
Design and Synthesis of Flavonoidal Ethers and Their Anti-Cancer Activity In Vitro

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Instruments and Materials
2.2. General Procedure for the Synthesis of Flavonoidal Ether Derivatives
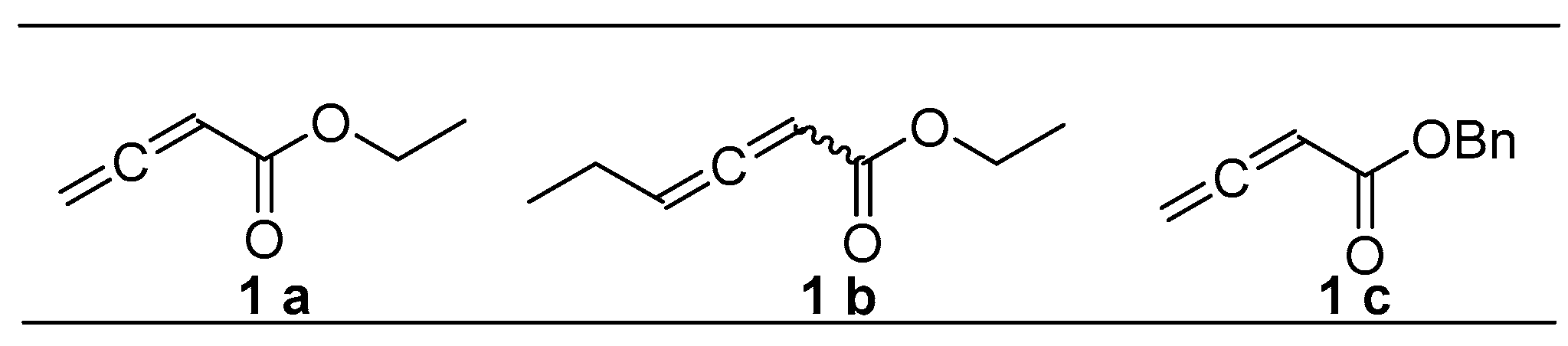
2.2.1. Synthesis of Allenes
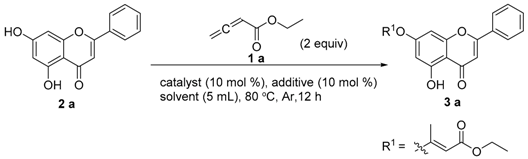
2.2.2. Optimization of Reaction Conditions
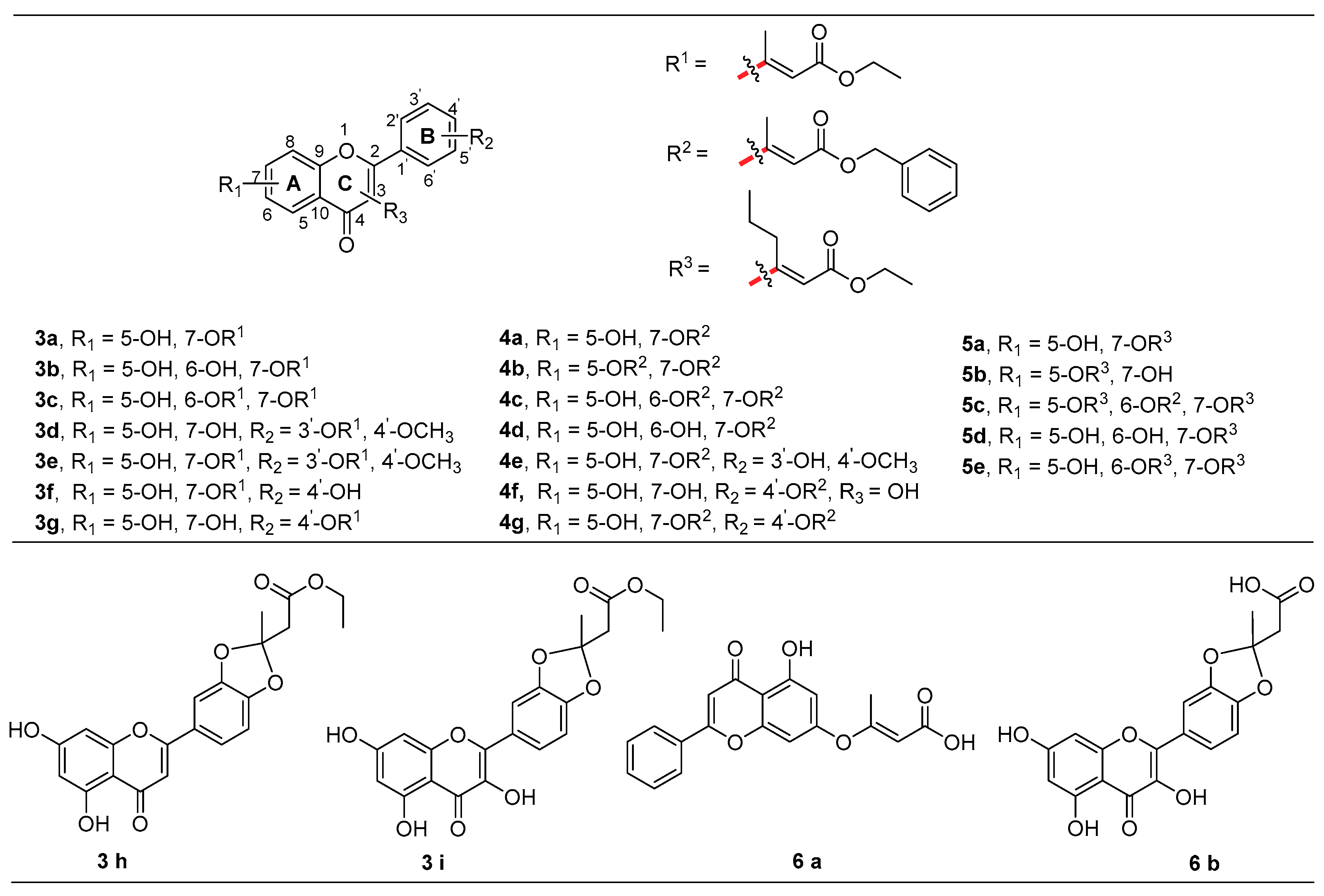
2.2.3. General Procedure for the Synthesis of Target Derivatives 3a–3i, 4a–4g, 5a–5e
2.2.4. General Synthetic Procedure for 6a–6b Target Derivatives
2.3. Anti-Cancer Activity Assay
2.3.1. Cell Lines and Cell Culture
2.3.2. Cytotoxic Activity Assay
2.3.3. Statistical Analysis
3. Results and Discussion
3.1. Synthesis
3.2. Anti-Cancer Activity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, X.; Liang, J.P.; Hao, B.C. Study on molecular modification and structure-activity relationship of flavonoids. J. Anhui Arg. Sci. 2015, 43, 57–61. [Google Scholar]
- Dhruti, B.; Ronak, S.; Gaurav, K.; Dashora, A. Synthesis and pharmacological activities of flavones: A review. Indo Am. J. Pharm. Res. 2016, 6, 4345–4363. [Google Scholar]
- Craig, W.J. Health-promoting properties of common herbs. Am. J. Clin. Nutr. 1999, 70, 491S–499S. [Google Scholar] [CrossRef] [PubMed]
- Kadarian, C.; Broussalis, A.M.; Mino, J.; Lopez, P.; Gorzalczany, S.; Ferraro, G.; Acevedo, C. Hepatoprotective activity of achyrocline satureioides (Lam) D. C. Pharmacol. Res. 2002, 45, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.E.; Slowing, K.; Carretero, E.; Sánchez Mata, D.; Villar, A. Lippia: Traditional uses, chemistry and pharmacology: A review. J. Ethnopharmacol. 2001, 76, 201–214. [Google Scholar] [CrossRef]
- Samuelsen, A.B. The traditional uses, chemical constituents and biological activities of Plantago major L. A review. J. Ethnopharmacol. 2000, 71, 1–21. [Google Scholar] [CrossRef]
- Tang, L.; Feng, Q.; Zhao, J.; Dong, L.; Liu, W.; Yang, C.; Liu, Z. Involvenment of UDP-glucuronosyltranferases and sulfotransferases in the liver and intestinal first- pass metabolism of seven flavones in C57 mice and humans in vitro. Food Chem. Toxicol. 2012, 50, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.M. Some typical advances in the synthetic applications of allenes. Chem. Rev. 2005, 105, 2829–2872. [Google Scholar] [CrossRef] [PubMed]
- Brasholz, M.; Reissig, H.U.; Zimmer, R. Sugars, alkaloids, and heteroaromatics: Exploring heterocyclic chemistry with alkoxyallenes. Acc. Chem. Res. 2009, 42, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pfrengle, F.; Reissig, H.U. Amino sugars and their mimetics Via 1,2-oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.; Ma, S.M. Allenes in catalytic asymmetric synthesis and natural product syntheses. Angew. Chem. Int. Ed. 2012, 51, 3074–3112. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.T.; Ma, S.M. Palladium-catalyzed cyclization reactions of allenes in the presence of unsaturated carbon-carbon bonds. Acc. Chem. Res. 2014, 47, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. Cyclization reactions of bis(allenes) for the synthesis of polycarbo(hetero)cycles. Chem. Soc. Rev. 2014, 43, 3106–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neff, R.K.; Frantz, D.E. Recent applications of chiral allenes in axial-to-central chirality transfer reactions. Tetrahedron 2015, 71, 7–18. [Google Scholar] [CrossRef]
- Jose, L.M.; Lvan, V.; Fernando, L. Allenes and derivatives in gold(I)- and platinum(II)-catalyzed formal cycloadditions. Acc. Chem. Res. 2019, 52, 465–479. [Google Scholar]
- Egle, M.B.; Gianluigi, B.; Michael, S.C.; Giofrè, S. Chapter one—Transition metal- catalyzed intramolecular amination and hydroammination reactions of allenes. Adv. Organomet. Chem. 2018, 69, 1–71. [Google Scholar]
- Campbell, K.A.; House, H.O.; Surber, B.W.; Trahanovsky, W.S. Enones with strained double bonds. 10. use of flash vacuum pyrolysis to obtain bicyclo[3.3.1]non-1-en-3-one. J. Org. Chem. 1987, 52, 2474–2481. [Google Scholar] [CrossRef]
- Rout, L.; Harned, A.M. Allene carboxylates as dipolarophiles in Rh-catalyzed carbonyl ylide cycloadditions. Chem. Eur. J. 2009, 15, 12926–12928. [Google Scholar] [CrossRef] [PubMed]
- Li, R.D.; Leng, P.L.; Liu, B.; Wang, X.; Ge, Z.; Li, R. Efficient and regioselective one-pot synthesis of S-vinyl dithiocarbamates from electron-deficient allenes, amines and CS2. Tetrahedron 2016, 72, 5707–5712. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Ligand | Solvent | Yield (%) | Entry | Catalyst | Base | Ligand | Solvent | Yield (%) |
| 1 | Pd(dba)2 | K2CO3 | PPh3 | DMA [a] | <5 | 10 | Pd(dba)2 | Cs2CO3 | PPh3 | DMF | 18.4 |
| 2 | Pd(dba)2 | K2CO3 | PPh3 | EtOH | 13.3 | 11 | PdCl2 | K2CO3 | - | DMF | 15.8 |
| 3 | Pd(dba)2 | K2CO3 | PPh3 | MeCN | 41.7 | 12 | Pd(OAc)2 | K2CO3 | - | DMF | 24.1 |
| 4 | Pd(dba)2 | K2CO3 | PPh3 | DCM | <5 | 13 | PdCl2(PPh3)2 | K2CO3 | - | DMF | 13.3 |
| 5 | Pd(dba)2 | K2CO3 | PPh3 | DMA [b] | 32.0 | 14 | - | K2CO3 | - | DMF | 14.9 |
| 6 | Pd(dba)2 | K2CO3 | PPh3 | DMF | 43.0 | 15 | Pd(OAc)2 | - | - | DMF | NO. |
| 7 | Pd(dba)2 | NaH | PPh3 | DMF | 14.2 | 16 | Pd(dba)2 | - | - | DMF | NO. |
| 8 | Pd(dba)2 | NH(iPr)2 | PPh3 | DMF | <5 | 17 | Pd(dba)2 | K2CO3 | PPh3 | DMF | 11.2[c] |
| 9 | Pd(dba)2 | Et3N | PPh3 | DMF | 17.2 | - | - | - | - | - | - |
| Compd | Inhibition Rate % (5 µM) | |||
|---|---|---|---|---|
| K562 | A549 | HEL | PC3 | |
| 3a | 34.1 ± 4.3 | −23.3 ± 2.1 | −2.5 ± 3.2 | 7.1 ± 3.2 |
| 3b | 28.4 ± 4.9 | −14.9 ± 2.8 | 27.5 ± 4.1 | 1.5 ± 3.4 |
| 3c | 23.1 ± 3.2 | 3.8 ± 1.7 | 21.2 ± 2.3 | 16.1 ± 1.2 |
| 3d | 14.0 ± 1.7 | −39.6 ± 2.9 | 2.7 ± 1.9 | 36.6 ± 3.6 |
| 3e | −45.0 ± 1.9 | −20.5 ± 2.5 | 10.5 ± 2.9 | 51.7 ± 3.9 ** |
| 3f | −13.7 ± 2.2 | 19.7 ± 2.6 | −24.3 ± 2.4 | 40.4 ± 2.7 |
| 3g | 26.2 ± 3.1 | 16.4 ± 2.4 | 24.9 ± 2.3 | 38.2 ± 2.5 |
| 3h | 16.3 ± 3.4 | −16.7 ± 2.3 | 58.6 ± 3.9 ** | −15.9 ± 2.6 |
| 3i | 26.2 ± 3.5 | −60.4 ± 2.7 | 24.9 ± 3.8 | 42.2 ± 2.1 |
| 4a | −33.6 ± 2.3 | −36.9±2.0 | 24.6 ± 2.9 | 8.9 ± 2.6 |
| 4b | −5.0 ± 1.7 | −2.7 ± 1.2 | −27.1 ± 2.7 | 56.2 ± 4.0 ** |
| 4c | −22.8 ± 2.9 | −6.6 ± 1.4 | 16.2 ± 2.6 | 51.0 ± 4.7 ** |
| 4d | 17.3 ± 1.3 | −1.9 ± 4.3 | 18.1 ± 2.5 | 28.4 ± 2.4 |
| 4e | 61.9 ± 4.2 ** | −3.7 ± 4.3 | 49.5 ± 2.4 | 21.9 ± 2.1 |
| 4f | −22.6 ± 2.9 | −17.5 ± 4.3 | 10.1 ± 2.1 | 28.8 ± 2.0 |
| 4g | −23.3 ± 2.5 | −14.5 ± 1.7 | 2.7 ± 2.3 | 36.6 ± 1.9 |
| 5a | −8.4 ± 1.2 | 19.5 ± 1.9 | 24.6 ± 2.8 | 21.9± 1.2 |
| 5b | 18.7 ± 3.5 | 14.0 ± 2.5 | 11.4 ± 1.9 | 32.9± 0.9 |
| 5c | 19.3 ± 3.1 | 15.9 ± 2.7 | 21.3 ± 3.0 | 31.8 ± 1.2 |
| 5d | −10.6 ± 3.0 | 17.5 ± 2.2 | 18.7 ± 3.2 | 30.6± 1.8 |
| 5e | −13.4 ± 4.2 | 21.8 ± 2.1 | 49.5 ± 2.0 ** | 39.5 ± 1.7 |
| 6a | 4.3 ± 1.6 | 11.7 ± 0.9 | 28.3 ± 2.2 | 41.1 ± 1.9 |
| 6b | 35.7 ± 1.7 | 38.2 ± 3.5 | 44.7 ± 1.7 ** | 35.2 ± 2.6 |
| Chrysin | 51.0 ± 4.1 | 37.5 ± 1.8 | 21.9 ± 1.3 | 39.2 ± 2.8 |
| Baicalein | 17.7 ± 2.2 | 29.3 ± 2.1 | 19.2 ± 3.5 | 28.3±1.8 |
| Quercetin | −5.0 ± 3.6 | 8.2 ± 3.0 | 10.2 ± 3.4 | 11.3 ± 1.9 |
| Diosmetin | 10.9 ± 1.8 | 7.3 ± 1.2 | 9.2 ± 3.0 | 9.5 ± 2.1 |
| Apigenin | −1.5 ± 1.9 | 5.6 ± 1.1 | 6.4 ± 2.1 | 8.3 ± 2.5 |
| Luteolin | 19.1 ± 1.7 | 14.7 ± 1.4 | 20.4 ± 1.7 | 16.5 ± 1.7 |
| Kaempferol | −25.3 ± 2.1 | 8.6 ± 1.3 | −24.0 ± 2.4 | 14.3 ± 2.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, L.; Wang, M.-L.; Lv, Y.; Zeng, X.-Y.; Chen, C.; Ren, H.; Luo, H.; Pan, W.-D. Design and Synthesis of Flavonoidal Ethers and Their Anti-Cancer Activity In Vitro. Molecules 2019, 24, 1749. https://doi.org/10.3390/molecules24091749
Jin L, Wang M-L, Lv Y, Zeng X-Y, Chen C, Ren H, Luo H, Pan W-D. Design and Synthesis of Flavonoidal Ethers and Their Anti-Cancer Activity In Vitro. Molecules. 2019; 24(9):1749. https://doi.org/10.3390/molecules24091749
Chicago/Turabian StyleJin, Lu, Meng-Ling Wang, Yao Lv, Xue-Yi Zeng, Chao Chen, Hai Ren, Heng Luo, and Wei-Dong Pan. 2019. "Design and Synthesis of Flavonoidal Ethers and Their Anti-Cancer Activity In Vitro" Molecules 24, no. 9: 1749. https://doi.org/10.3390/molecules24091749
APA StyleJin, L., Wang, M.-L., Lv, Y., Zeng, X.-Y., Chen, C., Ren, H., Luo, H., & Pan, W.-D. (2019). Design and Synthesis of Flavonoidal Ethers and Their Anti-Cancer Activity In Vitro. Molecules, 24(9), 1749. https://doi.org/10.3390/molecules24091749