
Probing the Effect of Bulky Lesion-Induced Replication Fork Conformational Heterogeneity Using 4-Aminobiphenyl-Modified DNA




Abstract
:1. Introduction
2. Results
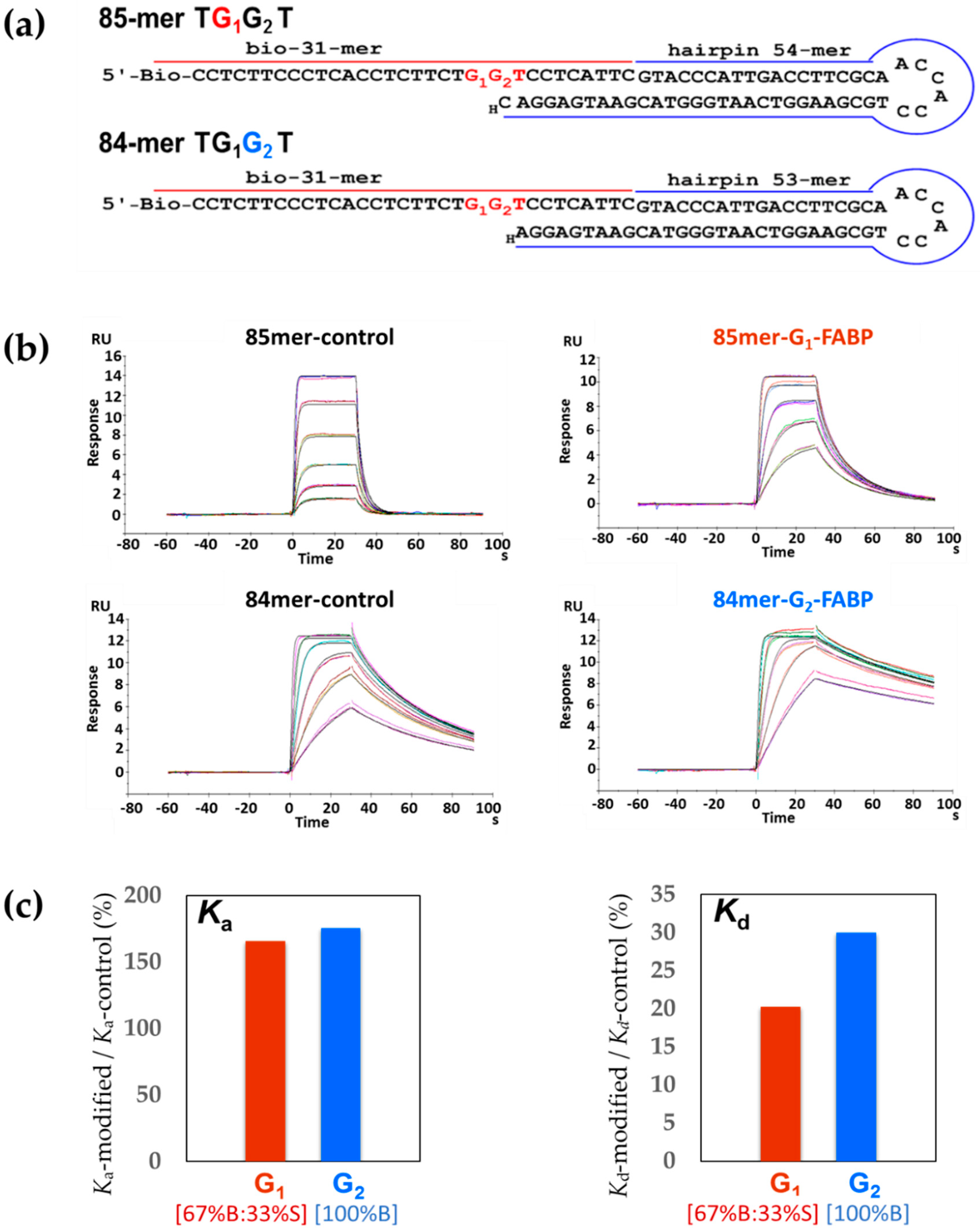
2.1. DNA Sequence Systems
2.2. Oligonucleotide Characterization by MALDI-TOF MS (Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometry)
2.3. HPLC-Based Steady-State Kinetics
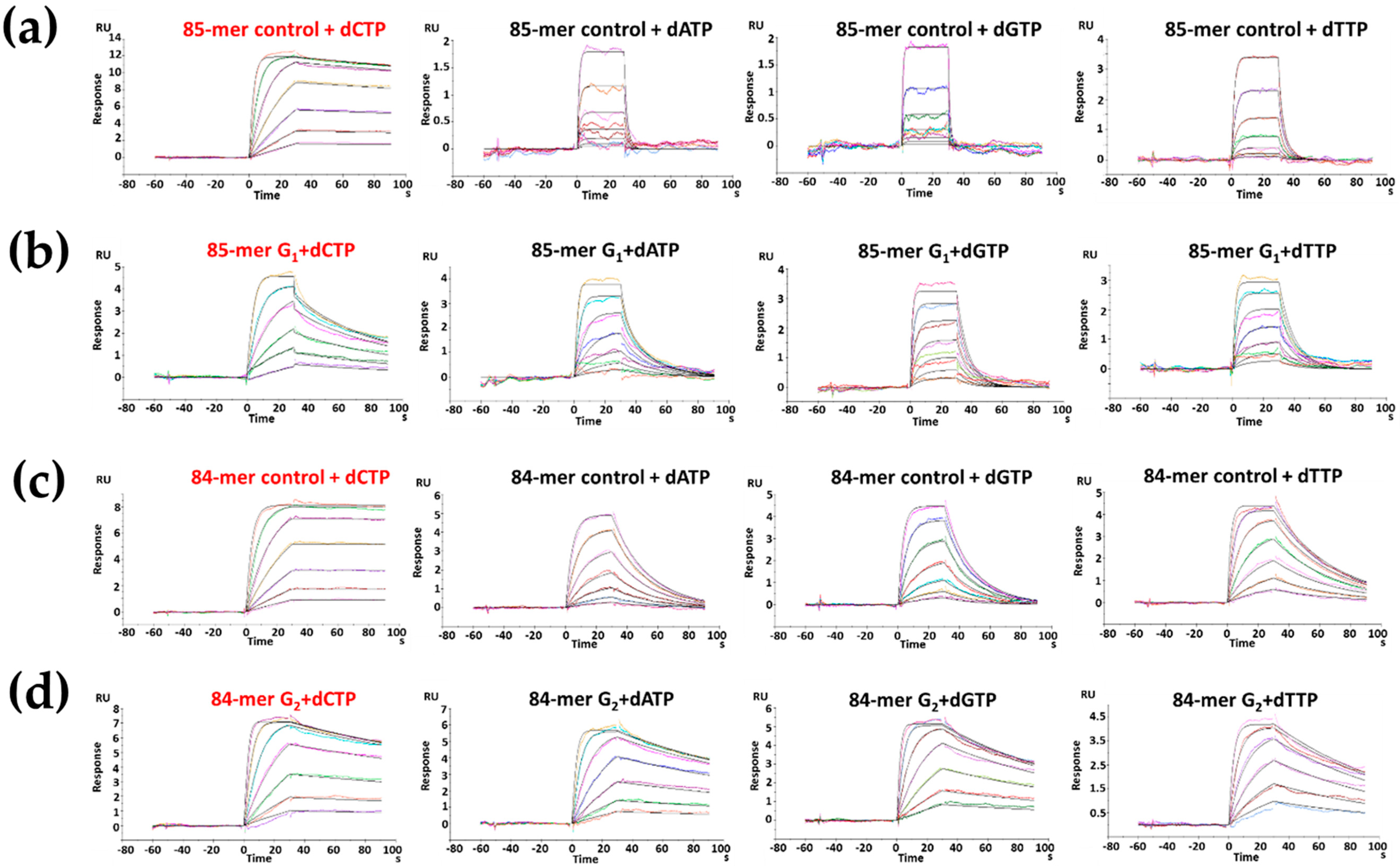
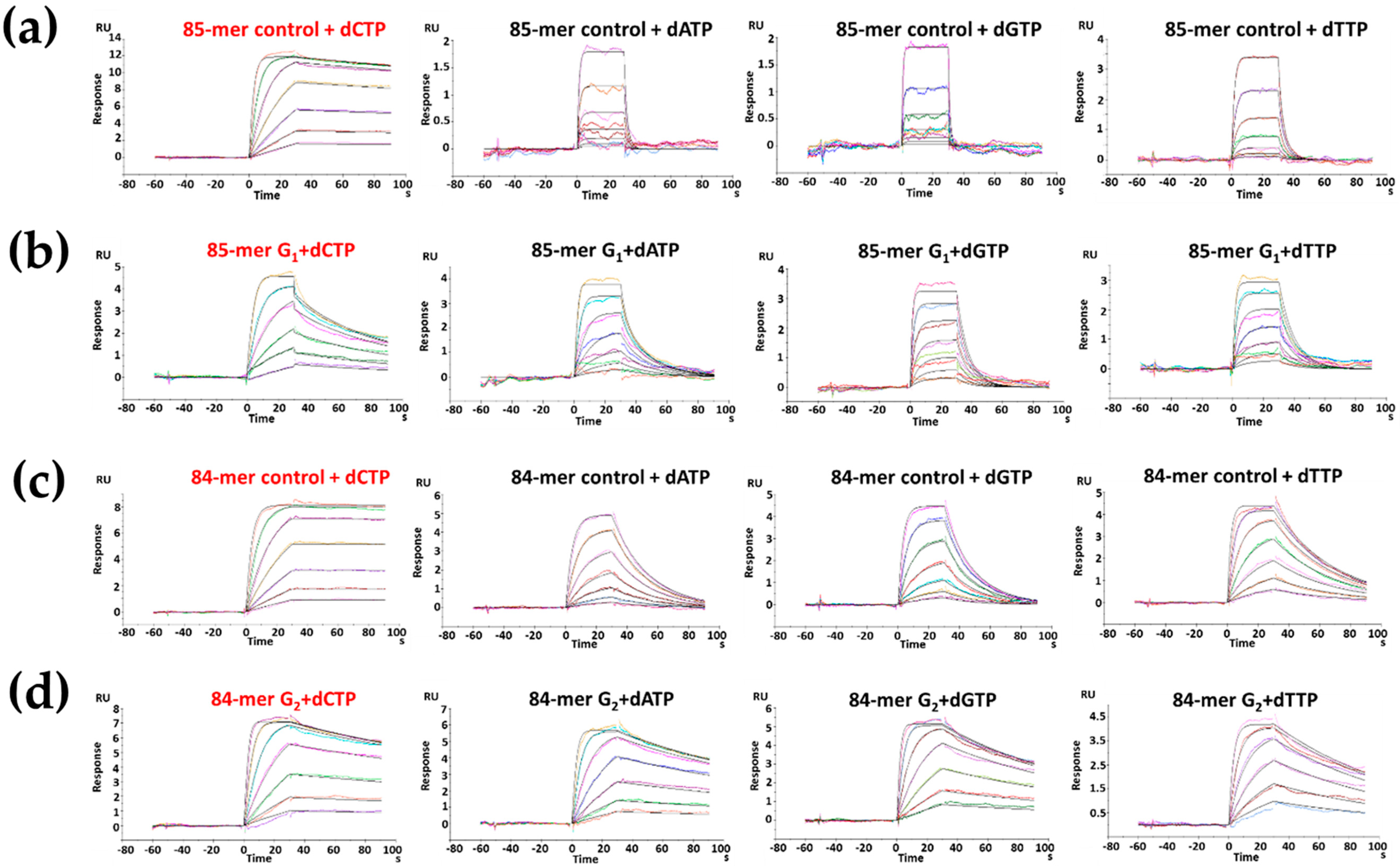
2.4. dCTP Incorporation
2.5. dATP Incorporation
2.6. Kf-exo− SPR Binding Kinetics
3. Discussion
3.1. Improvement on Model Hairpin Oligonucleotide Construction
3.2. Lesion and Sequence Effects on SPR Binding Affinities and Kinetics
4. Materials and Methods
4.1. Model Oligonucleotide DNA Sequences
4.2. HPLC-based Steady-State Kinetic Analysis
4.3. SPR Measurements
4.4. Real-Time Kinetic Analysis by SPR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Voulgaridou, G.-P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 13–27. [Google Scholar] [CrossRef]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Harris, C.C. Advances in Chemical Carcinogenesis: A Historical Review and Prospective. Cancer Res. 2008, 68, 6863–6872. [Google Scholar] [CrossRef] [Green Version]
- Poirier, M.C. Chemical-induced DNA damage and human cancer risk. Nat. Rev. Cancer 2004, 4, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.M.; Lee, M.S.; King, C.M. Mutagenesis by site-specific arylamine adducts in plasmid DNA: Enhancing replication of the adducted strand alters mutation frequency. Biochemistry 1990, 29, 6153–6161. [Google Scholar] [CrossRef] [PubMed]
- Hein, D.W.; Doll, M.A.; Rustan, T.D.; Gray, K.; Feng, Y.; Ferguson, R.J.; Grant, D.M. Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis 1993, 14, 1633–1638. [Google Scholar] [CrossRef]
- Fretland, A.J.; Doll, M.A.; Zhu, Y.; Smith, L.; Leff, M.A.; Hein, D.W. Effect of nucleotide substitutions in N-acetyltransferase-1 on N-acetylation (deactivation) and O-acetylation (activation) of arylamine carcinogens: Implications for cancer predisposition. Cancer Detect. Prev. 2002, 26, 10–14. [Google Scholar] [CrossRef]
- Cho, B.P.; Beland, F.A.; Marques, M.M. NMR structural studies of a 15-mer DNA duplex from a ras protooncogene modified with the carcinogen 2-aminofluorene: Conformational heterogeneity. Biochemistry 1994, 33, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.P. Dynamic conformational heterogeneities of carcinogen-DNA adducts and their mutagenic relevance. J. Environ. Sci. Health Part C 2004, 22, 57–90. [Google Scholar] [CrossRef]
- Patnaik, S.; Cho, B.P. Structures of 2-Acetylaminofluorene Modified DNA Revisited: Insight into Conformational Heterogeneity. Chem. Res. Toxicol. 2010, 23, 1650–1652. [Google Scholar] [CrossRef] [Green Version]
- Meneni, S.R.; Shell, S.M.; Gao, L.; Jurecka, P.; Lee, W.; Sponer, J.; Zou, Y.; Chiarelli, M.P.; Cho, B.P. Spectroscopic and Theoretical Insights into Sequence Effects of Aminofluorene-Induced Conformational Heterogeneity and Nucleotide Excision Repair. Biochemistry 2007, 46, 11263–11278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Rajabzadeh, M.; Traficante, D.D.; Cho, B.P. Conformational heterogeneity of arylamine-modified DNA: 19F NMR evidence. J. Am. Chem. Soc. 1997, 119, 5384–5389. [Google Scholar] [CrossRef]
- Cho, B.P.; Beland, F.A.; Marques, M.M. NMR structural studies of a 15-mer DNA sequence from a ras protooncogene, modified at the first base of codon 61 with the carcinogen 4-aminobiphenyl. Biochemistry 1992, 31, 9587–9602. [Google Scholar] [CrossRef]
- Liang, F.; Meneni, S.; Cho, B.P. Induced circular dichroism characteristics as conformational probes for carcinogenic aminofluorene− DNA adducts. Chem. Res. Toxicol. 2006, 19, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Tsuneoka, Y.; Dalton, T.P.; Miller, M.L.; Clay, C.D.; Shertzer, H.G.; Talaska, G.; Medvedovic, M.; Nebert, D.W. 4-aminobiphenyl-induced liver and urinary bladder DNA adduct formation in Cyp1a2(-/-) and Cyp1a2(+/+) mice. J. Natl. Cancer Inst. 2003, 95, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Hatcher, J.F. Identification of new DNA adducts in human bladder epithelia exposed to the proximate metabolite of 4-aminobiphenyl using 32P-postlabeling method. Chem. Biol. Interact. 2002, 139, 199–213. [Google Scholar] [CrossRef]
- Doerge, D.R.; Churchwell, M.I.; Marques, M.M.; Beland, F.A. Quantitative analysis of 4-aminobiphenyl-C8-deoxyguanosyl DNA adducts produced in vitro and in vivo using HPLC–ES-MS. Carcinogenesis 1999, 20, 1055–1061. [Google Scholar] [CrossRef]
- Beland, F.A.; Doerge, D.R.; Churchwell, M.I.; Poirier, M.C.; Schoket, B.; Marques, M.M. Synthesis, Characterization, and Quantitation of a 4-Aminobiphenyl− DNA Adduct Standard. Chem. Res. Toxicol. 1999, 12, 68–77. [Google Scholar] [CrossRef]
- Cho, B. Structure-function characteristics of aromatic amine-DNA adducts. Chem. Biol. DNA Damage 2010, 217–238. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Rom, W.N.; Beland, F.A.; Tang, M.-S. 4-aminobiphenyl is a major etiological agent of human bladder cancer: Evidence from its DNA binding spectrum in human p53 gene. Carcinogenesis 2002, 23, 1721–1727. [Google Scholar] [CrossRef]
- Yagi, T.; Fujikawa, Y.; Sawai, T.; Takamura-Enya, T.; Ito-Harashima, S.; Kawanishi, M. Error-prone and error-free translesion DNA synthesis over site-specifically created DNA adducts of aryl hydrocarbons (3-nitrobenzanthrone and 4-aminobiphenyl). Toxicol. Res. 2017, 33, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Wilson, K.A.; Patnaik, S.; Wetmore, S.D.; Cho, B.P. DNA base sequence effects on bulky lesion-induced conformational heterogeneity during DNA replication. Nucleic Acids Res. 2018, 46, 6356–6370. [Google Scholar] [CrossRef]
- Vaidyanathan, V.G.; Cho, B.P. Sequence effects on translesion synthesis of an aminofluorene-DNA adduct: Conformational, thermodynamic, and primer extension kinetic studies. Biochemistry 2012, 51, 1983–1995. [Google Scholar] [CrossRef]
- Eger, B.T.; Kuchta, R.D.; Carroll, S.S.; Benkovic, P.A.; Dahlberg, M.E.; Joyce, C.M.; Benkovic, S.J. Mechanism of DNA replication fidelity for three mutants of DNA polymerase I: Klenow fragment KF (exo+), KF (polA5), and KF (exo-). Biochemistry 1991, 30, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.; Chen, R.J.; Wilson, N.A.; Abu-Shumays, R.; Hurt, N.; Lieberman, K.R.; Deamer, D.W.; Dunbar, W.B.; Akeson, M. Sequence-specific detection of individual DNA polymerase complexes in real time using a nanopore. Nat. Nanotechnol. 2007, 2, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Vaidyanathan, V.G.; Cho, B.P. Real-Time Surface Plasmon Resonance Study of Biomolecular Interactions between Polymerase and Bulky Mutagenic DNA Lesions. Chem. Res. Toxicol. 2014, 27, 1796–1807. [Google Scholar] [CrossRef]
- Sandineni, A.; Lin, B.; MacKerell, A.D., Jr.; Cho, B.P. Structure and thermodynamic insights on acetylaminofluorene-modified deletion DNA duplexes as models for frameshift mutagenesis. Chem. Res. Toxicol. 2013, 26, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Cho, B.P. Conformational Insights into the Mechanism of Acetylaminofluorene-dG-Induced Frameshift Mutations in the Nar I Mutational Hotspot. Chem. Res. Toxicol. 2016, 29, 213–226. [Google Scholar] [CrossRef]
- Suzuki, N.; Yasui, M.; Santosh Laxmi, Y.; Ohmori, H.; Hanaoka, F.; Shibutani, S. Translesion synthesis past equine estrogen-derived 2′-deoxycytidine DNA adducts by human DNA polymerases η and κ. Biochemistry 2004, 43, 11312–11320. [Google Scholar] [CrossRef]
- Sakamoto, K.; Gouzu, H.; Komiya, K.; Kiga, D.; Yokoyama, S.; Yokomori, T.; Hagiya, M. Molecular computation by DNA hairpin formation. Science 2000, 288, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- LaBean, T.H.; Yan, H.; Kopatsch, J.; Liu, F.; Winfree, E.; Reif, J.H.; Seeman, N.C. Construction, analysis, ligation, and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 2000, 122, 1848–1860. [Google Scholar] [CrossRef]
- Vastenhout, K.J.; Tornberg, R.H.; Johnson, A.L.; Amolins, M.W.; Mays, J.R. High-performance liquid chromatography-based method to evaluate kinetics of glucosinolate hydrolysis by Sinapis alba myrosinase. Anal. Biochem. 2014, 465, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Klingaman, C.A.; Wagner, M.J.; Brown, J.R.; Klecker, J.B.; Pauley, E.H.; Noldner, C.J.; Mays, J.R. Synthesis and spectral characterization of 2,2-diphenylethyl glucosinolate and HPLC-based reaction progress curve data for the enzymatic hydrolysis of glucosinolates by Sinapis alba myrosinase. Data Brief 2017, 10, 151–181. [Google Scholar] [CrossRef] [Green Version]
- Miller, H.; Grollman, A.P. Kinetics of DNA polymerase I (Klenow fragment exo-) activity on damaged DNA templates: Effect of proximal and distal template damage on DNA synthesis. Biochemistry 1997, 36, 15336–15342. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Hilton, B.; Lin, B.; Jain, A.; MacKerell, A.D., Jr.; Zou, Y.; Cho, B.P. Structural and thermodynamic insight into Escherichia coli UvrABC-mediated incision of cluster diacetylaminofluorene adducts on the NarI sequence. Chem. Res. Toxicol. 2013, 26, 1251–1262. [Google Scholar] [CrossRef]
- Vrzheshch, P.V. Steady-state kinetics of bifunctional enzymes. Taking into account kinetic hierarchy of fast and slow catalytic cycles in a generalized model. Biochemistry 2007, 72, 936–943. [Google Scholar] [CrossRef]
- Kuriyan, J.; Konforti, B.; Wemmer, D. The Molecules of Life: Physical and Chemical Principles; Garland Science: New York, NY, USA; Milton Park, Abingdon, UK, 2012. [Google Scholar]
- Vaidyanathan, V.G.; Xu, L.; Cho, B.P. Binary and ternary binding affinities between exonuclease-deficient Klenow fragment (Kf-exo(-)) and various arylamine DNA lesions characterized by surface plasmon resonance. Chem. Res. Toxicol. 2012, 25, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, V.G.; Liang, F.; Beard, W.A.; Shock, D.D.; Wilson, S.H.; Cho, B.P. Insights into the conformation of aminofluorene-deoxyguanine adduct in a DNA polymerase active site. J. Biol. Chem. 2013, 288, 23573–23585. [Google Scholar] [CrossRef]
- Dzantiev, L.; Romano, L.J. Interaction of Escherichia coli DNA polymerase I (Klenow fragment) with primer-templates containing N-acetyl-2-aminofluorene or N-2-aminofluorene adducts in the active site. J. Biol. Chem. 1999, 274, 3279–3284. [Google Scholar] [CrossRef]
- Jain, N.; Li, Y.; Zhang, L.; Meneni, S.R.; Cho, B.P. Probing the sequence effects on NarI-induced -2 frameshift mutagenesis by dynamic 19F NMR, UV, and CD spectroscopy. Biochemistry 2007, 46, 13310–13321. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.; Chen, F.; Humulock, Z.T.; Tang, Q.; Li, D. Copper Inhibits the AlkB Family DNA Repair Enzymes under Wilson’s Disease Condition. Chem. Res. Toxicol. 2017, 30, 1794–1796. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Krüger, S.; Meier, C. Synthesis of Site-Specific Damaged DNA Strands by 8-(Acetylarylamino)-2′-deoxyguanosine Adducts and Effects on Various DNA Polymerases. Eur. J. Org. Chem. 2013, 2013, 1158–1169. [Google Scholar] [CrossRef]
Sample Availability: FABP modified DNA sequences are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Km (µM) | kcat (min−1) | kcat/Km (µM−1·min−1) | * fins |
|---|---|---|---|---|
| FABP-G1 | 23.4 ± 0.01 | 29.7 ± 0.63 | 1.27 | 0.33 |
| Control-G1 | 7.4 ± 0.10 | 28.8 ± 0.96 | 3.87 | 1.00 |
| FABP-G2 | 5.7 ± 0.01 | 5.5 ± 0.11 | 0.97 | 0.24 |
| Control-G2 | 5.8 ± 0.11 | 23.2 ± 0.70 | 4.00 | 1.00 |
| Sequence | Binary | KD of Ternary (nM) | |||||
|---|---|---|---|---|---|---|---|
| ka (1/Ms) × 107 | kd (1/s) | KD (nM) | dCTP | dATP | dGTP | dTTP | |
| 85-mer TG1*G2T | 15.90 (0.17) # | 0.170 (0.002) | 1.050 (0.050) | 0.200 (0.060) | 1.17 (0.04) | 1.08 (0.07) | 1.08 (0.08) |
| 85-mer control | 9.59 (0.05) | 0.840 (0.004) | 8.740 (0.030) | 0.022 (0.001) | 14.90 (5.00) | 11.40 (4.20) | 5.14 (0.74) |
| 84-mer TG1G2*T | 11.10 (0.07) | 0.009 (0.000) | 0.086 (0.001) | 0.045 (0.006) | 0.09 (0.00) | 0.11 (0.00) | 0.09 (0.01) |
| 84-mer control | 6.33 (0.04) | 0.030 (0.002) | 0.480 (0.030) | 0.004 (0.000) | 1.39 (0.02) | 1.25 (0.01) | 0.39 (0.04) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, A.; Bian, K.; Chen, F.; Tang, Q.; Carley, R.; Li, D.; Cho, B.P. Probing the Effect of Bulky Lesion-Induced Replication Fork Conformational Heterogeneity Using 4-Aminobiphenyl-Modified DNA. Molecules 2019, 24, 1566. https://doi.org/10.3390/molecules24081566
Cai A, Bian K, Chen F, Tang Q, Carley R, Li D, Cho BP. Probing the Effect of Bulky Lesion-Induced Replication Fork Conformational Heterogeneity Using 4-Aminobiphenyl-Modified DNA. Molecules. 2019; 24(8):1566. https://doi.org/10.3390/molecules24081566
Chicago/Turabian StyleCai, Ang, Ke Bian, Fangyi Chen, Qi Tang, Rachel Carley, Deyu Li, and Bongsup P. Cho. 2019. "Probing the Effect of Bulky Lesion-Induced Replication Fork Conformational Heterogeneity Using 4-Aminobiphenyl-Modified DNA" Molecules 24, no. 8: 1566. https://doi.org/10.3390/molecules24081566
APA StyleCai, A., Bian, K., Chen, F., Tang, Q., Carley, R., Li, D., & Cho, B. P. (2019). Probing the Effect of Bulky Lesion-Induced Replication Fork Conformational Heterogeneity Using 4-Aminobiphenyl-Modified DNA. Molecules, 24(8), 1566. https://doi.org/10.3390/molecules24081566