Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
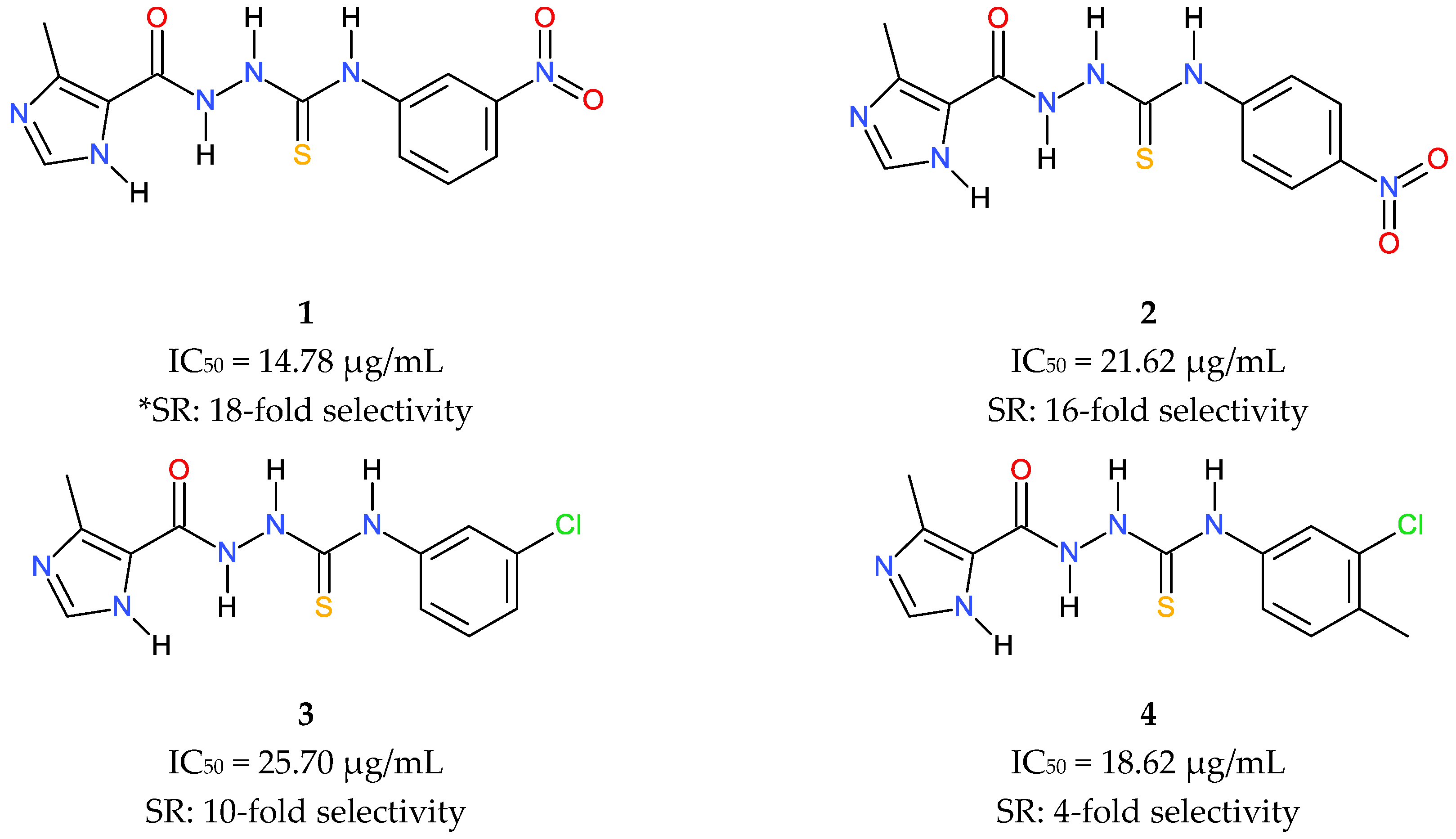
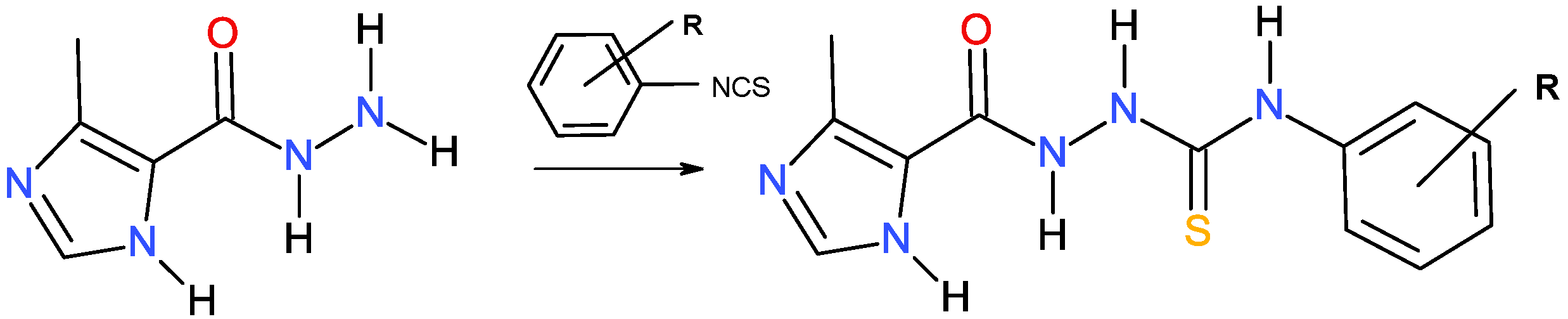
2.1. Molecular Design and Synthesis
2.2. Cytotoxicity against L929 Cells
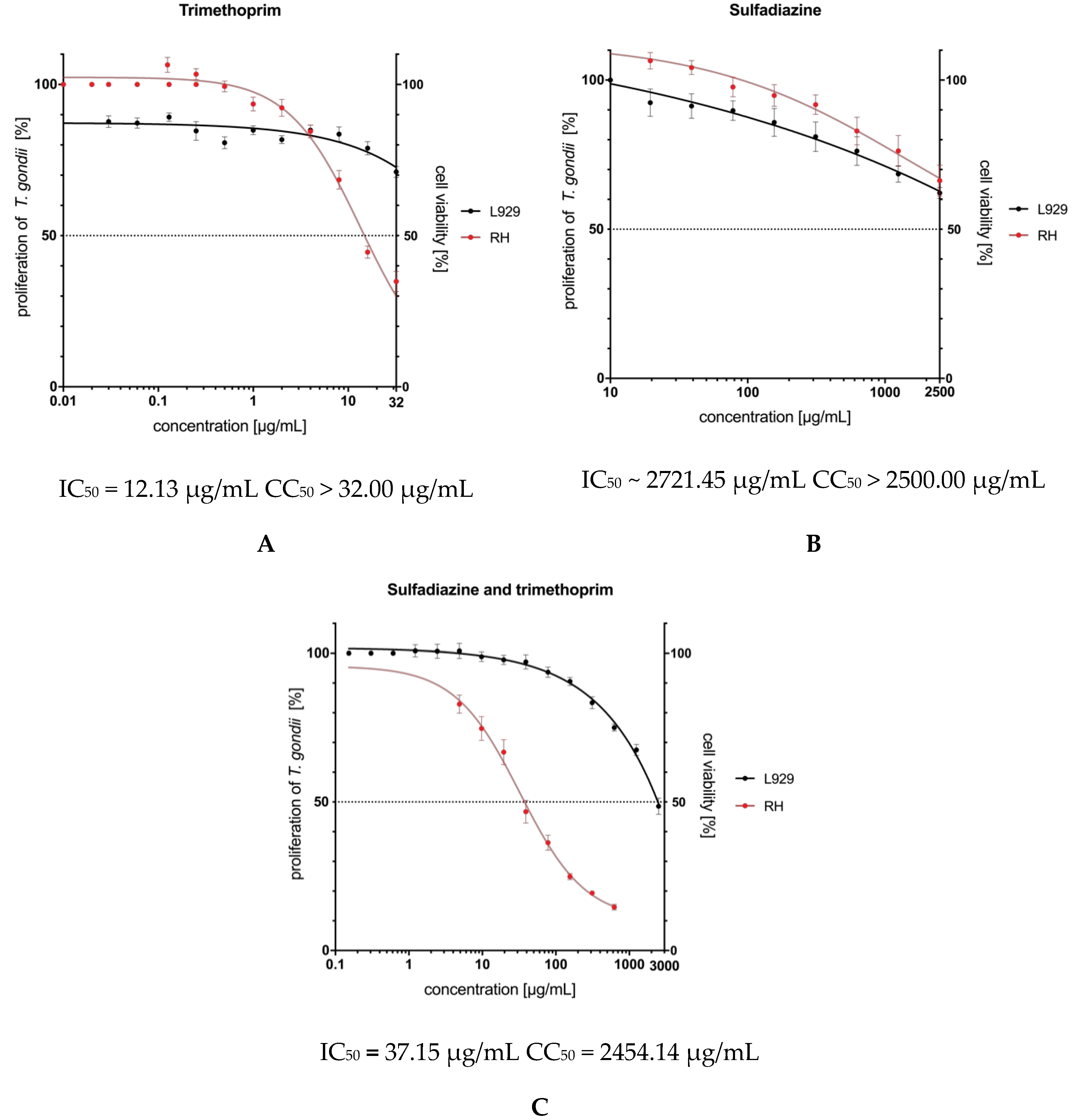
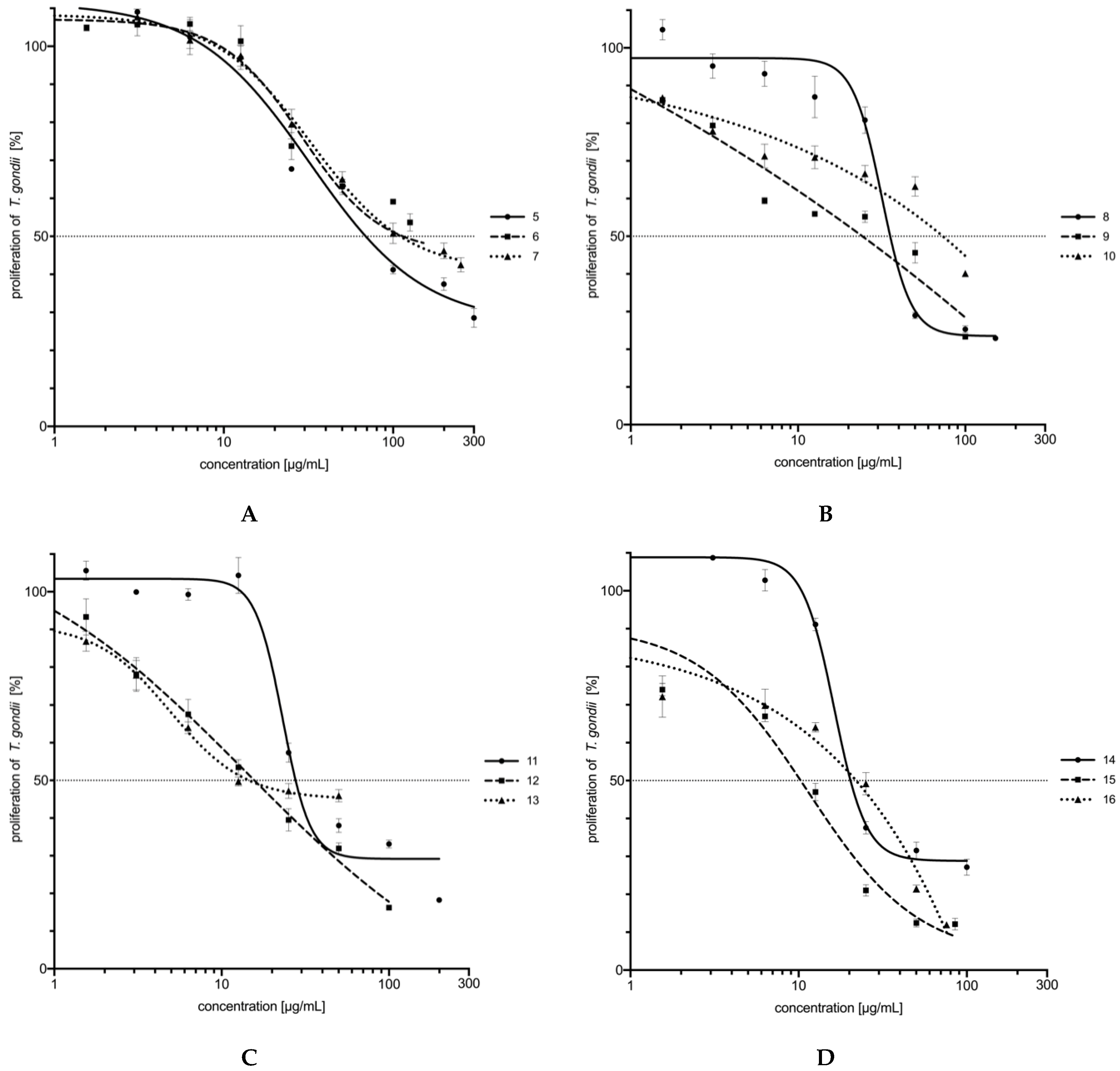
2.3. In Vitro Activity and Cytotoxicity
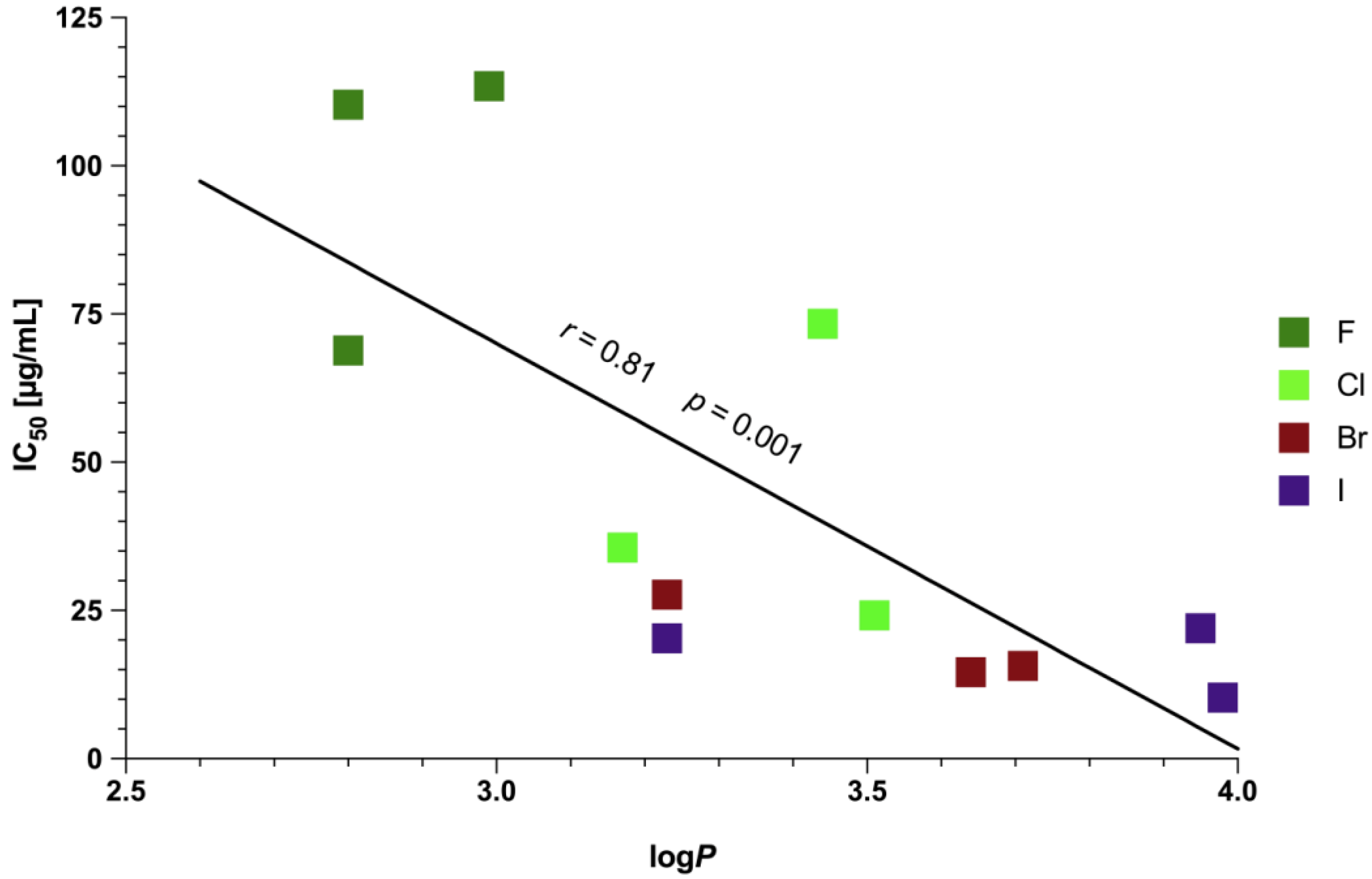
2.4. Physicochemical Characterization of 5–16
3. Materials and Methods
3.1. Chemistry
3.2. Procedure for Synthesis of the Imidazole-Thiosemicarbazides 5–16
3.3. Assay In Vitro for Anti-T. gondii Activity
3.4. Cytotoxic Assay
3.5. Graphs and Statistical Analyses
3.6. Lipophilicity Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Jeffers, V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol. Rev. 2012, 36, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Luft, B.J.; Remington, J.S. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 1992, 15, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Israelski, D.M.; Remington, J.S. Toxoplasmosis in the non-AIDS immunocompromised host. Curr. Clin. Top Infect. Dis. 1993, 13, 322–356. [Google Scholar]
- Carlier, Y.; Truyens, C.; Deloron, P.; Peyron, F. Congenital parasitic infections: A review. Acta Trop. 2012, 121, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.P.; Jones, J.L. Toxoplasma gondii infection in humans and animals in the United States. Int. J. Parasitol. 2008, 38, 1257–1278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, L.; Fangli, L. Current treatment of ocular toxoplasmosis in immunocompetent patients: A network meta-analysis. Acta Trop. 2018, 185, 52–62. [Google Scholar] [CrossRef]
- Dubey, J.P.; Speer, C.A.; Shen, S.K.; Kwok, O.C.H.; Blixt, J.A. Oocyst-induced murine Toxoplasmosis: Life cycle, pathogenicity, and stage conversion in mice fed Toxoplasma gondii oocysts. J. Parasitol. 1997, 83, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.X.; Wei, S.S.; Lindsay, D.S.; Peng, H.J. A systematic review and meta-analysis of the efficacy of anti-Toxoplasma gondii medicines in humans. PLoS ONE 2015, 10, e0138204. [Google Scholar] [CrossRef]
- Kaplan, J.E.; Benson, C.; Holmes, K.K.; Brooks, J.T.; Pau, A.; Masur, H. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents. Recommendations from CDC, the National Institutes of Health and the HIV Medicine Association of the Infectious Diseases Society of America. CDC MMWR 2009, 58, 1–198. [Google Scholar] [PubMed]
- Jacobson, J.M.; Davidian, M.; Rainey, P.M.; Hafner, R.; Raasch, R.H.; Luft, B.J. Pyrimethamine pharmacokinetics in human immunodeficiency virus-positive patients seropositive for Toxoplasma gondii. Antimicrob. Agents Chemother. 1996, 40, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Ben-Harari, R.R.; Goodwin, E.; Casoy, J. Adverse event profile of pyrimethamine-based therapy in toxoplasmosis: A systematic review. Drugs R & D 2017, 17, 523–544. [Google Scholar]
- Hernandez, A.V.; Thota, P.; Pellegrino, D.; Pasupuleti, V.; Benites-Zapata, V.A.; Deshpande, A.; Penalva de Oliveira, A.C.; Vidal, J.E. A systematic review and meta-analysis of the relative efficacy and safety of treatment regimens for HIV-associated cerebral toxoplasmosis: Is trimethoprim-sulfamethoxazole a real option? HIV Med. 2017, 18, 115–124. [Google Scholar] [CrossRef] [PubMed]
- McCabe, R.E. Antitoxoplasma chemotherapy. In Toxoplasmosis: A Comprehensive Clinical Guide; Joynson, D.H.M., Wreghitt, T.G., Eds.; Cambridge University Press: Cambridge, UK, 2001; pp. 319–359. [Google Scholar]
- Kieffer, F.; Wallon, M. Congenital toxoplasmosis. Handb. Clin. Neurol. 2013, 112, 1099–1101. [Google Scholar] [PubMed]
- Kortagere, S. Novel molecules to treat chronic central nervous system Toxoplasmosis. J. Med. Chem. 2017, 60, 9974–9975. [Google Scholar] [CrossRef] [PubMed]
- Piper, J.R.; Johnson, C.A.; Krauth, C.A.; Carter, R.L.; Hosmer, C.A.; Queener, S.F.; Borotz, S.E.; Pfefferkorn, E.R. Lipophilic antifolates as agents against opportunistic infections. 1. Agents superior to trimetrexate and piritrexim against Toxoplasma gondii and Pneumocystis carinii in in vitro evaluations. J. Med. Chem. 1996, 39, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Węglińska, L.; Bekier, A.; Stefaniszyn, E.; Wujec, M.; Trotsko, N.; Dzitko, K. Systematic identification of thiosemicarbazides for inhibition of Toxoplasma gondii. Molecules 2019, 24, 614. [Google Scholar] [CrossRef]
- Buchini, S.; Buschiazzo, A.; Withers, S.G. A new generation of specific Tryponosoma cruzi trans-sialidase inhibitors. Angew. Chem. Int. Ed. 2008, 47, 2700–2703. [Google Scholar] [CrossRef]
- Bonnefous, C.; Payne, J.E.; Roppe, J.; Zhuang, H.; Chen, X.; Symons, K.T.; Nguyen, P.M.; Sablad, M.; Rozenkrants, N.; Zhang, Y.; et al. Discovery of inducible nitric oxide synthase (iNOS) inhibitor development candidate KD7332, part 1: Identification of a novel, potent, and selective series of quinolinone iNOS dimerization inhibitors that are orally active in rodent pain models. J. Med. Chem. 2009, 52, 3047–3062. [Google Scholar] [CrossRef]
- Leite, A.C.L.; de M. Moreira, D.R.; de O. Cardoso, M.V.; Hernandes, M.Z.; Pereira, V.R.A.; Silva, R.O.; Kiperstok, A.C.; da S. Lima, M.; Soares, M.B.P. Synthesis, cruzain docking, and in vitro studies of aryl-4-oxothiazolylhydrazones against Trypanosoma cruzi. ChemMedChem 2009, 2, 1339–1345. [Google Scholar] [CrossRef]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of drugs enhances membrane binding and permeation. ChemBioChem 2004, 5, 676–684. [Google Scholar] [CrossRef]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood–brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Plech, T.; Wujec, M.; Siwek, A.; Kosikowska, U.; Malm, A. Synthesis and antimicrobial activity of thiosemicarbazides, s-triazoles and their Mannich bases bearing 3-chlorophenyl moiety. Eur. J. Med. Chem. 2011, 46, 241–248. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a drug’s membrane permeability: A computational model validated with in vitro permeability assay data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.J. Substituent Constant for Correlation Analysis in Chemistry and Biology; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Elsevier: Oxford, UK, 2008; p. 45. [Google Scholar]
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic permeability efficiency reconciles the opposing roles of lipophilicity in membrane permeability and aqueous solubility. J. Med. Chem. 2018, 61, 11169–11182. [Google Scholar] [CrossRef]
- Siwek, A.; Wujec, M.; Dobosz, M.; Wawrzycka-Gorczyca, I. Study of direction of cyclization of 1-azolyl-4-aryl/alkyl-thiosemicarbazides. Heteroatom Chem. 2010, 21, 521–532. [Google Scholar] [CrossRef]
- Liesen, A.P.; de Aquino, T.M.; Carvalho, C.S.; Lima, V.T.; de Araújo, J.M.; de Lima, J.G.; de Faria, A.R.; de Melo, E.J.; Alves, A.J.; Alves, E.W.; et al. Synthesis and evaluation of anti-Toxoplasma gondii and antimicrobial activities of thiosemicarbazides, 4-thiazolidinones and 1,3,4-thiadiazoles. Eur. J. Med. Chem. 2010, 45, 3685–3691. [Google Scholar] [CrossRef]
- OECD. Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method, OECD Guidelines for the Testing of Chemicals, Section 1; OECD Publishing: Paris, France, 2004. [Google Scholar]
Sample Availability: Samples of the compounds 5–16 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration [µg/mL] | CC50 [µg/mL] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1000.00 | 500.00 | 250.00 | 125.00 | 50.00 | 25.00 | 5.00 | 0.00 | ||
| 5 | 45.78 | 64.14 | 69.30 | 86.95 | 74.82 | 97.27 | 98.12 | 99.61 | 454.78 |
| 0.70 | 1.28 | 1.38 | 1.01 | 1.26 | 1.24 | 1.75 | 0.54 | ||
| 6 | 35.36 | 52.30 | 62.37 | 64.78 | 75.39 | 97.35 | 96.34 | 99.61 | 884.71 |
| 1.80 | 1.10 | 2.02 | 0.98 | 3.74 | 1.55 | 3.09 | 0.54 | ||
| 7 | 48.70 | 61.44 | 69.93 | 83.12 | 89.17 | 103.25 | 103.16 | 99.61 | 977.01 |
| 2.32 | 1.64 | 1.36 | 3.80 | 2.24 | 4.60 | 3.88 | 0.54 | ||
| 8 | 39.48 | 57.08 | 64.80 | 69.43 | 78.47 | 86.28 | 89.94 | 99.61 | 629.80 |
| 2.16 | 2.12 | 1.96 | 2.08 | 1.12 | 2.45 | 2.08 | 0.54 | ||
| 9 | 8.47 | 30.19 | 56.66 | 64.67 | 75.29 | 88.49 | 93.91 | 99.61 | 272.83 |
| 0.23 | 0.11 | 2.48 | 3.06 | 4.89 | 6.68 | 5.10 | 0.54 | ||
| 10 | 13.60 | 23.56 | 56.60 | 63.59 | 70.31 | 96.34 | 102.71 | 99.61 | 230.99 |
| 1.52 | 1.98 | 3.23 | 4.47 | 2.51 | 3.54 | 2.60 | 0.54 | ||
| 11 | 29.77 | 47.10 | 70.02 | 76.29 | 87.53 | 92.97 | 94.72 | 99.61 | 495.34 |
| 2.89 | 3.60 | 2.49 | 2.82 | 3.00 | 3.29 | 1.01 | 0.54 | ||
| 12 | 7.52 | 20.25 | 56.79 | 65.74 | 74.43 | 88.99 | 95.27 | 99.61 | 237.79 |
| 0.81 | 1.10 | 1.60 | 3.04 | 1.05 | 4.15 | 2.46 | 0.54 | ||
| 13 | 30.76 | 42.14 | 51.09 | 56.60 | 64.10 | 81.98 | 93.51 | 99.61 | 223.67 |
| 1.96 | 4.06 | 4.75 | 2.56 | 2.68 | 0.22 | 1.08 | 0.54 | ||
| 14 | 25.01 | 26.12 | 44.49 | 65.41 | 83.73 | 92.75 | 95.23 | 99.61 | 198.84 |
| 2.70 | 0.45 | 4.67 | 2.71 | 2.81 | 1.66 | 1.18 | 0.54 | ||
| 15 | 8.49 | 8.53 | 54.05 | 66.20 | 70.79 | 87.30 | 96.65 | 99.61 | 197.38 |
| 0.20 | 0.55 | 1.77 | 2.56 | 1.30 | 2.37 | 1.61 | 0.54 | ||
| 16 | 13.54 | 16.28 | 49.91 | 68.09 | 66.23 | 84.37 | 96.74 | 99.61 | 194.98 |
| 1.20 | 0.61 | 1.03 | 0.91 | 0.95 | 2.74 | 0.41 | 0.54 | ||
| Compound | Compound Structure | CC50 [µg/mL] | IC50 [µg/mL] | SR | LogP |
|---|---|---|---|---|---|
| 5 |  | 454.78 | 68.83 | 6.61 | 2.80 |
| 6 |  | 884.71 | 113.45 | 7.79 | 2.99 |
| 7 |  | 977.01 | 110.31 | 8.86 | 2.80 |
| 8 |  | 629.80 | 35.61 | 17.69 | 3.17 |
| 9 |  | 272.83 | 24.15 | 11.30 | 3.51 |
| 10 |  | 230.99 | 73.37 | 3.15 | 3.44 |
| 11 |  | 495.34 | 27.65 | 17.91 | 3.23 |
| 12 |  | 237.79 | 15.64 | 15.20 | 3.71 |
| 13 |  | 223.67 | 14.57 | 15.35 | 3.64 |
| 14 |  | 198.84 | 20.31 | 9.79 | 3.23 |
| 15 |  | 197.38 | 10.30 | 19.16 | 3.98 |
| 16 |  | 194.98 | 22.01 | 8.86 | 3.95 |
| sulfadiazine |  | >2500.00 | ~2721.45 | <0.92 | n.d. |
| trimethoprim |  | >32.00 | 12.13 | >2.64 | n.d. |
| sulfadiazine trimethoprim ratio 5:1 | 2454.14 | 37.15 | 66.06 | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paneth, A.; Węglińska, L.; Bekier, A.; Stefaniszyn, E.; Wujec, M.; Trotsko, N.; Hawrył, A.; Hawrył, M.; Dzitko, K. Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design. Molecules 2019, 24, 1618. https://doi.org/10.3390/molecules24081618
Paneth A, Węglińska L, Bekier A, Stefaniszyn E, Wujec M, Trotsko N, Hawrył A, Hawrył M, Dzitko K. Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design. Molecules. 2019; 24(8):1618. https://doi.org/10.3390/molecules24081618
Chicago/Turabian StylePaneth, Agata, Lidia Węglińska, Adrian Bekier, Edyta Stefaniszyn, Monika Wujec, Nazar Trotsko, Anna Hawrył, Miroslaw Hawrył, and Katarzyna Dzitko. 2019. "Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design" Molecules 24, no. 8: 1618. https://doi.org/10.3390/molecules24081618
APA StylePaneth, A., Węglińska, L., Bekier, A., Stefaniszyn, E., Wujec, M., Trotsko, N., Hawrył, A., Hawrył, M., & Dzitko, K. (2019). Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design. Molecules, 24(8), 1618. https://doi.org/10.3390/molecules24081618