Optical Resolution of Rimantadine

 ,
,
Abstract
:
1. Introduction
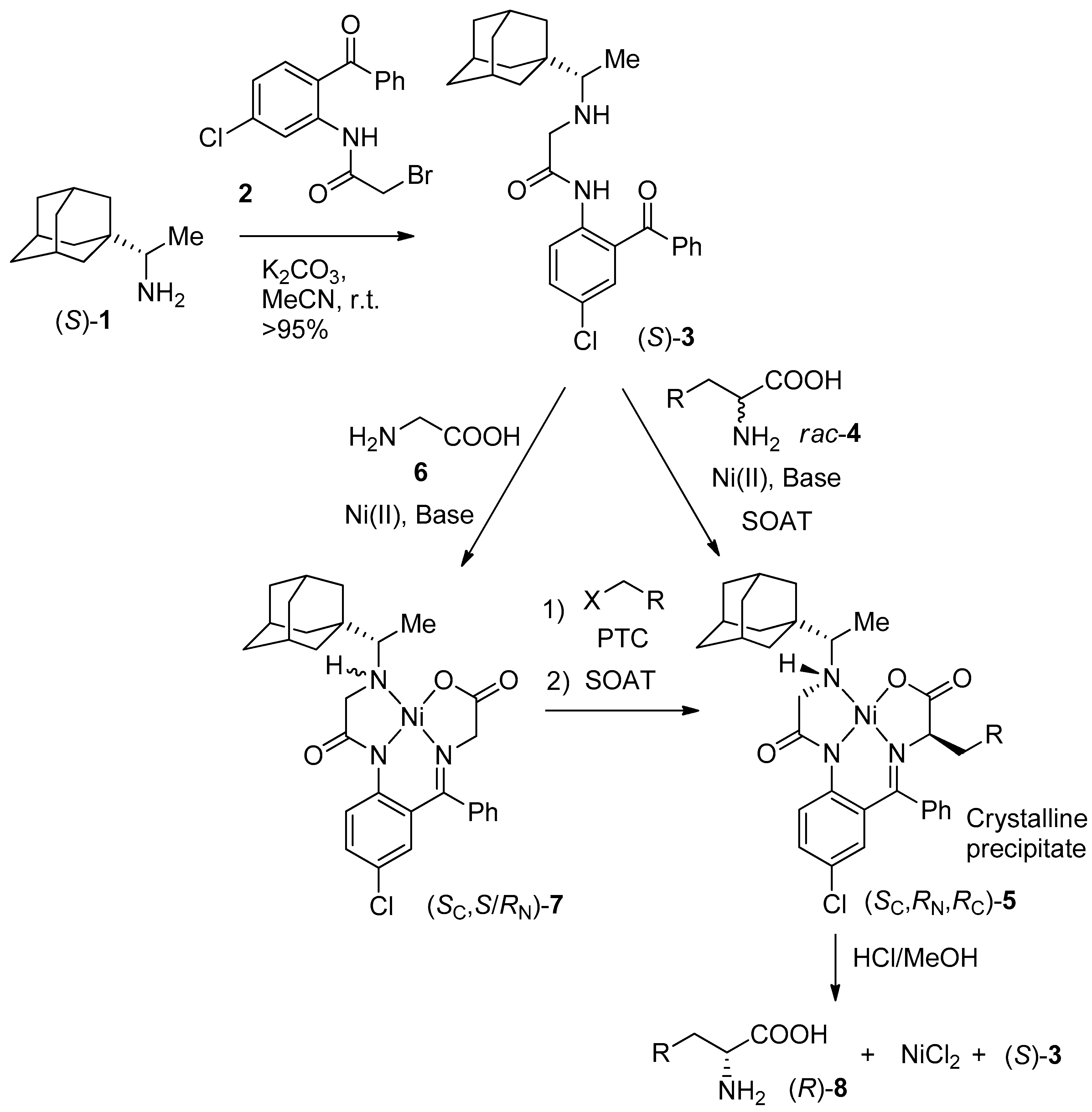
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Transformation of Rimantadine HCl Salt 9 to Free Amine 10
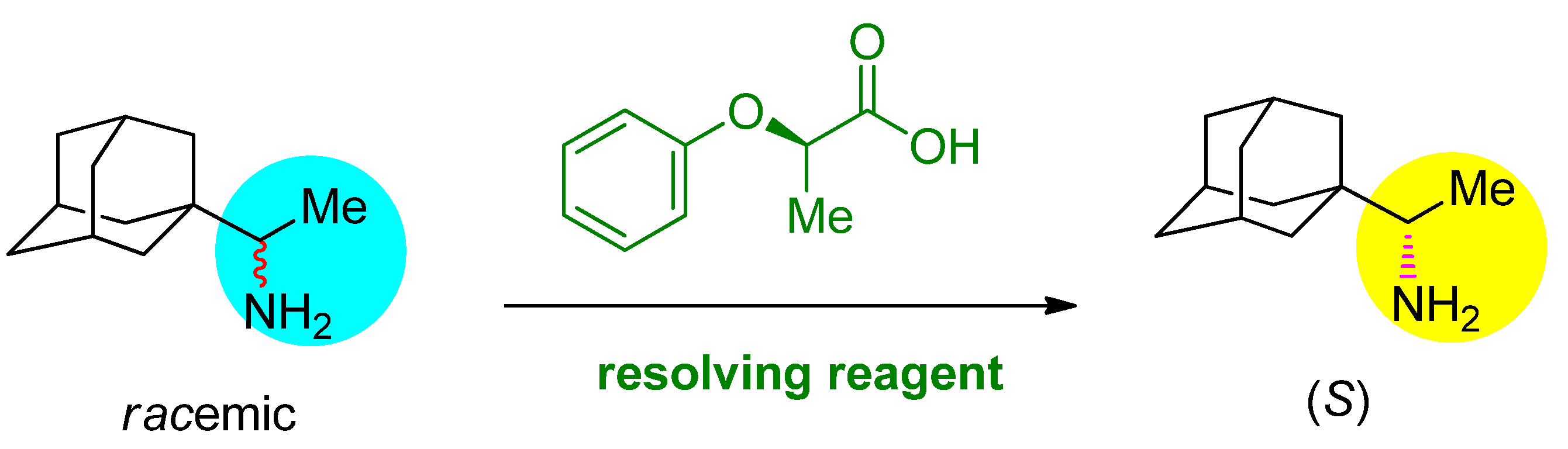
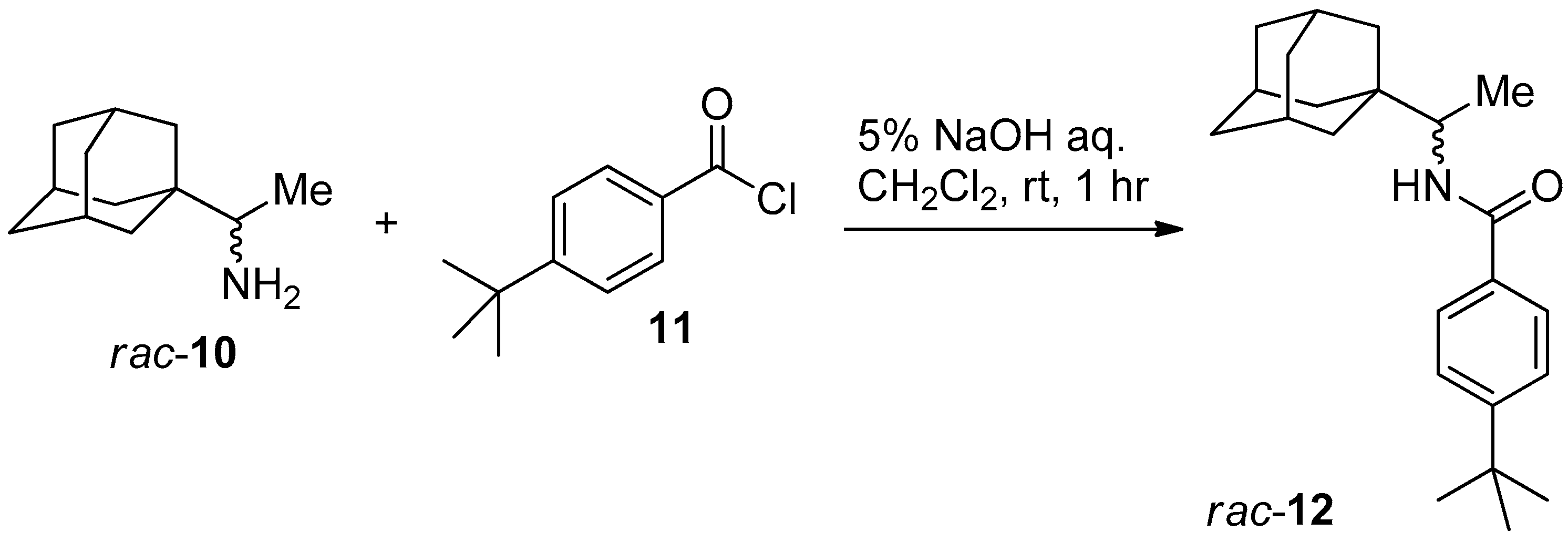
3.3. General Procedure for Resolution
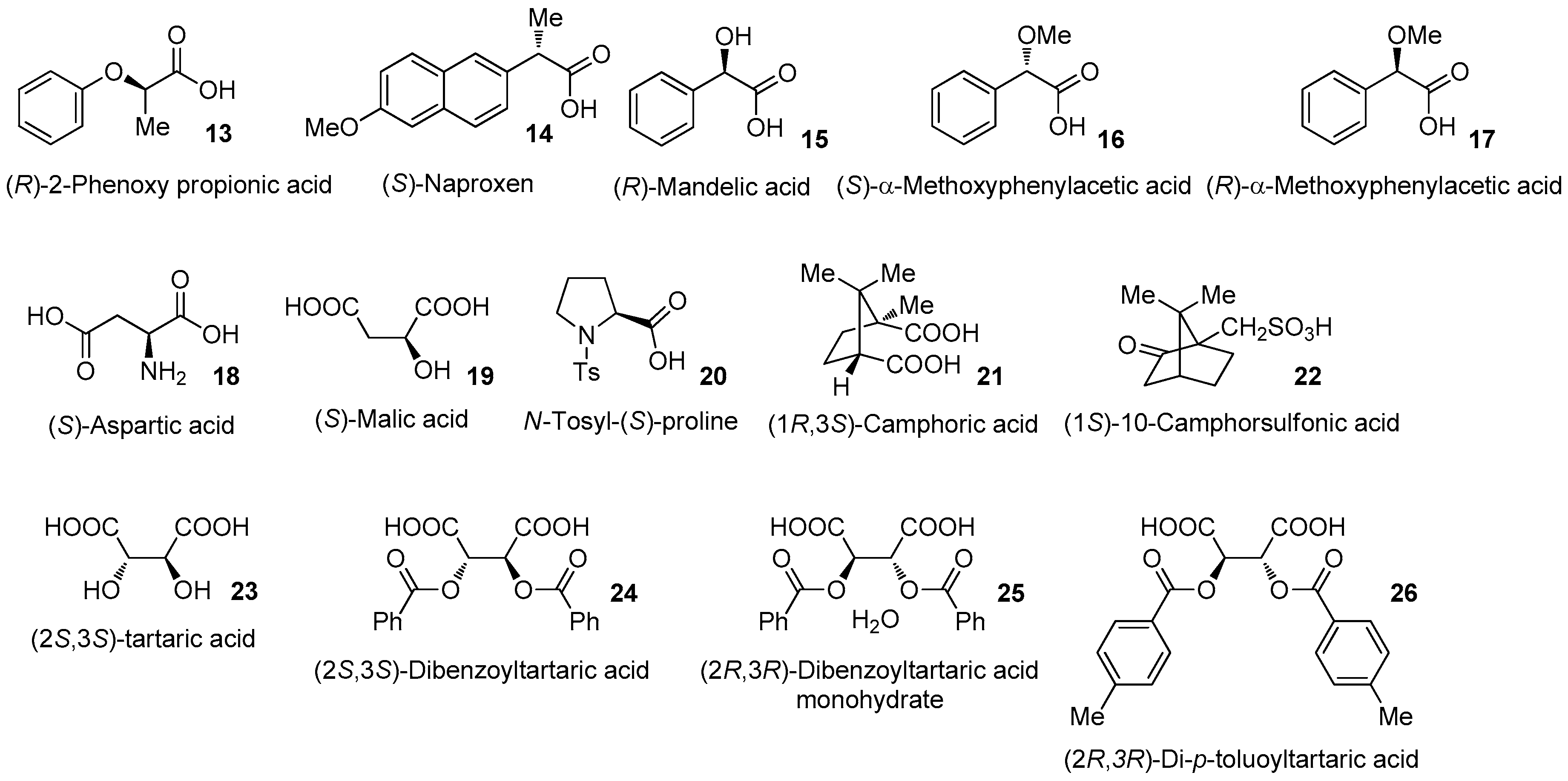
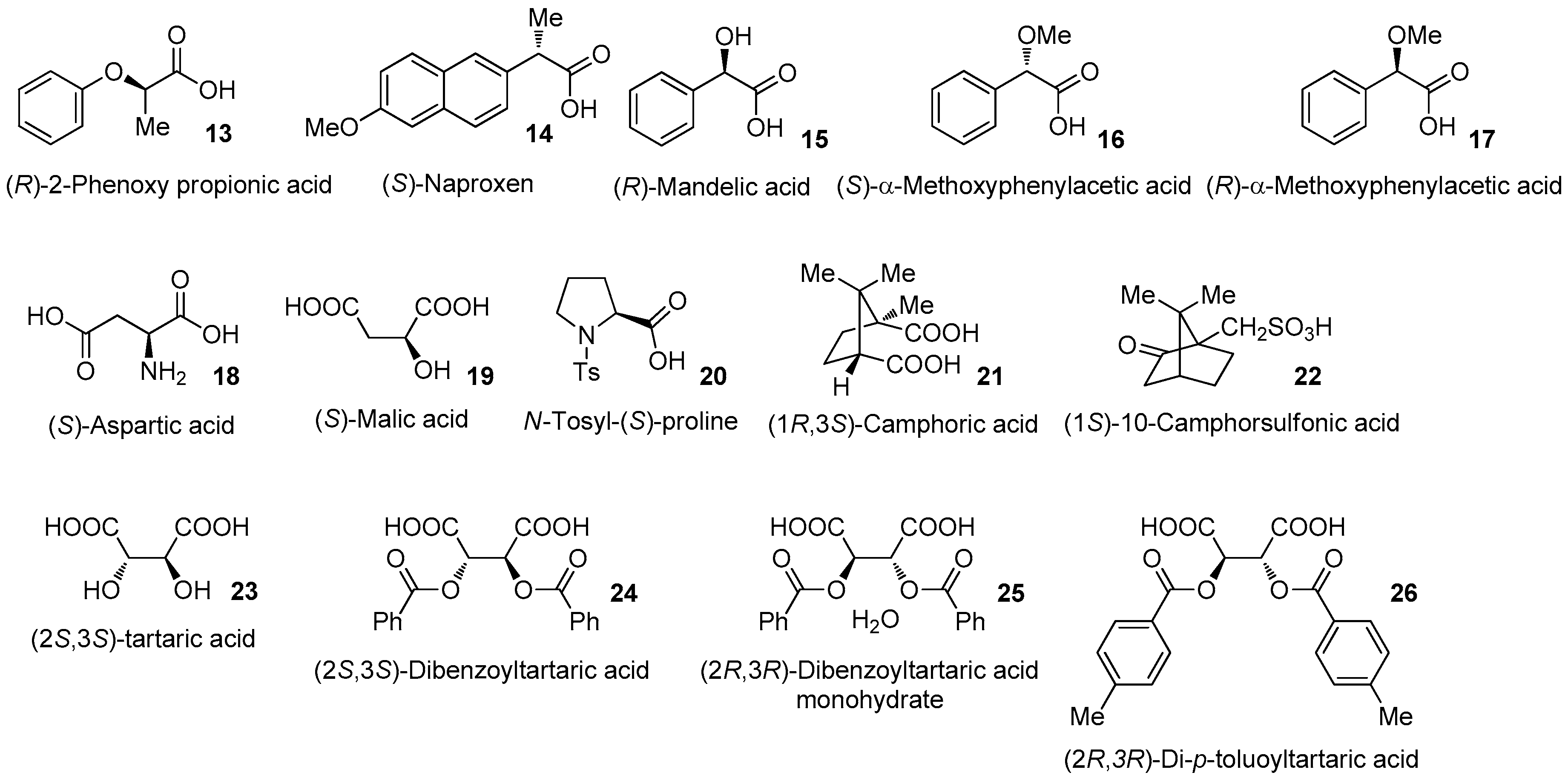
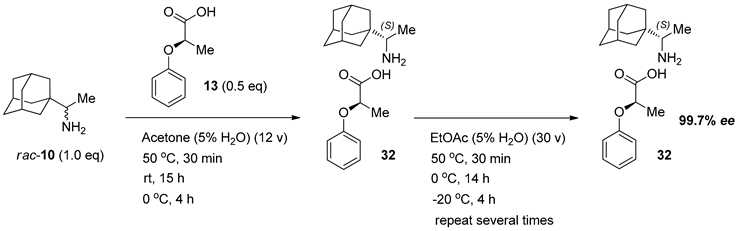
3.3.1. Rimantadine (10) (R)-2-Phenoxy Propionic Acid (13) 0.5 Eq. Salt (99.7% ee)
3.3.2. Rimantadine (10) (S)-Naproxen (14) 0.5 Eq. Salt
3.3.3. Rimantadine (10) (R)-Mandelic Acid (15) 0.5 Eq. Salt
3.3.4. Rimantadine (10) (S)-α-Methoxyphenylacetic Acid (16) 0.5 Eq. Salt
3.3.5. Rimantadine (10) (R)-α-Methoxyphenylacetic Acid (17) 0.5 Eq. Salt
3.3.6. Rimantadine (10) (S)-Aspartic Acid (18) 0.5 Eq. Salt
3.3.7. Rimantadine (10) (S)-Aspartic acid (18) 0.25 Eq. Salt (1:1 Salt)
3.3.8. Rimantadine (10) (S)-Malic Acid (19) 1.0 Eq. Salt
3.3.9. Rimantadine (10) (S)-Malic Acid (19) 0.25 Eq. Salt
3.3.10. Rimantadine (10) N-Tosyl-(S)-proline (20) 0.5 Eq. Salt
3.3.11. Rimantadine (10) (1R,3S)-Camphoric Acid (21) 0.5 Eq. Salt
3.3.12. Rimantadine (10) (1R,3S)-Camphoric Acid (21) 0.25 Eq. Salt
3.3.13. Rimantadine (10) (1S)-10-Camphorsulforic Acid (22) 1.0 Eq. Salt
3.3.14. Rimantadine (10) (2S,3S)-Tartaric Acid (23) 0.5 Eq. Salt
3.3.15. Rimantadine (10) (2S,3S)-Tartaric Acid (23) 0.25 Eq. Salt
3.3.16. Rimantadine (10) (2S,3S)-Dibenzoyltartaric Acid (24) 0.5 Eq. Salt (2:1 Salt)
3.3.17. Rimantadine (10) (2S,3S)-Dibenzoyltartaric Acid (24) 0.25 Eq. Salt
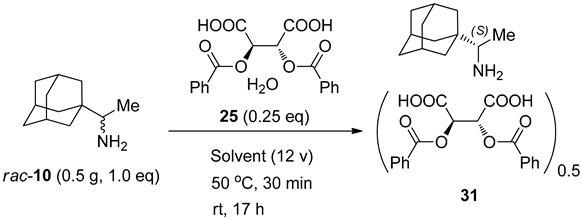
3.3.18. Rimantadine (10) (2R,3R)-Dibenzoyltartaric Acid Monohydrate (25) 0.25 Eq. Salt (94.8% ee)
3.3.19. Rimantadine (10) (2R,3R)-Di-p-toluoyltartaric Acid (26) 1.0 Eq. Salt
3.3.20. Rimantadine (10) (2R,3R)-Di-p-toluoyltartaric Acid (26) 0.25 Eq. Salt
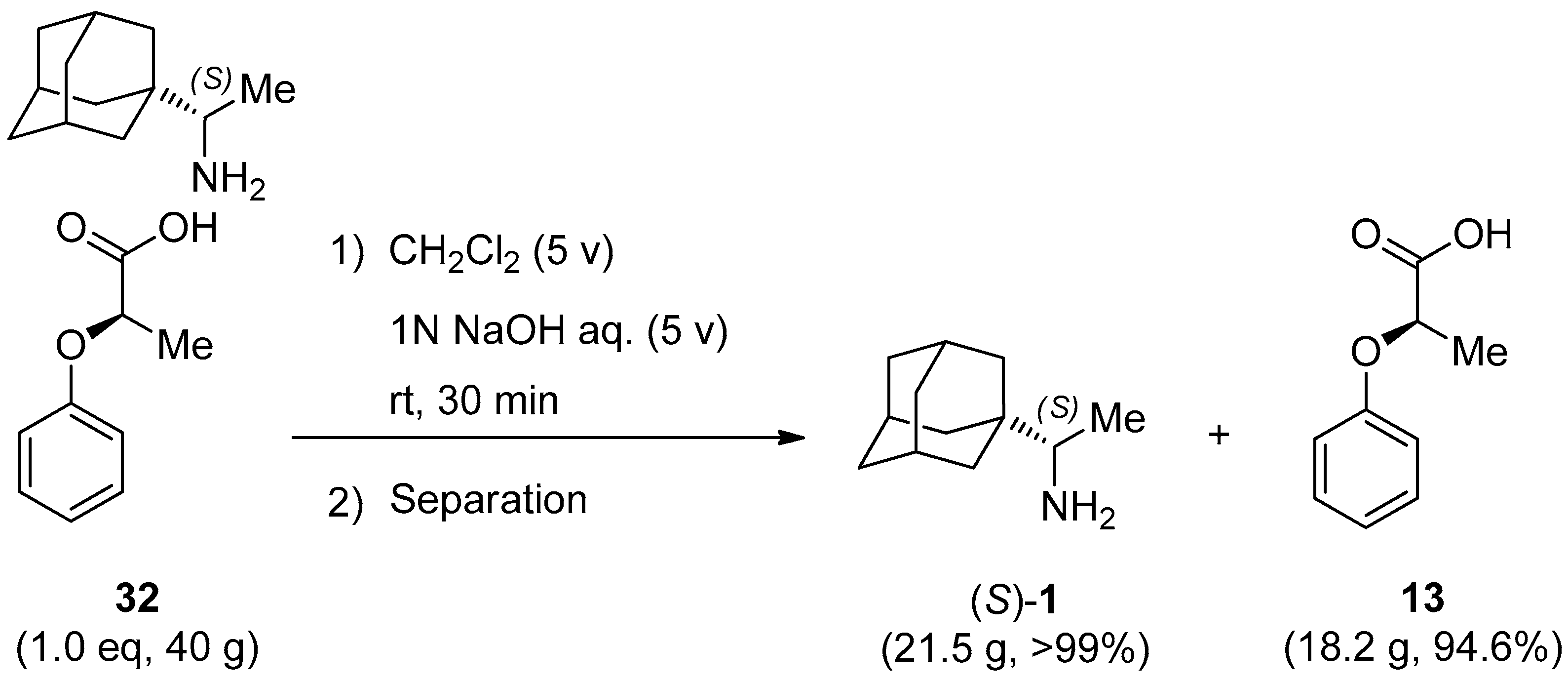
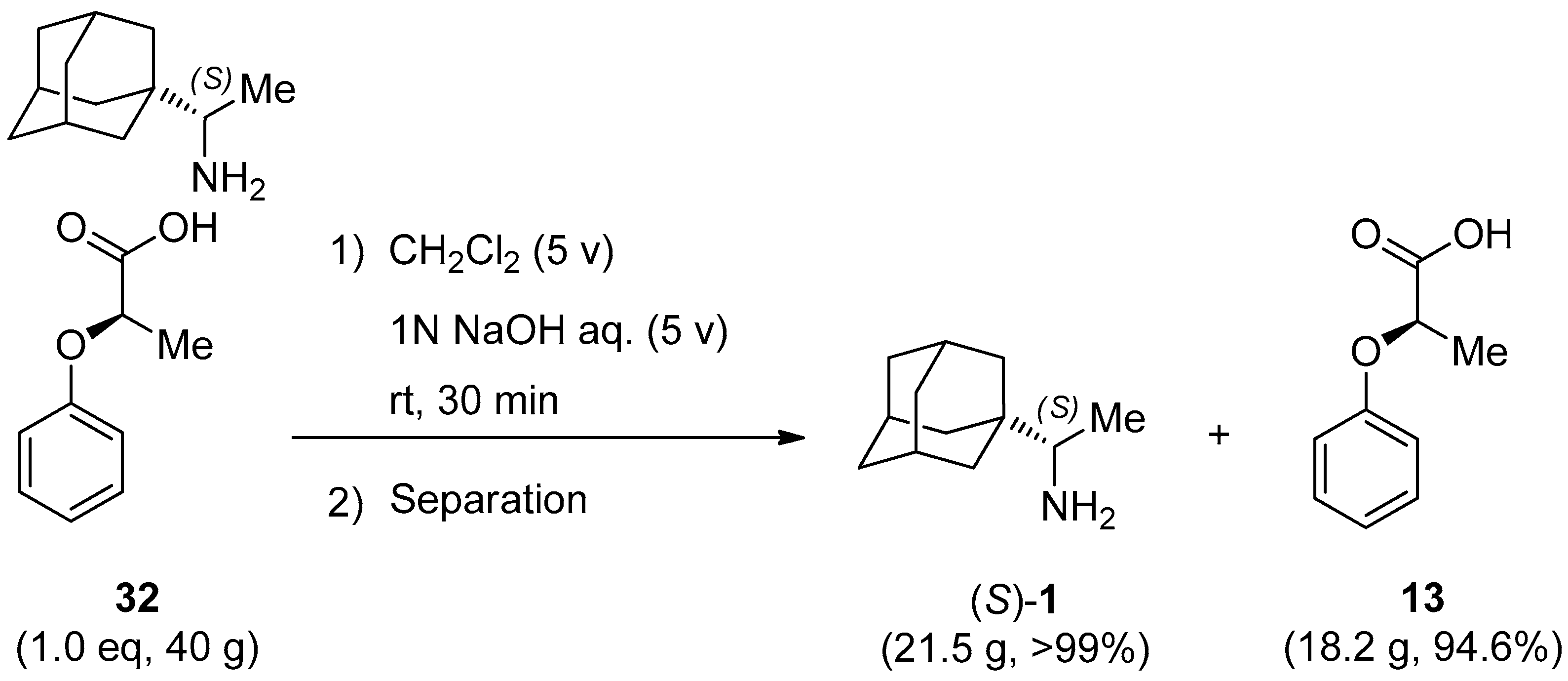
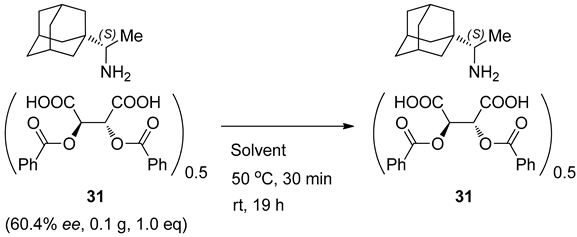
3.4. Isolation of Enantiomerically Pure (99.7% ee) Rimantadine (S)-1 from Salt 32
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- An Efficient Asymmetric Synthesis of (2S,3S)-3-Methyl- and -3-Trifluoromethylpyroglutamic Acids. Tetrahedron 1999, 55, 12031–12044. [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Ma, J.S. Unnatural amino acids in drug discovery. Chim. Oggi 2003, 21, 65–68. [Google Scholar]
- Hodgson, D.R.W.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Izawa, K.; Aceña, J.L.; Liu, H.; Soloshonok, V.A. Tailor-Made α-Amino Acids in the Pharmaceutical Industry: Synthetic Approaches to (1R, 2S)-1-Amino-2-vinylcyclopropane-1-carboxylic Acid (Vinyl-ACCA). Eur. J. Org. Chem. 2016, 2757–2774. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Izawa, K. (Eds.) Asymmetric Synthesis and Application of alpha-Amino Acids; ACS Symposium Series #1009; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Kukhar, V.P.; Sorochinsky, A.E.; Soloshonok, V.A. Practical synthesis of fluorine containing alpha- and beta-amino acids: recipes from Kiev, Ukraine. Future Med. Chem. 2009, 1, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 2319–2344. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, M.J. Dynamic kinetic resolution of amines and amino acids by enzyme–metal cocatalysis. ChemCatChem 2011, 3, 271–277. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of fluorinated beta-amino acids. Synthesis 2011, 3045–3079. [Google Scholar]
- Wang, J.; Zhang, L.; Jiang, H.; Chen, K.; Liu, H. Application of Nickel (II) Complexes to the Efficient Synthesis of α- or β-Amino Acids. Chimia 2011, 65, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Popkov, A.; De Spiegeleer, B. Chiral nickel (II) complexes in the preparation of 11 C-and 18 F-labelled enantiomerically pure α-amino acids. Dalton Trans. 2012, 41, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- So, S.M.; Kim, H.; Mui, L.; Chin, J. Mimicking Nature to Make Unnatural Amino Acids and Chiral Diamines. Eur. J. Org. Chem. 2012, 2012, 229–241. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Recent Advances in the Asymmetric Synthesis of α-(Trifluoromethyl)-Containing α-Amino Acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Cerioli, L.; Servi, S.; Viani, F.; Tessaroa, D. Synergy between catalysts: enzymes and bases. DKR of non-natural amino acids derivatives. Catal. Sci. Technol. 2012, 2, 1606–1616. [Google Scholar]
- D’Arrigo, P.; Cerioli, L.; Fiorati, A.; Servi, S.; Viani, F.; Tessaro, D. Naphthyl-L-α-amino acids via chemo-enzymatic dynamic kinetic resolution. Tetrahedron Asymmetry 2012, 23, 938–944. [Google Scholar]
- Periasamy, M.; Gurubrahamam, R.; Sanjeevakumar, N.; Dalai, M.; Alakonda, L.; Reddy, P.O.; Suresh, S.; Satishkumar, S.S.; Padmaja, M.; Reddy, M.N. Convenient methods for the synthesis of chiral amino alcohols and amines. Chimia 2013, 67, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine-containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluorine Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N.N. Asymmetric synthesis of quaternary α-amino acids and their phosphonate analogues. Asian J. Org. Chem. 2014, 3, 1234–1260. [Google Scholar]
- Metz, A.E.; Kozlowski, M.C. Recent advances in asymmetric catalytic methods for the formation of acyclic α, α-disubstituted α-amino acids. J. Org. Chem. 2015, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wang, B.; Nack, W.A.; Chen, G. Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C–H Functionalization. Acc. Chem. Res. 2016, 49, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic Transamination of α-Keto Perfluorocarboxylic Esters. An Efficient Preparative Synthesis of β,β,β-Trifluoroalanine. Tetrahedron 1997, 53, 8307–8314. [Google Scholar]
- Soloshonok, V.A.; Kirilenko, A.G.; Kukhar, V.P.; Resnati, G. Transamination of Fluorinated β-Keto Carboxylic Esters. A Biomimetic Approach to β-Polyfluoroalkyl-β-Amino Acids. Tetrahedron Lett. 1993, 34, 3621–3624. [Google Scholar]
- Shibata, N.; Nishimine, T.; Shibata, N.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Sorochinsky, A.E.; Soloshonok, V.A. Organic base-catalyzed stereodivergent synthesis of (R)- and (S)-3-amino-4,4,4-trifluorobutanoic acids. Chem. Commun. 2012, 48, 4124–4126. [Google Scholar] [CrossRef]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Belokon, Y.N.; Kuzmina, N.A.; Maleev, V.I.; Svistunova, N.Y.; Solodenko, V.A.; Kukhar, V.P. Asymmetric Synthesis of Phosphorus Analogs of Dicarboxylic α-Amino Acids. J. Chem. Soc. Perkin Trans. I 1992, 1525–1529. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Biitseva, A.V.; Zhersh, S. Amino sulfonic acids, peptidosulfonamides and other related compounds. Tetrahedron 2018, 74, 1355–1421. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J. A Practical Asymmetric Synthesis of Enantiomerically Pure 3-Substituted Pyroglutamic Acids and Related Compounds. Angew. Chem. Int. Ed. 2000, 39, 2172–2175. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Avilov, D.V.; Kukhar, V.P.; Meervelt, L.V.; Mischenko, N. An Efficient Asymmetric Synthesis of (2S,3S)-3-Trifluoromethylpyroglutamic Acid. Tetrahedron Lett. 1997, 38, 4903–4904. [Google Scholar] [CrossRef]
- Tang, X.; Soloshonok, V.A.; Hruby, V.J. Convenient Asymmetric Synthesis of Enantiomerically Pure 2’,6’-Dimethyltyrosine (DMT) via Alkylation of Chiral Nucleophilic Glycine Equivalent. Tetrahedron Asymmetry 2000, 11, 2917–2925. [Google Scholar] [CrossRef]
- Taylor, S.M.; Yamada, T.; Ueki, H.; Soloshonok, V.A. Asymmetric Synthesis of Enantiomerically Pure 4-Aminoglutamic Acids via Methylenedimerization of Chiral Glycine Equivalents with Dichloromethane under Operationally Convenient Conditions. Tetrahedron Lett. 2004, 45, 9159–9162. [Google Scholar] [CrossRef]
- Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V.A. Efficient asymmetric synthesis of the functionalized pyroglutamate core unit common to oxazolomycin and neooxazolomycin using Michael reaction of nucleophilic glycine Schiff base with α,β-disubstituted acrylate. Tetrahedron Asymmetry 2008, 19, 2789–2795. [Google Scholar] [CrossRef]
- Shevchuk, M.V.; Kukhar, V.P.; Roeschenthaler, G.V.; Bassil, B.S.; Kawada, K.; Soloshonok, V.A.; Sorochinsky, A.E. New Asymmetric Approach to β-Trifluoromethyl Isoserines. RSC Adv. 2013, 3, 6479–6484. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical purifications via Self-Disproportionation of Enantiomers by achiral chromatography; Case study of a series of α-CF3-containing secondary alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019. [Google Scholar] [CrossRef]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. RSC Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni (II) complexes of glycine Schiff bases; Part 1: alkyl halide alkylations. Amino Acids 2013, 45, 691–718. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 2: Aldol, Mannich addition reactions, deracemization and (S) to (R) interconversion of α-amino acids. Amino Acids 2013, 45, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar]
- Ellis, T.K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V.A. The Design, Synthesis and Evaluation of a New Generation of Modular Nucleophilic Glycine Equivalents for the Efficient Synthesis of Sterically Constrained α-Amino Acids. J. Org. Chem. 2006, 71, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Ueki, H.; Ellis, T.K.; Yamada, T.; Ohfune, Y. Application of Modular Nucleophilic Glycine Equivalents for Truly Practical Asymmetric Synthesis of β-Substituted Pyroglutamic Acids. Tetrahedron Lett. 2005, 46, 1107–1110. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V.A. Second-order asymmetric transformation and its application for the practical synthesis of α-amino acids. Org. Biomol. Chem. 2018, 16, 4968–4972. [Google Scholar] [CrossRef]
- Takeda, R.; Kawashima, A.; Yamamoto, J.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V.A. Tandem alkylation - second-order asymmetric transformation protocol for preparation of phenylalanine-type tailor-made α-amino acids. ACS Omega 2018, 3, 9729–9737. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ellis, T.K.; Ueki, H.; Ono, T. Resolution/Deracemization of Chiral α-Amino Acids Using Resolving Reagents with Flexible Stereogenic Centers. J. Am. Chem. Soc. 2009, 131, 7208–7209. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Chen, X.; Aceña, J.L.; Soloshonok, V.A.; Liu, H. Chemical Kinetic Resolution of Unprotected β-Substituted-β-Amino Acids Using Recyclable Chiral Ligands. Angew. Chem. Int. Ed. 2014, 53, 7883–7886. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J.L.; Soloshonok, V.A. Chemical Dynamic Kinetic Resolution and (S)/(R)-Interconversion of Unprotected α-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12214–12217. [Google Scholar] [CrossRef] [PubMed]
- Govorkova, E.A.; Fang, H.B.; Tan, M.; Webster, R.G. Neuraminidase Inhibitor-Rimantadine Combinations Exert Additive and Synergistic Anti-Influenza Virus Effects in MDCK Cells. Antimicrob. Agents Ch. 2004, 48, 4855–4863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreas, L.B.; Barnes, A.B.; Corzilius, B.; Chou, J.J.; Miller, E.A.; Caporini, M.; Rosay, M.; Griffin, R.G. Dynamic Nuclear Polarization Study of Inhibitor Binding to the M218–60 Proton Transporter from Influenza A. Biochemistry 2013, 52, 2774–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMenamy, R.H.; Oncley, J.L. The specific binding of L-tryptophan to serum albumin. J. Biol. Chem. 1958, 233, 1436–1447. [Google Scholar] [PubMed]
- Wright, A.K.; Batsomboon, P.; Dai, J.; Hung, I.; Zhou, H.X.; Dudley, G.B.; Cross, T.A. Differential Binding of Rimantadine Enantiomers to Influenza A M2 Proton Channel. J. Am. Chem. Soc. 2016, 138, 1506–1509. [Google Scholar] [CrossRef]
- Tanuwidjaja, J.; Peltier, H.M.; Ellman, J.A. One-Pot Asymmetric Synthesis of Either Diastereomer of tert-Butanesulfinyl-protected Amines from Ketones. J. Org. Chem. 2007, 72, 626–629. [Google Scholar]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef]
- Mei, H.; Xie, C.; Han, J.; Soloshonok, V.A. N-tert-Butanesulfinyl-(3,3,3)-trifluoroacetaldimine: Versatile Reagent for Asymmetric Synthesis of Trifluoromethyl-Containing Amines and Amino Acids of Pharmaceutical Importance, Eur. J. Org. Chem. 2016, 5917–5932. [Google Scholar]
- He, Q.; Peng, Q.F.; Rohani, S. Diastereomeric Resolution of p-Chloromandelic Acid with (R)-Phenylethylamine. Chirality 2010, 22, 16–23. [Google Scholar] [CrossRef]
- Peng, Y.; He, Q.; Rohani, S.; Jenkins, H. Resolution of 2-Chloromandelic Acid with (R)-(+)-N-Benzyl-1-Phenylethylamine: Chiral Discrimination Mechanism. Chirality 2012, 24, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B.; Faghihi, M.R.; Kazemi, F.; Yokomatsu, T. Resolution of Enantiomers of Novel C2-Symmetric Aminobisphosphinic Acids via Diastereomeric Salt Formation With Quinine. Chirality 2015, 27, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhao, Q.; Yang, K.; Liu, S.; Xia, F. Optical Resolution and Optimization of (R,S)-Propranolol Using Dehydroabietic Acid Via Diastereomeric Crystallization. Chirality 2015, 27, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, V.N.; Kozyrkov, Y.Y. A Simple Method for Resolution of Endo-/Exo-Monoesters of Trans-Norborn-5-Ene-2,3-Dicarboxylic Acids Into Their Enantiomers. Chirality 2015, 27, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Wataya, T.; Nunomura, S.; Takahashi, Y.; Fujii, I. Optically active amine having adamantly group or salt of the same, method for producing the same, diastereomers salt and method for separating the diastereomers salt. Kokai Tokkyo Koho. JP2008024666.
- Siedlecka, R. Recent Developments in Optical Resolution. Tetrahedron 2013, 69, 6331–6363. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, racemates, and resolutions. Ber. Bunsen-Ges. Phys. Chem. 1981. [Google Scholar]
- Porter, W.H. Resolution of chiral drugs. Pure Appl. Chem. 1991, 63, 1119–1122. [Google Scholar] [CrossRef]
- Fogassy, E.; Nógrádi, M.; Kozma, D.; Egri, G.; Pálovicsc, E.; Kissa, V. Optical resolution methods. Org. Biomol. Chem. 2006, 4, 3011–3030. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
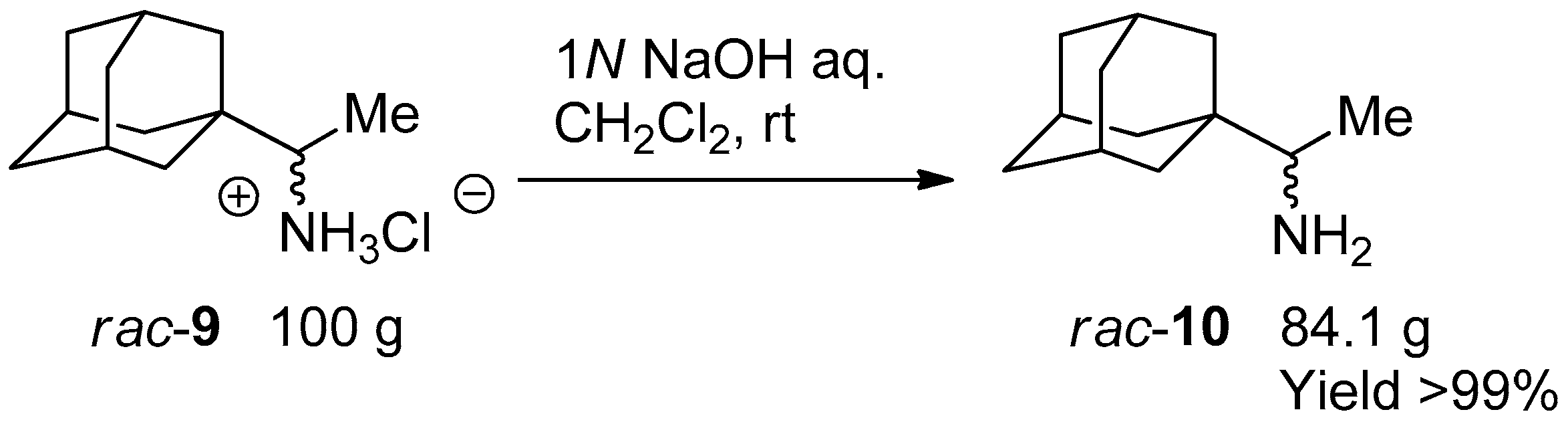{kind=link}
{kind=link}
{kind=link}

| Resolving Reagent | Equivalent of Resolving Reagent 2,3 | |||
|---|---|---|---|---|
| in Acetone (5% H2O) (12 v) 1 | 1.0 eq | 0.5 eq | 0.25 eq | |
| 13 | (R)-2-Phenoxy propionic acid | - | 0.48 g, 35.5% | - |
| 88.0% ee (S) | ||||
| 14 | (S)-Naproxen | 1.04 g, 91.2% | 0.51 g, 44.8% | - |
| 11.0% ee4 (R) | 35.3% ee (R) | |||
| 15 | (R)-Mandelic acid | 0.86 g, 93.3% | 0.39 g, 42.0% | - |
| 2.82% ee (S) | 7.32% ee (S) | |||
| 16 | (S)-α-Methoxyphenylacetic acid | 0.83 g, 86.1% | 0.35 g, 36.1% | - |
| 0.88% ee (S) | 1.34% ee (S) | |||
| 17 | (R)-α-Methoxyphenylacetic acid | 0.84 g, 86.7% | 0.39 g, 40.1% | - |
| 0.08% ee (S) | 0.08% ee (S) | |||
| 18 | (S)-Aspartic acid | 0.80 g, 91.3% | 0.36 g, 52.0% | 0.17 g, 24.6% |
| 4.86% ee (S) | 7.44% ee (S) | 7.20% ee (S) | ||
| 19 | (S)-Malic acid | 0.52 g, 59.2% | - | 0.35 g, 50.6% |
| 0.20% ee (S) | 0.26% ee (S) | |||
| 20 | N-Tosyl-(S)-proline | 0.35 g, 28.3% | 0.08 g, 6.4% | - |
| 94.96% ee (R) | 95.46% ee (R) | |||
| 21 | (1R,3S)-Camphoric acid | 1.03 g, 97.5% | 0.78 g, >98% | 0.13 g, 16.8% |
| 1.36% ee (S) | 0.72% ee (S) | 0.38% ee (S) | ||
| 22 | (1S)-10-Camphorsulforic acid | 0.81 g, 70.9% | - | - |
| 5.22% ee (R) | ||||
| 23 | (2S,3S)-Tartaric acid | 0.92 g, 95.0% | 0.73 g, >98% | 0.35 g, 49.0% |
| 0.08% ee (S) | 1.38% ee (S) | 23.24% ee (S) | ||
| 24 | (2S,3S)-Dibenzoyltartaric acid | 1.41 g, 94.2% | 0.87 g, 86.6% | 0.48 g, 48.4% |
| 0.28% ee (S) | 16.38% ee (R) | 62.72% ee (R) | ||
| 25 | (2R,3R)-Dibenzoyltartaric acid monohydrate | - | - | 0.47 g, 46.8% |
| 60.44% ee (S) | ||||
| 26 | (2R,3R)-Di-p-toluoyltartaric acid | 1.612 g, >98% | 0.53 g, 50.6% | |
| 0.38% ee (R) | 14.12% ee (S) | |||
| Entry | Solvent (12 v) | Results 1 |
|---|---|---|
| 1 | Acetone (5% H2O) | 0.47 g, 46.8% |
| 60.4% ee (S) | ||
| 2 | THF2 | 0.32 g, 31.8% |
| 36.8% ee (S) | ||
| 3 | IPE2 | 0.52 g, 52.4% |
| 36.2% ee (S) | ||
| 4 | Acetone | 0.51 g, 51.1% |
| 32.9% ee (S) | ||
| 5 | EtOAc | 0.50 g, 50.4% |
| 28.4% ee (S) | ||
| 6 | MIBK2 | 0.50 g, 50.1% |
| 67.4% ee (S) | ||
| 7 | MTBE2 | 0.46 g, 46.1% |
| 42.7% ee (S) | ||
| 8 | Toluene | 0.13 g, 13.2% |
| 88.0% ee (S) | ||
| 9 | MeCN | 0.048 g, 4.8% |
| 36.3% ee (S) |
| Entry | Solvent | Results 1 |
|---|---|---|
| 1 | Acetone (5% H2O)/60 v | 0.084 g, 84% |
| 73.8% ee (S) | ||
| 2 | EtOAc (5% H2O)/60 v | 0.090 g, 90% |
| 65.3% ee (S) | ||
| 3 | THF/30 v | 0.036 g, 36% |
| 92.8% ee (S) |
| 3rd Batch (100 g) | ||
|---|---|---|
| 1st salt formation | Yield | 66.9 g, 34.7% |
| Acetone (5% H2O) | Optical purity 1 | 88.4% ee |
| 2nd recrystallization | Yield | 56.7 g, 87.3% |
| EtOAc (5% H2O) | Optical purity 1 | 98.7% ee |
| 3rd recrystallization | Yield | 51.1 g, 92.9% |
| EtOAc (5% H2O) | Optical purity 1 | 99.7% ee |
| Overall yield | 28.1% | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Takeda, R.; Sato, T.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. Optical Resolution of Rimantadine. Molecules 2019, 24, 1828. https://doi.org/10.3390/molecules24091828
Han J, Takeda R, Sato T, Moriwaki H, Abe H, Izawa K, Soloshonok VA. Optical Resolution of Rimantadine. Molecules. 2019; 24(9):1828. https://doi.org/10.3390/molecules24091828
Chicago/Turabian StyleHan, Jianlin, Ryosuke Takeda, Tatsunori Sato, Hiroki Moriwaki, Hidenori Abe, Kunisuke Izawa, and Vadim A. Soloshonok. 2019. "Optical Resolution of Rimantadine" Molecules 24, no. 9: 1828. https://doi.org/10.3390/molecules24091828
APA StyleHan, J., Takeda, R., Sato, T., Moriwaki, H., Abe, H., Izawa, K., & Soloshonok, V. A. (2019). Optical Resolution of Rimantadine. Molecules, 24(9), 1828. https://doi.org/10.3390/molecules24091828