1. Introduction
The demand for alternative approaches to the classical chemotherapy as principal treatment of cancer has led to the development of advanced techniques, and to the identification of new targets [
1]. Among these, G-quadruplex DNA (G4 DNA) has emerged as a promising target as it is over-expressed in the promoter regions of a wide number of oncogenes as well as telomeres [
2,
3,
4]. DNA sequences that are sufficiently rich in guanine (G) have the ability to self-assemble into four stranded G-quadruplex (G4) structures [
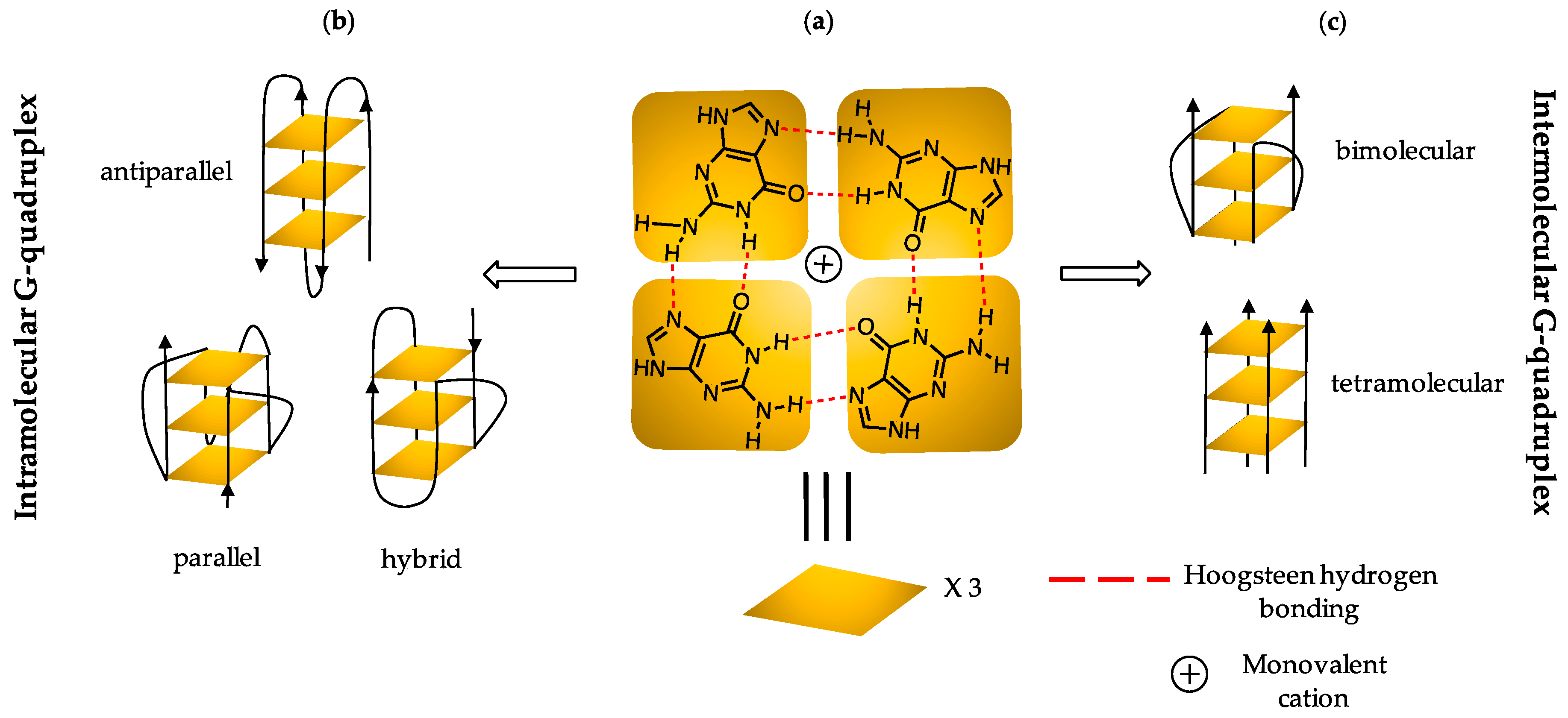
4]. The main structural component of G4 DNA is a planar arrangement of four guanines held together by cyclic Hoogsteen hydrogen bonding networks [
5] (
Figure 1). The directionality of the strands defines the folding topology as parallel, antiparallel, or a hybrid conformation [
6], as illustrated in
Figure 1. The type of conformation adopted by sequences is driven by a monovalent cation, which co-ordinates to the O6 oxygen of each G and thus stabilizes the G4 [
4,
7,
8]. G4 DNA governs several vital biological processes such as transcription, translation and replication [
9,
10], which makes it a promising target for anticancer therapies.
There is direct evidence that small ligands can suppress gene transcription, and therefore induce apoptosis [
11]. However, the structural and topological diversity of quadruplexes and ligand-binding specificity remain key challenges in developing effective and selective G4 DNA-binding ligands [
12]. A considerable number of small ligands have been developed as potential G4 binders, including Telomestatin
®, porphyrinoids, quinolones, alkaloids, carbazoles, acridine derivatives [
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26], and recently, reductive-activated G4-binders [
27]. Besides ligands that have a planar surface for interaction with G4 DNA, molecules containing bi-aryl linkages have also emerged recently as promising candidates [
28,
29,
30,
31]. Representative examples include: phenanthroline-bisoxazoles [
31], pentaheteroaryls [
28], oxazoles [
29], and oligomeric pyridyl-oxazoles [
30] ligands. However, only one candidate—Quarfloxin
®—has advanced to Phase II clinical trials albeit unsuccessfully [
32].
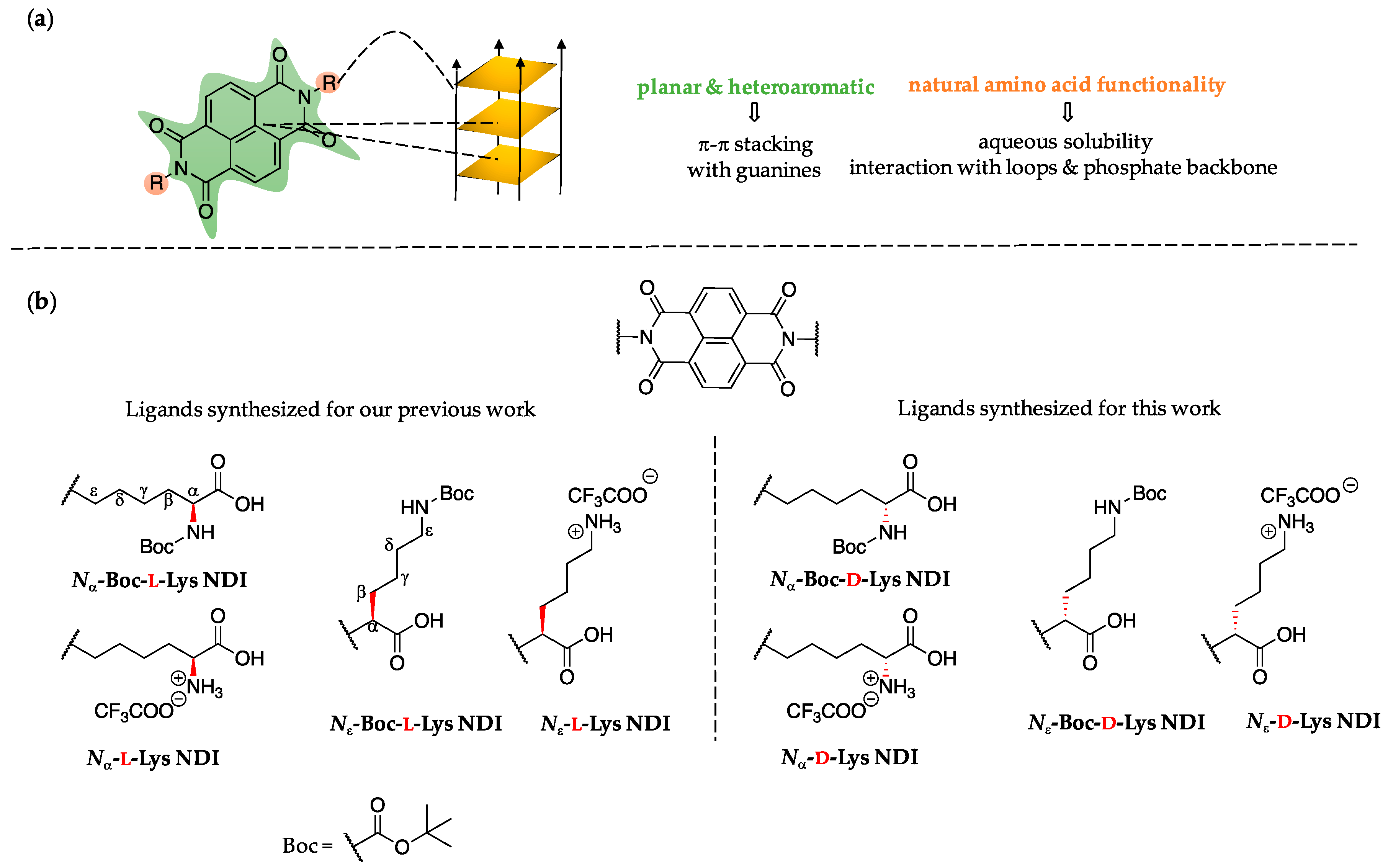
A scaffold that has attracted interest as potential G4 DNA-binding ligand is naphthalenediimide (NDI). NDI-based molecules consist of a large, planar and heteroaromatic surface that can interact with G4 DNA via aromatic π-π stacking [
33,
34]. The literature reports di-, tri-, and tetra-substituted NDI ligands with high affinity for G4 [
8,
9,
35,
36,
37,
38,
39,
40,
41]. We have recently reported the synthesis of symmetrical di-substituted NDIs bearing amino acids as peripheral substituents [
35]. One of the ligands (
Nε-Boc-
l-Lys NDI, see
Figure 2) has exhibited a highly discriminating nature by stabilizing only the oncogene promoter c-KIT2 [
35].
One of the key features of the series of NDIs we have previously reported is their chiral nature, which was not discussed in detail in our previous study. Chirality is ubiquitous in nature and plays crucial roles in drug design [
42,
43], thus we decided to explore the influence of chirality on the interaction of our NDI-based ligands with G4 DNA. It is widely known that enantiomers can display different biological activity on the basis of toxicology, pharmacology, pharmacokinetics and metabolism [
42]. To the best of our knowledge, all chiral G4-ligands explored so far are metal-containing, and can be categorized into either metallo-supramolecular assemblies [
44,
45,
46] or complexes with transition metals (Ru, Cu, Ni, Fe, Zn, Pt) [
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57]. A recently reported study on metallohelices and enantiomeric G-quadruplex DNA showed an interesting mirror-image dependence of ligands binding to
l- or
d-DNA [
58]. Although point chirality is present in Telomestatin
® (a natural product) and Quarfloxin
® [
32,
51,
59], no account or consideration of the significance of point chirality/enantiomeric forms is given in the literature.
The current study comes as a complementary work on the series of
l-NDIs we have recently reported [
35], thus references to this will be made throughout this manuscript. This work highlights the significance of point chirality toward stabilization of G4 within telomeres (h-TELO) and oncogene promoter regions k-RAS, c-KIT and BCL-2. The k-RAS sequence is dis-regulated in pancreatic cancer [
9], while c-KIT2 is upregulated up to 80% in breast malignancies [
60]. BCL-2 is highly expressed in androgen-independent tumors in advanced states of prostate cancer [
61,
62]. BCL-2 is also dis-regulated in pancreatic cancer; therefore, BCL-2 has emerged as an important target for both prostate and pancreatic cancers therapies [
61,
62]. We have changed the chirality from
l- to
d-enantiomers of lysine-functionalized NDIs and have explored their potential as G4 DNA as well as dsDNA binders. The interaction with the latter must be very small so that any G4 DNA potential ligands do not affect the dsDNA present in healthy cells. This work paves the way for understanding the importance of chiral recognition in designing efficient ligands for G4 DNA.
3. Materials and Methods
The oligonucleotides used in this study were purchased from Invitrogen®, having the following sequences (from 5′ to 3′): c-KIT1: GGG AGG GCG CTG GGA GGA GGG; c-KIT2: GGG CGG GCG CGA GGG AGG GG; k-RAS: AGG GCG GTG TGG GAA GAG GGA AGA GGG GGA GG; h-TELO: AGG GTT AGG GTT AGG GTT AGG GT; BCL-2: GGG CGC GGG AGG AAG GGG GCG GG; dsDNA: TAT AGC TAT A Heg TA TAG CTA. The sequences were used as received without further purification.
The oligonucleotide solutions were annealed in pH 7.4 PBS (phosphate-buffered saline) that contained KH2PO4/K2HPO4 (10 mM) and potassium fluoride (KF, 100 mM). The oligonucleotide solutions were placed in a dry-block heater at 95 °C for exactly 5 min, before being allowed to cool to room temperature and stored at 4 °C for at least 24 h prior to use. The samples were annealed to 10 μM, then diluted to desired concentration (3–10 μM range) and used without further purification. The exact concentration of each oligonucleotide after annealing (and the corresponding diluted batches) was determined by measuring the absorbance for the peak corresponding to λmax. The extinction coefficients were provided by the manufacturer. The following values were used (given in L mol−1 cm−1): c-KIT2: 199,100; c-KIT1: 213,000; k-RAS: 341,000; BCL-2: 221,000; h-TELO: 237,000; dsDNA: 6600.
Ultrapure water from a Milli-Q
® water system was used in all CD experiments. All other reagents and solvents were supplied by either Sigma-Aldrich (Gillingham, UK), VWR (Lutterworth, UK) or TCI (Oxford, UK) and used as received. Microwave-assisted reactions were conducted in a CEM
® microwave reactor. Samples for NMR analyses were prepared using dimethylsulfoxide (DMSO-
d6). The
1H and
13C NMR spectra were acquired at 500 and 126 MHz, respectively, using an Agilent ProPulse spectrometer (Santa Clara, CA, USA). The data was recorded at 298 K, and the spectra were referenced to the residual solvent peak. Coupling constants are reported in Hertz (Hz), and signal multiplicity is denoted as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), multiplet (m). Chemical shifts are reported in parts per million (ppm). The nanospray ionization (NSI) spectra (negative or positive ion, as specified) were recorded on an LTQ Orbitrap XL hybrid FTMS instrument. The synthetic route for all NDIs followed a previously reported procedure [
35].
3.1. Synthesis of Nα-Boc-d-Lys and Nε-Boc-d-Lys NDIs
An 8-mL microwave tube was charged with 1,4,5,8-naphthalenetetracarboxylic dianhydride (NDA) and dissolved in dry DMF (5 mL). The corresponding amino acid was added to the formed suspension along with dry Et3N (0.2 mL). The mixture was sonicated until it became a homogeneous solution. The reaction mixture was heated under microwave irradiation for 5 min at 140 °C. The solvent was removed under reduced pressure to yield a brown residue, which was further suspended in minimum amount of acetone. The suspension was added dropwise to a vigorously stirred solution of 1 M HCl(aq.) (200 mL). The resulting precipitate was filtered off and dried in vacuum to yield a light brown colored solid.
Nα-Boc-d-Lys NDI: The reaction was performed on 1 equivalent of NDA (200 mg, 0.746 mmol) and 2.1 equivalents of Nα-Boc-d-lysine (386 mg, 1.567 mmol) by using the general procedure. Yield: 524 mg, 92%; 1H NMR (500 MHz, DMSO-d6): δ 12.40 (br s, 2H), 8.67 (s, 4H), 7.03 (d, J(H,H) = 8.1 Hz, 2H), 4.05 (t, J(H,H) = 7.4 Hz, 4H), 3.88–3.79 (m, 2H), 1.76–1.50 (m, 8H), 1.36–1.30 (m, 22H); 13C NMR (126 MHz, DMSO-d6): δ 174.6, 163.0, 156.0, 130.8, 126.6, 126.5, 78.3, 53.7, 36.2, 31.9, 28.6, 27.5, 23.6; FTMS-NSI: m/z calcd for C36H44N4O12: 723.2883 [C36H44N4O12-H]−, found: 723.2873.
Nε-Boc-d-Lys NDI: The reaction was performed on 1 equivalent of NDA (200 mg, 0.746 mmol) and 2.1 equivalents of Nε-Boc-d-lysine (386 mg, 1.567 mmol) by using the general procedure. Yield: 554 mg, 98%; 1H NMR (500 MHz, DMSO-d6): δ 12.85 (br s, 2H), 8.75 (s, 4H), 6.67 (t, J(H,H) = 6.0 Hz, 2H), 5.53 (dd, J(H,H) = 9.4, 5.0 Hz, 2H), 2.85–2.79 (m, 4H), 2.27–2.17 (m, 4H), 2.10–2.01 (m, 4H), 1.21 (s, 22H); 13C NMR (126 MHz, DMSO-d6): δ 171.1, 162.8, 155.9, 131.7, 126.8, 126.4, 77.6, 54.0, 36.3, 31.2, 29.8, 28.6, 23.7; FTMS-NSI: m/z calcd for C36H44N4O12: 723.2883 [C36H44N4O12-H]−, found: 723.2874.
3.2. Synthesis of Nα-d-Lys and Nε-d-Lys NDIs
The deprotected derivatives (Nα-d-Lys and Nε-d-Lys NDIs) were obtained from Nα-Boc-d-Lys and Nε-Boc-d-Lys NDIs via the following general procedure: a 25-mL round-bottomed flask equipped with a stirrer bar was charged with starting material (NDI) suspended in CH2Cl2 (5 mL) and trifluoroacetic acid (5 mL) was added to the suspension. The reaction mixture was stirred at room temperature for 3 h. The solvents were removed under reduced pressure, and the residue was subsequently washed with diethyl ether (2 × 30 mL), filtered, and vacuum dried to yield a light brown solid in both cases.
Nα-d-Lys NDI: The aforementioned general procedure was followed using Nα-Boc-d-Lys NDI (394 mg). Yield: 331 mg, 85%; 1H NMR (500 MHz, DMSO-d6): δ 13.05 (br s, 2H), 8.66 (s, 4H), 8.16 (br s, 6H), 4.04 (t, J(H,H) = 7.6 Hz, 4H), 3.92–3.84 (m, 2H), 1.88–1.72 (m, 8H), 1.72–1.60 (m, 4H); 13C NMR (126 MHz, DMSO-d6): δ 171.5, 163.1, 158.9, 158.4, 130.9, 126.8, 126.6, 52.3, 50.2, 30.2, 27.4, 24.2, 22.4; FTMS-NSI: m/z calcd for C26H29N4O8: 525.1993 [C26H29N4O8-H]+, found: 525.1980.
Nε-d-Lys NDI: The aforementioned general procedure was following using Nε-Boc-d-Lys NDI (275 mg). Yield: 176 mg, 62%; 1H NMR (500 MHz, DMSO-d6): δ 12.39 (br s, 2H), 8.74 (s, 4H), 7.59 (br s, 6H), 5.53 (dd, J(H,H) = 8.9, 5.3 Hz, 2H), 3.37 (t, J(H,H) = 7.0 Hz, 4H), 2.31–2.23 (m, 4H), 2.08–2.00 (m, 4H), 1.08 (q, J(H,H) = 7.0 Hz, 4H); 13C NMR (126 MHz, DMSO-d6): δ 171.1, 162.9, 131.7, 126.9, 126.5, 53.8, 39.1, 36.2, 28.5, 27.3, 23.4. The MS data could not be acquired because of the aggregation issue.
3.3. Variable-Temperature CD Studies
CD and UV-vis experiments were performed on an Applied Photophysics Chirascan Circular Dichroism Spectrophotometer (Leatherhead, UK) equipped with a Peltier temperature controller. The following parameters were used for full spectra measurements: wavelength scanning range 220–420 nm; temperature 5.0 °C; scanning increments 1 nm; monochromator bandwidth 2.5 nm; sampling time-per-point 0.5 s; pathlength cuvette 1 mm.
In all variable-temperature (VT) studies, the ligand (10 equivalents) was added to the DNA sequence solution previously annealed. Any samples with a maximum ellipticity <4.0 mdeg were transferred to a quartz cuvette with a longer pathlength (2, 5 and 10 mm cuvettes were used). For VT experiments, a single-wavelength spectrum was recorded using the wavelength with the maximum ellipticity (λ
max) with the following parameters: temperature range 5–95 °C with 1 °C increments; temperature slope 1 °C min
−1; temperature equilibration time 45 s; monochromator bandwidth 2.5 nm; sampling time-per-point 2.0 s. The VT CD data was processed using QtiPlot
® (version 0.9.8.9, București, Romania), and fitted to the Boltzmann equation (the spectra are given in the
Supplementary Materials).
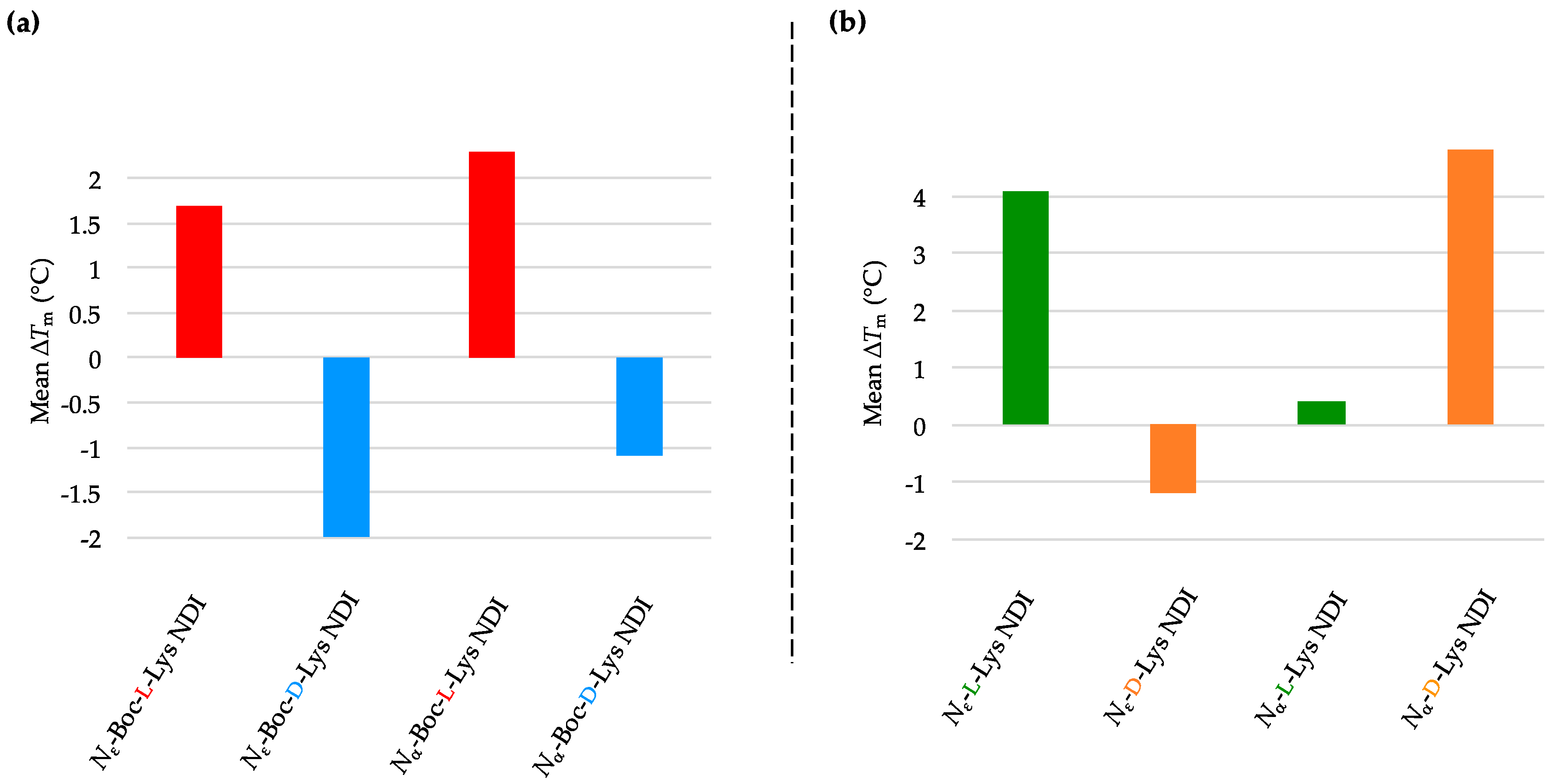
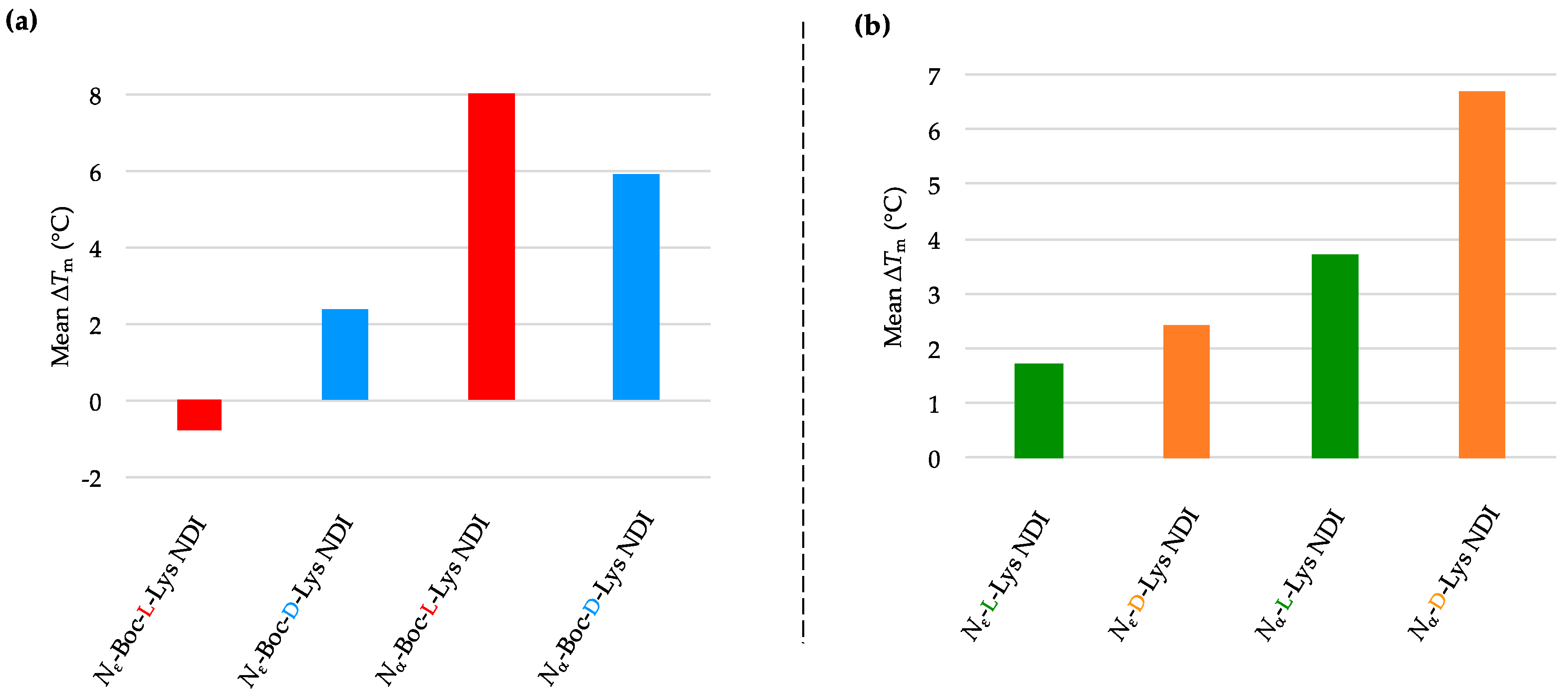
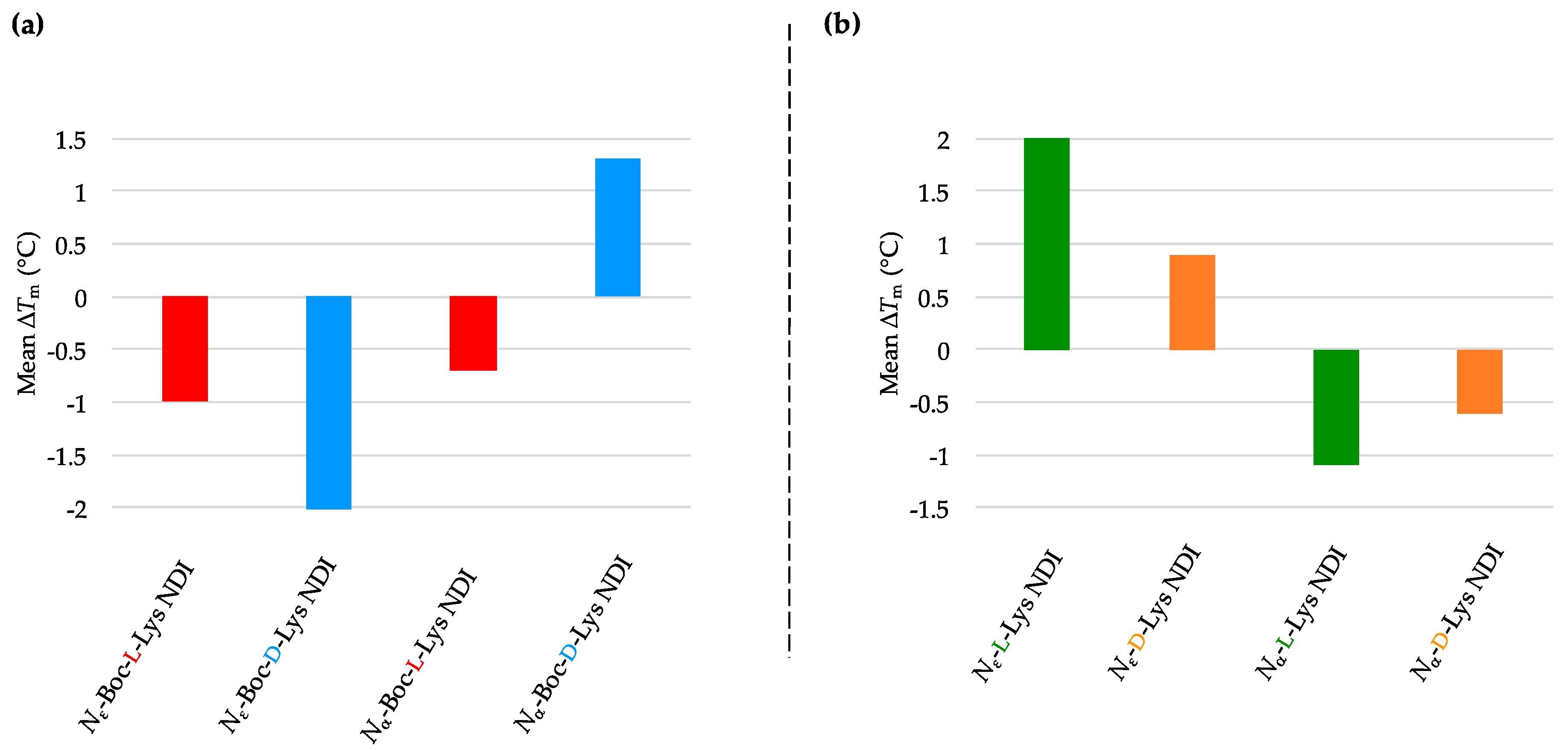
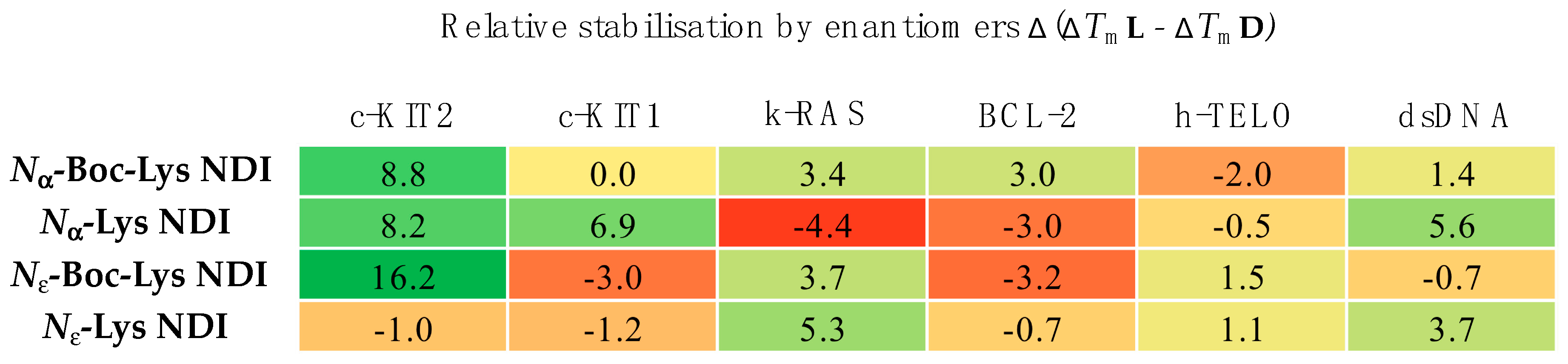
4. Conclusions
We have synthesized the
d-enantiomers of
l-Lys NDIs (with chiral center proximal and distal from the polyaromatic unit) and screened them against G4-forming sequences in telomeres (h-TELO) and the oncogene promoters c-KIT1, c-KIT2, k-RAS and BCL-2. To the best of our knowledge, this is the first study to explore the effect of point chirality in metal-free ligands and chiral recognition toward stabilizing G4 DNA sequences. The heat map table in
Figure 8 summarizes the potential of
l-NDIs (a) vs.
d-NDIs (b) as G4 and dsDNA binders.
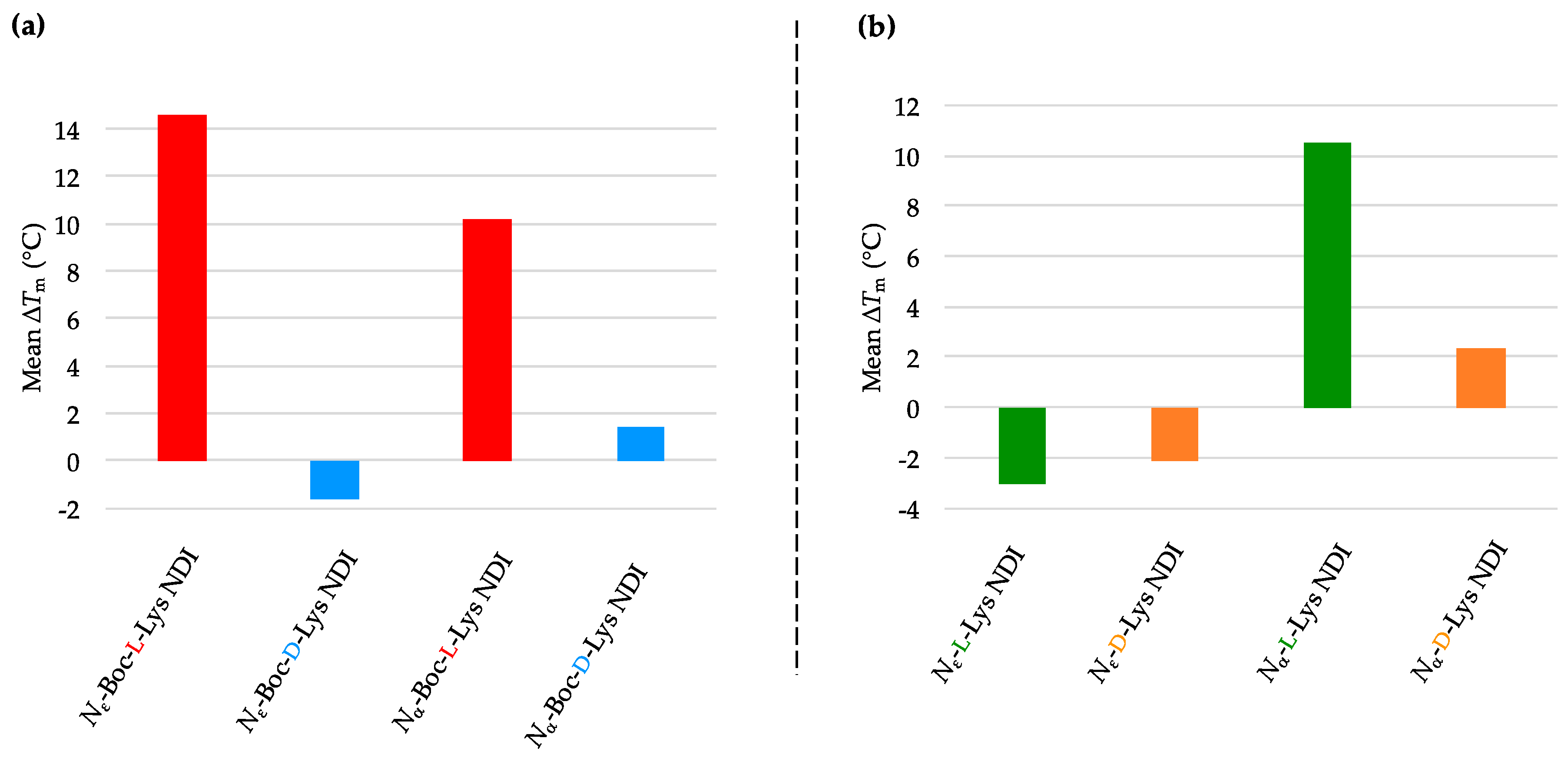
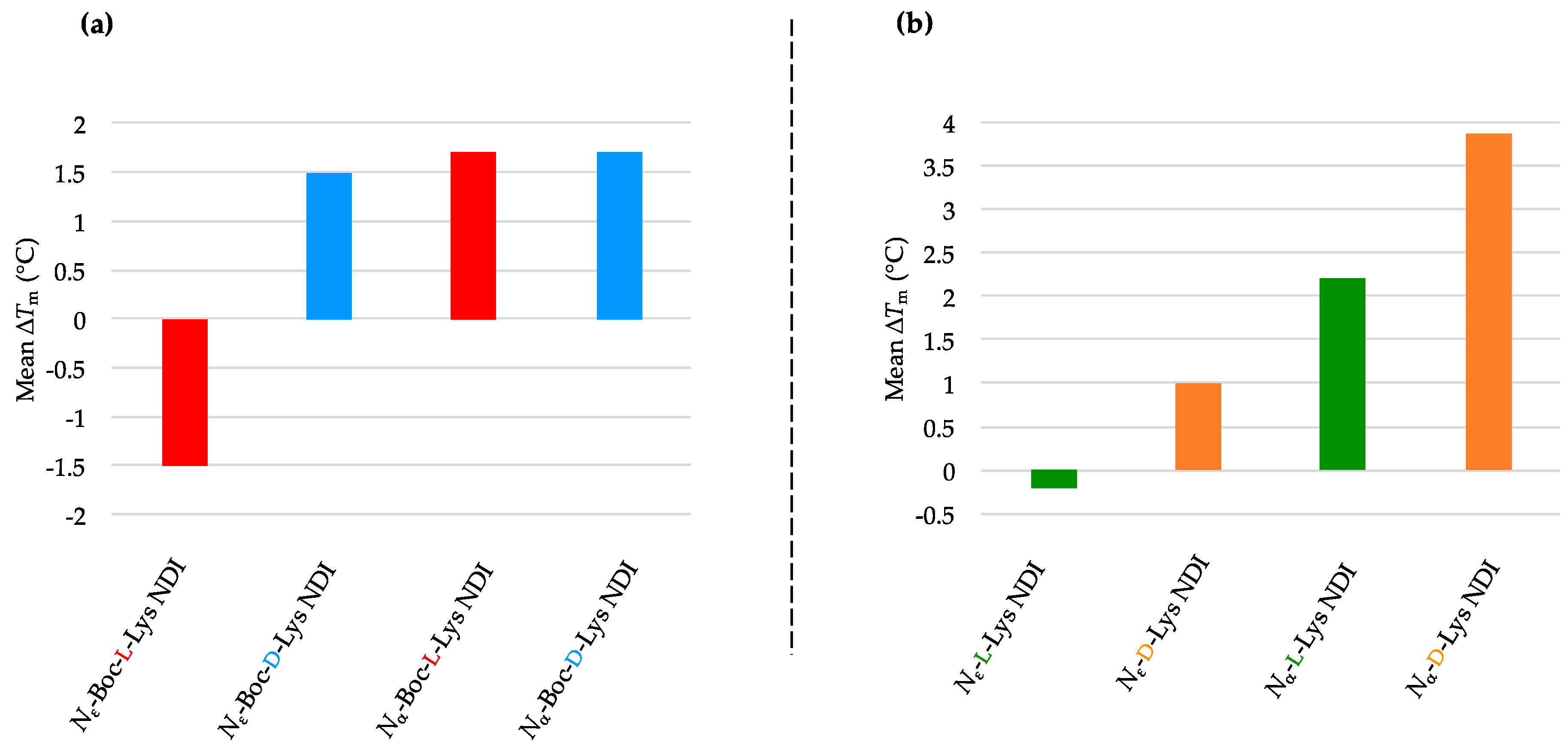
Enantioselectivity was observed for certain ligand/quadruplex pairs adopting parallel or hybrid conformations. The biggest stabilization difference between the enantiomers was observed for
Nε-Boc-Lys NDI interacting with c-KIT2, where the
l-enantiomer stabilizes the quadruplex sequence 16 °C more than the
d-enantiomer; the latter has a mild destabilizing effect on the c-KIT2 oligonucleotide (
Table 1). The protected and deprotected
Nα-Lys NDI enantiomers showed differential stabilization toward this quadruplex in favor of the
l-enantiomer. All of the NDI ligands displayed enantiomeric differentiation in the interaction with c-KIT1, k-RAS and BCL-2 with the exception of
Nα-Boc-Lys NDI and c-KIT1, and
Nε-Lys NDI with both c-KIT1 and BCL-2. h-TELO does not have a strong interaction with any of the NDI ligands studied.
The enantioselectivity observed cannot be explained by the aromatic π-π stacking of the NDI core on the terminal G-tetrads, thus implying that the sidechain interaction with the loops is the cause of this selectivity. This study paves the way toward further exploration of chiral ligands for G4 DNA with the hope of developing a new generation of G-quadruplex sequence selective anticancer drugs.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}