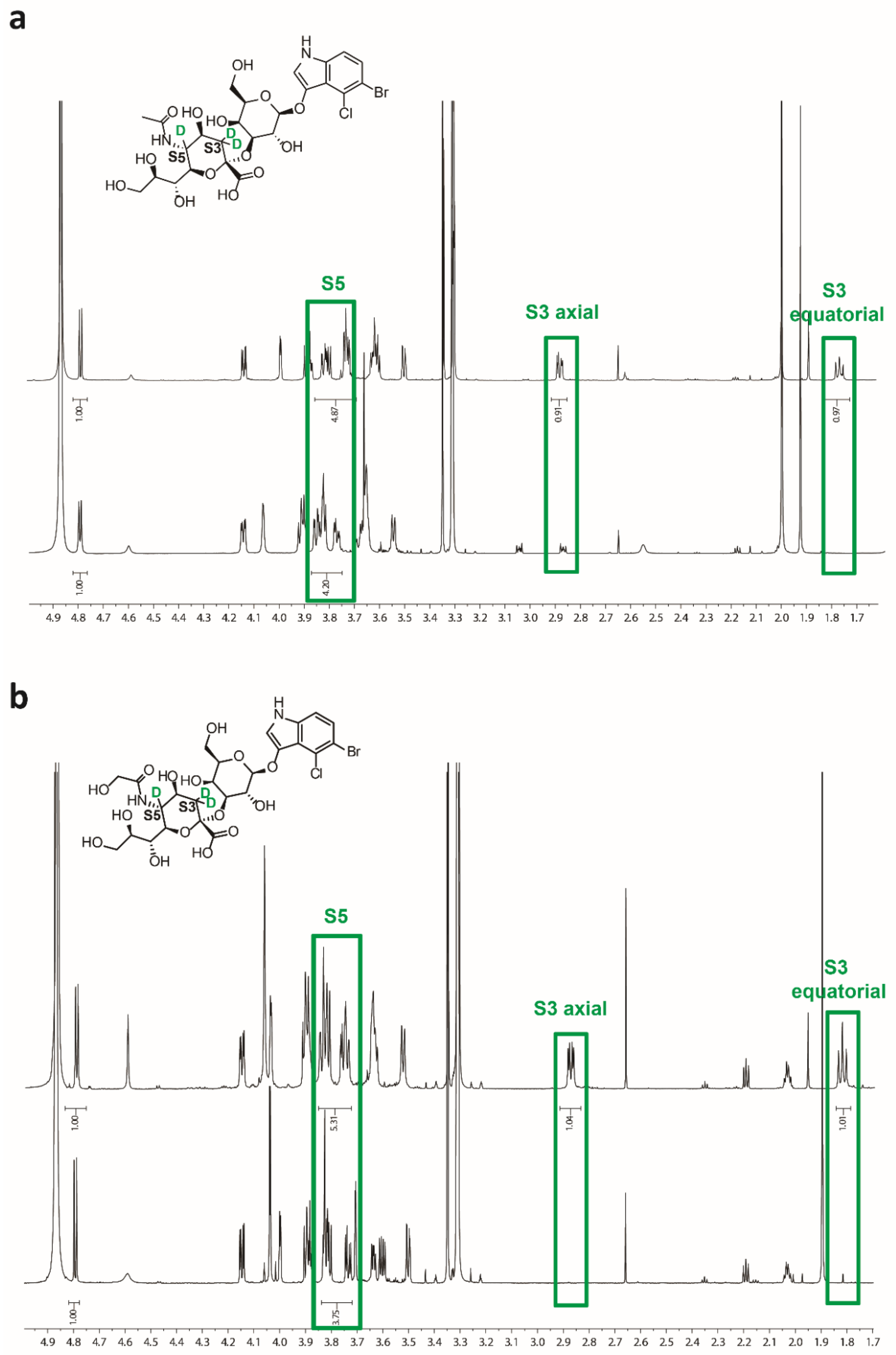
2.3. Characterization of Sialosides by NMR
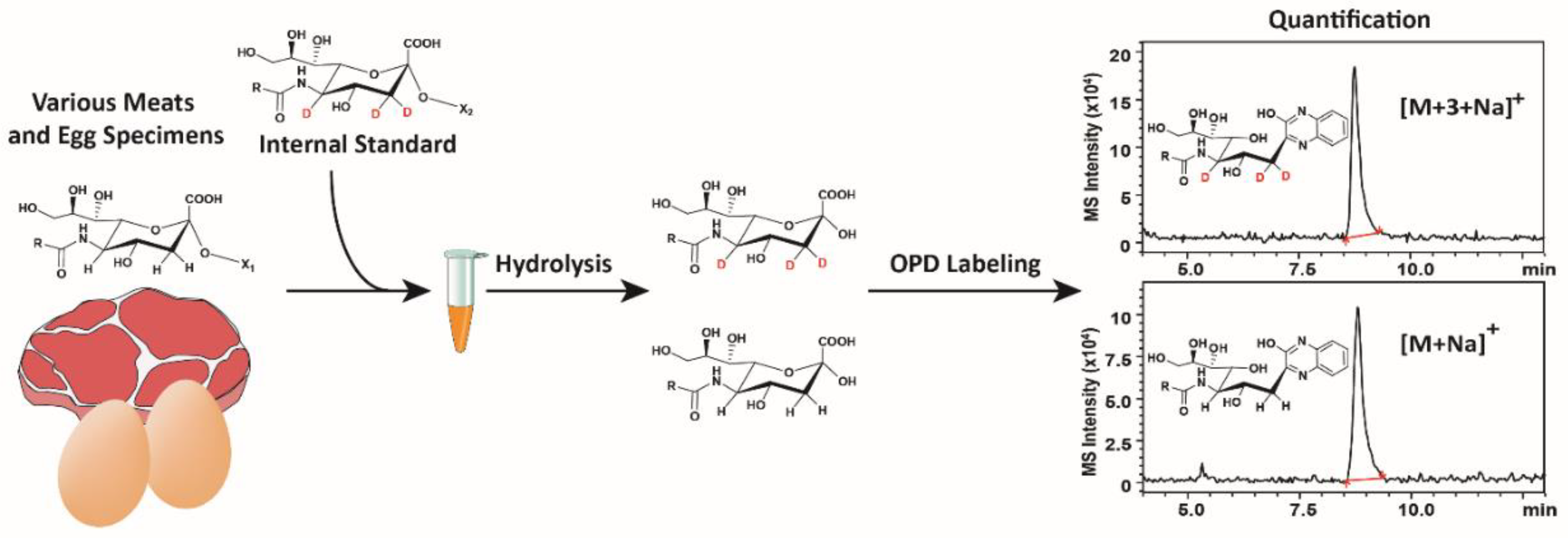
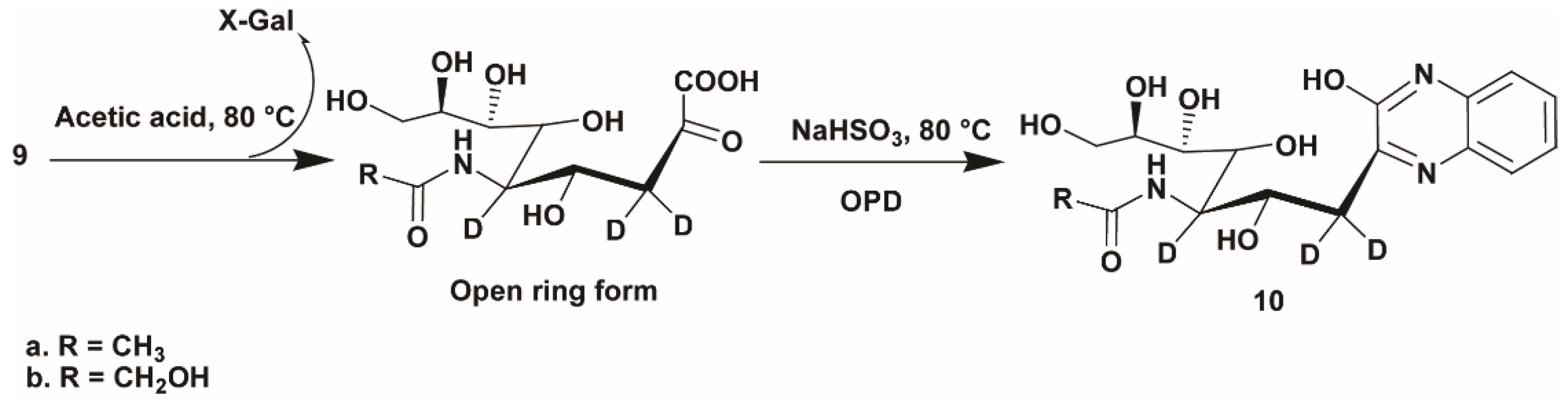
2.4. Application of d3-Sialosides as Internal Standards
3. Discussion
3.1. Synthesis and Structure Determination of d3-Sialosides
3.2. Sialic acid Quantification
4. Materials and Methods
4.1. Reagents
4.2. Chemical Synthesis of N-Acetyl-D-Glucosamine (GlcNAc) and N-Glycolyl-D-Glucosamine (GlcNGc)
4.3. One-Pot Four-Enzyme Synthesis of d3-Sialosides
4.4. Analysis of the Sialosides by NMR Spectroscopy
4.5. Analysis of the Sialosides by HPLC-ESI-MS
4.6. Acid Hydrolysis of d3-Sialosides and OPD Labeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Spichtig, V.; Michaud, J.; Austin, S. Determination of sialic acids in milks and milk-based products. Anal. Biochem. 2010, 405, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xia, L.; Liu, L.; Qu, F.; Sun, Z.; Zhao, X.; You, J. A sensitive and efficient method for determination of N-acetylhexosamines and N-acetylneuraminic acid in breast milk and milk-based products by high-performance liquid chromatography via UV detection and mass spectrometry identification. J. Chromatogr. B 2016, 1011, 14–23. [Google Scholar]
- Weerapana, E.; Imperiali, B. Asparagine-linked protein glycosylation: From eukaryotic to prokaryotic systems. Glycobiology 2006, 16, 91R–101R. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R. Chemistry, Metabolism, and biological functions of sialic acids. Adv. Carbohydr. Chem. Biochem. 1982, 40, 131–234. [Google Scholar] [PubMed]
- Drake, P.M.; Nathan, J.K.; Stock, C.M.; Chang, P.V.; Muench, M.O.; Nakata, D.; Reader, J.R.; Gip, P.; Golden, K.P.; Weinhold, B. Polysialic acid, a glycan with highly restricted expression, is found on human and murine leukocytes and modulates immune responses. J. Immunol. 2008, 181, 6850–6858. [Google Scholar] [CrossRef]
- Nadano, D.; Iwasaki, M.; Endo, S.; Kitajima, K.; Inoue, S.; Inoue, Y. A naturally occurring deaminated neuraminic acid, 3-deoxy-d-glycero-d-galacto-nonulosonic acid (KDN). Its unique occurrence at the nonreducing ends of oligosialyl chains in polysialoglycoprotein of rainbow trout eggs. J. Biol. Chem. 1986, 261, 11550–11557. [Google Scholar]
- Varki, A. Diversity in the sialic acids. Glycobiology 1992, 2, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Zanetta, J.-P.; Pons, A.; Iwersen, M.; Mariller, C.; Leroy, Y.; Timmerman, P.; Schauer, R. Diversity of sialic acids revealed using gas chromatography/mass spectrometry of heptafluorobutyrate derivatives. Glycobiology 2001, 11, 663–676. [Google Scholar] [CrossRef]
- Irie, A.; Koyama, S.; Kozutsumi, Y.; Kawasaki, T.; Suzuki, A. The Molecular Basis for the Absence of N-Glycolylneuraminic Acid in Humans. J. Biol. Chem. 1998, 273, 15866–15871. [Google Scholar] [CrossRef] [PubMed]
- Tangvoranuntakul, P.; Gagneux, P.; Diaz, S.; Bardor, M.; Varki, N.; Varki, A.; Muchmore, E. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc. Natl. Acad. Sci. USA 2003, 100, 12045–12050. [Google Scholar] [CrossRef]
- Alisson-Silva, F.; Kawanishi, K.; Varki, A. Human risk of diseases associated with red meat intake: Analysis of current theories and proposed role for metabolic incorporation of a non-human sialic acid. Mol. Asp. Med. 2016, 51, 16–30. [Google Scholar] [CrossRef]
- Varki, A. Uniquely human evolution of sialic acid genetics and biology. Proc. Natl. Acad. Sci. USA 2010, 107, 8939–8946. [Google Scholar] [CrossRef]
- Jourdian, G.W.; Dean, L.; Roseman, S. The Sialic Acids: XI. A periodate-resorcinol method for the quantitative estimation of free sialic acids and their glycosides. J. Biol. Chem. 1971, 246, 430–435. [Google Scholar]
- Warren, L. The Thiobarbituric Acid Assay of Sialic Acids. J. Biol. Chem. 1959, 234, 1971–1975. [Google Scholar]
- Kamerling, J.P.; Vliegenthart, J.F.G.; Versluis, C.; Schauer, R. Identification of O-acetylated N-acylneuraminic acids by mass spectrometry. Carbohydr. Res. 1975, 41, 7–17. [Google Scholar] [CrossRef]
- Che, F.Y.; Shao, X.X.; Wang, K.Y.; Xia, Q.C. Characterization of derivatization of sialic acid with 2-aminoacridone and determination of sialic acid content in glycoproteins by capillary electrophoresis and high performance liquid chromatography-ion trap mass spectrometry. Electrophoresis 1999, 20, 2930–2937. [Google Scholar] [CrossRef]
- Rohrer, J.S.; Thayer, J.; Weitzhandler, M.; Avdalovic, N. Analysis of the N-acetylneuraminic acid and N-glycolylneuraminic acid contents of glycoproteins by high-pH anion-exchange chromatography with pulsed amperometric detection (HPAEC/PAD). Glycobiology 1998, 8, 35–43. [Google Scholar] [CrossRef]
- Ota, T.; Yasuda, M.; Iijima, R.; Yui, S.; Fukuuchi, T.; Yamaoka, N.; Mawatari, K.-I.; Kaneko, K.; Nakagomi, K. Development of a fluorescence analysis method for N-acetylneuraminic acid and its oxidized product ADOA. J. Chromatogr. B 2013, 932, 152–157. [Google Scholar] [CrossRef]
- Ito, M.; Ikeda, K.; Suzuki, Y.; Tanaka, K.; Saito, M. An Improved Fluorometric High-Performance Liquid Chromatography Method for Sialic Acid Determination: An Internal Standard Method and Its Application to Sialic Acid Analysis of Human Apolipoprotein E. Anal. Biochem. 2002, 300, 260–266. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, L.; Liu, N.; Troy, F.A.; Wang, B. LC–MS/MS quantification of N-acetylneuraminic acid, N-glycolylneuraminic acid and ketodeoxynonulosonic acid levels in the urine and potential relationship with dietary sialic acid intake and disease in 3-to 5-year-old children. Br. J. Nutr. 2014, 111, 332–341. [Google Scholar] [CrossRef]
- Wang, F.; Xie, B.; Wang, B.; Troy, F.A., 2nd. LC-MS/MS glycomic analyses of free and conjugated forms of the sialic acids, Neu5Ac, Neu5Gc and KDN in human throat cancers. Glycobiology 2015, 25, 1362–1374. [Google Scholar] [CrossRef]
- Yao, H.L.; Conway, L.P.; Wang, M.M.; Huang, K.; Liu, L.; Voglmeir, J. Quantification of sialic acids in red meat by UPLC-FLD using indoxylsialosides as internal standards. Glycoconj. J. 2016, 33, 219–226. [Google Scholar] [CrossRef]
- Both, P.; Riese, M.; Gray, C.J.; Huang, K.; Pallister, E.G.; Kosov, I.; Conway, L.P.; Voglmeir, J.; Flitsch, S.L. Applications of a highly alpha2, 6-selective pseudosialidase. Glycobiology 2018, 28, 261–268. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Laborda, P.; Lu, A.-M.; Duan, X.-C.; Ma, H.-Y.; Liu, L.; Voglmeir, J. N-acetylglucosamine 2-epimerase from Pedobacter heparinus: First experimental evidence of a deprotonation/reprotonation Mechanism. Catalysts 2016, 6, 212. [Google Scholar] [CrossRef]
- Daniels, A.D.; Campeotto, I.; van der Kamp, M.W.; Bolt, A.H.; Trinh, C.H.; Phillips, S.E.; Pearson, A.R.; Nelson, A.; Mulholland, A.J.; Berry, A. Reaction mechanism of N-acetylneuraminic acid lyase revealed by a combination of crystallography, QM/MM simulation, and mutagenesis. ACS Chem. Biol. 2014, 9, 1025–1032. [Google Scholar] [CrossRef]
- Furlong, M.T.; Wujcik, C.E.; Ji, C.; Su, Y. Identifying and overcoming bioanalytical challenges associated with chlorine-containing dehydrogenation metabolites. Rapid Commun. Mass Spectrom. 2010, 24, 3092–3102. [Google Scholar] [CrossRef]
- Echeverria, B.; Etxebarria, J.; Ruiz, N.; Hernandez, A.; Calvo, J.; Haberger, M.; Reusch, D.; Reichardt, N.C. Chemo-Enzymatic Synthesis of (13)C Labeled Complex N-Glycans As Internal Standards for the Absolute Glycan Quantification by Mass Spectrometry. Anal. Chem. 2015, 87, 11460–11467. [Google Scholar] [CrossRef]
- Schauer, R. Achievements and challenges of sialic acid. Glycoconj. J. 2000, 7, 485–499. [Google Scholar] [CrossRef]
- Juneja, L.R.; Koketsu, M.; Nishimoto, K.; Kim, M.; Yamamoto, T.; Itoh, T. Large-scale preparation of sialic acid from chalaza and egg-yolk membrane. Carbohydr. Res. 1991, 214, 179–186. [Google Scholar] [CrossRef]
- Koketsu, M.; Juneja, L.R.; Kawanami, H.; Kim, M.; Yamamoto, T. Preparation of N-acetylneuraminic acid from delipidated egg yolk. Glycoconj. J. 1992, 9, 70–74. [Google Scholar] [CrossRef]
- Lapidot, Y.; Rappoport, S.; Wolman, Y. Use of esters of N-hydroxysuccinimide in the synthesis of N-acylamino acids. J. Lipid Res. 1967, 8, 142–145. [Google Scholar]
- Huang, K.; Wang, M.M.; Kulinich, A.; Yao, H.L.; Ma, H.Y.; Martínez, J.E.R.; Duan, X.C.; Chen, H.; Cai, Z.P.; Flitsch, S.L.; et al. Biochemical characterisation of the neuraminidase pool of the human gut symbiont Akkermansia muciniphila. Carbohydr. Res. 2015, 415, 60–65. [Google Scholar] [CrossRef]
- Cao, C.; Wang, W.J.; Huang, Y.Y.; Yao, H.L.; Conway, L.P.; Liu, L.; Voglmeir, J. Determination of Sialic Acids in Liver and Milk Samples of Wild-type and CMAH Knock-out Mice. J. Vis. Exp. 2017, 125, e56030. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Content (µg Neu5Gc/g Meat) |
|---|---|
| Beef | 11.8 ± 3.8 |
| Pork | 5.7 ± 1.1 |
| Wild boar | 8.0 ± 0.8 |
| Tibetan mutton | 8.3 ± 1.0 |
| Xu Zhou mutton | 7.4 ± 2.3 |
| Mongolian mutton | 22.3 ± 4.0 |
| Donkey | 7.6 ± 6.0 |
| Yak | 8.2 ± 0.48 |
| Species | Content (mg Neu5Ac/g Egg White and Egg Yolk) | |
|---|---|---|
| Egg White | Egg Yolk | |
| Chicken | 0.40 ± 0.10 | 1.37 ± 0.15 |
| Duck | 0.18 ± 0.02 | 1.34 ± 0.13 |
| Grey goose | 0.05 ± 0.00 | 0.55 ± 0.06 |
| Pigeon | 1.50 ± 0.16 | 2.25 ± 0.13 |
| Quail | 0.23 ± 0.02 | 0.96 ± 0.11 |
| Silkie chicken | 0.26 ± 0.02 | 0.55 ± 0.05 |
| Turkey | 0.82 ± 0.11 | 1.15 ± 0.12 |
| Guinea fowl | 0.38 ± 0.06 | 0.98 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Z.-P.; Conway, L.P.; Huang, Y.Y.; Wang, W.J.; Laborda, P.; Wang, T.; Lu, A.M.; Yao, H.L.; Huang, K.; Flitsch, S.L.; et al. Enzymatic Synthesis of Trideuterated Sialosides. Molecules 2019, 24, 1368. https://doi.org/10.3390/molecules24071368
Cai Z-P, Conway LP, Huang YY, Wang WJ, Laborda P, Wang T, Lu AM, Yao HL, Huang K, Flitsch SL, et al. Enzymatic Synthesis of Trideuterated Sialosides. Molecules. 2019; 24(7):1368. https://doi.org/10.3390/molecules24071368
Chicago/Turabian StyleCai, Zhi-P., Louis P. Conway, Ying Y. Huang, Wen J. Wang, Pedro Laborda, Ting Wang, Ai M. Lu, Hong L. Yao, Kun Huang, Sabine L. Flitsch, and et al. 2019. "Enzymatic Synthesis of Trideuterated Sialosides" Molecules 24, no. 7: 1368. https://doi.org/10.3390/molecules24071368
APA StyleCai, Z.-P., Conway, L. P., Huang, Y. Y., Wang, W. J., Laborda, P., Wang, T., Lu, A. M., Yao, H. L., Huang, K., Flitsch, S. L., Liu, L., & Voglmeir, J. (2019). Enzymatic Synthesis of Trideuterated Sialosides. Molecules, 24(7), 1368. https://doi.org/10.3390/molecules24071368