Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–6

 ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
,  , , ,
, , ,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,  ,
, 
 ,
,  ,
,  ,
,  ,
,  and
and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
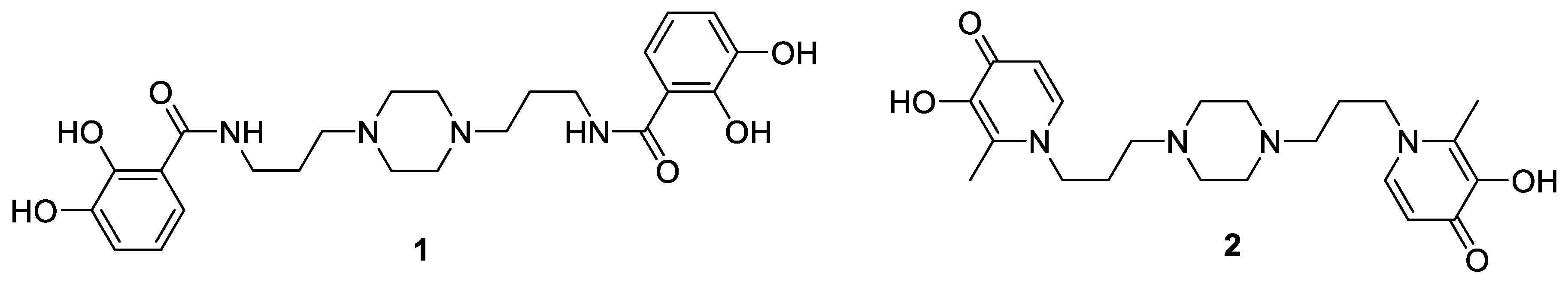
2. Siderophores, Did You Say Siderophores?
Highlighted by Jean Jacques Vanden Eynde
3. A Combined Neuroprotective Approach for Dementia
Highlighted by Arduino A. Mangoni
4. Prodrug Strategy for Multiple Charged Drugs
Highlighted by Jarkko Rautio
5. A SOX9-Derived Peptide Inhibits the Growth of Colorectal Cancer Tumors
Highlighted by Jérôme Leprince
6. Ustekinumab, an Anti-IL-12/23 p40 Monoclonal Antibody, as a Molecularly-Targeted Therapy for the Treatment of Crohn’s Disease
Highlighted by Yasu-Taka Azuma
7. A Stab in the Back of TB: Recovering Mycobacterium tuberculosis Fumarate Hydratase Through a Non-Conserved Allosteric Site
Highlighted by Alfonso T. García-Sosa
8. New Hopes for 3rd Generation Castration Resistant Prostate Cancer Drugs: Arv7 Splice Variant Binders
Highlighted by Christopher Hulme
9. Multi-Target Drugs for Rheumatoid Arthritis Management
Highlighted by Josef Jampilek
10. Sodium Oligomannate—A Novel Drug for the Treatment of Mild to Moderate Alzheimer’s Disease
Highlighted by Rafik Karaman
11. New Strategies to Reduce Off-Target Toxicity and Identify True Targets to Improve Clinical Successes
Highlighted by Wei Li
12. Crossing a Formidable Barrier: Seven Molecular Descriptors for BBB Permeability Prediction
Highlighted by Paula A. C. Gomes
13. Design, Synthesis and Biological Evaluation of Second-Generation NLRP3 Inflammasome Inhibitors
Highlighted by Dimitra Hadjipavlou-Litina
14. Sphingosine-1-Phosphate Signaling Is Involved in Cigarette Smoke-Induced Airway Pathology
Highlighted by Raffaele Capasso
15. New Class of Antifungals against Aspergillus fumigatus
Highlighted by Athina Geronikaki
16. Conditional Oligonucleotide Aptamers as a Powerful Tool for Targeted Cancer Therapy
Highlighted by Laura Cerchia
17. DNA-Based Nanostructures Open the Way to the Discovery of ‘Novel’ Antivirals with Appropriate 3-D Arrangements and Binding Properties
Highlighted by Jean-Marc Sabatier
18. Towards the Synthesis of Highly Effective Treatment of Autoimmune Diseases through Inhibition of Tyrosine Kinase 2
Highlighted by Rino Ragno
19. A Bayesian Machine Learning Model Developed using Diverse Data Types: A Promising Approach in the Target-Fishing Field
Highlighted by Tiziano Tuccinardi
20. First-in-Class Clinical O-GlcNAcase Inhibitors as a Therapeutic Agent for the Treatment of Tauopathies
Highlighted by Andrea Trabocchi
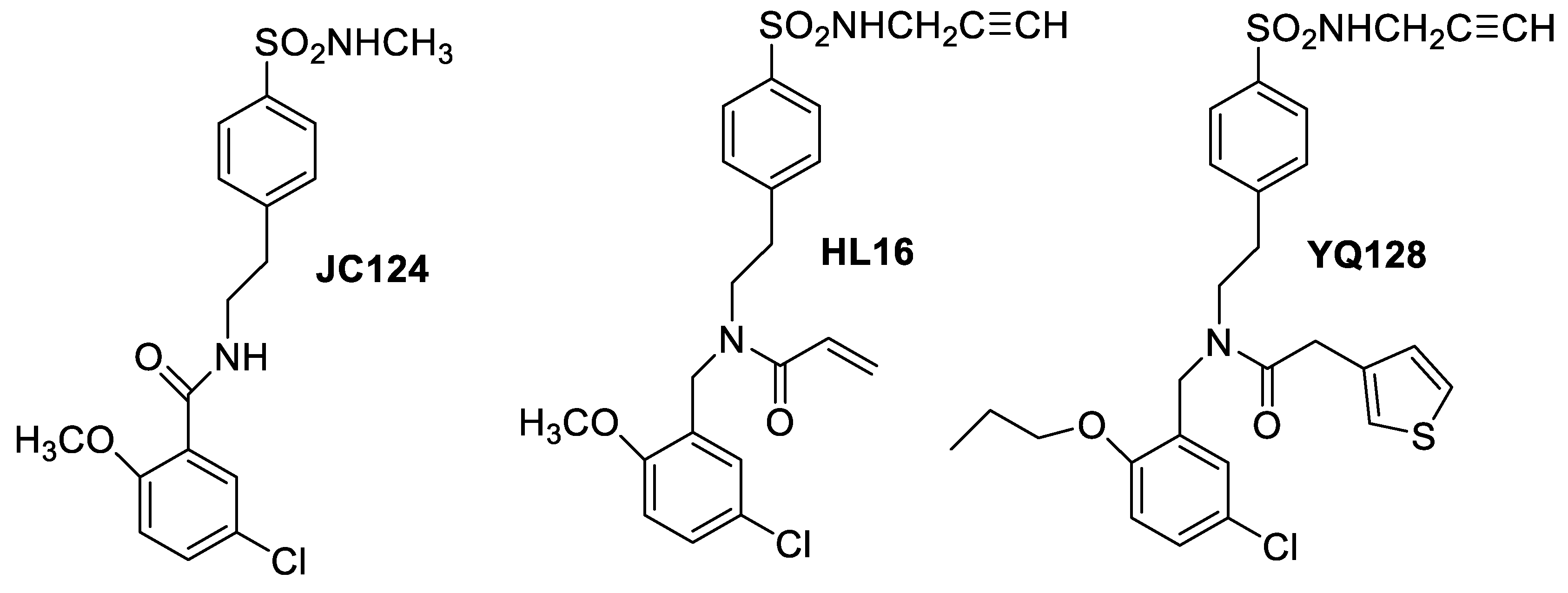
21. 2H-Benzo[e][1,2,4]thiadiazin-3(4H)-one 1,1-dioxide Scaffold as New Chemotype for Selectively Targeting the Tumor-Associated Carbonic Anhydrases IX and XII
Highlighted by Jean-Yves Winum
22. Expanding the Human Proteome with Functionalized Enantiomeric Probes
Highlighted by F. Javier Luque
23. Targeting IL-11 to Halt and Reverse Fibrosis and Inflammation in the Lung
Highlighted by Katalin Prokai-Tatrai
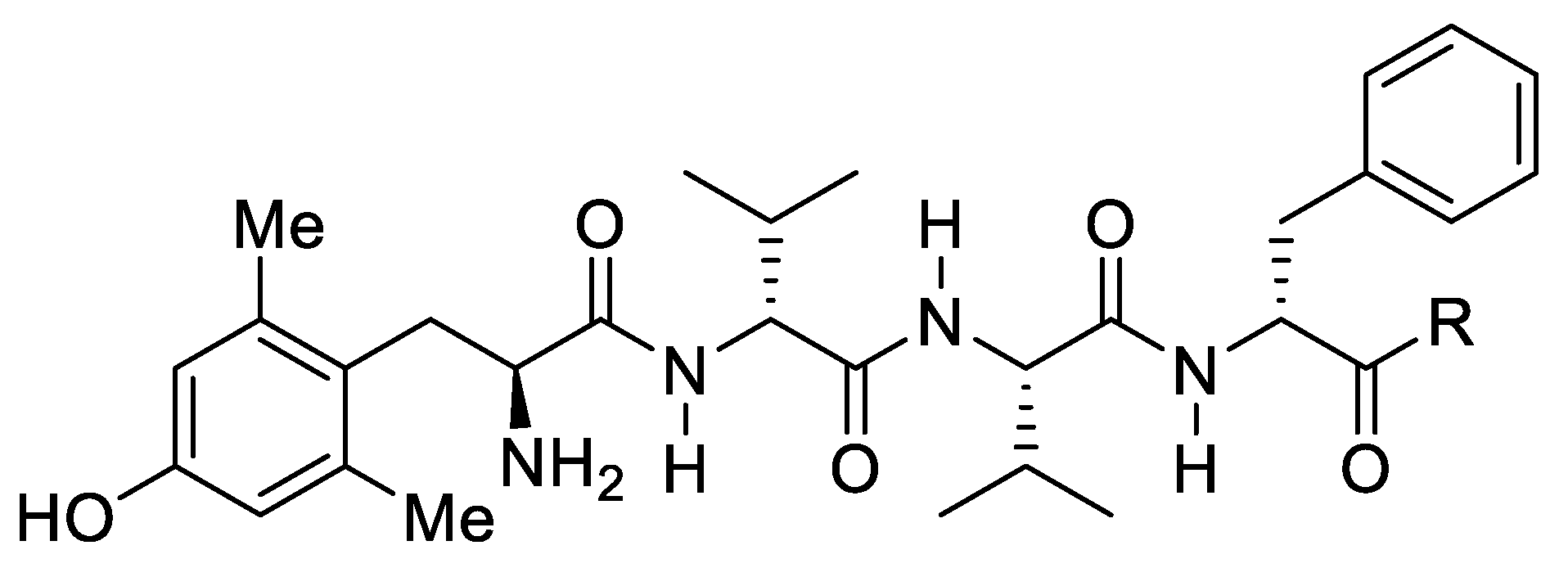
24. Unique Tetrapeptides from a Penicillium Fungus as Biased Analgesics Targeting the µ-Opioid Receptor
Highlighted by Mariana Spetea
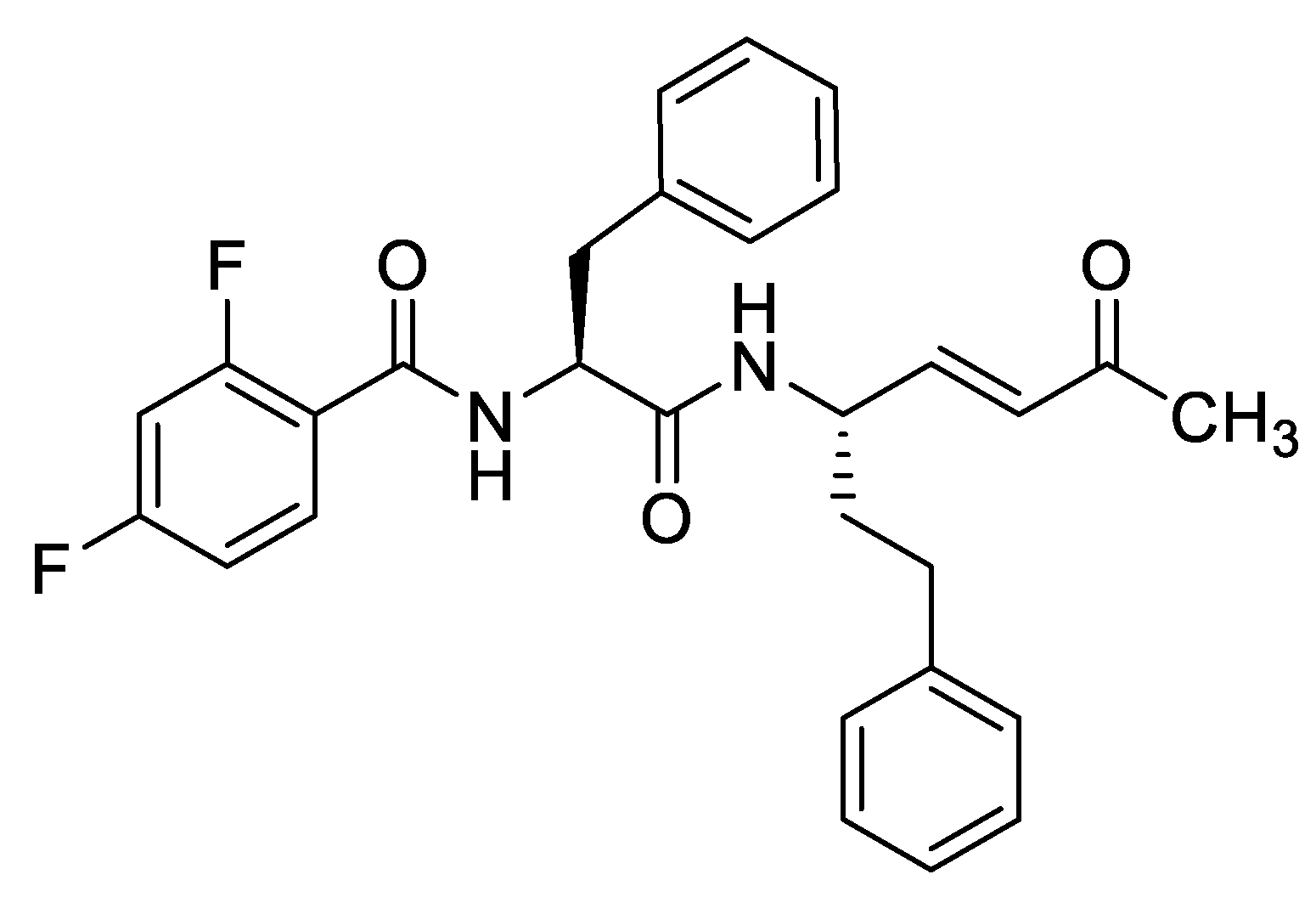
25. Optimized Peptidic Michael Acceptors for the Treatment of Human African Trypanosomiasis
Highlighted by Michael Gütschow
26. Inhibition of Virulence Factors, Sporulation and Cellular Damage of Auranofin in a Mouse Model Infected with Clostridioides difficile
Highlighted by Ivan Kosalec
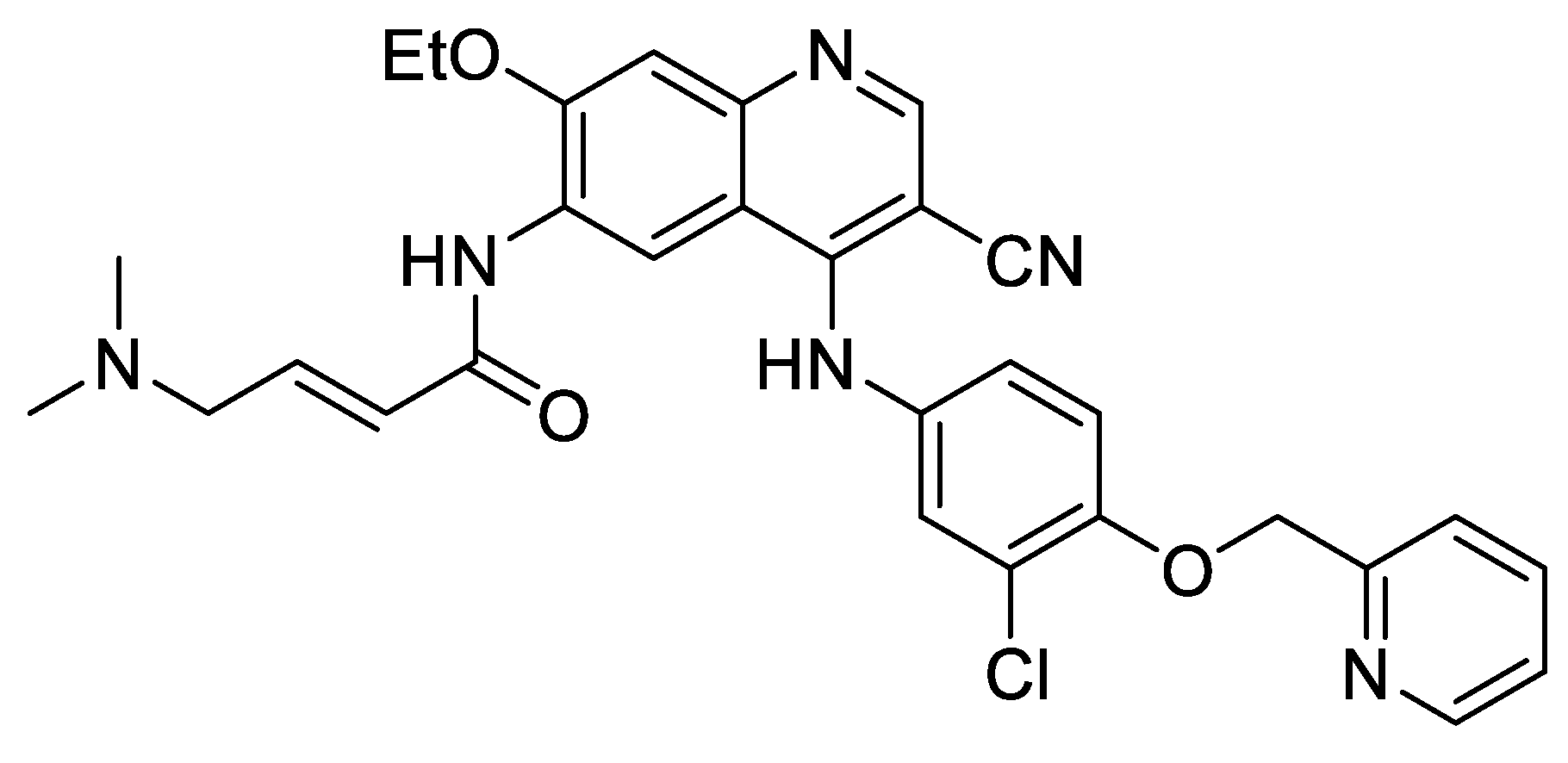
27. A New Efficient Positive Allosteric Modulator of α7 Nicotinic Acetylcholine Receptor. New Hope for the Treatment of Patients with Mild to Moderate Alzheimer’s Disease
Highlighed by Catherine Guillou
28. CD9 Is a Marker and a Potential Therapeutic Target for Pancreatic Ductal Adenocarcinoma Tumor-Initiating Cells
Highlighed by M. Helena Vasconcelos
29. The FDA-Approved Drug Neratinib Protects Pancreatic Beta Cells in Diabetes
Highlighted by George Kokotos
30. Human Asparagine Synthetase is a Promising Anticancer Drug Target
Highlighted by Giulio Rastelli
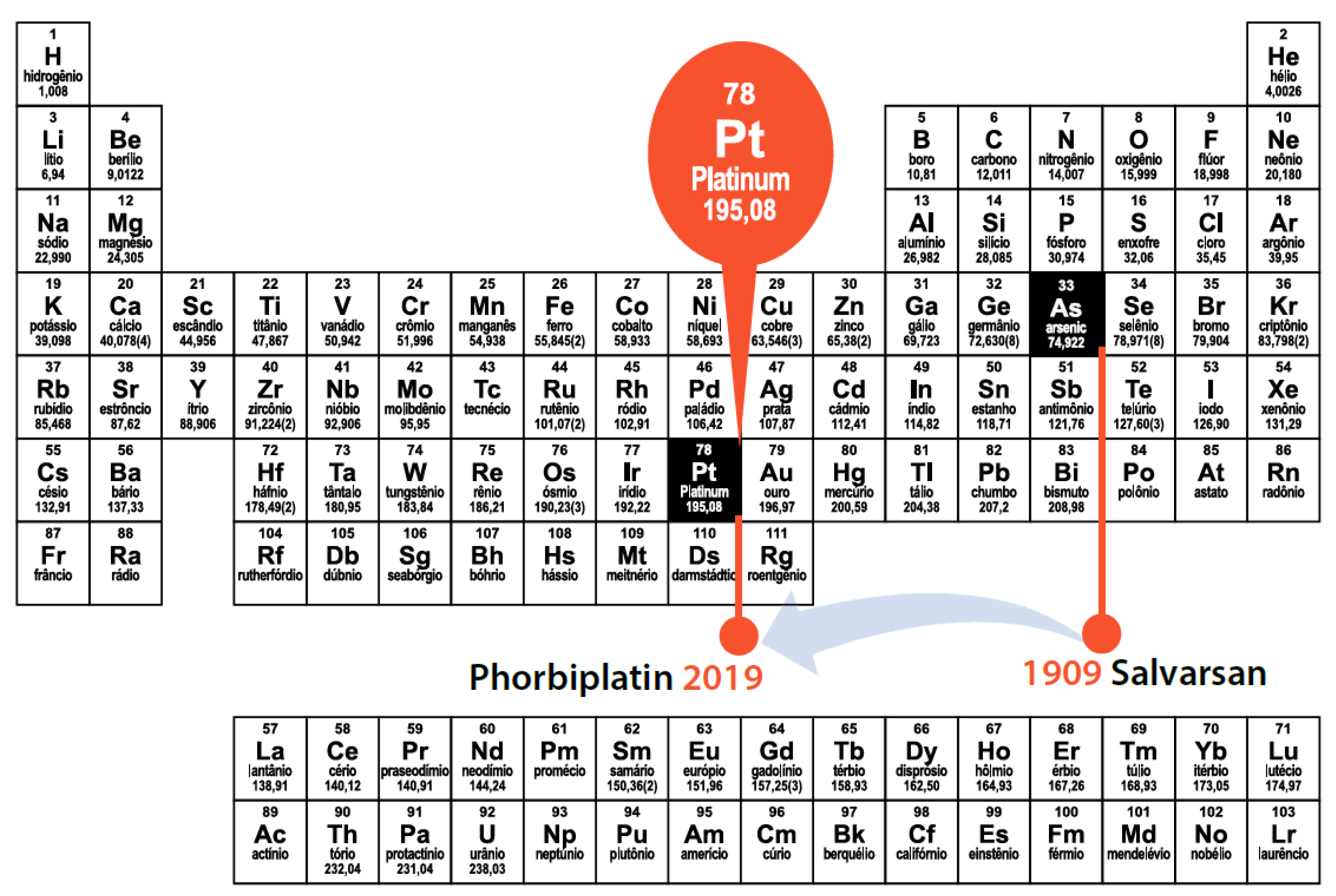
31. Visible Light Photoactivatable Prodrugs: Design of the Platinum Magic Bullet
Highlighted by Maria Emília de Sousa
32. Allosteric Modulators of Cannabinoid Receptor 1: Developing Compounds for Improved Functional Diversity
Highlighted by Clementina Manera
33. Inhibition of Trypanothione Reductase by 4,15-iso-Atriplicolide Tiglate: Mode of Inhibition and Structural Requirements
Highlighted by Sandra Gemma
34. The Pseudomonas aeruginosa Importer PfeA-Fe3+-Enterobactin Complex Indicates New Ways to Overcome Bacterial Resistance to Antibiotics
Highlighted by Stefano Mangani
35. Anticancer Phosphonodiesters Containing Nature Inspired Molecular Scaffolds
Highlighted by Carlo Siciliano
36. A Bioinspired Technology as an Antitumor Drug Carrier: Enzymatic Self-Assemblies
Highlighted by Stefania Galdiero
37. MOST: A New Strategy with High Precision to Pry into the Vasculature-Dependent Pathogenesis of Alzheimer’s Disease
Highlighted by Hong Liu
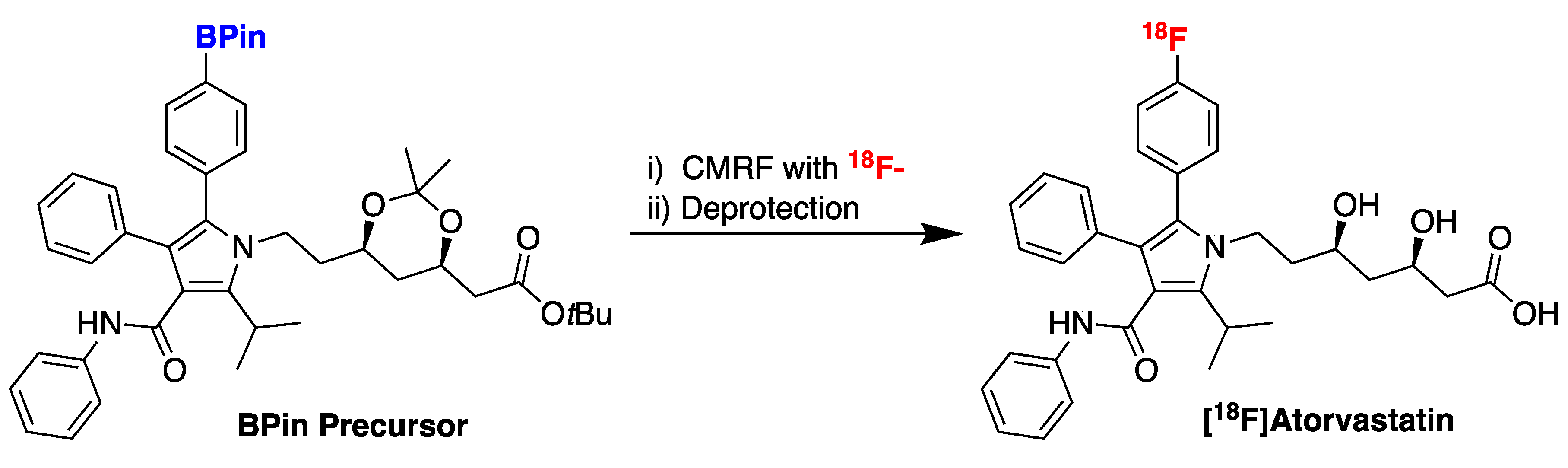
38. Accessing New PET Radiotracers with Copper-Mediated Radiofluorination: Radiosynthesis of [18F]Atorvastatin
Highlighted by Peter J. H. Scott
39. Fighting Hypoxic Tumors with Photoactivated Organometallics
Highlighted by Cristóbal de los Ríos
40. Enzymatic Preparation of 2’3’-Cyclic Dinucleotides and Their Binding to STING Adaptor Protein
Highlighted by Luigi A. Agrofoglio
41. Pteridine-Reductase-1 Inhibitors as a New Weapon against Trypanosoma brucei Infections
Highlighted by Simona Collina
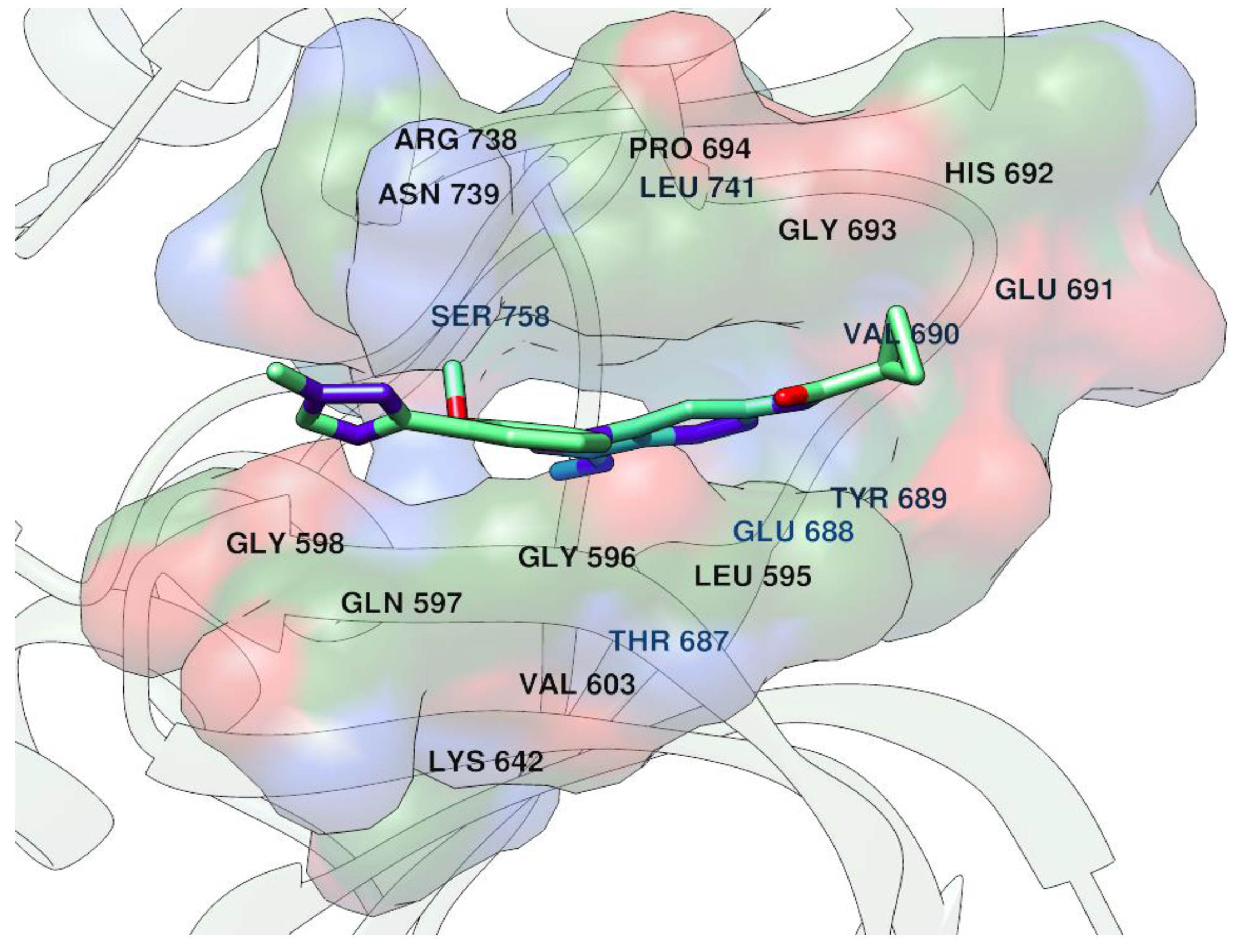
42. Exploring and Targeting Protein–Protein Druggable Cavities
Highlighted by Rita C. Guedes
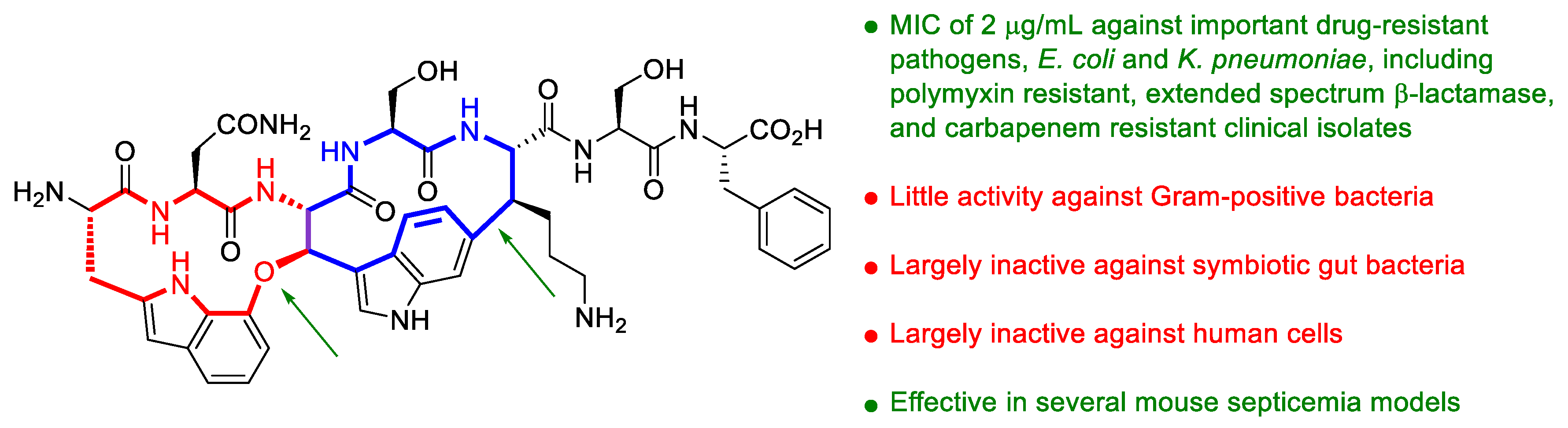
43. The Enemy of My Enemy Is My Friend: A New Antibiotic Selective for Gram-Negative Pathogens Isolated from Nematode Microbiome Symbionts
Highlighted by Diego Muñoz-Torrero
Conflicts of Interest
References
- Loupias, P.; Dechamps-Olivier, I.; Dupont, L.; Vanlemmens, P.; Mullié, C.; Taudon, N.; Bouchut, A.; Dassonville-Klimpt, A.; Sonnet, P. Study of iron piperazine-based chelators as potential siderophore mimetic. Pharmaceuticals 2019, 12, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, M.; Dolles, D.; Gunesch, S.; Hoffmann, M.; Nabissi, M.; Marinelli, O.; Naldi, M.; Bartolini, M.; Petralla, S.; Poeta, E.; et al. Dual-acting cholinesterase-human cannabinoid receptor 2 ligands show pronounced neuroprotection in vitro and overadditive and disease-modifying neuroprotective effects In Vivo. J. Med. Chem. 2019, 62, 9078–9102. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Dash, R.P.; Tichý, T.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced oral bioavailability of 2-(phosphonomethyl)-pentanedioic acid (2-PMPA) from its (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-based prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef]
- Blache, P.; Canterel-Thouennon, L.; Busson, M.; Verdié, P.; Subra, G.; Ychou, M.; Prévostel, C. A short SOX9 peptide mimics SOX9 tumor suppressor activity and is sufficient to inhibit colon cancer cell growth. Mol. Cancer Ther. 2019, 18, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Li, K.; Friedman, J.R.; Chan, D.; Pollack, P.; Yang, F.; Jacobstein, D.; Brodmerkel, C.; Gasink, C.; Feagan, B.G.; Sandborn, W.J.; et al. Effects of ustekinumab on histologic disease activity in patients with Crohn’s disease. Gastroenterology 2019, 157, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, A.J.; Libardo, M.D.J.; Kasbekar, M.; Brear, P.D.; Fischer, G.; Thomas, C.J.; Barry, C.E., III; Boshoff, H.I.M.; Coyne, A.G.; Abell, C. Targeting of fumarate hydratase from Mycobacterium tuberculosis using allosteric inhibitors with a dimeric-binding mode. J. Med. Chem. 2019, 62, 10586–10604. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, D.; Johnsoni, J.K.; Pascal, L.E.; Takubo, K.; Avula, R.; Chakka, A.B.; Zhou, J.; Chen, W.; Zhong, M.; et al. A novel small molecule targets androgen receptor and its splice variants in castration-resistant prostate cancer. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef] [Green Version]
- Rheumatoid Arthritis, National Institutes of Health. Available online: https://www.niams.nih.gov/health-topics/rheumatoid-arthritis (accessed on 22 November 2019).
- Micheli, L.; Bozdag, M.; Akgul, O.; Carta, F.; Guccione, C.; Bergonzi, M.C.; Bilia, A.R.; Cinci, L.; Lucarini, E.; Parisio, C.; et al. Pain relieving effect of-NSAIDs-CAIs hybrid molecules: Systemic and intra-articular treatments against rheumatoid arthritis. Int. J. Mol. Sci. 2019, 20, E1923. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.J.; Hong, J.H. An overview of carbonic anhydrases and membrane channels of synoviocytes in inflamed joints. J. Enzyme Inhib. Med. Chem. 2019, 34, 1615–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bua, S.; Lucarini, L.; Micheli, L.; Menicatti, M.; Bartolucci, G.; Selleri, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Masini, E.; Carta, F.; et al. Bioisosteric development of multitarget nonsteroidal anti-inflammatory drug-carbonic anhydrases inhibitor hybrids for the management of rheumatoid arthritis. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wand, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Mignani, S.; Rodrigues, J.; Tomas, H.; Jalal, R.; Singh, P.P.; Majoral, J.P.; Vishwakarma, R.A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: How far can they be simplified? Drug Discov. Today 2018, 23, 605–615. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The blood-brain barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Fulp, J.; He, L.; Toldo, S.; Jiang, Y.; Boice, A.; Guo, C.; Li, X.; Rolfe, A.; Sun, D.; Abbate, A.; et al. Structural insights of benzenesulfonamide analogues as Nlrp3 inflammasome inhibitors: Design, synthesis, and biological characterization. J. Med. Chem. 2018, 61, 5412–5423. [Google Scholar] [CrossRef]
- Jiang, Y.; He, L.; Green, J.; Blevins, H.; Guo, C.; Harsiddhbhai Patel, S.; Halquist, M.S.; McRae, M.P.; Venitz, J.; Wang, X.Y.; et al. Discovery of second-generation NLRP3 inflammasome inhibitors: Design, synthesis, and biological characterization. J. Med. Chem. 2019, 62, 9718–9731. [Google Scholar] [CrossRef]
- De Cunto, G.; Brancaleone, V.; Riemma, M.A.; Cerqua, I.; Vellecco, V.; Spaziano, G.; Cavarra, E.; Bartalesi, B.; D’Agostino, B.; Lungarella, G.; et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; De Cunto, G.; Sundar, I.K.; Lungarella, G. Vulnerability and genetic susceptibility to cigarette smoke-induced emphysema in mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Altose, M.D.; Connett, J.E.; Kanner, R.E.; Lee, W.W.; Wise, R.A. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. Am. J. Respir. Crit. Care Med. 1996, 153, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Kritsi, E.; Matsoukas, M.-T.; Potamitis, C.; Detsi, A.; Ivanov, M.; Sokovic, M.; Zoumpoulakis, P. Novel hit compounds as putative antifungals: The case of Aspergillus fumigatus. Molecules 2019, 24, 3853. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Fu, T.; Huang, Q.; Kuai, H.; Mo, L.; Liu, H.; Wang, Q.; Peng, Y.; Han, D.; Zhao, Z.; et al. Hypoxia-activated PEGylated conditional aptamer/antibody for cancer imaging with improved specificity. J. Am. Chem. Soc. 2019, 141, 18421–18427. [Google Scholar] [CrossRef]
- Kwon, P.S.; Ren, S.; Kwon, S.J.; Kizer, M.E.; Kuo, L.; Xie, M.; Zhu, D.; Zhou, F.; Zhang, F.; Kim, D.; et al. Designer DNA architecture offers precise and multivalent spatial pattern-recognition for viral sensing and inhibition. Nat. Chem. 2020, 12, 26–35. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Pitts, W.J.; Tokarski, J.; Strnad, J.; Shuster, D.; et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: Discovery of the allosteric inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. [Google Scholar] [CrossRef]
- Moslin, R.; Zhang, Y.; Wrobleski, S.T.; Lin, S.; Mertzman, M.; Tokarski, J.S.; Strnad, J.; Gillooly, K.; McIntyre, K.W.; Zupa-Fernandez, A.; et al. Identification of N-methyl nicotinamide and N-methyl pyridazine-3-carboxamide pseudokinase domain ligands as highly selective inhibitors of tyrosine kinase 2 (TYK2). J. Med. Chem. 2019, 62, 8953–8972. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef] [Green Version]
- Selnick, H.G.; Hess, J.F.; Tang, C.; Liu, K.; Schachter, J.B.; Ballard, J.E.; Marcus, J.; Klein, D.J.; Wang, X.; Pearson, M.; et al. Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. J. Med. Chem. 2019, 62, 10062–10097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T.; Alterio, V.; Di Fiore, A.; D’ Ambrosio, K.; Carta, F.; Monti, S.M.; De Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef] [PubMed]
- Bua, S.; Lomelino, C.L.; Murray, A.B.; Osman, S.M.; Alothman, Z.A.; Bozdag, M.; Aziz, H.A.A.; Eldehna, W.M.; McKenna, R.; Nocentini, A.; et al. “A sweet combination”: Developing saccharin and acesulfame K structures for selectively targeting the tumor-associated carbonic anhydrases IX and XII. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.G.; Galmozzi, A.; Wang, Y.; Correia, B.E.; Sasaki, K.; Joslyn, C.M.; Kim, A.S.; Cavallaro, C.L.; Lawrence, R.M.; Johnson, S.R.; et al. Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dix, M.M.; Biando, G.; Remsberg, J.R.; Lee, H.-Y.; Kalocsay, M.; Gygi, S.P.; Forli, S.; Vite, G.; Lawrence, R.M.; et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 2019, 11, 1113–1123. [Google Scholar] [CrossRef]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Trans. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef]
- Dekan, Z.; Sianati, S.; Yousuf, A.; Sutcliffe, K.J.; Gillis, A.; Mallet, C.; Singh, P.; Jin, A.H.; Wang, A.M.; Mohammadi, S.A.; et al. A tetrapeptide class of biased analgesics from an Australian fungus targets the µ-opioid receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 22353–22358. [Google Scholar] [CrossRef] [Green Version]
- Ettari, R.; Previti, S.; Maiorana, S.; Amendola, G.; Wagner, A.; Cosconati, S.; Schirmeister, T.; Hellmich, U.A.; Zappalà, M. Optimization strategy of novel peptide-based Michael acceptors for the treatment of human African trypanosomiasis. J. Med. Chem. 2019, 62, 10617–10629. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. The damage-response framework of microbial pathogenesis. Nat. Microbiol. Rev. 2003, 1, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.L.; Pehlivanoglu, H.; Vidor, C.J.; James, M.L.; Thomson, M.J.; Lyras, D. Repurposing auranofin as a Clostridioides difficile therapeutic. J. Antimicrob. Chemother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Karche, N.P.; Verma, M.K.; Walunj, S.S.; Nigade, P.B.; Jana, G.; Kurhade, S.P.; Hajare, A.K.; Tilekar, A.R.; Jadhav, G.R.; et al. Discovery of novel, potent, brain-permeable, and orally efficacious positive allosteric modulator of α7 nicotinic acetylcholine receptor [4-(5-(4-chlorophenyl)-4-methyl-2-propionylthiophen-3-yl)benzenesulfonamide]: Structure−activity relationship and preclinical characterization. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Wang, V.M.-Y.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Zaw Thin, M.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Podergajs, N.; Motaln, H.; Rajčević, U.; Verbovšek, U.; Koršič, M.; Obad, N.; Espedal, H.; Vittori, M.; Herold-Mende, C.; Miletic, H.; et al. Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 2016, 7, 593–609. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Xu, C.W.; Naito, M.; Nishida, H.; Okamoto, T.; Ghani, F.I.; Iwata, S.; Inukai, T.; Sugita, K.; Morimoto, C. Regulation of cancer stem cell properties by CD9 in human B-acute lymphoblastic leukemia. Biochem. Biophys. Res. Commun. 2011, 409, 14–21. [Google Scholar] [CrossRef]
- Ardestani, A.; Li, S.; Annamalai, K.; Lupse, B.; Geravandi, S.; Dobrowolski, A.; Yu, A.; Zhu, S.; Baguley, T.D.; Surakattula, M.; et al. Neratinib protects pancreatic beta cells in diabetes. Nat. Commun. 2019, 10, 5015. [Google Scholar] [CrossRef] [Green Version]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Radadiya, A.; Bisson, C.; Wenzel, S.; Nordin, B.E.; Martínez-Márquez, F.; Imasaki, T.; Sedelnikova, S.E.; Coricello, A.; Baumann, P.; et al. High-resolution crystal structure of human asparagine synthetase enables analysis of inhibitor binding and selectivity. Commun. Biol. 2019, 2, 345. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of recent strategic advances in medicinal chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Cheng, S.-C.; Xu, K.; Deng, Z.; Chen, S.; Xu, Z.; Xie, K.; Tse, M.-K.; Shi, P.; et al. Phorbiplatin, a highly potent Pt(IV) antitumor prodrug that can be controllably activated by red light. Chem 2019, 5, 3151–3165. [Google Scholar] [CrossRef]
- Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Leo, L.; Brandt, A.L.; Zagzoog, A.; Black, T.; Lin, X.; Hurst, D.P.; Janero, D.R.; et al. Application of fluorine- and nitrogen-walk approaches: Defining the structural and functional diversity of 2-phenylindole class of CB1 receptor positive allosteric modulators. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Reggio, P.H.; Thakur, G.A. Identification of CB1 receptor allosteric sites using force-biased MMC simulated annealing and validation by structure-activity relationship studies. ACS Med. Chem. Lett. 2019, 10, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Da Costa, F.B.; Lopes, N.P.; Kaiser, M.; Brun, R. In silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 2014, 58, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, M.; Krauth-Siegel, R.L.; Schmidt, T.J. Natural sesquiterpene lactones of the 4,15-iso-atriplicolide type are inhibitors of trypanothione reductase. Molecules 2019, 24, 3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-dependent transporters: Regulation, structure, and function. Annu. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalde-Rico, M.; Hernando-Amado, S.; Blanco, P.; Martínez, J.L. Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front. Microbiol. 2016, 7, 1483. [Google Scholar] [CrossRef] [Green Version]
- Schalk, I.J.; Mislin, G.L.A. Bacterial iron uptake pathways: Gates for the import of bactericide compounds. J. Med. Chem. 2017, 60, 4573–4576. [Google Scholar] [CrossRef]
- Moynié, L.; Milenkovic, S.; Mislin, G.L.A.; Gasser, V.; Malloci, G.; Baco, E.; McCaughan, R.P.; Page, M.G.P.; Schalk, I.J.; Ceccarelli, M.; et al. The complex of ferric-enterobactin with its transporter from Pseudomonas aeruginosa suggests a two-site model. Nat. Commun. 2019, 10, 3673. [Google Scholar] [CrossRef]
- Gibadullina, E.; Nguyen, T.T.; Strelnik, A.; Sapunova, A.; Voloshina, A.; Sudakov, I.; Vyshtakalyuk, A.; Voronina, J.; Pudovik, M.; Burilov, A. New 2,6-diaminopyridines containing a sterically hindered benzylphosphonate moiety in the aromatic core as potential antioxidant and anti-cancer drugs. Eur. J. Med. Chem. 2019, 184, 111735. [Google Scholar] [CrossRef]
- Gong, Z.; Liu, X.; Dong, J.; Zhang, W.; Jiang, Y.; Zhang, J.; Feng, W.; Chen, K.; Bai, J. Transition from vesicles to nanofibres in the enzymatic self-assemblies of an amphiphilic peptide as an antitumour drug carrier. Nanoscale 2019, 11, 15479–15486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, X.; Zhang, J.; Li, A.; Gong, H.; Luo, Q.; Zhang, H.; Gao, Z.; Jiang, H. High-resolution mapping of brain vasculature and its impairment in the hippocampus of Alzheimer’s disease mice. Natl. Sci. Rev. 2019. [Google Scholar] [CrossRef]
- Elsinga, P.H.; van Waarde, A.; Paans, A.M.J.; Dierckx, R.A.J.O. Trends on the Role of PET in Drug Development; World Scientific Pub Co Inc.: Singapore, 2012. [Google Scholar]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Clemente, G.S.; Zarganes-Tzitzikas, T.; Dömling, A.; Elsinga, P.H. Late-stage copper-catalyzed radiofluorination of an arylboronic ester derivative of atorvastatin. Molecules 2019, 24, 4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Banerjee, S.; Qiu, K.; Zhang, P.; Blacque, O.; Malcomson, T.; Paterson, M.J.; Clarkson, G.J.; Staniforth, M.; Stavros, V.G.; et al. Targeted photoredox catalysis in cancer cells. Nat. Chem. 2019, 11, 1041–1048. [Google Scholar]
- Novotna, B.; Vaneková, L.; Zavřel, M.; Buděšínský, M.; Dejmek, M.; Smola, M.; Gutten, O.; Tehrani, Z.A.; Polidarová, M.P.; Brázdová, A.; et al. Enzymatic preparation of 2’-5’,3’-5’-cyclic dinucleotides, their binding properties to stimulator of interferon genes adaptor protein, and structure/activity correlations. J. Med. Chem. 2019, 62, 10676–10690. [Google Scholar] [CrossRef] [PubMed]
- Landi, G.; Linciano, P.; Borsari, C.; Bertolacini, C.P.; Moraes, C.B.; Cordeiro-da-Silva, A.; Gul, S.; Witt, G.; Kuzikov, M.; Costi, M.P.; et al. Structural insights into the development of cycloguanil derivatives as Trypanosoma brucei pteridine-reductase-1 inhibitors. ACS Infect. Dis. 2019, 5, 1105–1114. [Google Scholar] [CrossRef]
- Da Silva, F.; Bret, G.; Teixeira, L.; Gonzalez, C.F.; Rognan, D. Exhaustive repertoire of druggable cavities at protein–protein interfaces of known three-dimensional structure. J. Med. Chem. 2019, 62, 9732–9742. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanden Eynde, J.J.; Mangoni, A.A.; Rautio, J.; Leprince, J.; Azuma, Y.-T.; García-Sosa, A.T.; Hulme, C.; Jampilek, J.; Karaman, R.; Li, W.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–6. Molecules 2020, 25, 119. https://doi.org/10.3390/molecules25010119
Vanden Eynde JJ, Mangoni AA, Rautio J, Leprince J, Azuma Y-T, García-Sosa AT, Hulme C, Jampilek J, Karaman R, Li W, et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–6. Molecules. 2020; 25(1):119. https://doi.org/10.3390/molecules25010119
Chicago/Turabian StyleVanden Eynde, Jean Jacques, Arduino A. Mangoni, Jarkko Rautio, Jérôme Leprince, Yasu-Taka Azuma, Alfonso T. García-Sosa, Christopher Hulme, Josef Jampilek, Rafik Karaman, Wei Li, and et al. 2020. "Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–6" Molecules 25, no. 1: 119. https://doi.org/10.3390/molecules25010119
APA StyleVanden Eynde, J. J., Mangoni, A. A., Rautio, J., Leprince, J., Azuma, Y.-T., García-Sosa, A. T., Hulme, C., Jampilek, J., Karaman, R., Li, W., Gomes, P. A. C., Hadjipavlou-Litina, D., Capasso, R., Geronikaki, A., Cerchia, L., Sabatier, J.-M., Ragno, R., Tuccinardi, T., Trabocchi, A., ... Muñoz-Torrero, D. (2020). Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–6. Molecules, 25(1), 119. https://doi.org/10.3390/molecules25010119