
Coumarins and P450s, Studies Reported to-Date


Abstract
1. Introduction
1.1. Cytochrome P450s Enzymes
1.1.1. Basic Enzyme Structure
1.1.2. Catalytic Activity Cycle
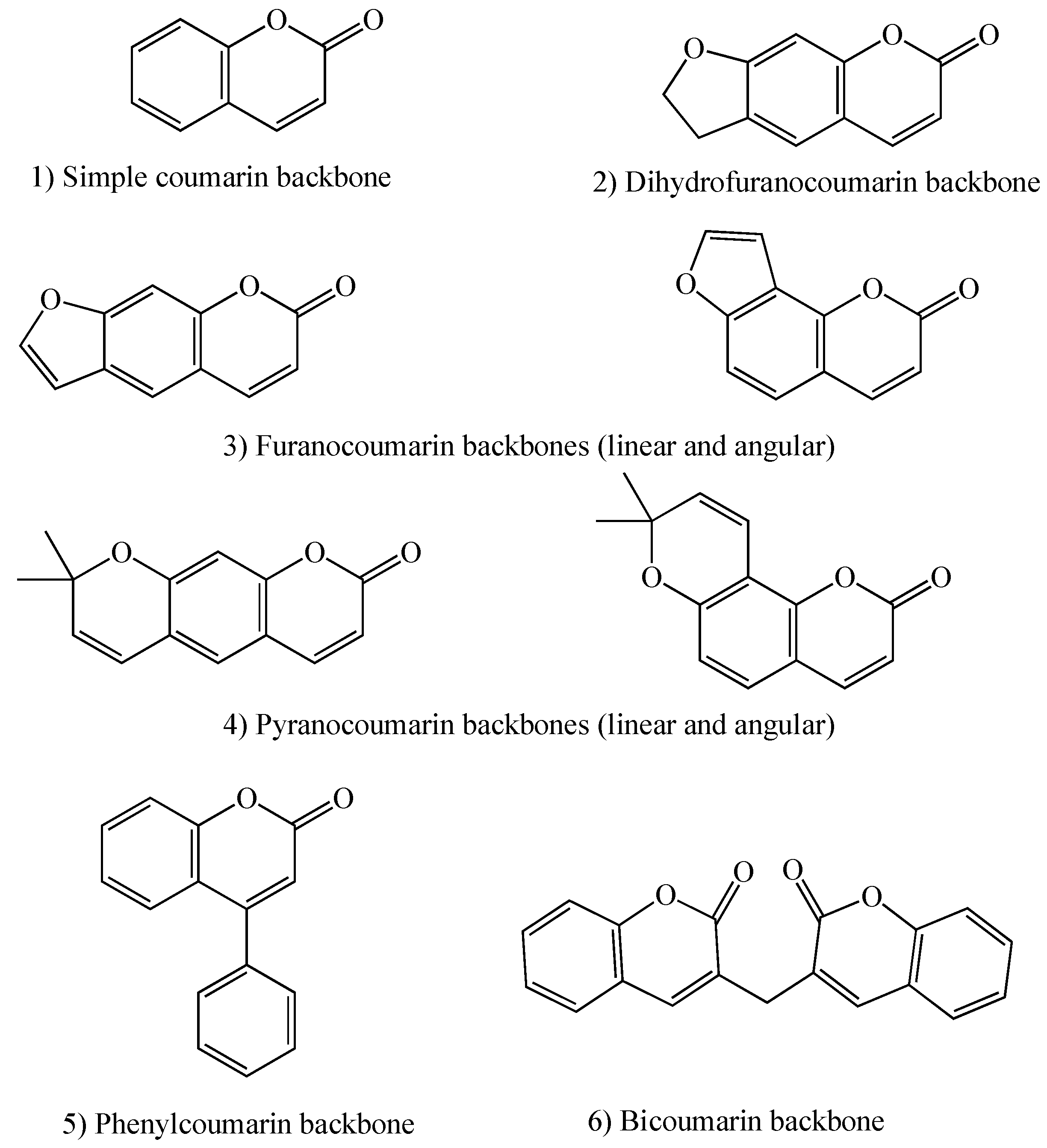
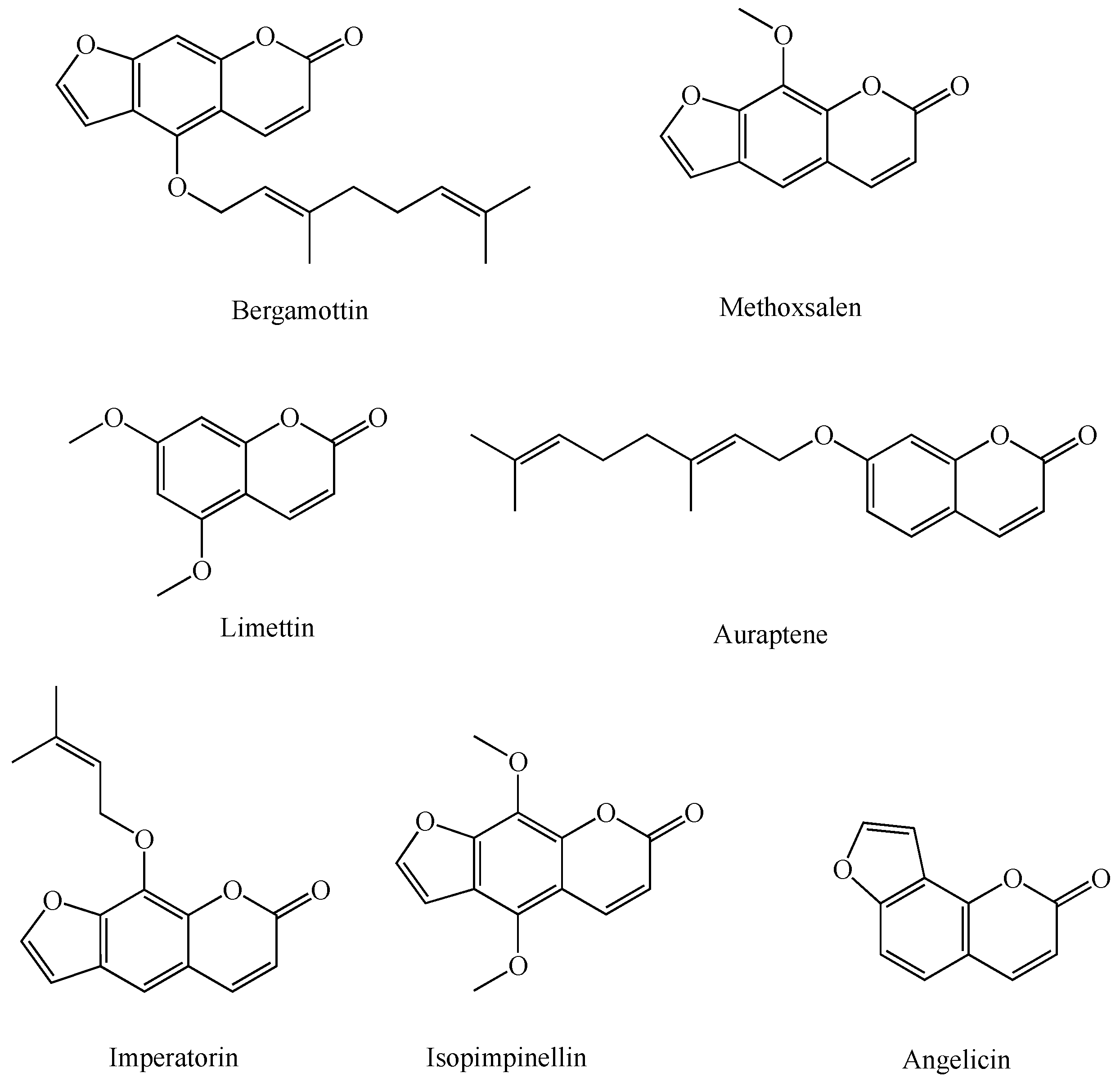
1.2. Coumarins
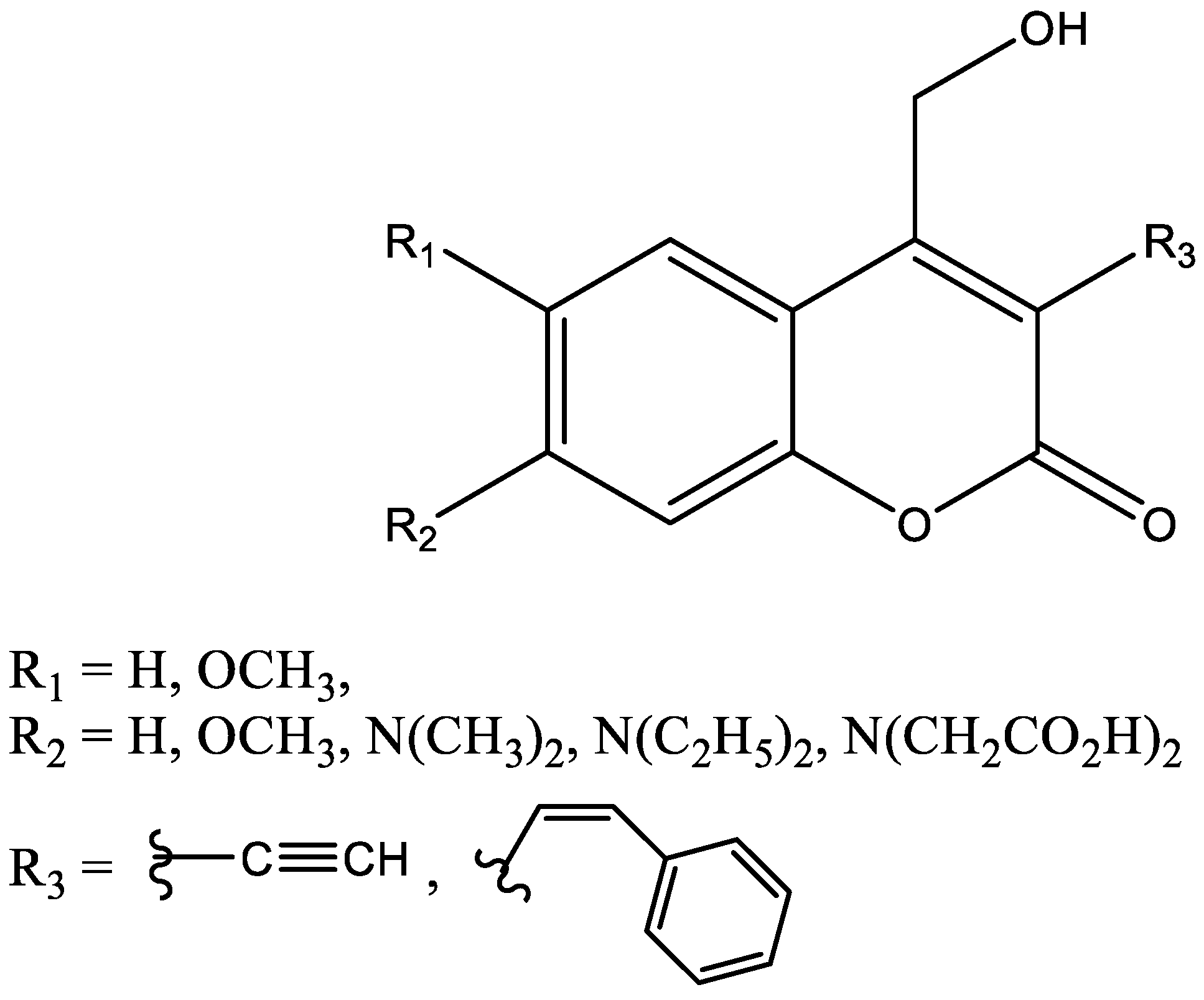
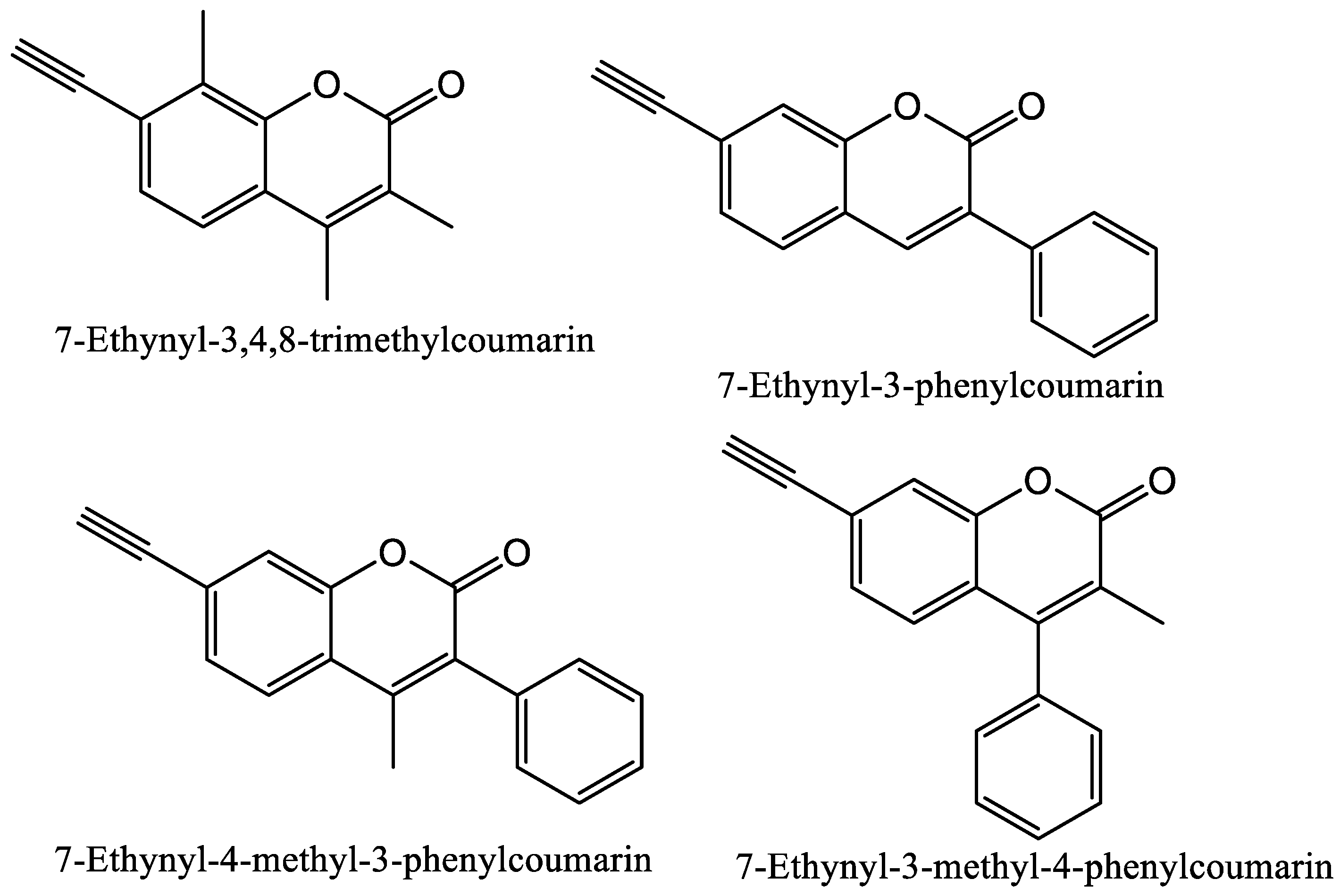
1.2.1. Coumarins and P450s
1.2.2. Coumarin Metabolism by P450s
1.2.3. P450 Inhibition Mechanism by Coumarins
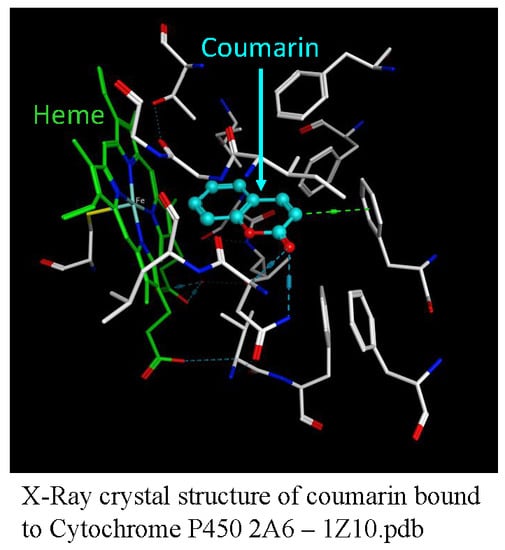
1.2.4. Computational Molecular Modeling Studies
2. Conclusions
Funding
Conflicts of Interest
References
- Guengerich, F.P. Cytochrome P450 Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2005; pp. 377–463. [Google Scholar]
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Curr. Biol. 1995, 3, 41–62. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Zeldin, D.; Makita, K.; Karara, A.; Falck, J.R. Cytochrome P450 Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2005; pp. 531–545. [Google Scholar]
- Williams, P.A.; Cosme, J.; Sridhar, V.; Johnson, E.F.; McRee, D.E. Mammalian microsomal cytochrome P450 monooxygenase: Structural adaptations for membrane binding and functional diversity. Mol. Cell 2000, 5, 121–131. [Google Scholar] [CrossRef]
- Schenkman, J.B. Cytochrome P450 Handbook of Experimental Pharmacology; Greim, H., Ed.; Springer: Berlin, Germany, 1993; Volume 105, pp. 1–739. [Google Scholar]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUC /AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Kim, D.-H.; Iwasaki, M. Enzymatic oxidation of ethyl carbamate to vinyl carbamate and its role as an intermediate in the formation of 1, N6-ethenoadenosine. Chem. Res. Toxicol. 1991, 4, 168–179. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, Y.; Prakash, C. Xenobiotic-metabolizing enzymes in human lung. Curr. Drug Metab. 2006, 7, 939–948. [Google Scholar] [CrossRef]
- Shimada, T.; Hayes, C.L.; Yamazaki, H.; Amin, S.; Hecht, S.S.; Guengerich, F.P.; Sutter, T.R. Activation of chemically diverse procarcinogens by human cytochrome P450 1B1. Cancer Res. 1996, 56, 2979–2984. [Google Scholar]
- Shimada, T.; Gillam, E.M.; Sandhu, P.; Guo, Z.; Tukey, R.H.; Guengerich, F.P. Activation of procarcinogens by human cytochrome P450 enzymes expressed in Escherichia coli. Simplified bacterial systems for genotoxicity assays. Carcinogenesis 1994, 15, 2523–2529. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Oda, Y.; Gillam, E.M.J.; Guengerich, F.P.; Inoue, K. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochromes P450 1A1 and P450 1B1 allelic variants and other human cytochromes P450 in Salmonella typhimurium NM2009. Drug Metab. Dispos. 2001, 29, 1176–1182. [Google Scholar] [PubMed]
- Nelson, D.R. Progress in tracing the evolutionary paths of cytochrome P450. Biochim. Biophys. Acta BBA Proteins Proteom. 2011, 1814, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Taylor, S.F.; Dupart, P.S.; Arnold, C.L.; Sridhar, J.; Jiang, Q.; Wang, Y.; Skripnikova, E.V.; Zhao, M.; Foroozesh, M. Pyranoflavones: A group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 2013, 56, 4082–4092. [Google Scholar] [CrossRef]
- Liu, J.; Sridhar, J.; Foroozesh, M. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules 2013, 18, 14470–14495. [Google Scholar] [CrossRef]
- Sridhar, J.; Liu, J.; Foroozesh, M.; Klein Stevens, C.L. Inhibition of cytochrome P450 enzymes by quinones and anthraquinones. Chem. Res. Toxicol. 2012, 25, 357–365. [Google Scholar] [CrossRef]
- Sridhar, J.; Liu, J.; Komati, R.; Schroeder, R.; Jiang, Q.; Tram, P.; Riley, K.; Foroozesh, M. Ortho-methylarylamines as time-dependent inhibitors of cytochrome P450 1A1 enzyme. Drug Metab. Lett. 2016, 10, 270–277. [Google Scholar] [CrossRef]
- Sridhar, J.; Liu, J.; Foroozesh, M.; Stevens, C.L. Insights on cytochrome P450 enzymes and inhibitors obtained through QSAR studies. Molecules 2012, 17, 9283–9305. [Google Scholar] [CrossRef]
- Sridhar, J.; Ellis, J.; Dupart, P.; Liu, J.; Stevens, C.L.; Foroozesh, M. Development of flavone propargyl ethers as potent and selective inhibitors of cytochrome P450 enzymes 1A1 and 1A2. Drug Metab. Lett. 2012, 6, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Foroozesh, M.; Stevens, C.L. QSAR models of cytochrome P450 enzyme 1A2 inhibitors using CoMFA, CoMSIA and HQSAR. SAR QSAR Environ. Res. 2011, 22, 681–697. [Google Scholar] [CrossRef]
- Sridhar, J.; Jin, P.; Liu, J.; Foroozesh, M.; Stevens, C.L. In silico studies of polyaromatic hydrocarbon inhibitors of cytochrome P450 enzymes 1A1, 1A2, 2A6, and 2B1. Chem. Res. Toxicol. 2010, 23, 600–607. [Google Scholar] [CrossRef]
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nguyen, T.T.; Dupart, P.S.; Sridhar, J.; Zhang, X.; Zhu, N.; Stevens, C.L.; Foroozesh, M. 7-ethynylcoumarins: Selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 2012, 25, 1047–1057. [Google Scholar] [CrossRef]
- Sridhar, J.; Goyal, N.; Liu, J.; Foroozesh, M. Review of ligand specificity factors for CYP1A subfamily enzymes from molecular modeling studies reported to-date. Molecules 2017, 22, 1143. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Wang, A.; Savas, U.; Hsu, M.-H.; Stout, C.D.; Johnson, E.F. Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J. Biol. Chem. 2012, 287, 10834–10843. [Google Scholar] [CrossRef] [PubMed]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef]
- Yun, C.-H.; Miller, G.P.; Guengerich, F.P. Oxidations of p-alkoxyacylanilides catalyzed by human cytochrome P450 1A2: Structure-activity relationships and simulation of rate constants of individual steps in catalysis. Biochemistry 2001, 40, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Hsu, M.-H.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 2005, 12, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.B.; Liu, J.; Huo, L.; Zhang, Q.; Dearing, M.D.; Wilderman, P.R.; Szklarz, G.D.; Stout, C.D.; Halpert, J.R. Structure-function analysis of mammalian CYP2B enzymes using 7-substituted coumarin derivatives as probes: Utility of crystal structures and molecular modeling in understanding xenobiotic metabolism. Mol. Pharmacol. 2016, 89, 435–445. [Google Scholar] [CrossRef]
- Pereira, T.M.; Franco, D.P.; Votorio, F.; Kummerle, A.E. Coumarin compounds in medicinal chemistry: Some important examples from the last years. Curr. Top. Med. Chem. 2018, 18, 124–148. [Google Scholar] [CrossRef]
- Poumale, H.M.; Hamm, R.; Zang, Y.; Shiono, Y.; Kuete, V. Medicinal Plant Research in Africa, Pharmacology and Chemistry; Kuete, V., Ed.; Elsevier: London, UK, 2013; pp. 261–300. [Google Scholar]
- Bone, K.; Mills, S. Principles and Practice of Phytotherapy; Churchill Livingstone: Toronto, ON, Canada, 2013; pp. 968–969. [Google Scholar]
- Aoyama, Y.; Katayama, T.; Yamamoto, M.; Tanaka, H.; Kon, K. A new antitumor antibiotic product, demethylchartreusin. Isolation and biological activities. J. Antibiot. 1992, 45, 875–878. [Google Scholar] [CrossRef][Green Version]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. BioMed Res. Int. 2013, 2013, 963248. [Google Scholar] [CrossRef] [PubMed]
- Born, S.L.; Caudill, D.; Filter, K.L.; Purdonm, M.P. Identification of the cytochromes P450 that catalyze coumarin 3, 4-epoxidation and 3-hydroxylation. Drug Metab. Dispos. 2002, 30, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Sridar, C.; Kent, U.M.; Noon, K.; McCall, A.; Alworth, W.; Foroozesh, M.; Hollenberg, P.F. Differential inhibition of cytochromes P450 3A4 and 3A5 by the newly synthesized coumarin derivatives 7-coumarin propargyl ether and 7-(4-trifluoromethyl) coumarin propargyl ether. Drug Metab. Dispos. 2008, 36, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Ito, Y.; Lake, B.G. Metabolism of coumarin by human P450s: A molecular modelling study. Toxicol. In Vitro 2006, 20, 256–264. [Google Scholar] [CrossRef]
- Lake, B.G. Coumarin metabolism, toxicity and carcinogenicity: Relevance for human risk assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]
- Bourgaud, F.; Hehn, A.; Larbat, R.; Doerper, S.; Gontier, E.; Kellner, S.; Matern, U. Biosynthesis of coumarins in plants: A major pathway still to be unraveled for cytochrome P450 enzymes. Phytochem. Rev. 2006, 5, 293–308. [Google Scholar] [CrossRef]
- He, K.; Lyer, K.R.; Hayes, R.N.; Sinz, M.W.; Woolf, T.F.; Hollenberg, P.F. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem. Res. Toxicol. 1998, 11, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Kent, U.M.; Hollenberg, P.F. The grapefruit juice effect is not limited to cytochrome P450 (P450) 3A4: Evidence for bergamottin-dependent inactivation, heme destruction, and covalent binding to protein in P450s 2B6 and 3A5. J. Pharmacol. Exp. Ther. 2005, 313, 154–164. [Google Scholar] [CrossRef]
- Hung, W.-L.; Suh, J.H.; Wang, Y. Chemistry and health effects of furanocoumarins in grapefruit. J. Food Drug Anal. 2017, 25, 71–83. [Google Scholar] [CrossRef]
- Kleiner, H.E.; Xia, X.; Sonoda, J.; Zhang, J.; Pontius, E.; Abey, J.; Evans, R.M.; Moore, D.D.; DiGiovanni, J. Effects of naturally occurring coumarins on hepatic drug-metabolizing enzymes in mice. Toxicol. Appl. Pharmacol. 2008, 232, 337–350. [Google Scholar] [CrossRef]
- Tine, M.; Belghit, J.; Descatoire, V.; Amouyal, G.; Letteron, P.; Geneve, J.; Larrey, D.; Pessayre, D. Inactivation of human liver cytochrome P-450 by the drug methoxsalen and other psoralen derivatives. Biochem. Pharmacol. 1987, 36, 951–955. [Google Scholar] [CrossRef]
- Palacharla, R.C.; Molgara, P.; Panthangi, H.R.; Boggavarapu, R.K.; Manoharan, A.K.; Ponnamaneni, R.K.; Ajjala, D.R.; Nirogi, R. Methoxsalen as an in vitro phenotyping tool in comparison with 1-aminobenzotriazole. Xenobiotica 2017, 49, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Campbell, C.T.; Robertson, T.A.; Wells, A.J.; Kleiner, H.E. Naturally occurring coumarins inhibit 7, 12-dimethylbenz [a] anthracene DNA adduct formation in mouse mammary gland. Carcinogenesis 2006, 27, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Born, S.L.; Hu, J.K.; Lehman-McKeeman, L.D. O-Hydroxyphenylacetaldehyde is a hepatotoxic metabolite of coumarin. Drug Metab. Dispos. 1999, 8, 218–223. [Google Scholar]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Crespi, C.L.; W-Penman, B.; Steimel, D.T.; Smith, T.; Yang, C.S.; Sutter, T.R. Development of a human lymphoblastoid cell line constitutively expressing human CYP1B1 cDNA: Substrate specificity with model substrates and promutagens. Mutagenesis 1997, 12, 83–89. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yun, C.-H.; Kim, K.-H.; Calcutt, M.W.; Guengerich, F.P. Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J. Biol. Chem. 2005, 280, 12279–12291. [Google Scholar] [CrossRef] [PubMed]
- Miwa, G.T.; Lu, A.Y.H. Kinetic isotope effects and “metabolic switching” in cytochrome P450-catalyzed reactions. BioEssays 1987, 7, 215–219. [Google Scholar] [CrossRef]
- Miwa, G.T.; Walsh, J.S.; Lu, A.Y.H. Kinetic isotope effects on cytochrome P-450-catalyzed oxidation reactions: The oxidative O-dealkylation of 7-ethoxycoumarin. J. Biol. Chem. 1984, 259, 3000–3004. [Google Scholar] [PubMed]
- Liu, J.; Shah, M.B.; Zhang, Q.; Stout, C.D.; Halpert, J.R.; Wilderman, P.R. Coumarin derivatives as substrate probes of mammalian cytochromes P450 2B4 and 2B6: Assessing the importance of 7-alkoxy chain length, halogen substitution, and non-active site mutations. Biochemistry 2016, 55, 1997–2007. [Google Scholar] [CrossRef]
- Scott, E.E.; White, M.A.; He, Y.A.; Johnson, E.F.; Stout, C.D.; Halpert, J.R. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-Chlorophenyl)imidazole at 1.9-Å resolution. J. Biol. Chem. 2004, 279, 27294–27301. [Google Scholar] [CrossRef]
- Shah, M.B.; Pascual, J.; Zhang, Q.; Stout, C.D.; Halpert, J.R. Structures of cytochrome P450 2B6 bound to 4-benzylpyridine and 4-(4-nitrobenzyl)pyridine: Insight into inhibitor binding and rearrangement of active site side chains. Mol. Pharmacol. 2011, 80, 1047–1055. [Google Scholar] [CrossRef]
- Nahar, R.; Dube, D.P.; Parakh, R.; Deb, R.; Saxena, R.S.; Singh, T.P.; Verma, I.C. Implication of novel CYP2C9*57 (p. Asn204His) variant in coumarin hypersensitivity. Thromb. Res. 2013, 131, 535–539. [Google Scholar] [CrossRef]
- Ngui, J.S.; Chen, Q.; Shou, M.; Wang, R.W.; Stearns, R.A.; Baillie, T.A.; Tang, W. In vitro stimulation of warfarin metabolism by quinidine: Increases in the formation of 4′- and 10-hydroxywarfarin. Drug Metab. Dispos. 2001, 29, 877–886. [Google Scholar]
- Zanger, U.M.; Fischer, J.; Raimundo, S.; Stüven, T.; Evert, B.O.; Schwab, M. Comprehensive analysis of the genetic factors determining expression and function of hepatic CYP2D6. Pharmacogenetics 2001, 11, 573–585. [Google Scholar] [CrossRef]
- Venhorst, J.; Laak, A.M.; Commandeur, J.N.; Hiroi, T.; Vermeulen, N.P. Homology modeling of rat and human cytochrome P450 2D (CYP2D) isoforms and computational rationalization of experimental ligand-binding specificities. J. Med. Chem. 2003, 46, 74–86. [Google Scholar] [CrossRef]
- Yamazaki, H.; Inoue, K.; Mimura, M.; Oda, Y.; Guengerich, F.P.; Shimada, T. 7-Ethoxycoumarin O-deethylation catalyzed by cytochromes P450 1A2 and 2E1 in human liver microsomes. BioChem. Pharmacol. 1996, 51, 313–319. [Google Scholar] [CrossRef]
- Wei, Y.; Wu, H.; Li, L.; Liu, Z.; Zhou, X.; Zhang, Q.-Y.; Weng, Y.; D’Agostino, J.; Ling, G.; Zhang, X.; et al. Generation and Characterization of a CYP2A13/2B6/2F1- Transgenic Mouse Model. Drug Metab. Dispos. 2012, 40, 1144–1150. [Google Scholar] [CrossRef]
- Mooiman, K.D.; Maas-Bakkar, R.F.; Hendrikx, J.J.M.A.; Bank, P.C.D.; Rosing, H.; Beijnen, J.H.; Schellens, J.H.M.; Meijerman, I. The effect of complementary and alternative medicines on CYP3A4—Mediated metabolism of three different substrates: 7-Benzyloxy-4-trifluoromethyl--coumarin, midazolam and docetaxel. J. Pharm. Pharmacol. 2014, 66, 865–874. [Google Scholar] [CrossRef]
- Rwei, A.Y.; Wang, W.; Kohane, D.S. Photoresponsive nanoparticles for drug delivery. Nano Today 2015, 10, 451–467. [Google Scholar] [CrossRef]
- Wagner, N.; Stephan, M.; Hoglinger, D.; Nadler, A. A click case: Organelle-specific uncaging of lipid messengers. Angew. Chem. Int. Ed. 2018, 57, 13339–13343. [Google Scholar] [CrossRef]
- Yang, L.; Tang, H.; Sun, H. Progress in photo-responsive polypeptide derived nano-assemblies. Micromachines 2018, 9, 296. [Google Scholar] [CrossRef]
- Schmidt, R.; Geissler, D.; Hagen, V.; Bendig, J. Mechanism of photocleavage of (coumarin-4-yl) methyl esters. J. Phys. Chem. A 2007, 111, 5768–5774. [Google Scholar] [CrossRef]
- Lin, Q.; Bao, C.; Cheng, S.; Yang, Y.; Ji, W.; Zhu, L. Target-activated coumarin phototriggers specifically switch on fluorescence and photocleavage upon binding to thiol-bearing protein. J. Am. Chem. Soc. 2012, 134, 5052–5055. [Google Scholar] [CrossRef]
- Lin, Q.; Yang, L.; Wang, Z.; Hua, Y.; Zhang, D.; Bao, B.; Bao, C.; Gong, X.; Zhu, L. Coumarin photocages modified with an electron-rich styryl moiety at the 3-position: Long wavelength excitation, rapid photolysis and photobleaching. Angew. Chem. Int. Ed. 2019, 57, 3722–3726. [Google Scholar] [CrossRef] [PubMed]
- Ueng, Y.F.; Jan, W.C.; Lin, L.C.; Chen, T.L.; Guengerich, F.P.; Chen, C.F. The alkaloid rutaecarpine is a selective inhibitor of cytochrome p450 1a in mouse and human liver microsomes. Drug Metab. Dispos. 2002, 30, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.W.; Britt, S.G.; Pohl, L.R. Inactivation of cytochrome p-450 by 2-isopropyl-4-pentenamide and other xenobiotics leads to heme-derived protein adducts. Chem. Biol. Interact. 1986, 58, 345–352. [Google Scholar] [CrossRef]
- Halpert, J. Further studies of the suicide inactivation of purified rat liver cytochrome p-450 by chloramphenicol. Mol. Pharmacol. 1982, 21, 166–172. [Google Scholar]
- Liu, J.; Pham, P.T.; Skripnikova, E.V.; Zheng, S.; Lovings, L.J.; Wang, Y.; Goyal, N.; Bellow, S.M.; Mensah, L.M.; Chatters, A.J.; et al. A ligand-based drug design. Discovery of 4-trifluoromethyl-7, 8-pyranocoumarin as a selective inhibitor of human cytochrome P450 1A2. J. Med. Chem. 2015, 58, 6481–6493. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Tocixol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef]
- Shimada, T. Inhibition of Carcinogen-activating cytochrome P450 enzymes by xenobiotic chemicals in relation to antimutagenicity and anticarcinogenecity. Toxicol. Res. 2017, 33, 79–96. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Akimoto, I.; Motegi, K.; Yoshimura, T.; Wada, K.; Nishizono, N.; Oda, K. Synthetic models related to methoxalen and menthofuran-cytochrome P450 (CYP) 2A6 interactions. benzofuran and coumarin derivatives as potent and selective inhibitors of CYP2A6. Chem. Pharm. Bull. 2013, 61, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Meineke, I.; Desel, H.; Kahl, R.; Kahl, G.F.; Gundert-Remy, U. Determination of 2-hydroxyphenylacetic acid (2HPAA) in urine after oral and parenteral administration of coumarin by gas-liquid chromatography with flame-ionization detection. J. Pharm. Biomed. Anal. 1998, 17, 487–492. [Google Scholar] [CrossRef]
- Pelkonen, O.; Rautio, A.; Raunio, H.; Pasanen, M. CYP2A6: A human coumarin 7-hydroxylase. Toxicology 2000, 144, 139–147. [Google Scholar] [CrossRef]
- Rautio, A.; Kraul, H.; Koji, A.; Salmela, E.; Pelkonen, O. Interindividual variability of coumarin 7-hydroxylase in healthy volunteers. Pharmacogenetics 1992, 2, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Van Iersel, M.L.P.S.; Henderson, C.J.; Walters, D.G.; Price, R.J.; Wolf, C.R.; Lake, B.G. Metabolism of [3-14C] coumarin by human liver microsomes. Xenobiotica 1994, 24, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Gu, J.; Zhang, Q.-Y.; Spink, D.C.; Kaminsky, L.S.; Ding, X. Biotransformation of coumarin by rodent and human cytochromes P450: Metabolic basis of tissue-selective toxicity in olfactory mucosa of rats and mice. J. Pharmacol. Exp. Ther. 1999, 288, 463–471. [Google Scholar] [PubMed]
- Lewis, D.F.V.; Lake, B.G.; Dickins, M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: The importance of compound lipophilicity. J. Enzym. Inhib. Med. Chem. 2007, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Ito, Y.; Lake, B.G. Quantitative structure-activity relationships (QSARs) for inhibitors and substrates of CYP2B enzymes: Importance of compound lipophilicity in explanation of potency differences. J. Enzym. Inhib. Med. Chem. 2010, 25, 679–684. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Shen, J.; Hu, W.Y.; Ding, X.; Lu, A.Y.; Hong, J.Y. Identification of Val117 and Arg372 as critical amino acid residues for the activity difference between human CYP2A6 and CYP2A13 in coumarin 7-hydroxylation. Arch. Biochem. Biophys. 2004, 427, 143–153. [Google Scholar] [CrossRef]
- Von Weymarn, L.B.; Murphy, S.E. CYP2A13-catalysed coumarin metabolism: Comparison with CYP2A5 and CYP2A6. Xenobiotica 2003, 33, 73–81. [Google Scholar] [CrossRef]
- DeVore, N.M.; Smith, B.D.; Wang, J.L.; Lushington, G.H.; Scott, E.E. Key residues controlling binding of diverse ligands to human cytochrome P450 2A enzymes. Drug Metab. Dispos. 2009, 37, 1319–1327. [Google Scholar] [CrossRef]
- Leong, M.K.; Chen, Y.M.; Chen, T.-H. Prediction of human cytochrome P450 2B6-substrate interactions using Hierarchical Support Vector Regression approach. J. Comput. Chem. 2009, 30, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar] [PubMed]
- Rendic, S.; DiCarlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foroozesh, M.; Sridhar, J.; Goyal, N.; Liu, J. Coumarins and P450s, Studies Reported to-Date. Molecules 2019, 24, 1620. https://doi.org/10.3390/molecules24081620
Foroozesh M, Sridhar J, Goyal N, Liu J. Coumarins and P450s, Studies Reported to-Date. Molecules. 2019; 24(8):1620. https://doi.org/10.3390/molecules24081620
Chicago/Turabian StyleForoozesh, Maryam, Jayalakshmi Sridhar, Navneet Goyal, and Jiawang Liu. 2019. "Coumarins and P450s, Studies Reported to-Date" Molecules 24, no. 8: 1620. https://doi.org/10.3390/molecules24081620
APA StyleForoozesh, M., Sridhar, J., Goyal, N., & Liu, J. (2019). Coumarins and P450s, Studies Reported to-Date. Molecules, 24(8), 1620. https://doi.org/10.3390/molecules24081620