1. Introduction
Rhenium(VII) compounds, like the commonly known organic catalyst methyltrioxorhenium (MTO), may be applied in various catalytic processes as well as used to prepare active catalytic matrices. NH
4ReO
4, Al(ReO
4)
3 and HReO
4 are used for the preparation of heterogeneous catalysts of the Re
2O
7 type, which is embedded on solid carriers, usually Al
2O
3 or more rarely SiO
2, or another support with a highly developed specific surface area like SiO
2-Al
2O
3, Al
2O
3-B
2O
3. The most important Re
2O
7/Al
2O
3 system is characterized by high catalytic activity and selectivity in low a temperature (20–100°C), which is an important advantage. It is used in the metathesis of gaseous and/or liquid alkenes (also these with various functional groups) and acyclic alkenes with functional groups [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19]. The Re
2O
7/Al
2O
3 system was also used in large-scale technologies, e.g., FEAST [
20,
21] and META-4
® [
21,
22,
23]. For a long time it was considered that perrhenates used as catalysts are inactive, e.g., in oxidation reactions. Further research revealed that organic-inorganic compounds that contain a group, or more specifically ReO
4− anion, are promoters of olefins epoxidation [
24,
25], sulphide oxidation [
26], hydrolysis of cellulose [
27] or oxidative esterification of benzaldehydes with alcohols [
28]. A major advantage of these catalytic systems compared to metaloorganic catalysts, is their higher stability. They normally constitute a catalytic system component based on ionic liquids, which are formed by an organic part–e.g., imidazole as the ionic liquid (cation) and an inorganic part—a ReO
4− (anion) from a perrhenate (AgReO
4, NH
4ReO
4) [
24,
25,
26,
27,
28,
29] (
Figure 1).
Pyridinium and ammonium perrhenates were also investigated as potential catalysts for the deoxydehydration of diols to afford alkenes [
30].
N-alkylammonium perrhenate salts [N(R
1)
3R
2]
+[ReO
4]
− with varying substituents R
1 (C
4H
9, CH
3 or C
8H
17) and R
2 (C
4H
9, CH
3, C
8H
17, C
12H
25 or C
16H
33) are catalysts in olefin epoxidation processes [
31], while [N(hexyl)
4][ReO
4] catalyzes the reduction of organic carbonyls and carbon dioxide reactions in the presence of primary and secondary hydrosilanes [
32]. All mentioned rhenium(VII) organic salts are usually obtained from easily available ammonium perrhenate and/or perrhenic acid. NH
4ReO
4 and NaReO
4 were also examined as catalysts for the deoxydehydration of glycols in the presence of a reducing agent—sodium sulfite—in organic solvents [
33,
34] and NH
4ReO
4 has found application as a glycol deoxydehydration catalyst with alcohols as reducing agents in high boiling point organic solvents, producing alkene derivatives in high yield [
35].
Rhenium in the +7 oxidation state has ability to create many complex compounds, however, in this paper only one ammonium complex, i.e., hexaamminecobalt(III) perrhenate was discussed. Ammonium complex compounds containing metals like: copper(II), cobalt(II), cobalt(III), zinc(II), nickel(II), cadmium(II) and have been already described in the literature [
36,
37,
38,
39,
40]. The history of these compounds dates back to 1933, when synthesis methods and selected properties were reported by Wilke-Dorfurt and Gunzert [
37]. One of these compounds was hexaamminecobalt(III) perrhenate produced by the reaction of hexaamminecobalt(III) chloride with an excess of perrhenic acid [
37]. Another cobalt complex, namely [Co(NH
3)
5(ReO
4)](ReO
4)
2, was described by Lenz and Murmann. Their procedure was composed of two stages: in the first, [Co(NH
3)
5H
2O](ClO
4)
3 was dissolved in water and exposed to HReO
4, resulting in the formation of [Co(NH
3)
5(H
2O)](ReO
4)
3·2H
2O, which in the second stage was dried initially under vacuum for 3 h at 50–60 °C to produce a pink [Co(NH
3)
5(H
2O)](ReO
4)
3 complex, followed by further drying at 95–130 °C for 2–5 h to obtain the violet [Co(NH
3)
5(ReO
4)](ReO
4)
2 [
38].
In the literature syntheses of tetraamine complexes of zinc and cadmium can be found as well. These are obtained by adding concentrated ammonia solutions to suitable metal perrhenates [
37,
39]. Thermal gravimetric analysis (TGA) for both complexes indicated the loss of ammonia units with the increase in temperature, which can be ascribed to the formation of [M(NH
3)
2](ReO
4)
2 and M(ReO
4)
2 (where M is: Zn, Cd). Zinc(II)-diamine perrhenate is formed in the 150–95 °C temperature range, while anhydrous zinc(II) perrhenate is formed at 230–275 °C. For the formation of cadmium(II)-diamine perrhenate and cadmium(II) perrhenate the temperature ranges are 100–150 °C and 180–230 °C, respectively [
39].
It was found using Raman spectroscopy that the ReO
4− anion is present in ionic form and is not coordinated with the metal atom, while X-ray diffraction showed the isostructurality of the following compounds: [Zn(NH
3)
4](ReO
4)
2, [Cd(NH
3)
4](ReO
4)
2 and [Co(NH
3)
4](ReO
4)
2 [
39].
Preparation procedures of perrhenates described in the literature do not however pay enough attention to their environmental and economic aspects. Rhenium is a rare and valuable metal, with an annual output of ca. 60 tonnes. Application of suitable auxiliary operations could allow minimization of rhenium losses and maximize the purity of the compounds.
This publication presents production methods for aluminum and copper(II) perrhenates, and the ammonia complex of cobalt(III) perrhenate. These compounds are precursors for catalytic system preparation or are constituents of such systems. All the described technologies are complete, including management of generated wastes, and could be easily applied in industry. They were scaled up and tested so it is possible to implement them even in a continuous production line. Currently, these technologies constitute an offer made by the industrial partner Syntal Sp.z.o.o (Gliwice, Poland) for the Institute of Non-Ferrous Metals (IMN, Gliwice, Poland).
3. Experimental Section
3.1. Materials
Perrhenic acid of high purity, produced at the IMN using extraction as well as ion-exchange method (rhenium concentrations above 300.0 g/dm
3), was used as the source of rhenium in all experiments. Perrhenic acid contained 50-900g/dm
3 Re and <20 ppm ammonium ions, <2 ppm As; <2 ppm Bi, <3 ppm Zn, <3 ppm Mg, <3 ppm Cu, <5 ppm Mo, <5 ppm Ni, <5 ppm Pb, <10 ppm K, <3 ppm Ca, <3 ppm Fe, <3 ppm Al, <5 ppm Co [
41,
42,
43,
44]. High purity nitric acid (HNO
3) and 30% solution of hydrogen peroxide (POCH, Gliwice, Poland) were also used. Cobalt complex was synthesized from ammonium perrhenate (KGHM Polska Miedź S.A., Głogów, Poland) and cobalt(II) perrhenate (produced based on technology developed at IMN) [
45,
46,
47,
48]. In ion-exchange experiments two acidic resins, namely, C160 and PFC 100 × 10 (Purolite, Gdańsk, Poland) were used. In the ammonium complex production, [Co(NH
3)
6]Cl
3 (Acros Organics, Trenton, NJ, USA) was used. Additionally, aluminum, copper and cobalt salts and oxides (Acros Organics) were also used. Purification was performed using anhydrous extra purity acetone (POCH, Gliwice, Poland).
3.2. Various Metals Perrhenates–Ion-Exchange Methods
Ion-exchange method was used for nickel(II), cobalt(II), cesium, rubidium, copper(II), chromium(III) and aluminum perrhenates production, where sorption of nickel, cobalt, cesium, rubidium, copper, chromium and aluminum cations occurred. In the next step, these cations were eluted with aqueous solutions of perrhenic acid. The production method of perrhenic acid depends on the planned further use of the mentioned perrhenates. It can be obtained either by ion-exchange method (for superalloy precursors production) [
41,
42,
43] or by solvent extraction [
44] (for catalyst preparation). Solutions from the elution step were mixed with the other one, e.g., obtained from the washing after elution, and directed for the evaporation (in case of Ni, Co, Cr, Cs, Rb, Al, Cu) or crystallization (in case of
nano-Rb,
nano-Cs). Produced perrhenates were washed and dried to obtain their final form. Production methods of perrhenates of selected metals, i.e., nickel(II), cobalt(II), chromium(III) were presented in details in previous publications of the main author [
45,
46,
47,
48,
49,
50,
51]. It has to be mentioned that the important techno-economical factor of these technologies is attributed to the applied recycles that influence product purity and technology efficiency. The production methodologies of copper(II) perrhenate and aluminum perrhenate were described.
Parameters for sorption process were: sorption efficiency (described as the mass ratio of Cu or Al sorbed in ionite to their mass in initial solutions times 100%), and ionite saturation ratio (described as mass ratio of Cu or Al in the solution directed to neutralization to mass of the components sorbed in ionite times 100%). Elution efficiency was calculated as the ratio of metal mass in the solution before neutralization to metal mass sorbed in the ionite, multiplied by 100%.
3.2.1. Copper(II) Perrhenate
The first preparation of copper(II) perrhenate hydrates from copper(II) carbonate and perrhenic acid was reported in 1931 by Briscoe et al. [
36]. In this paper synthesis of copper(II) perrhenate using copper-containing aqueous nitrate solutions was described. Research conducted in dynamic and static conditions allowed us to identify C160 ionite in the hydrogenated state as the most effective resin. Preliminary conditions for each process step were chosen during experiments too. Consequently, it was assumed that the ionite saturation ratio required to conduct process properly needs to be between 2.0 and 4.0%. It was considered that the sorption of copper(II) ions should be performed in such way that the final concentration of copper in the solution does not exceed 0.5g/dm
3. Research on a large-laboratory scale under dynamic conditions were performed with 1.0 kg of the ionite placed inside an ion-exchange column. The ratio of the column’s height to its diameter was above 8.5. Due to the cyclic character of the process and ability to verify the results only after conducting few cycles it was decided to do ten cycles. Experiments were conducted until the ionite saturation ratio with copper ions was 2.0% in two consecutive cycles. Ionite after cycle X was regenerated with 32% nitric acid solution and tested for another five cycles. Sorption was performed using a solution containing 5.0 g/dm
3 Cu from the top of the column, at room temperature with the flow rate of 5.0 dm
3/h. Ionite after the sorption was washed with water from the top of the column, using about 1.5 dm
3 of water on each 1 kg of ionite, with a flow rate of 7.5 dm
3/h. Solutions resulting from washing were mixed with that from sorption, and managed. Washed resin was eluted with aqueous solution of perrhenic acid 80–120 g/dm
3 Re, using above 6.0 g of rhenium on each 1 g of copper absorbed in the ionite, with addition of 0.05–0.10 dm
3 concentrated nitric acid and 0.025–0.050 dm
3 30% solution of hydrogen peroxide on each 1 dm
3 of eluting agent (perrhenic acid). Elution was performed from the top to the bottom of the column with a flow rate of 2.5 dm
3/h. Solutions from the elution were stored in two portions–1/3 of the total volume, which was mixed with solutions resulting from purification, was recycled to the eluting agent preparation step, and 2/3 of the total volume was mixed with solutions resulting from washing after elution and directed to neutralization. The eluted ionite bed was washed with water using about 3.0 dm
3 of water for each 1 kg of ionite with flow rate of 3.0–4.0 dm
3/h. Solutions from washing after elution were collected and then mixed with qcsecond portion of solution resulting from elution. Such a mixture was treated with copper(II) oxide and/or copper(II) hydroxide in order to obtain a solution of stoichiometric composition of copper to rhenium, which is favorable for extraction of the final product–copper(II) perrhenate. Solution neutralization was performed at 80 °C until the pH between 3.5–7.2 was achieved. Post-neutralization solution was filtered at elevated temperature to remove solid impurities. Concentrating of the neutralized solution was performed at temperature up to 80 °C with vigorous mixing until completely dried. Three temperatures, i.e., 20, 60 and 80 °C and two neutralizing agents (copper(II) oxide and freshly precipitated copper(II) hydroxide) were tested. During neutralization and concentration steps, 30% solution of hydrogen peroxide was added to prevent the reduction of copper(II) ions. Obtained precipitates were dried in air, and then purified in two steps: firstly with 15% aqueous solution of hydrogen peroxide at ≤5 °C (using 0.002 dm
3 of agent on each 1.0 g of precipitate) and then anhydrous acetone washing at ≤10 °C (using 0.0005 dm
3 of acetone per each 1.0 g of precipitate). Aqueous solution from the washing was mixed with the first portion of solution resulting from elution and then recycled to eluting agent preparation step. Same portion of acetone was used in all fifteen cycles. Precipitate after purification was dried at the temperature below 140 °C until constant mass was obtained. The obtained anhydrous copper(II) perrhenate with stoichiometric composition was then analyzed for the content of selected impurities.
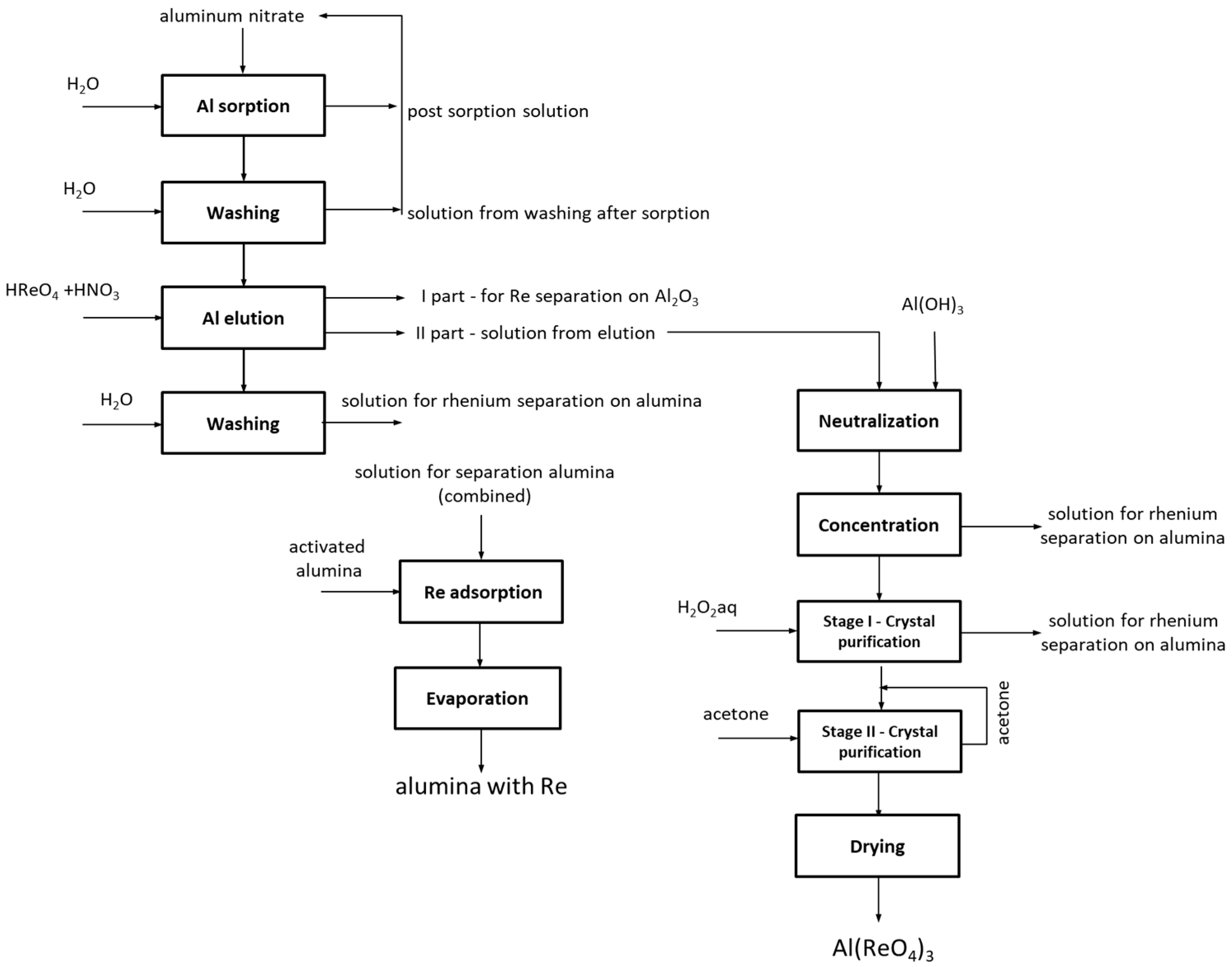
3.2.2. Aluminum Perrhenate
The history of Al(ReO
4)
3 dates back to 1968 when it was first synthesized by Varfolomeev et al. [
52]. Since then this compound is produced in aluminum salt (either nitrate or carbonate) reactions with perrhenic acid. The production of anhydrous aluminum perrhenate, which was described in this publication, was conducted with an aqueous solution of aluminum nitrate as the metal source. Based on results from dynamic and static conditions on various scales PFC 100 × 10 in hydrogenated form was the most effective resin. In these experiments preliminary parameters of each operations were chosen. It was assumed that the ionite saturation ratio must be above 2.0% for the process to proceed properly. It was considered that the sorption of aluminum ions should be continued to the final aluminum concentration below 1.0 g/dm
3. Large-laboratory scale research was conducted under dynamic conditions using an ion-exchange column, with a height to diameter ratio above 10. The column was loaded with 1 kg of ionite. Due to the cyclical nature of the process and ability to verify results only after performing a few cycles ten cycles of ion-exchange were done. Research was conducted until the ionite saturation ratio with aluminum ions was below 1.0% in two consecutive cycles. Ionite after cycle X was regenerated with 32% nitric acid solution and tested for another five cycles. Sorption was performed using solution containing 5.0 g/dm
3 Al, from the top to the bottom of the column, at room temperature with a flow rate of 3.0 dm
3/h. After sorption the ionite was washed with water from the top to the bottom of the column, using about 1.5 dm
3 of water on each 1.0 kg of ionite with a flow rate of 3.0 dm
3/h. Solutions resulting from washing after sorption were mixed with solutions resulting from sorption and used to dissolve aluminum nitrate. Washed resin was eluted with aqueous solution of perrhenic acid of 200 g/dm
3 Re concentration, using 21.0 g of rhenium on each 1.0 g of aluminum, with addition of 0.15 dm
3 nitric acid on each 1.0 dm
3 of eluting agent (perrhenic acid). Elution was performed from the top to the bottom with a flow rate of 1.5 dm
3/h. Solutions resulting from elution were collected in two portions: first one-bed volume was mixed with solution resulting from washing after elution, while the second 2-5 bed volumes (BV) was neutralized and crystallized. Eluted ionite bed was washed using 2.0–5.0 dm
3 of water on each 1 dm
3 of ionite with a flow rate of 3.0 dm
3/h. Solutions obtained from washing after elution were collected and mixed with the first portion of solution from elution and then sent to rhenium extraction on aluminum oxide. The second portion from elution (addressed to neutralization, concentration and separation), which contained an excess of rhenium vs aluminum, was treated with freshly precipitated aluminum hydroxide in a stoichiometric amount to the excess of rhenium in the solution. It was fed as aqueous solution washed from ammonium ions to the level <0.1%. This operation was performed at the temperature below 80 °C until all the aluminum oxide reacted, i.e., until a pH in the range from 3.5 to 4.4 was obtained. After neutralization, the solution was filtered at elevated temperatures to remove solid impurities. The solution was neutralized and concentrated at a temperature below 80 °C with intensive mixing until completely dry. Obtained precipitate was purified in two steps: washing with demineralized water at temperature ≤10 °C (using 0.001–0.020 dm
3 of water on each 1 g of precipitate), and then with anhydrous acetone at temperature ≤10 °C (using 0.0005–0.0050 dm
3 of acetone on each 1 g of precipitate). Solutions resulting from the purification step were mixed together with the solution obtained from washing after elution and that from the first part of elution. Solid residue after purification was dried at 120–160 °C to constant mass of the aluminum perrhenate with a stoichiometric composition. Next thus was dried at 160–200 °C to obtain a constant mass of its anhydrous form. Solutions from mixing washings after elution and the first part of elution, were treated with active aluminum oxide in such amount to obtain up to 80 wt.% of rhenium in final precipitate (product B) and then directed to complete drying with intensive mixing at the temperature below 80 °C. Obtained precipitate that contained up to 80 wt.% of rhenium was sent to the calcination and activation. It should be mentioned that both materials (products A and B) are suitable substrates for rhenium catalyst production.
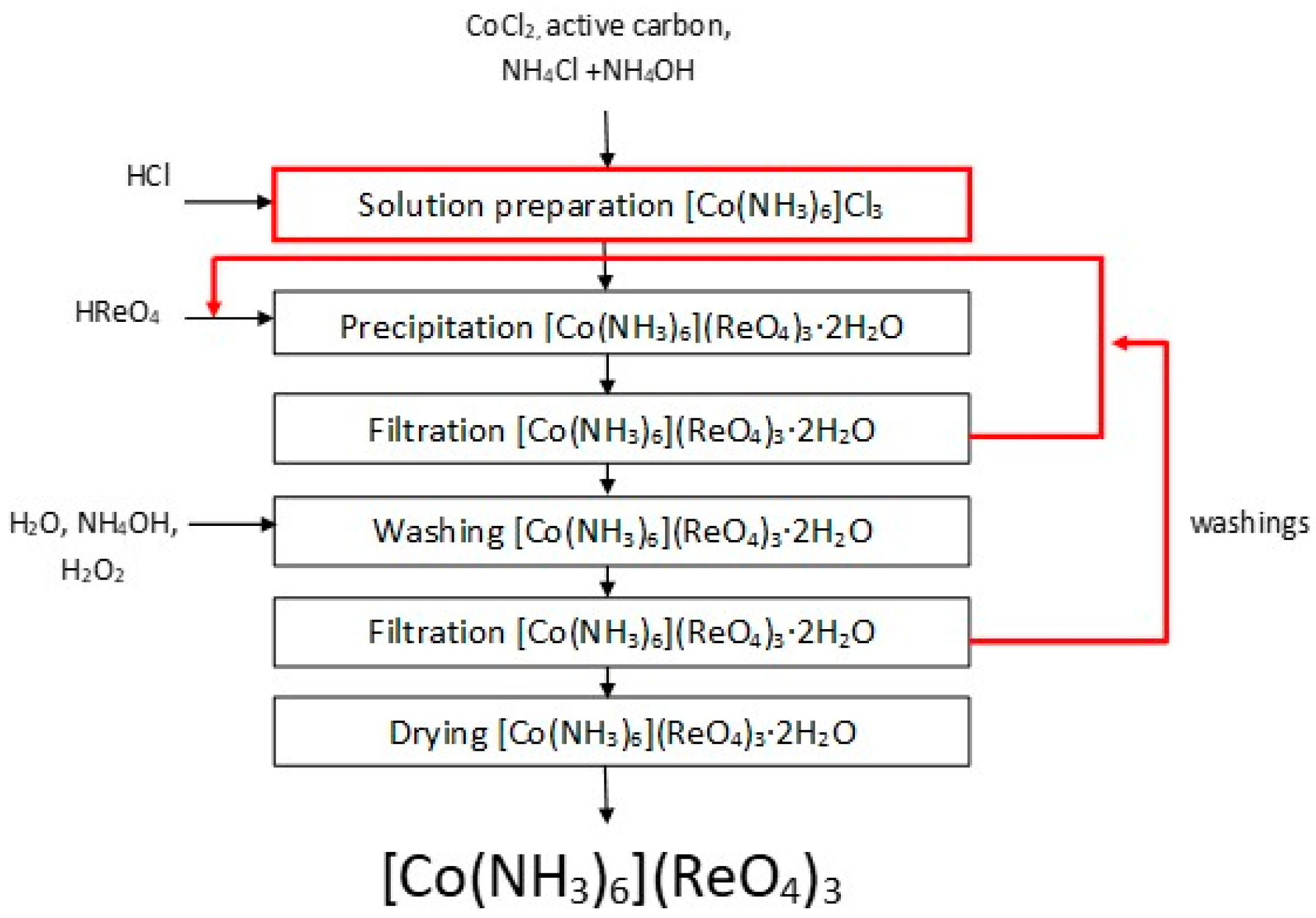
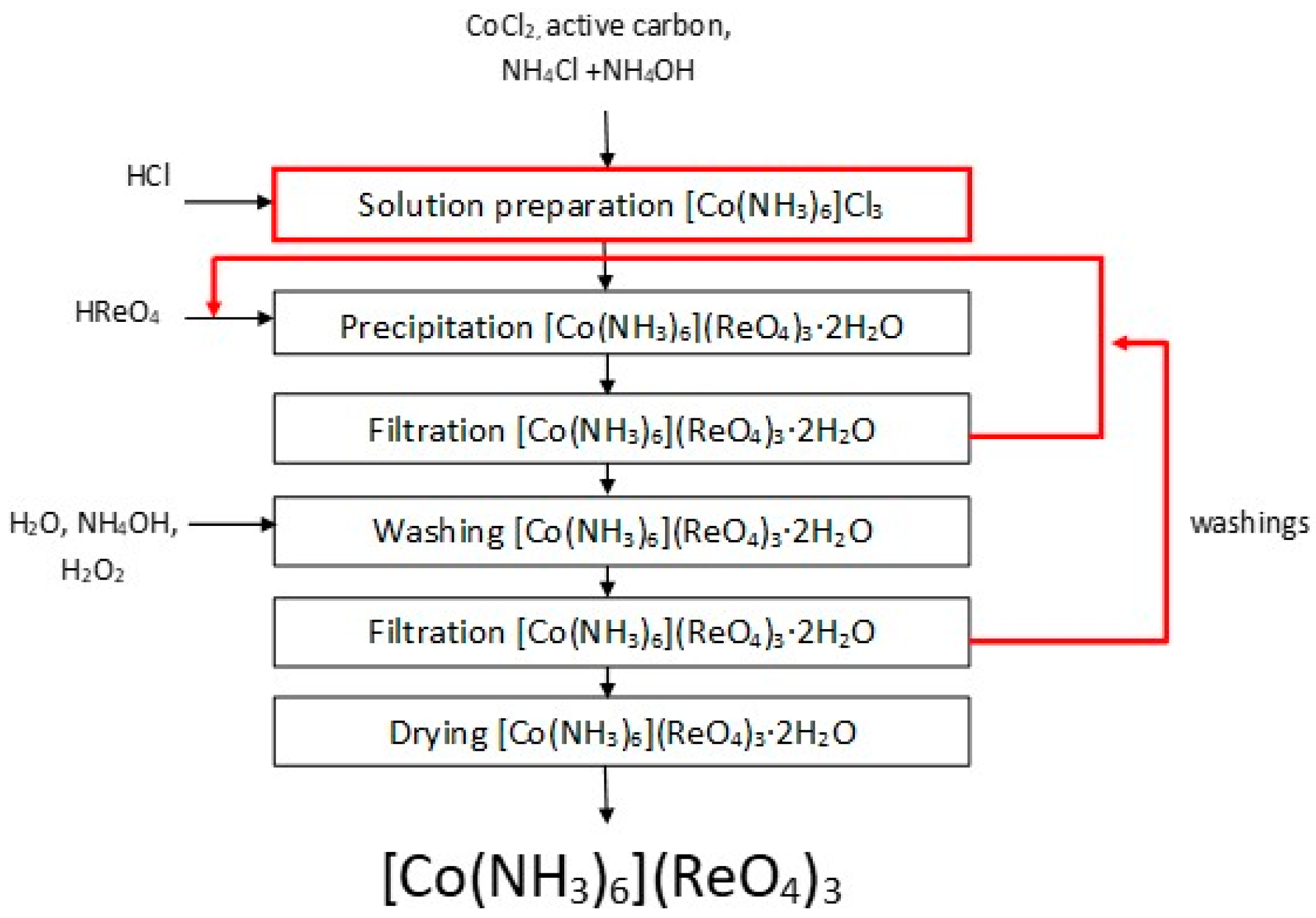
3.3. Hexamminecobalt(III) Perrhenate
Ammonia complexes of perrhenate are compounds of ML
x(ReO
4)
y type, containing coordinated ReO
4− ions, where M is either nickel or cobalt, and L is a coordinated NH
3 group. A method of anhydrous hexaamminecobalt(III) perrhenate production based on reaction (1) with perrhenic acid and hexaamminecobalt(III) chloride, was described in this paper:
Possible replacement of commercial product by obtained hexaamminecobalt(III) chloride and ammonia solution containing cobalt was checked. Moreover, the influence of selected factors on efficiency and composition of obtained [Co(NH3)6](ReO4)3·2H2O, i.e., hexaamminecobalt(III) chloride solution concentration, process temperature, reaction time, excess of perrhenic acid, and concentration of perrhenic acid, were investigated. The possibilities to replace perrhenic acid by other precipitating agents like ammonium perrhenate or anhydrous cobalt(II) perrhenate to produce [Co(NH3)6](ReO4)3·were investigated. The effect of hexaamminecobalt(III) perrhenate concentration were conducted based on the following procedure: 0.1 dm3 of 5.0–50.0 g/dm3 hexamminecobalt(III) chloride solution was magnetically mixed with aqueous solution of perrhenic acid (670.0 g/dm3 Re with 30 or 60% excess of rhenium to cobalt) at room temperature for half an hour. Phases were separated, and cobalt content in solution was analyzed, while the precipitate was analyzed with respect to the cobalt, ammonium and rhenium ions.
Effect of temperature was examined in the range from 10 to 80 °C. The reaction time was tested in the range from 0.5 to 5.0 h. The excess of rhenium was investigated as follows: 0.1 dm3 of 20.0 g/dm3 hexaamminecobalt(III) chloride solution was magnetically mixed with solution containing 569.0 g/dm3 of perrhenic acid, satisfying an excess of rhenium vs cobalt ranging from 10 to 200%. A trial with stochiometric amount of rhenium and cobalt was performed as well. Experiments were conducted at room temperature, intensively mixing with precipitate for 30 min.
The influence of perrhenic acid concentration on process efficiency was investigated as follows: 0.1 dm3 of 50.0 g/dm3 hexaamminecobalt(III) chloride solution was mixed with aqueous solution of perrhenic acid of concentration 107.0 to 508.0 g/dm3 Re with 60% excess of rhenium to cobalt, at room temperature intensively mixing with precipitate for 30 min.
The type of rhenium substrate was tested using the following procedure: 0.1 dm3 of 30.0 g/dm3 hexaamminecobalt(III) chloride solution was mixed with ammonium perrhenate either in solid state or as water solution, or with cobalt(II) perrhenate in solid state. Research was conducted at room temperature intensively mixing with precipitate for half an hour. An 60% excess of rhenium to cobalt was used.
Experiments concerning possible substrate change were also an important part of the research. These were done by mixing 50.0 g of cobalt(II) chloride hexahydrate and 33.0 g of ammonium chloride in 0.3 dm3 of water. Then 10.0 g of activated carbon and 0.45 dm3 of ammonia solution were added. After cooling in ice bath, 0.04 dm3 of 30% hydrogen peroxide was carefully added drop by drop, to prevent the temperature from exceeding 10 °C. The resulting mixture was heated to 60 °C by 30 min and then cooled again in an ice bath. Crystallized product and activated carbon were separated from the solution, and dissolved in 0.4 dm3 of hot water with 0.01 dm3 of HCl. Next the mixture was heated and activated carbon was separated from the solution. The resulting solution was divided into two parts. The first part was used in the synthesis of hexaamminecobalt(III) perrhenate, while the second was placed in an ice bath to crystallize hexaamminecobalt(III) chloride. Crystallized product was separated from the filtrate, washed with water containing ice and then dried in the air. After drying (>120 °C) the content of chlorine, cobalt and ammonium ions, were analyzed. Crystallized product was dissolved in water to obtain hexaamminecobalt(III) chloride solution of 30.0 g/dm3 concentration. To 0.1 dm3 of such solution, 664 g/dm3 of Re as perrhenic acid solution was added with 30% excess of rhenium to cobalt. Trials were performed at room temperature with intensive mixing of the solution with precipitate for 30 min. Precipitate and filtrate were analyzed regarding cobalt content. Additionally in the case of precipitates the content of rhenium and ammonia was also analyzed. Similar test was performed for ammonia solution containing cobalt obtained directly after separation from activated carbon. The presented methodology was also performed on a 10-times larger scale.
3.4. Analytical Methods
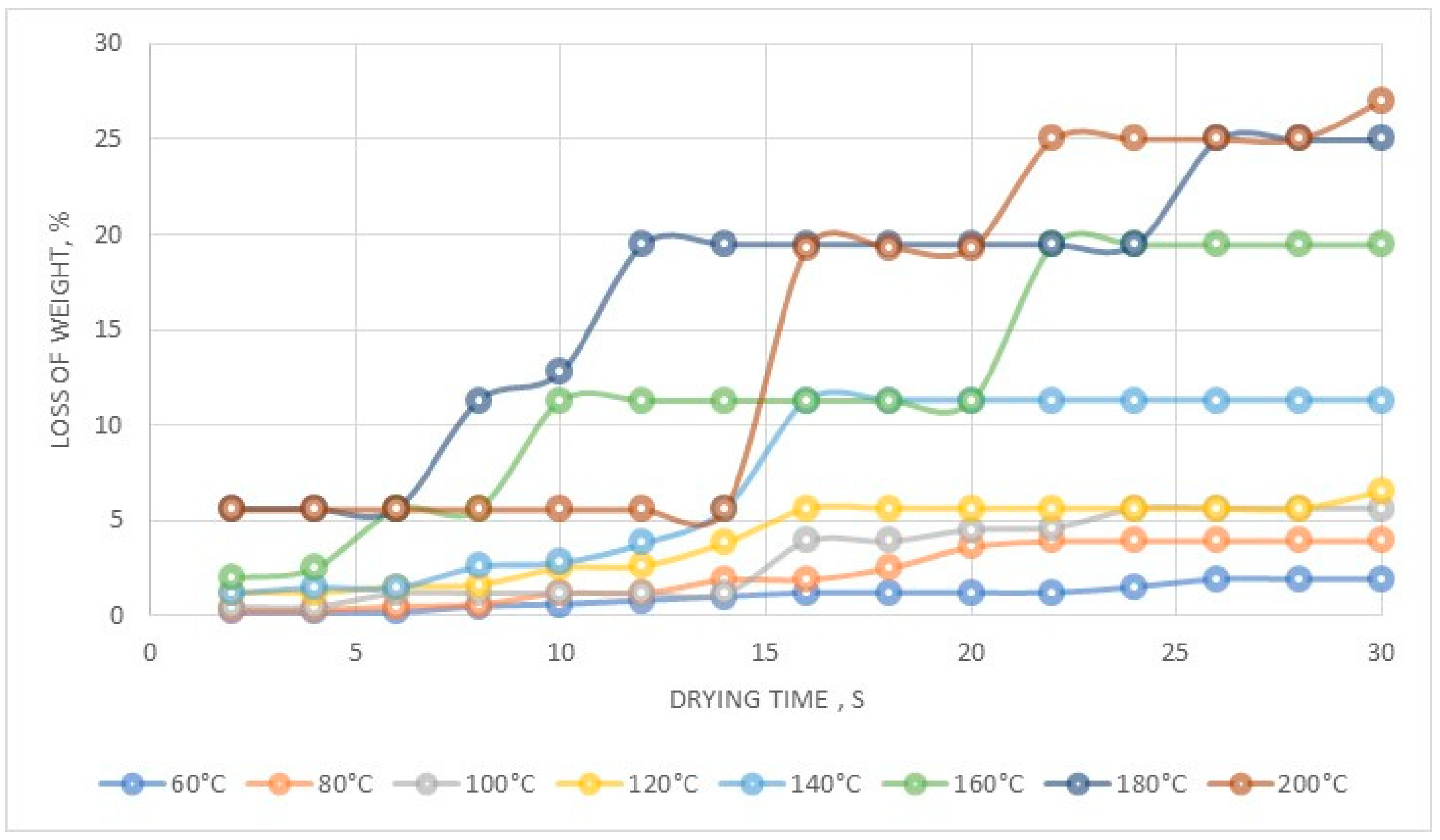
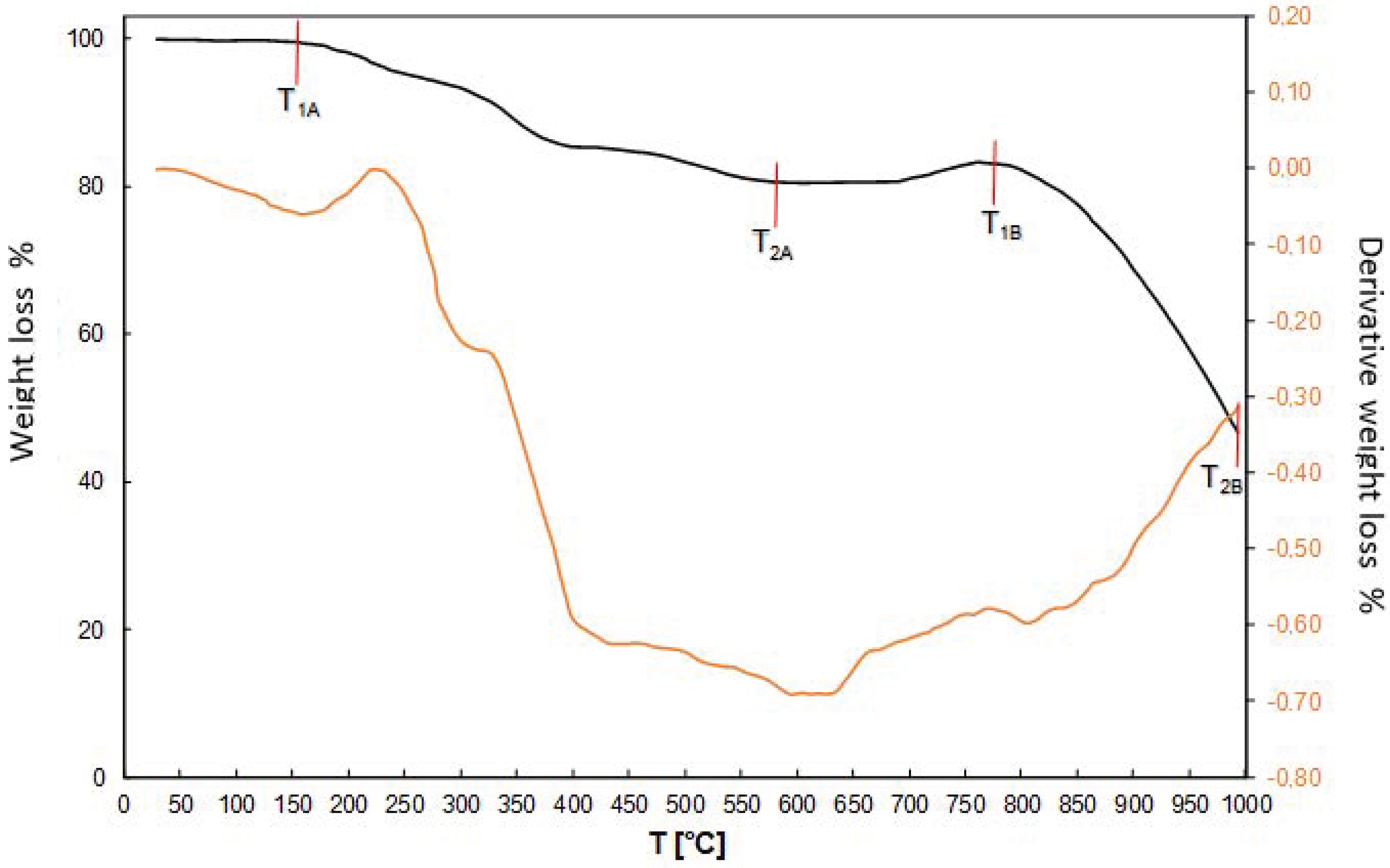
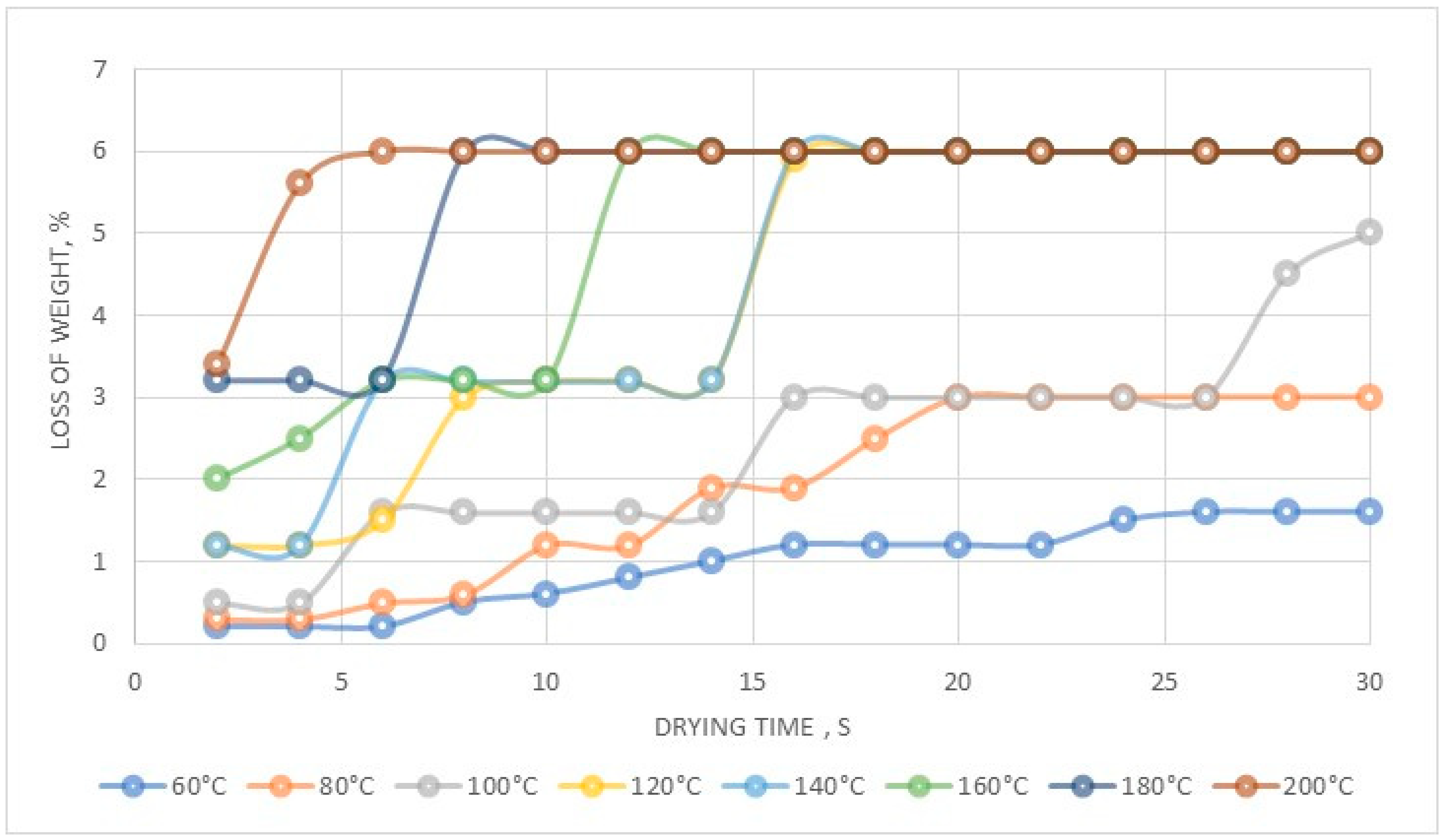
The Department of Analytical Chemistry at IMN was responsible for all the necessary analysis. Rhenium content in product was analyzed using a gravimetric method with tetraphenylarsonium chloride (TPAC) as the precipitating agent, while aluminum and copper(II) were analyzed using flame atomic emission spectroscopy (FAES, spectrophotometer AAS novAA400, Persee, Auburn, AL, USA). Ammonia was analyzed with gravimetric method prior to NH3 distillation (error ± 0.05 g/dm3), while chlorine by potentiometric titration. Solutions were characterized with respect to Re, Co, Ni and Cr content by flame atomic absorption spectroscopy (FAAS, SOLAAR S4, THERMO, Waltham, MA, USA) equipped with flame module and deuterium background correction with measurement error ±0.2%. Analysis of most important contaminations i.e., Mo, Na, Ni, Co, Pb, was performed using ICP-MS (inductively coupled plasma mass spectrometry, ICP MS NexION, PerkinElmer, Waltham, MA, USA), while Ca, Fe, K, Mg, Zn by ICP-OES (inductively coupled plasma—optical emission spectrometer, ULTIMA 2, HORIBA Jobin-Ivon, Kyoto, Japan), and graphite furnace atomic absorption spectroscopy (GFAAS, Z-2000, HITACHI, Tokyo, Japan) with graphite cells Thermal stability analysis was performed using thermobalance HG63, Mettler Toledo (Columbus, OH, USA), at the temperature range from 60 to 200 °C. Thermal analysis was performed in a broader range from 30 to 1000 °C, at heating rate 10 °C/min, in an Ar atmosphere with a flow rate 150 cm3/min using a Netzsch STA 409 C/CD with DTA/TG (Selb, Germany). X-ray powder diffraction analysis was carried out using Co Kα radiation in 2θ range 10°–100° (XRD 7, Seifert-FPM, Freiberg, Germany).
4. Conclusions
It was determined that aluminum and copper(II) perrhenates of high purity may be produced by the ion-exchange method using the strongly acidic cation-exchange resins PFC 100x10 and C160, respectively. The developed technologies produce high purity perrhenates of the following compositions: in case of Al(ReO4)3–71.85% Re, 3.48% Al and <5 ppm Ni; 8 ppm Co; 15 ppm Ca; 10 ppm K; <2 ppm Mg; 15 ppm Na; <5 ppm Pb <5 ppm Mo and 10 ppm Zn, while for Cu(ReO4)2–66.02% Re, 11.27% Cu and <10 ppm Ni; <5 ppm Co; <3 ppm Ca; <10 ppm K; <2 ppm Mg; <10 ppm Na; <5 ppm Pb <5 ppm Mo and <3 ppm Zn. In (ReO4)3 technology the product B, which was Re adsorbed on Al2O3 from 73.2% to 79.6% being the proper heterogeneous catalyst precursor, was additionally produced. The produced [Co(NH3)6](ReO4)3 contained 61.3% Re, 6.5% Co, 11.2% NH3, <20 ppm Cl, <5 ppm Na, <5 ppm Ca, <5 ppm Mg, <5 ppm Pb, <5 ppm Fe, <5 ppm Mo, <5 ppm Cr, <5 ppm Ni, <10 ppm K, <3 ppm Zn. All developed technologies are practically waste-free processes thanks to a proper recycling of spent solutions. Metal losses, i.e., Al, Cu, Re and Co for each of the developed technology were limited to a minimum and did not exceed 0.1%. The purity of produced compounds may enable their use in catalysis.

 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}