
Analysis of Cellulose and Lignocellulose Materials by Raman Spectroscopy: A Review of the Current Status

Abstract
:1. Introduction
2. Cellulose Materials
2.1. Characteristic Bands
2.2. Allomorphs of Cellulose
2.3. Cellulose Crystallinity Measurements
2.4. Characteristics of Supramolecular Structures
2.5. Nanocelluloses
2.6. Cellulose Nanocomposites
3. Lignocellulose Materials
3.1. Lignin Bands
3.2. Spectral Domination by the Conjugated Structures in Lignins
3.3. Differentiation between G-, S-, and H-Lignins
3.4. %S Content and S/G Ratio
4. Hemicellulose Contributions
5. New Methods of Spectral (Data) Analysis
6. New Raman Techniques (Instrumentation)
7. Quantum Chemical Calculations
8. Conclusions and Outlook
Funding
Conflicts of Interest
References
- Blackwell, J.; Vasko, P.D.; Koeing, J.L. Infrared and Raman Spectra of the Cellulose from the Cell Wall of Valonia ventricosa. J. Appl. Phys. 1970, 41, 4375. [Google Scholar] [CrossRef]
- Atalla, R.H.; Nagel, S.C. Laser-induced Fluorescence in Cellulose. Chem. Commun. 1972, 19, 1049. [Google Scholar] [CrossRef]
- Atalla, R.H.; Nagel, S.C. Cellulose: Its Regeneration in the Native Lattice. Science 1974, 185, 522. [Google Scholar] [CrossRef]
- Atalla, R.H.; Dimick, B.E. Raman-spectral Evidence for Differences Between the Conformations of Cellulose I and Cellulose II. Carbohydr. Polym. 1975, 39, C1. [Google Scholar] [CrossRef]
- Atalla, R.H.; Agarwal, U.P. Raman Microprobe Evidence for Lignin Orientation in the Cell Wails of Native Woody Tissue. Science 1985, 227, 636. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, U.P.; Atalla, R.H. In-situ Raman Microprobe Studies of Plant Cell Walls: Macromolecular organization and compositional variability in the secondary wall of Picea mariana (Mill.) BSP. Planta 1986, 169, 325. [Google Scholar] [CrossRef]
- Atalla, R.H.; Agarwal, U.P. Recording Raman Spectra from Plant Cell Walls. J. Raman Spectrosc. 1986, 17, 229. [Google Scholar] [CrossRef]
- Agarwal, U.P. An overview of Raman Spectroscopy as Applied to Lignocellulosic Materials. In Advances in Lignocellulosics Characterization; Argyropoulos, D.S., Ed.; TAPPI Press: Atlanta, GA, USA, 1999; pp. 209–225. [Google Scholar]
- Agarwal, U.P.; Atalla, R.H. Using Raman Spectroscopy to Identify Chromophores in Lignin-lignocellulosics. In Lignin: Historical, Biological, and Material Perspectives; Glasser, W.G., Northey, R.A., Eds.; ACS Symposium Series 742; American Chemical Society: Washington, DC, USA, 2000; pp. 250–264. [Google Scholar]
- Agarwal, U.P. Raman spectroscopic Characterization of Wood and Pulp Fibers. In Characterization of Lignocellulosic Materials; Hu, T.Q., Ed.; Blackwell Publishing: Oxford, UK, 2008; pp. 17–35. [Google Scholar]
- Agarwal, U.P.; Atalla, R.H. Vibrational Spectroscopy. In Lignin and Lignans: Advances in Chemistry; Heitner, C., Dimmel, D.R., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 103–136. [Google Scholar]
- Gierlinger, N.; Keplinger, T.; Harrington, M. Imaging of plant cell walls by confocal Raman microscopy. Nat. Protocols 2012, 7, 1694. [Google Scholar] [CrossRef]
- Agarwal, U.P. 1064 nm FT-Raman spectroscopy for Investigations of Plant Cell Walls and Other Biomass Materials. Front. Plant Sci. 2014, 5, 490. [Google Scholar] [CrossRef]
- Lupoi, J.S.; Gjersing, E.; Davis, M.F. Evaluating Lignocellulosic Biomass, its Derivatives, and Downstream Products with Raman Spectroscopy. Front. Bioeng. Biotechnol. 2015, 3, 50. [Google Scholar] [CrossRef] [PubMed]
- Gierlinger, N. New Insights into Plant Cell Walls by Vibrational Microspectroscopy. Appl. Spectrosc. Rev. 2018, 53, 517. [Google Scholar] [CrossRef]
- Wiley, J.H.; Atalla, R.H. Band Assignments in the Raman Spectra of Celluloses. Carbohydr. Res. 1987, 160, 113. [Google Scholar] [CrossRef]
- Makarem, M.; Lee, C.M.; Kafle, K.; Huang, S.; Chae, I.; Yang, H.; Kubicki, J.D.; Kim, S.H. Probing Cellulose Structures with Vibrational Spectroscopy. Cellulose 2019, 26, 35. [Google Scholar] [CrossRef]
- Edwards, H.G.M.; Farwell, D.W.; Williams, A.C. FT-Raman Spectrum of Cotton: A polymeric biomolecular analysis. Spectrochim. Acta Part A Mol. Spectrosc. 1994, 50, 807. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Ralph, S.A. FT-Raman Spectroscopy of Wood: Identifying contributions of lignin and carbohydrate polymers in the spectrum of black spruce (Picea mariana). Appl. Spectrosc. 1997, 51, 1648. [Google Scholar] [CrossRef]
- Agarwal, U.P. Raman Spectroscopy in the Analysis of Cellulose Nanomaterials. In Nanocelluloses: Their Preparation, Properties and Applications; Agarwal, U.P., Atalla, R.H., Isogai, A., Eds.; American Chemical Society: Washington, DC, USA, 2017; pp. 75–90. [Google Scholar]
- Chundawat, S.P.S.; Donohoe, B.S.; Sousa, L.C.; Elder, T.; Agarwal, U.P.; Lu, F.; Ralph, J.; Himmel, M.E.; Balan, V.; Dale, B.E. Multi-scale Visualization and Characterization of Lignocellulosic Plant Cell Wall Deconstruction during Thermochemical Pretreatment. Energy Environ. Sci. 2011, 4, 973. [Google Scholar] [CrossRef]
- Schenzel, K.; Fischer, S.; Brendler, E. New Method for Determining the Degree of Cellulose I Crystallinity by Means of FT Raman Spectroscopy. Cellulose 2005, 12, 223. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Reiner, R.S.; Ralph, S.A. Cellulose I Crystallinity Determination Using FT–Raman Spectroscopy: Univariate and multivariate methods. Cellulose 2010, 17, 721. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Ralph, S.A.; Reiner, R.S.; Baez, C. New Cellulose Crystallinity Estimation Method that Differentiates between Organized and Crystalline Phases. Carbohydr. Polym. 2018, 190, 262. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Ralph, S.A.; Reiner, R.S.; Baez, C. Probing Crystallinity of Never-dried Wood Cellulose with Raman Spectroscopy. Cellulose 2016, 23, 125. [Google Scholar] [CrossRef]
- Foster, E.J.; Moon, R.J.; Agarwal, U.P.; Bortner, M.J.; Bras, J.; Camarero-Espinosa, S.; Chan, K.J.; Clift, M.J.D.; Cranston, E.D.; Eichhorn, S.J.; et al. Current Characterization Methods for Cellulose Nanomaterials. Chem. Soc. Rev. 2018, 47, 2609. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Q.; Hirth, K.; Baez, C.; Agarwal, U.P.; Zhu, J.Y. Tailoring the Yield and Characteristics of Wood Cellulose Nanocrystals (CNC) using Concentrated Acid Hydrolysis. Cellulose 2015, 22, 1753. [Google Scholar] [CrossRef]
- Qing, Y.; Sabo, R.; Zhu, J.Y.; Agarwal, U.; Cai, Z.; Wu, Y. A Comparative Study of Cellulose Nanofibrils Disintegrated via Multiple Processing Approaches. Carbohydr. Polym. 2013, 97, 226. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Agarwal, U.P.; Hirth, K.C.; Matuana, L.M.; Sabo, R.C.; Stark, N.M. Chemical Modification of Nanocellulose with Canola Oil Fatty Acid Methyl Ester. Carbohydr. Polym. 2017, 169, 108. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Renneckar, S. Supramolecular Structure Characterization of Molecularly Thin Cellulose I Nanoparticles. Biomacromolecules 2011, 12, 650. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, U.P.; Sabo, R.; Reiner, R.S.; Clemons, C.M.; Rudie, A.W. Spatially Resolved Characterization of Cellulose Nanocrystal–polypropylene Composite by Confocal Raman Microscopy. Appl. Spectrosc. 2012, 66, 750. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, U.P. Raman Spectroscopy of CNC- and CNF-Based Nanocomposites. In Handbook of Nanocellulose and Cellulose Nanocomposites; Kargarzadeh, H., Ahmad, I., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2017; pp. 609–625. [Google Scholar]
- Lewandowska, A.E.; Inai, N.H.; Ghita, O.R.; Eichhorn, S.J. Quantitative Analysis of the Distribution and Mixing of Cellulose Nanocrystals in Thermoplastic Composites using Raman Chemical Imaging. RSC Adv. 2018, 8, 35831. [Google Scholar] [CrossRef]
- Sturcova, A.; Davies, G.R.; Eichhorn, S.J. Elastic Modulus and Stress-transfer Properties of Tunicate Cellulose Whiskers. Biomacromolecules 2005, 6, 1055. [Google Scholar] [CrossRef]
- Rusli, R.; Shanmuganathan, K.; Rowan, S.J.; Weder, C.; Eichhorn, S.J. Stress-transfer in Anisotropic and Environmentally Adaptive Cellulose Whisker Nanocomposites. Biomacromolecules 2010, 11, 762. [Google Scholar] [CrossRef]
- Peng, J.; Ellingham, T.; Sabo, R.; Turng, L.-S.; Clemons, C.M. Short Cellulose Nanofibrils as Reinforcement in Polyvinyl Alcohol Fiber. Cellulose 2014, 21, 4287. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Weinstock, I.A.; Atalla, R.H. FT-Raman Spectroscopy for Direct Measurement of Lignin Concentrations in Kraft Pulps. Tappi J. 2003, 2, 22. [Google Scholar]
- Segmehl, J.S.; Keplinger, T.; Krasnobaev, A.; Berga, J.K.; Will, C.; Burgert, I. Facilitated Delignification in CAD Deficient Transgenic Poplar Studied by Confocal Raman Spectroscopy Imaging. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 206, 177. [Google Scholar] [CrossRef]
- Li, Y.-J.; Li, H.-Y.; Cao, X.-F.; Sun, S.N.; Sun, R.-C. Understanding the Distribution and Structural Feature of Eucalyptus Lignin Isolated by γ-Valerolactone/Water/Acid System. ACS Sustain. Chem. Eng. 2018, 6, 12124. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Ramaswamy, S.; Kim, Y.S.; Xu, F. Obtaining Pure Spectra of Hemicellulose and Cellulose from Poplar Cell Wall Raman Imaging Data. Cellulose 2017, 24, 4671. [Google Scholar] [CrossRef]
- Kanbayashi, T.; Kataoka, Y.; Ishikawa, A.; Matsunaga, M.; Kobayashi, M.; Kiguchi, M. Depth Profiling of Photodegraded Wood Surfaces by Confocal Raman Microscopy. J. Wood Sci. 2018, 64, 169. [Google Scholar] [CrossRef]
- Lucas, M.; Wagner, G.L.; Nishiyama, Y.; Hanson, L.; Samayam, I.P.; Schall, C.A.; Langan, P.; Rector, K.D. Reversible Swelling of the Cell Wall of Poplar Biomass by Ionic Liquid at Room Temperature. Bioresour Technol. 2011, 102, 4518. [Google Scholar] [CrossRef] [PubMed]
- Kanbayashi, T.; Miyafuji, H. Raman Microscopic Analysis of Wood after Treatment with the Ionic Liquid, 1-Ethyl-3-methylimidazolium Chloride. Holzforschung 2015, 69, 273. [Google Scholar] [CrossRef]
- Kanbayashi, T.; Miyafuji, H. Topochemical and Morphological Characterization of Wood Cell Wall Treated with the Ionic Liquid, 1-Ethylpyridinium Bromide. Planta 2015, 242, 509. [Google Scholar] [CrossRef]
- Agarwal, U.P.; McSweeny, J.D.; Ralph, S.A. FT–Raman Investigation of Milled-wood Lignins: Softwood, hardwood, and chemically modified black spruce lignins. J. Wood Chem. Technol. 2011, 17, 1. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Reiner, R.S.; Pandey, A.K.; Ralph, S.A.; Hirth, K.C.; Atalla, R.H. Raman Spectra of Lignin Model Compounds. In Proceedings of the 13th International Symposium on Wood Fiber Pulping Chemistry, Auckland, New Zealand, 16–19 May 2005; Appita Inc.: Carlton, Australia, 2005; Volume 1, pp. 1–8. [Google Scholar]
- Saariaho, A.-M.; Jaaskelainen, A.-S.; Nuopponen, M.; Vuorinen, T. Ultra Violet Resonance Raman Spectroscopy in Lignin Analysis: Determination of Characteristic Vibrations of p-Hydroxyphenyl, Guaiacyl, and Syringyl Lignin Structures. Appl. Spectrosc. 2003, 57, 58. [Google Scholar] [CrossRef]
- Larsen, K.L.; Barsberg, S. Theoretical and Raman Spectroscopic Studies of Phenolic Lignin Model Monomers. J. Phys. Chem. B 2010, 114, 8009. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Bergström, B.; Gref, R.; Ericsson, A. Detection of Pinosylvins in Solid Wood of Scots Pine Using Fourier Transform Raman and Infrared Spectroscopy. J. Wood Chem. Technol. 1999, 1–2, 139. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Terashima, N. FT-Raman Study of Dehydrogenation Polymer (DHP) Lignins. In Proceedings of the 12th International Symposium on Wood, Fiber, and Pulping Chemistry, Madison, WI, USA, 9–12 June 2003; University of Wisconsin-Madison, Department of Forest Ecology and Management: Madison, WI, USA, 2003; Volume III, pp. 123–126. [Google Scholar]
- Ona, T.; Sonoda, T.; Ito, K.; Shibata, M.; Katayama, T.; Kato, T.; Ootake, Y. Nondestructive Determination of Lignin Syringyl/Guaiacyl Monomeric Composition in Native Wood by Fourier-Transform Raman Spectroscopy. J. Wood Chem. Technol. 1998, 18, 43. [Google Scholar] [CrossRef]
- Sun, L.; Varanasi, P.; Yang, F.; Loque, D.; Simmons, B.A.; Singh, S. Rapid Determination of Syringyl:Guaiacyl Ratios using FT-Raman Spectroscopy. Biotechnol. Bioeng. 2012, 109, 647. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Ralph, S.A.; Padmakshan, D.; Liu, S.; Foster, C.E. Estimation of Syringyl Units in Wood Lignins by FT-Raman Spectroscopy. J. Agric. Food Chem. 2019, in press. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, Z.; Zhou, X.; Ma, J.F.; Hu, Y.H.; Xu, F. Method for Automatically Identifying Spectra of Different Wood Cell Wall Layers in Raman Imaging Data Set. Anal. Chem. 2015, 87, 1344. [Google Scholar] [CrossRef]
- Gierlinger, N. Revealing Changes in Molecular Composition of Plant Cell Walls on the Micron-level by Raman Mapping and Vertex Component Analysis (VCA). Front. Plant Sci. 2014, 5, 306. [Google Scholar] [CrossRef] [PubMed]
- Mateu, B.P.; Felhofer, M.; Felhofer, M.; Juan, A.D.; Gierlinger, N. Multivariate Unmixing Approaches on Raman Images of Plant Cell Walls: New insights or over interpretation of results? Plant Methods 2018, 14, 52. [Google Scholar] [CrossRef]
- Saariaho, A.-M.; Jaaskelainen, A.-S.; Matousek, P.; Towrie, M.; Parker, A.W.; Vuorinen, T. Resonance Raman Spectroscopy of Highly Fluorescing Lignin Containing Chemical Pulps: Suppression of fluorescence with an optical Kerr gate. Holzforschung 2004, 58, 82. [Google Scholar] [CrossRef]
- Lignin Radicals in the Plant Cell Wall Probed by Kerr-Gated Resonance Raman Spectroscopy. Biophys. J. 2006, 90, 2978. [CrossRef]
- Jin, K.; Liu, X.; Wang, K.; Jiang, Z.; Tian, G.; Yang, S.; Shang, L.; Ma, J. Imaging the dynamic deposition of cell wall polymer in xylem and phloem in Populus × euramericana. Planta 2018, 248, 849. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Reiner, R.S. Near-IR surface-enhanced Raman Spectrum of Lignin. J. Raman Spectrosc. 2009, 40, 1527. [Google Scholar] [CrossRef]
- Saar, B.G.; Zeng, Y.; Freudiger, C.W.; Liu, Y.-S.; Himmel, M.E.; Xie, X.S.; Ding, S.-Y. Label-Free, Real-Time Monitoring of Biomass Processing with Stimulated Raman Scattering Microscopy. Angew. Chem. 2010, 32, 5608. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhao, S.; Wei, H.; Tucker, M.P.; Himmel, M.E.; Mosier, N.S.; Meilan, R.; Ding, S.-Y. In situ Micro-spectroscopic Investigation of Lignin in Poplar Cell Walls Pretreated by Maleic Acid. Biotechnol. Biofuels 2015, 8, 126. [Google Scholar] [CrossRef]
- Zeng, Y.; Yarbrough, J.M.; Mittal, A.; Tucker, M.P.; Vinzant, T.B.; Decker, S.R.; Himmel, M.E. In Situ Label-free Imaging of Hemicellulose in Plant Cell Walls using Stimulated Raman Scattering Microscopy. Biotechnol. Biofuels 2016, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Saar, B.G.; Friedrich, M.G.; Chen, F.; Liu, Y.-S.; Dixon, R.A.; Himmel, M.E.; Xie, X.S.; Ding, S.-Y. Imaging Lignin-Downregulated Alfalfa Using Coherent Anti-Stokes Raman Scattering Microscopy. BioEnergy Res. 2010, 3, 272. [Google Scholar] [CrossRef]
- Zimmerley, M.; Younger, R.; Valenton, T.; Oertel, D.C.; Ward, J.L.; Potma, E.O. Molecular Orientation in Dry and Hydrated Cellulose Fibers: A Coherent Anti-Stokes Raman Scattering Microscopy Study. J. Phys. Chem. B 2010, 114, 10200. [Google Scholar] [CrossRef]
- Lee, C.M.; Mohamed, N.M.A.; Watts, H.D.; Kubicki, J.D.; Kim, S.H. Sum-frequency-generation vibration spectroscopy and density functional theory calculations with dispersion corrections (DFT-D2) for cellulose Iα and Iβ. J. Phys. Chem. B 2013, 117, 6681. [Google Scholar]
- Lee, C.M.; Kubicki, J.D.; Fan, B.; Zhong, L.; Jarvis, M.C.; Kim, S.H. Hydrogen-bonding Network and OH Stretch Vibration of Cellulose: Comparison of computational modeling with polarized IR and SFG spectra. J. Phys. Chem. B 2015, 119, 15138. [Google Scholar] [CrossRef]







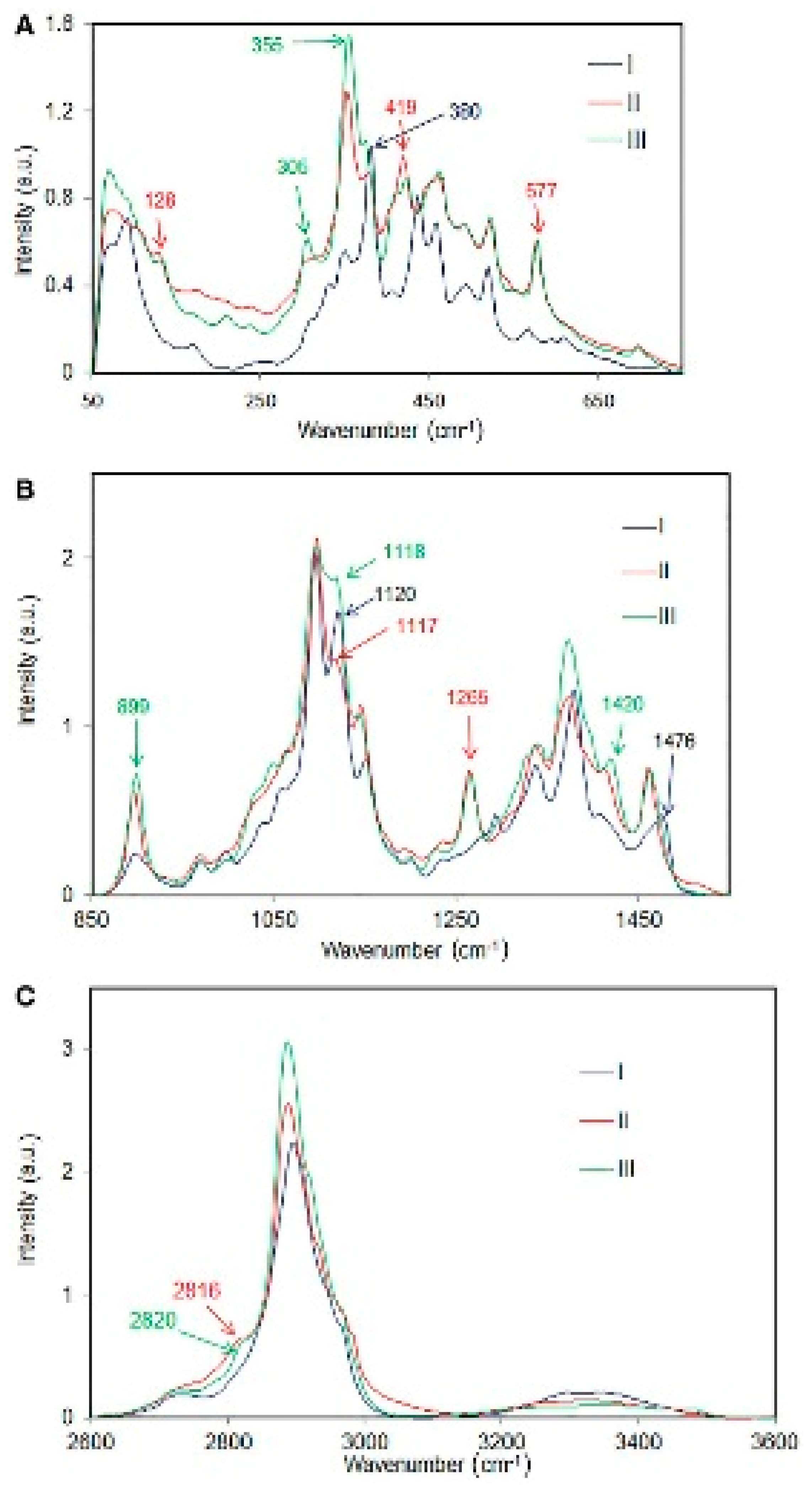{kind=link}
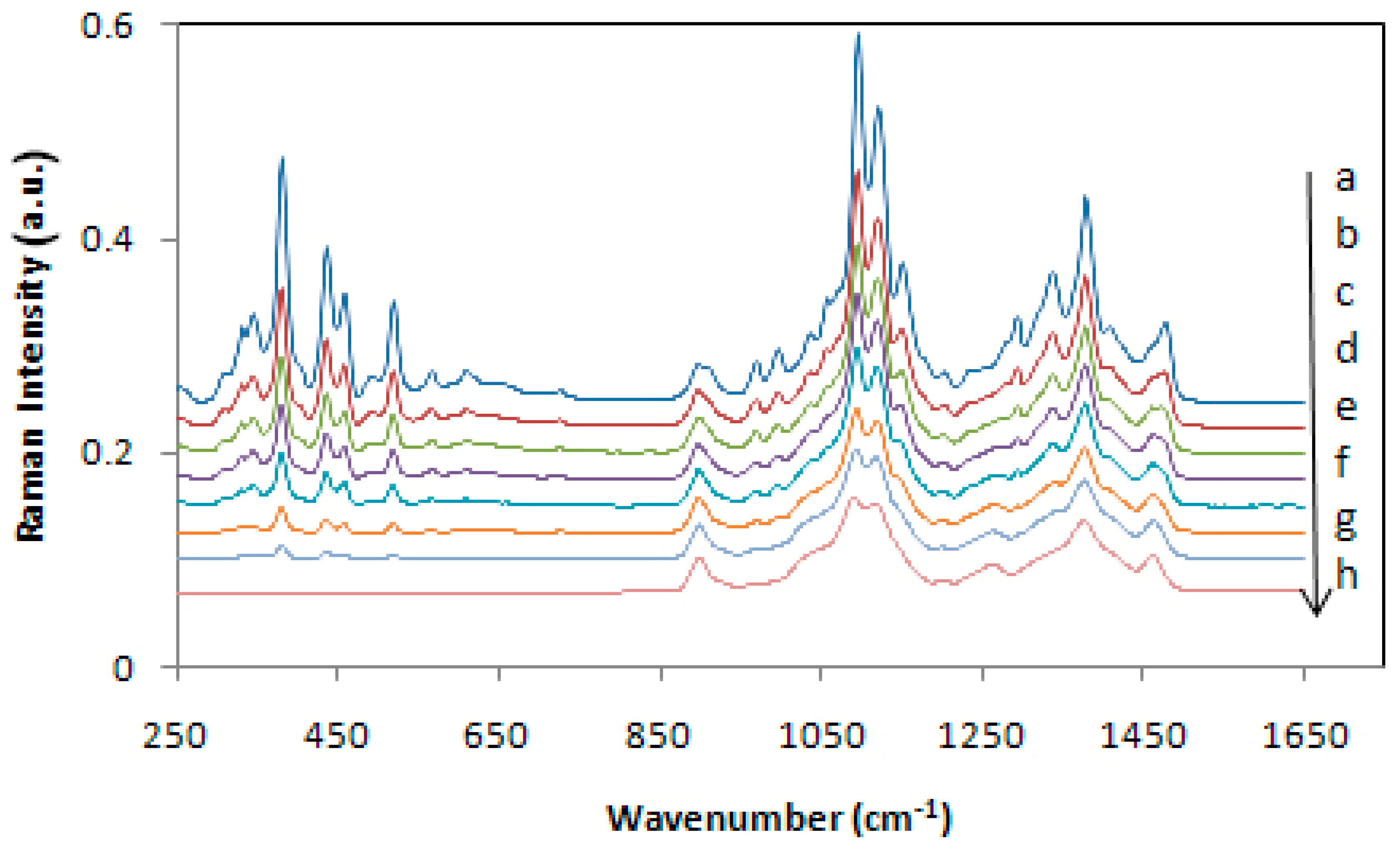{kind=link}
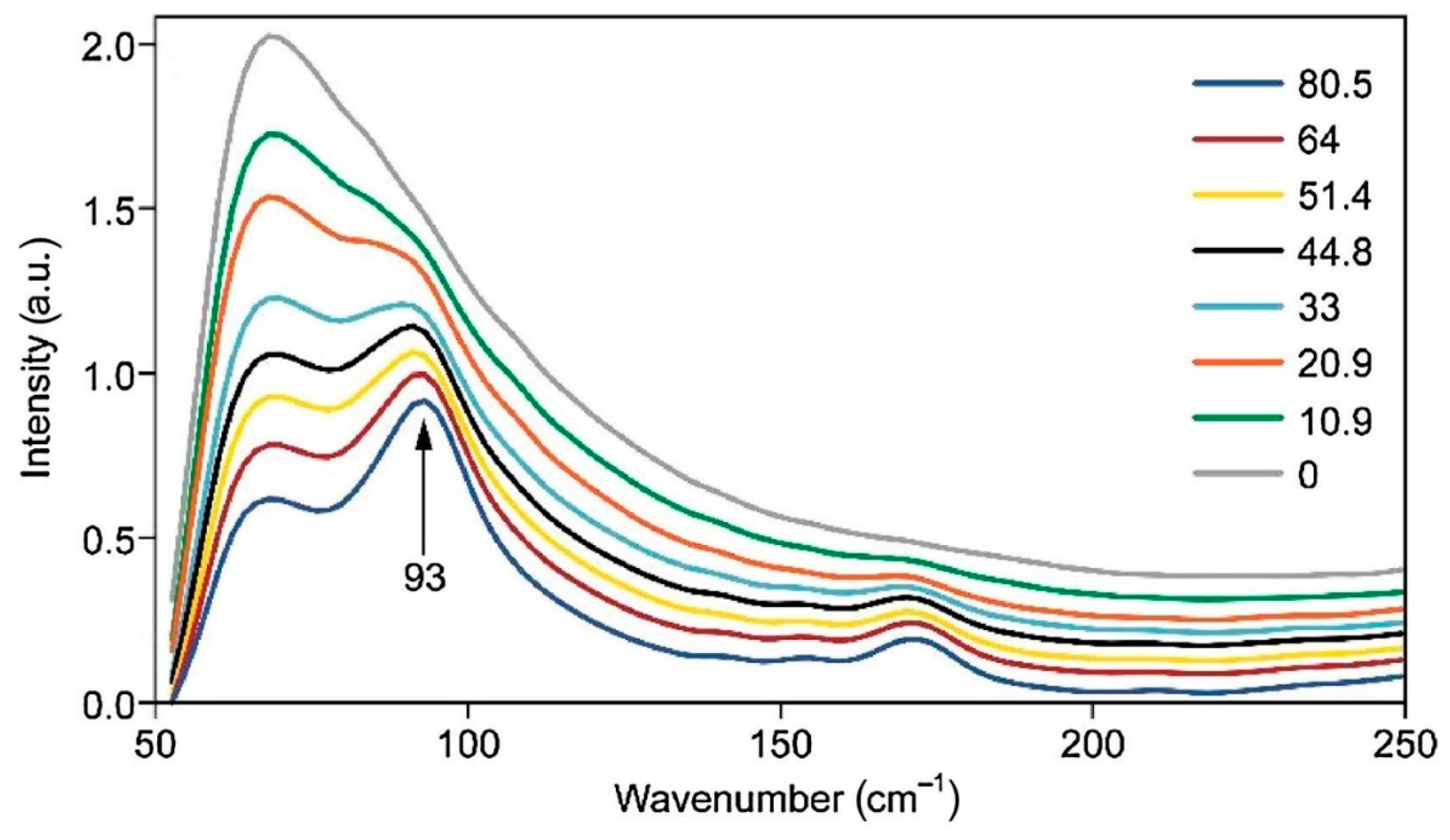{kind=link}
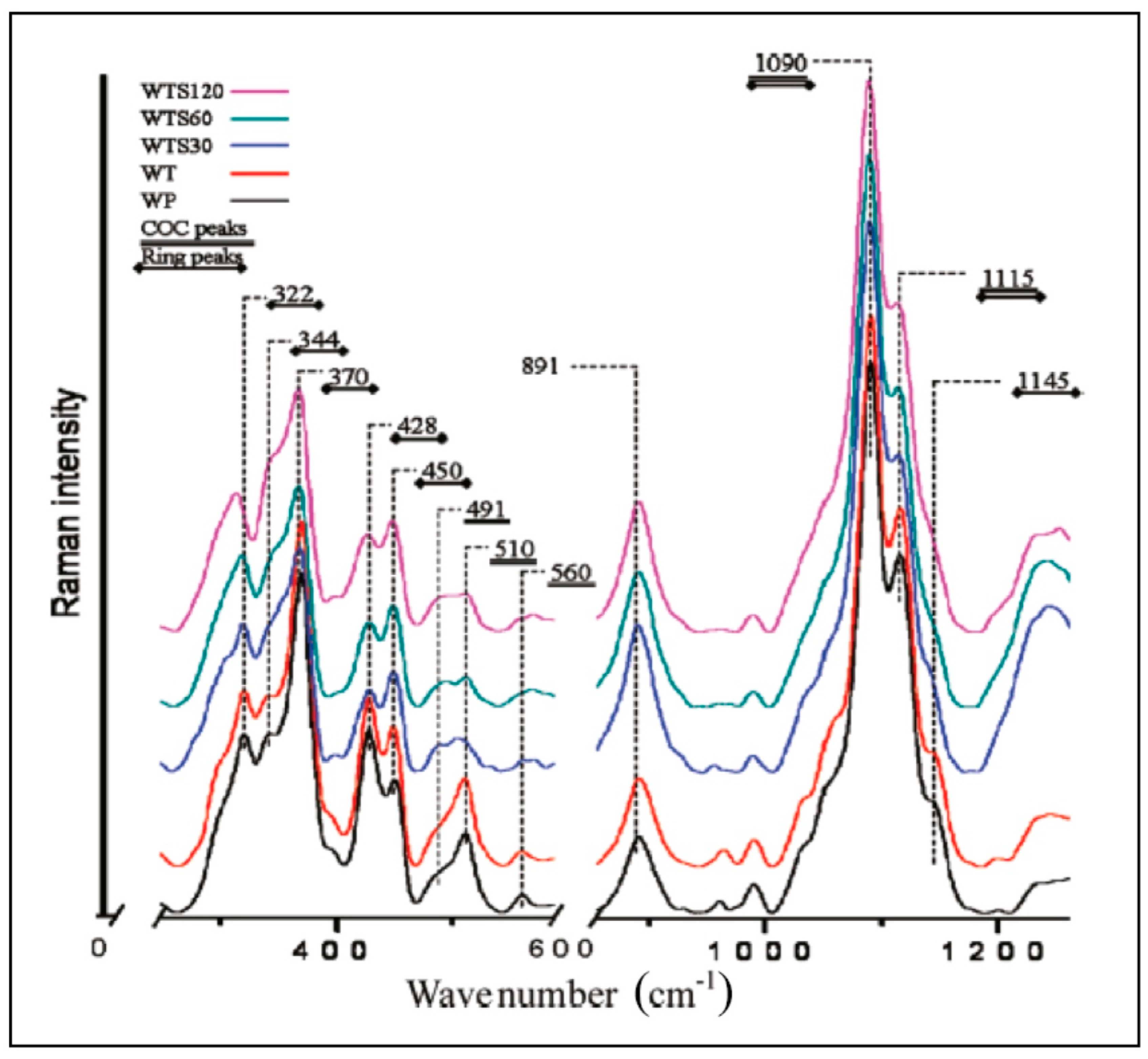{kind=link}
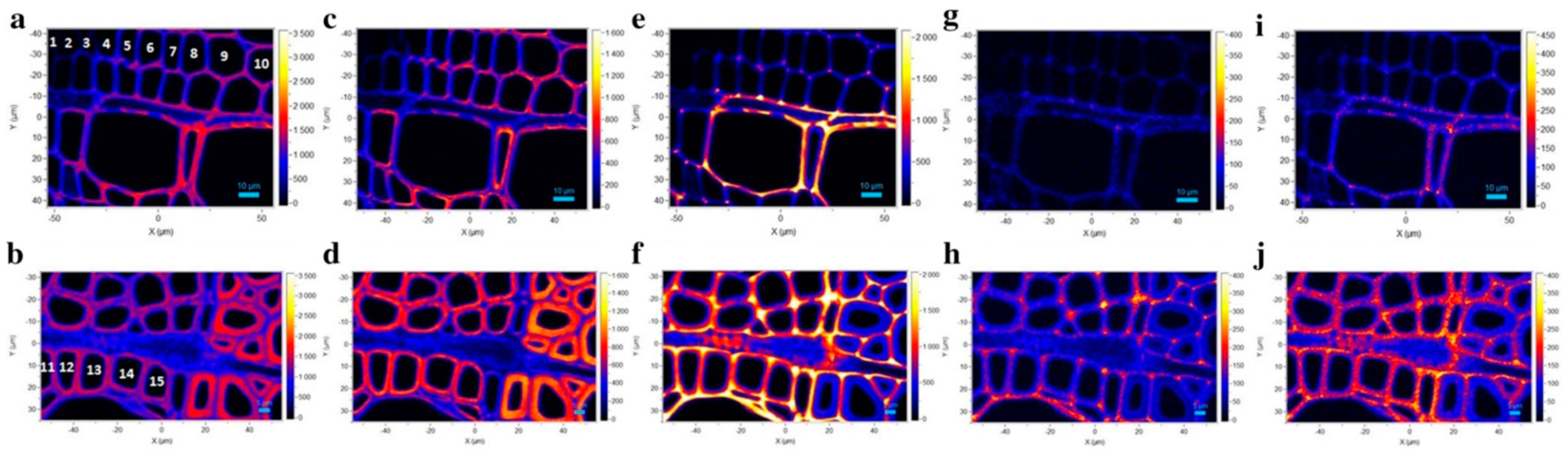{kind=link}
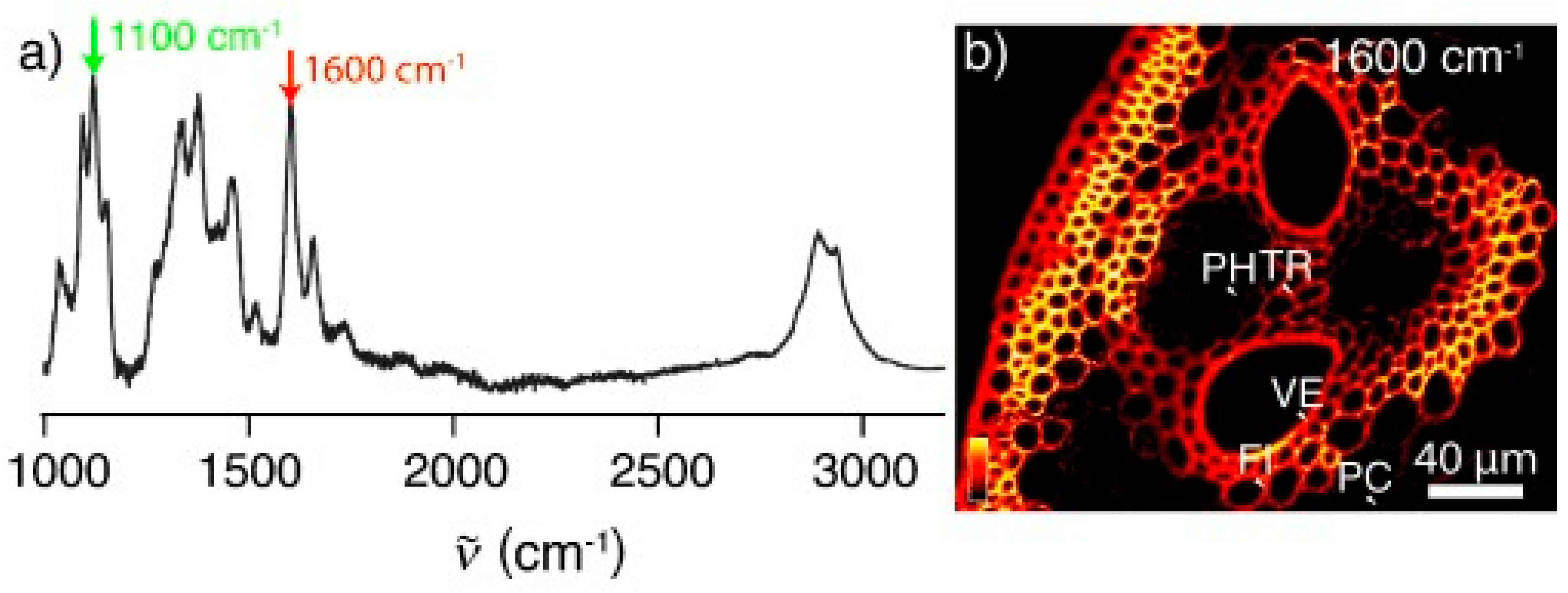{kind=link}
| Untreated | H2O2 bl. (% Δ Int.) | Hydro. (% Δ Int.) | Acet. (% Δ Int.) | Methy. (% Δ Int.) |
|---|---|---|---|---|
| 3071 m a | 3070 (sc b) | 3065 (sc) | 3073 (na) | 3071 (na) |
| 3008 sh | 3008 (sc b) | 3002 (sc) | 3010 (nc) | 3008 (nc) |
| 2940 m | 2940 (na c) | 2937 (na) | 2939 (–36) | 2940 (–307) |
| 2890 sh | 2883 (sc b) | 2886 (sc) | - f (100) | 2890 (–298) |
| 2845 m | 2846 (nc d) | 2843 (nc) | 2846 (sc) | 2845 (nc) |
| 1662 s | 1656 e (86) | 1650 e (100) | 1674 e (53) | 1662 (94) |
| 1621 sh | - f (100) | - f (100) | 1622 (61) | 1621 (100) |
| 1597 vs | 1602 (67) | 1605 e (73) | 1600 d (59) | 1597 (65) |
| 1508 vw | 1508 | - f | 1508 | 1508 |
| 1453 m | 1453 (35) | 1453 (51) | 1454 (49) | 1453 (–111) |
| 1430 w | 1429 | 1435 e | 1426 | 1430 |
| 1392 sh | - f | - f | 1391 | 1392 |
| 1363 sh | 1360 | 1351 e | 1367 | 1363 |
| 1334 m | 1334 (69) | 1334 (57) | 1335 (52) | 1334 (50) |
| 1298 sh | - f | - f | 1300 | 1298 |
| 1272 m | 1271 (79) | 1273 (64) | 1274 (58) | 1272 (58) |
| 1226 vw | 1222 | 1224 | 1222 | 1226 |
| 1192 w | 1193 (33) | 1190 (50) | 1195 (36) | 1192 (38) |
| 1136 m | 1135 (88) | - f (92) | 1129 d,e (58) | 1136 (78) |
| 1089 w | 1079 d,e | - f | 1089 | 1089 |
| 1033 w | 1032 (35) | 1032 (47) | 1034 (54) | 1033 (–84) |
| 975 vw | - f | - f | 972 | 975 |
| 928 vw | 927 | 929 | 936 d,e | 928 |
| 895 vw | 884 d,e | 895 | 907 d,e | 895 |
| 811 sh | - f | 811 | - f | 811 |
| 787 w | 781 e (46) | 789 (34) | 792 e (70) | 787 (54) |
| 731 w | 731 (41) | 731 (14) | 730 (44) | 731 (–67) |
| 637 vw | 638 | 641 | 641 | 637 |
| 588 vw | - f | - f | 607 e | 588 |
| 557 vw | 562 e | 561 | 548 e | 557 |
| 534 vw | 535 | - f | 531 | 534 |
| 491 vw | 484 e vw | - f | 490 | 491 |
| 457 vw | 458 | 465 e | 461 | 457 |
| 361 w | 368 e (42) | 370 e (51) | 366 (60) | 361 e (73) |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, U.P. Analysis of Cellulose and Lignocellulose Materials by Raman Spectroscopy: A Review of the Current Status. Molecules 2019, 24, 1659. https://doi.org/10.3390/molecules24091659
Agarwal UP. Analysis of Cellulose and Lignocellulose Materials by Raman Spectroscopy: A Review of the Current Status. Molecules. 2019; 24(9):1659. https://doi.org/10.3390/molecules24091659
Chicago/Turabian StyleAgarwal, Umesh P. 2019. "Analysis of Cellulose and Lignocellulose Materials by Raman Spectroscopy: A Review of the Current Status" Molecules 24, no. 9: 1659. https://doi.org/10.3390/molecules24091659
APA StyleAgarwal, U. P. (2019). Analysis of Cellulose and Lignocellulose Materials by Raman Spectroscopy: A Review of the Current Status. Molecules, 24(9), 1659. https://doi.org/10.3390/molecules24091659