1. Introduction
Peptides are used as therapeutic agents for controlling diseases related to peptide functions. However, the use of native peptides for clinical applications has been hampered mainly by their rapid degradation by proteases, poor oral bioavailability, difficult transportation through cell membranes and nonselective receptor binding [
1]. These limitations of peptides have led to the synthesis of peptidomimetics through numerous modifications of peptide structures [
2,
3,
4]. Among the several peptide modifications, isosteric replacement of a peptide bond represents a viable approach in the rational design of peptidomimetics. Peptidomimetics in which the peptide amide bond is replaced by ethylene (
1a)-, methyleneimino (
1b)-, methyleneoxy (
1c)- and methylenethio (
1d)-groups (
Figure 1) were found to exhibit potential pharmaceutical application [
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. Such modifications alter the physical and chemical properties of the native peptide, reducing its peptide character and leading to peptide analogues with increased resistance to proteolytic enzymes, flexibility and lipophilicity, while they also confer diverse electrostatic properties and new secondary conformations on the peptidomimetic chain, often resulting in improved pharmacokinetic properties [
2,
16,
17].
Solid-phase synthesis (SPS) is a common technique for peptide synthesis. The classical step-by-step solid-phase peptide synthesis (SPPS) is a well-established methodology for the synthesis of small to medium-sized peptides [
18]. Besides SPPS, the convergent solid-phase peptide synthesis (CSPPS) has been developed for the preparation of larger and/or complex peptides and proteins [
19,
20,
21,
22,
23,
24], based on the fact that no directional restrictions exist in CSPPS and the chain elongation can be performed with equal possibility of success to any direction. CSPPS methods include fragment condensation such as the solid- or solution-phase based protocols. Both methods consist of the rational and retro-synthetic detachment of segments in the native sequence, which in a synthetic flow would be connected in the appropriate manner in the solid- or in the solution-phase.
The very acid labile 2-chlorotrityl (Cltr) resin [
25] and the Fmoc/
tBu methodology [
23,
26] have been developed and widely used for SPPS and CSPPS of protected peptides. In general the sequential condensation of fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (
tBu) protected peptide fragments on a solid support to larger peptides (convergent synthesis, solid phase fragment condensation (SPFC)) is a simple method for the preparation of large or difficult peptides by convergent methods [
27,
28]. The main limitation of this method is that, in some cases, the
N-Fmoc deprotection of the resin-bound peptide and the subsequent condensation with the next fragment proceeds slowly and incomplete. A solution to this problem is provided with the cleavage of the protected peptide from the resin in order to perform the
N-Fmoc removal and the condensation in solution. This type of condensation is also advantageous from an economical point of view, since the C- and
N-components are used in equimolar amounts. The key step of this method is the esterification of the protected peptide, which is cleaved from the resin, at the C-terminal carboxyl function, and for this reason, the usefulness of 2-chlorotrityl chloride monomer as a temporary carboxy-protecting group has been studied in CSPPS [
24].
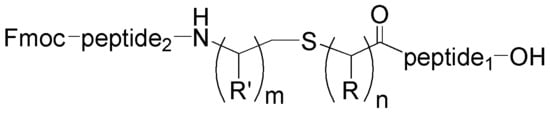
In this manuscript, we describe our efforts in the synthesis of peptidomimetics of type
2 (
Figure 1), by using simple methods of CSPPS. In particular, we describe three methods in the convergent synthesis of type
2d peptide isosteres, in which the amide bond is replaced by CH
2-S isostere bond (thioether bond), using methods of solid phase peptide fragment condensation or solution fragment condensation through the formation of an amide bond, and the condensation of two fragments through the formation of a thioether bond.
2. Results and Discussion
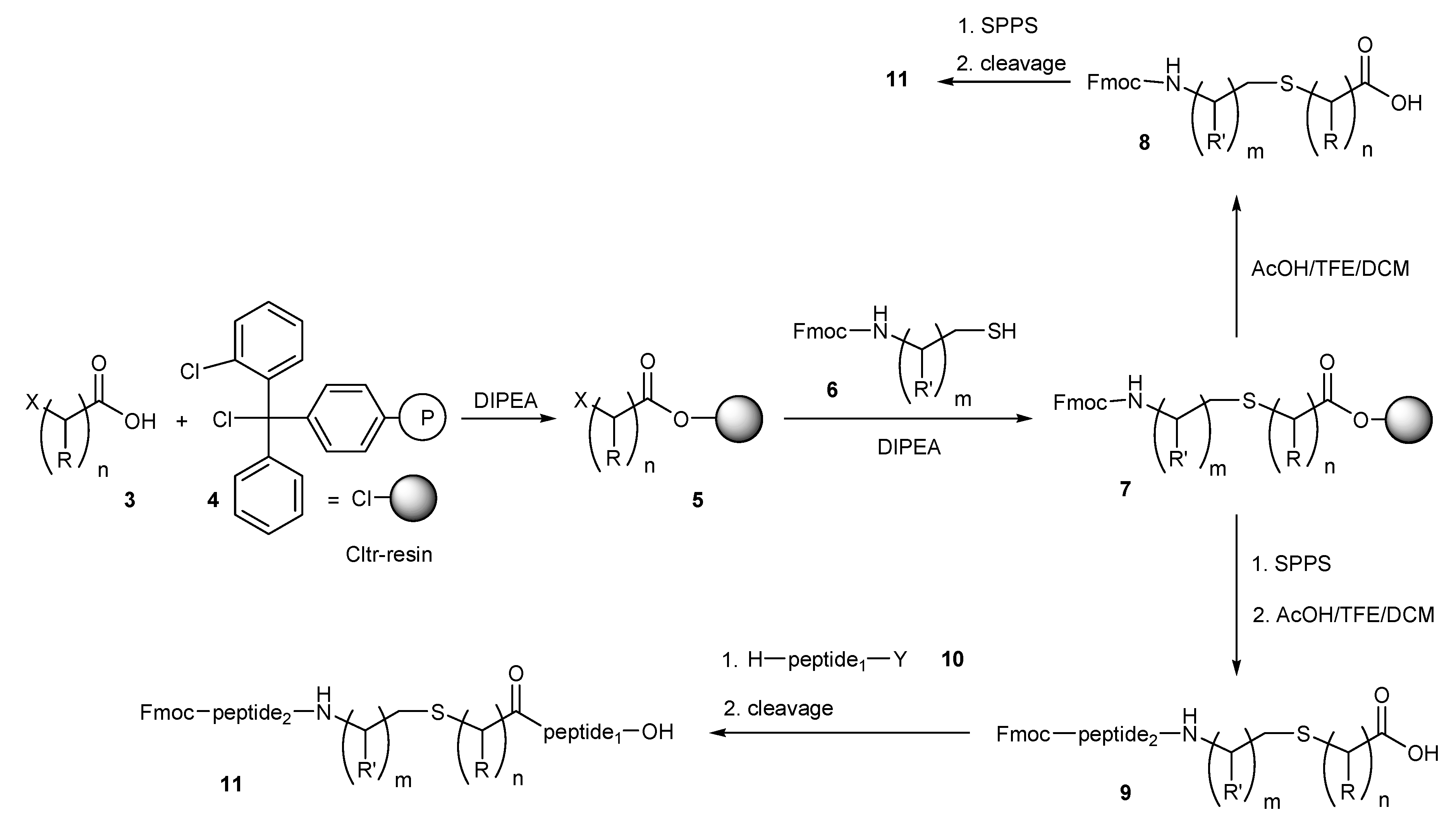
In an effort to apply standard methods of convergent solid phase peptide synthesis (CSPPS) [
21,
24,
26] in the preparation of the thioether containing peptides
11 (
Scheme 1), we esterified the halo acids
3 with 2-chlorotrityl(Cltr) resin
4 in dichloromethane (DCM) using
N,
N-diisopropylethylamine (DIPEA) as the hydrogen halide acceptor. The reaction proceeds fast independently from the bulkiness of
3 and resins containing 0.6–1.0 mmol
3/g were obtained. The loading of the resins
5 was determined by Gas Chromatography (GC) after cleavage of the haloacids by treatment of resin probes with 3% trifluoroacetic acid (TFA) in DCM for 30 min at room temperature. The obtained haloacid derivatives
5 were further reacted with a two-fold molar excess of the
N-fluorenylmethoxycarbonyl (Fmoc) aminothiols
6 [
29] and DIPEA in dimethylformamide (DMF) for 1–12 h at room temperature. Especially the more electrophilic α-haloacids, such as the bromoacetic acid, react very fast with the less hindered Fmoc-aminothiols
6 (in less than 30 min) to the resin-bound (
N-Fmoc aminothiol)carboxylic ester
7. Treatment of
7 with the cleavage mixture acetic acid (AcOH)/trifluoroethanol (TFE)/dichloromethane (DCM) (1:2:7) for 30 min at room temperature followed by extractive work up gave the key building blocks
8, which were applied in usual step by step solid phase peptide synthesis (SPPS), by Fmoc/tert-butyl (
tBu) methodology leading to the target peptides
11. Instead of using
8 in SPPS, direct peptide elongation on resin
7, by standard SPPS and cleavage from Cltr-resin by the cleavage mixture AcOH/TFE/DCM (1:2:7) gave the thiol-containing peptide
9. The obtained segment
9 can be condensed with the amino components of the peptide fragment
10 either in solution (in case of
O-Cltr protected peptide fragment
10a) [
24] by using solution fragment condensation strategy, or in solid phase (in case of resin-bound peptide fragments
10b). In the first case, fragment condensation of
9 with the
O-Cltr protected monomer
10a requires an equimolar amount of
9, while in the second case condensation of
9 with the
N-free amino group of peptides attached on the Cltr-resin
10b requires a two-fold molar excess of
9 to be completed.
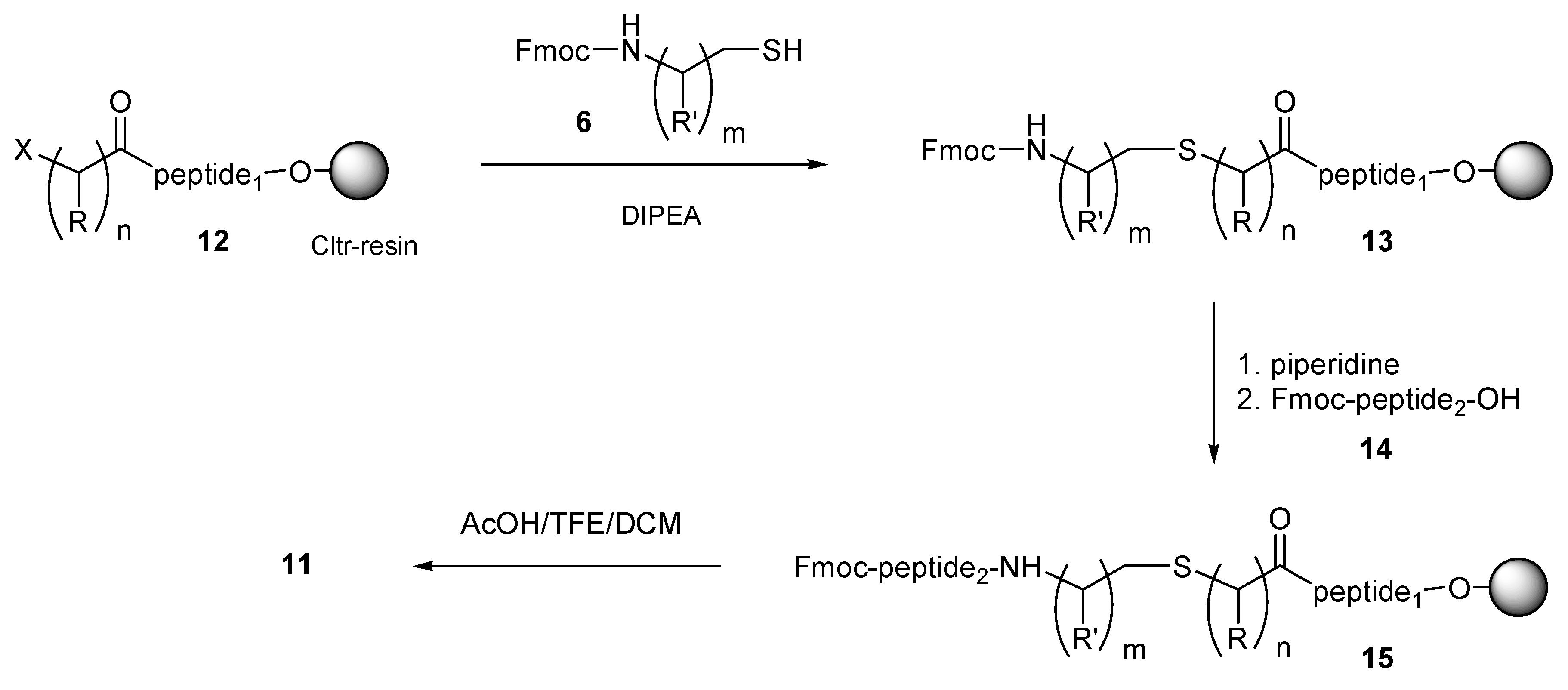
By applying a similar method, esterification of the halo acid
3 with the
N-free amino group of peptides elongated on Cltr-resin gave the resin-bound haloacylated peptide
12 (
Scheme 2). For this synthesis step by step SPPS (Fmoc/
tBu methodology) was used. The resin-bound haloacylated peptide
12 was then condensed with a two-fold molar excess of the
N-Fmoc aminothiol
6 and DIPEA in DMF for 1–12 h at room temperature to the resin-bound peptide
13. Removal of the
N-Fmoc group by piperidine liberated the amino group of the peptide chain, enabling the peptide elongation either by standard SPPS or by CSPPS using the
N-Fmoc protected peptide fragment
14. The obtained resin-bound thiol-containing peptide
15 was finally treated with the cleavage mixture AcOH/TFE/DCM (1:2:7) to the thioether containing peptide
11.
In order to evaluate the applicability of the above described methods in SPPS, we synthesized the simple thiolether containing peptide
16 (by using route A) and
17 (by using route B;
Figure 2). Their synthesis was done on Cltr-resin by using Fmoc/
tBu strategy, according to
Scheme 1 and
Scheme 2, respectively.
In brief, amino acid couplings were achieved by using 1-hydroxybenzotriazole/
N,
N′-diisopropylcarbodiimide (HOBt/DIC) in
N-methyl-2-pyrrolidone (NMP). As the
N-Fmoc aminothiols
6, the corresponding
N-Fmoc aminobutanethiol and
N-Fmoc aminopropanethiol derivatives were used. Both thioether containing peptides (
16 and
17) were prepared in high yield (87 and 90% yield, respectively) and purity (95 and 98%, respectively) as confirmed by hplc analysis (see
Figure S1 in Supplementary Materials section), revealing that these methods could be used for the preparation of larger thioether containing peptides. The correct mass of peptide analogues
16 and
17 was determined by ESI-MS analysis (
16: [M + H] calc.: 641.28; found: 641.42 and
17: [M + H] calc.: 712.31; found: 712.56). In this method, although the thioether formation between the
N-Fmoc aminothiol
6 and the haloacylated peptide
12 proceeds relatively fast, the fragment coupling of the resin-bound
N-Fmoc deprotected thiol-peptide
13 with the
N-Fmoc peptide fragment
14, proceeds slowly (6–12 h) and in some cases is incomplete, similarly to route A.
Since the condensation of the different fragments in both routes A (either by using 8 in SPPS, or by fragment coupling between 9 and 10) and B (fragment coupling between the N-Fmoc deprotected 13 and 14) proceeds through amide formation, which is relatively slow, we proposed a third method (route C), for the synthesis of thioether containing peptides 11, which enables the fast condensation reaction between two peptide fragments.
In this method (route C), the synthesis of thioether containing peptides
11 does not proceed through amide bond formation (as in routes A and B), but through thioether bond formation, by the nucleophilic attack of the
N-Fmoc protected thiol-peptide
20 with the resin-bound bromoacetylated peptide
12 (
Scheme 3). The first step in this procedure is the preparation of the thiol-peptide
20. For this, we used either aminothiols attached on 4-methoxytrityl(Mmt) resin through their thiol function
18 [
29], or resin-bound bis(aminoalkyl)dithiol attached on trityl(Trt) resin through their free amino group
19. Peptide elongation was achieved by using standard SPPS (Fmoc/
tBu strategy), while amino acid coupling was achieved by using HOBt/DIC in NMP. The derived resin bound peptides were then quantitatively cleaved with 1% TFA in presence of DCM/triethylsilane (TES) (95:5) (in case of peptides elongated on resins
18), and dithiothreitol (DTT) or TES (in case of peptides elongated on resins
19), to the
N-Fmoc thiol-peptides
20 (
Scheme 3). In the next step of the synthesis, the
N-Fmoc protected thiol-peptide fragments
20 were condensed with the resin-bound halo acylated peptides
12 in DMF and DIPEA and the obtained resin-bound thioether peptides were completely cleaved from the Cltr-resin by treatment with AcOH/TFE/DCM (1:2:7) for 15 min at room temperature. It was noticed that the condensation between the bromoacetylated peptide segments
12 and the
N-Fmoc thiol-peptides
20 proceeded with exceptional ease and in some cases very fast (10 min), as it is shown below, in the example of choice (thioether containing peptide
26).
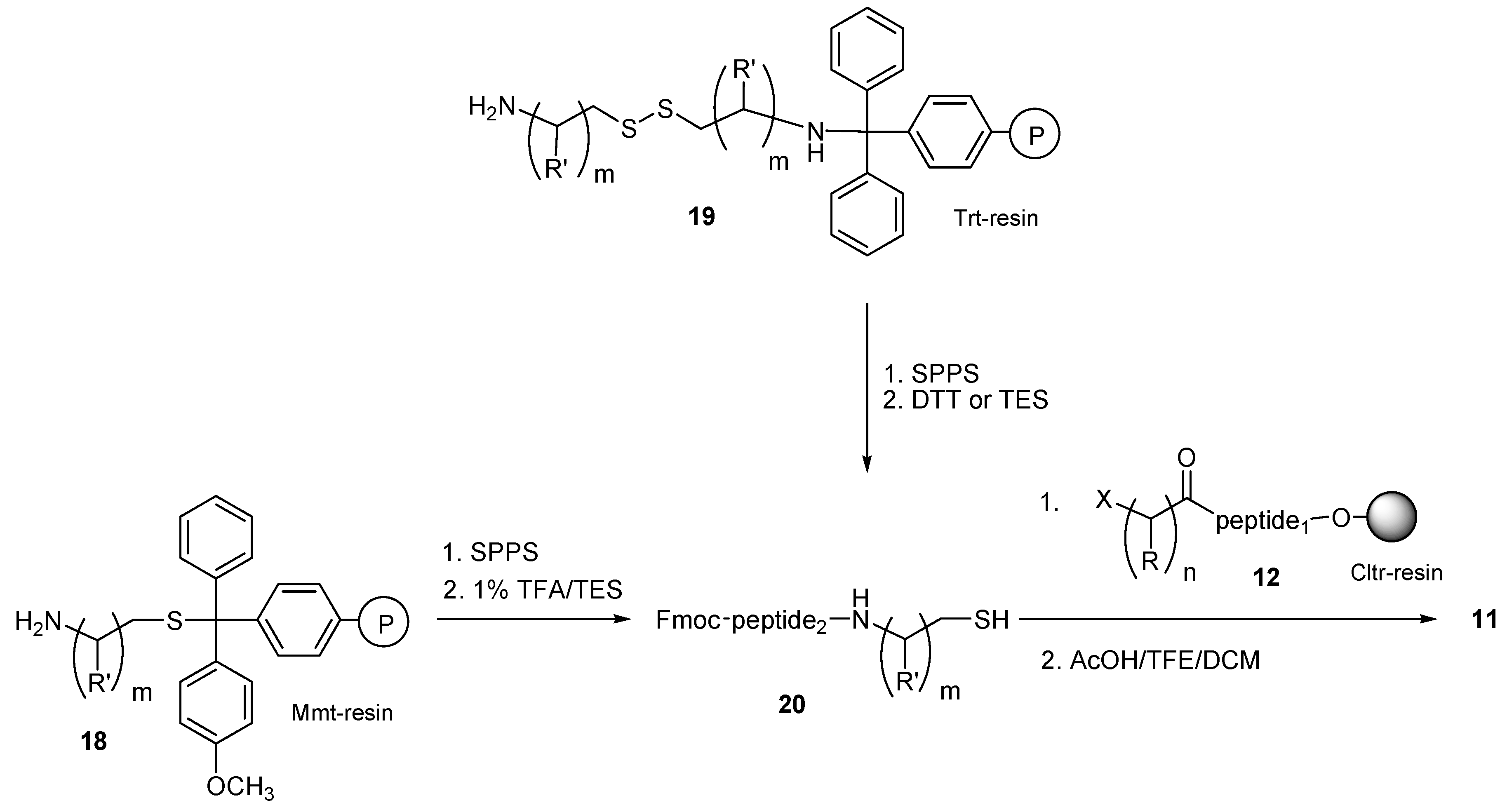
Although we have already published the advantages of resin bound aminothiols
18 (loaded through their thiol group on Mmt-resins) in the synthesis of
N-Fmoc thiol-containing peptides [
29], the use of bis(aminoalkyl)disulfides bound on Trt-resins
19 (through their amine group) in SPPS is described for the first time. These resins may offer several advantages in SPS, SPPS and CSPPS, based on their ability to form either thiol-peptides by reductive cleavage, or bis(aminoalkyl)thiol peptides by treatment of the resin with 1% TFA in presence of DCM/TES (95:5). The first case (reductive cleavage of thiol-peptides
20 from resins
19) can be used for the synthesis of the
N-Fmoc protected thiol-peptides
20, by their treatment with DTT or TES, which are considered as mild reducing agents. Thus, in an effort to optimize the cleavage of the thioether containing peptides
11 synthesized on Trt-resins
19, we compared the use of DTT or TES in terms of yield and purity. For this, the resin-bound bis(aminoalkyl)dithiol derivative (R′ = CH
3)
19 was coupled with
N-Fmoc-leucine by using HOBt/DIC as the carboxylic acid activator. The derived resin was treated with a 10 fold molar excess of DTT or TES. In both cases the expected Fmoc-Leu-alaninothiol was completely cleaved from the resin after 12 h reaction at room temperature. Hplc analysis of the crude product showed that the use of TES is superior in terms of product purity, as a purity of 67% was noticed for the crude product of Fmoc-Leu-alaninothiol cleaved by DTT, while a purity of 98% was noticed for Fmoc-Leu-alaninothiol cleaved by TES (see
Figure S2 in Supplementary Materials section). The correct mass of the released Fmoc-Leu-alaninothiol was determined by ESI-MS analysis ([M + H] calc.: 427.21; found: 427.32). For this reason, TES was used for the cleavage of the more complicated peptides that were prepared by this route.
As an example of the more complicated thiol-containing peptides that we prepared by the use of resin
19, we describe the synthesis of two thiol-peptide analogues:
21 (
Figure 3; synthesized on Trt-resin
19 where R′ = CH
3) and
22 (
Figure 3; synthesized on Trt-resin
19 where R′ = –CH
2–Ph).
The first product (peptide structure
21) is a thiol derivative of the prothymosin a peptide sequence (ProTa (69-75)) prepared in high yield (82%) and purity (>98%), as can be seen by the hplc analysis of the crude product (see
Figure S3A in Supplementary Materials section). Its correct mass was determined by ESI-MS analysis (see
Figure S3B in Supplementary Materials section): ([M + H] calc.: 1401.69; found: 1401.59). The second product (peptide structure
22) is a peptide derivative of hirudine (Hir (11-18)), which contains two differently protected cysteines: Cys14(
Mmt) and Cys16
(Acm). By using this resin, we were able to synthesize the thiol-peptide
22, after its cleavage from the Trt resin
19 by TES, without affecting the Cys14
(Mmt) protecting group. This product was also prepared in high yield (78%) and purity (90%), as can be seen by the hplc analysis (see
Figure S3C in Supplementary Materials section), while its correct mass was determined by ESI-MS: ([M + 2H] calc.: 1029.46; found: 1029.86 (see
Figure S3D in Supplementary Materials section)).
As an example of the final step of our strategy for the synthesis of thioether containing peptides
11 by route C, we present the synthesis of the cysteamine derivative
26, which is composed by two prothymosin a segments (ProTa (69-75)) linked by a thioether bond (
Scheme 4). For this synthesis, the resin-bound bromoacetylated peptide
23 was initially prepared on Cltr-resin by using standard SPPS methods (Fmoc/
tBu method and HOBt/DIC for the amino acid activation). Then a two-fold molar excess of
24 was condensed with
23 in DMF/DIPEA for 10 min at room temperature (
Scheme 4). The resin
25 was treated with AcOH/TFE/DCM (1:2:7) and the protected peptide
26 was obtained in 90% yield and 92% purity according to HPLC analysis (see
Figure S4 in Supplementary Materials section). The correct mass of the obtained peptide
26 was determined by ES-MS analysis ([M + 2H] calc.: 1191.60; found: 1191.80).
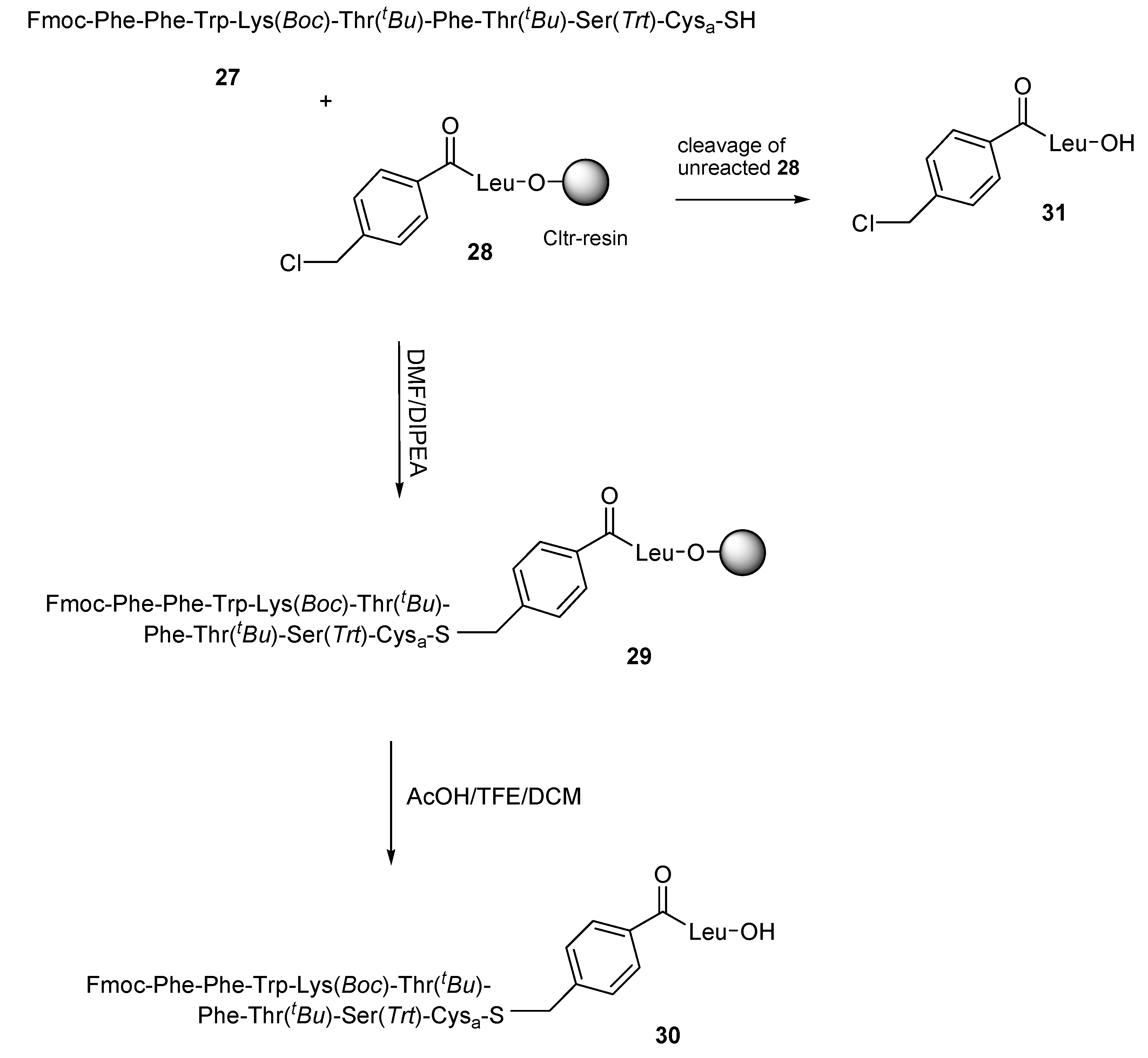
Ligation strategies on haloacylated peptides have been found to proceed very fast in case of bromoacylated peptides, while in case of chloroacylated peptides moderate reactivities were found [
30,
31]. In order to examine this parameter, we planned the fragment condensation of thiol-peptides to chloroacylated peptides. As an example we synthesized the somatostatin analogue (6-14) where Cys14 was replaced by cysteamine (Cys
a; 2-aminoethanethiol)
27. In this experiment, the thiol-peptide
27 contains the acid sensitive Ser
(Trt) (
O-Trt is cleaved by 1% TFA) and for this reason it was synthesized on Trt-resin
19. The thiol-peptide
27 was condensed with the 4-chloromethylbenzoyl-Leu-O-Cltr resin
28 (
Scheme 5) in a 1.5:1 molar ratio in DMF and DIPEA and the reaction process was followed by hplc analysis, after the treatment of resin probes with AcOH/TFE/DCM (1:2:7) for 15 min at room temperature, by which the desired product
30 and the un-reacted
31 were identified. Analysis of the hplc chromatograms (see
Figure S5 in Supplementary Materials section) showed a high percentage of un-reacted
31 (absorbance ratios of
30/31: 46/54) after 2 h reaction time, due to incomplete reaction of
27 with
28, while a significant percentage of
31 (absorbance ratios of
30/31: 82/18) was still observed even after 24 h reaction. It should be noted that, although the reaction progress was rather slow, no significant by-products were detected during the extended reaction time. Product
30 was identified by ESI-MS ([M − Trt + 2H] calc.: 902.46; found: 903.38; (see
Figure S5 in Supplementary Materials section).
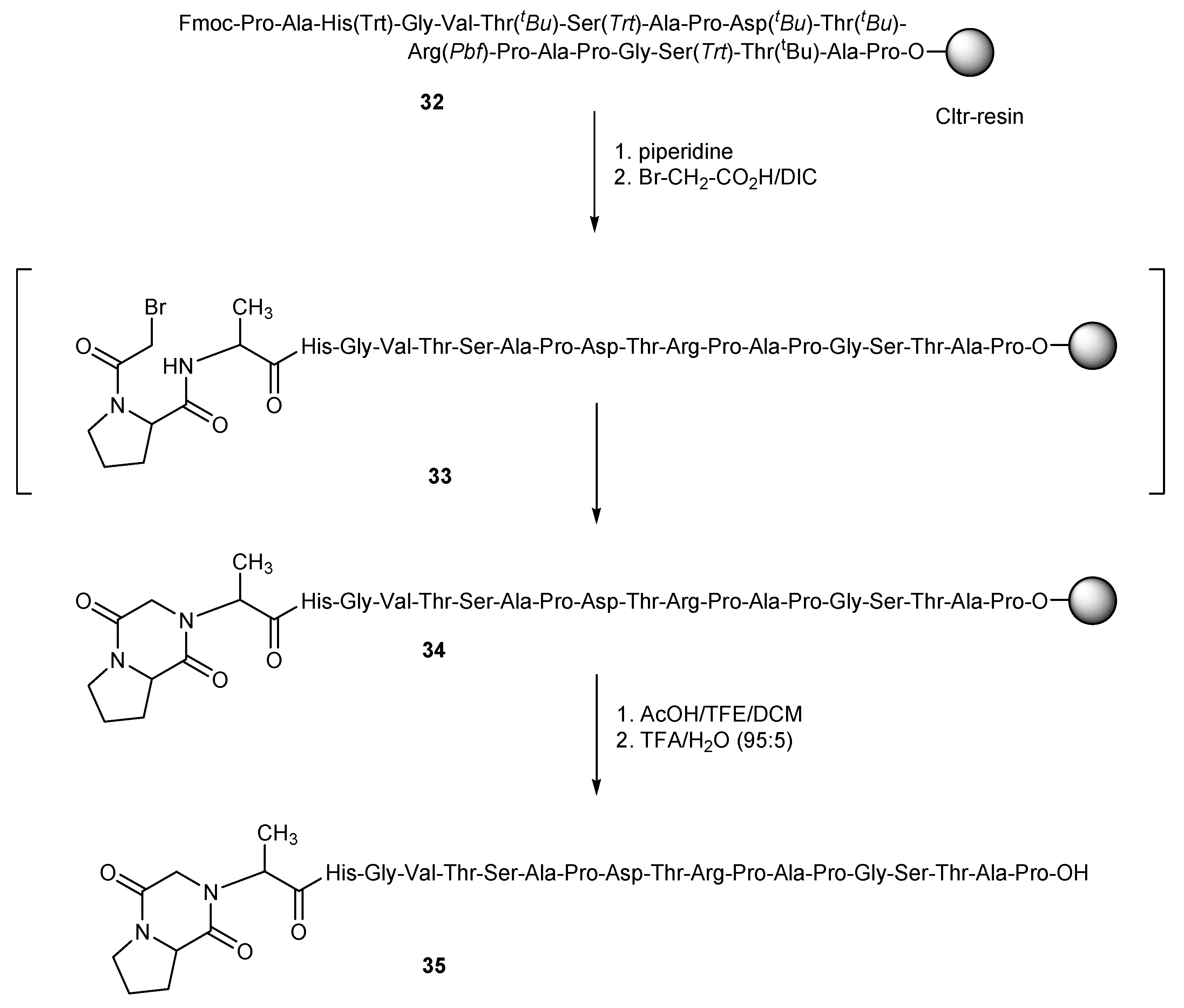
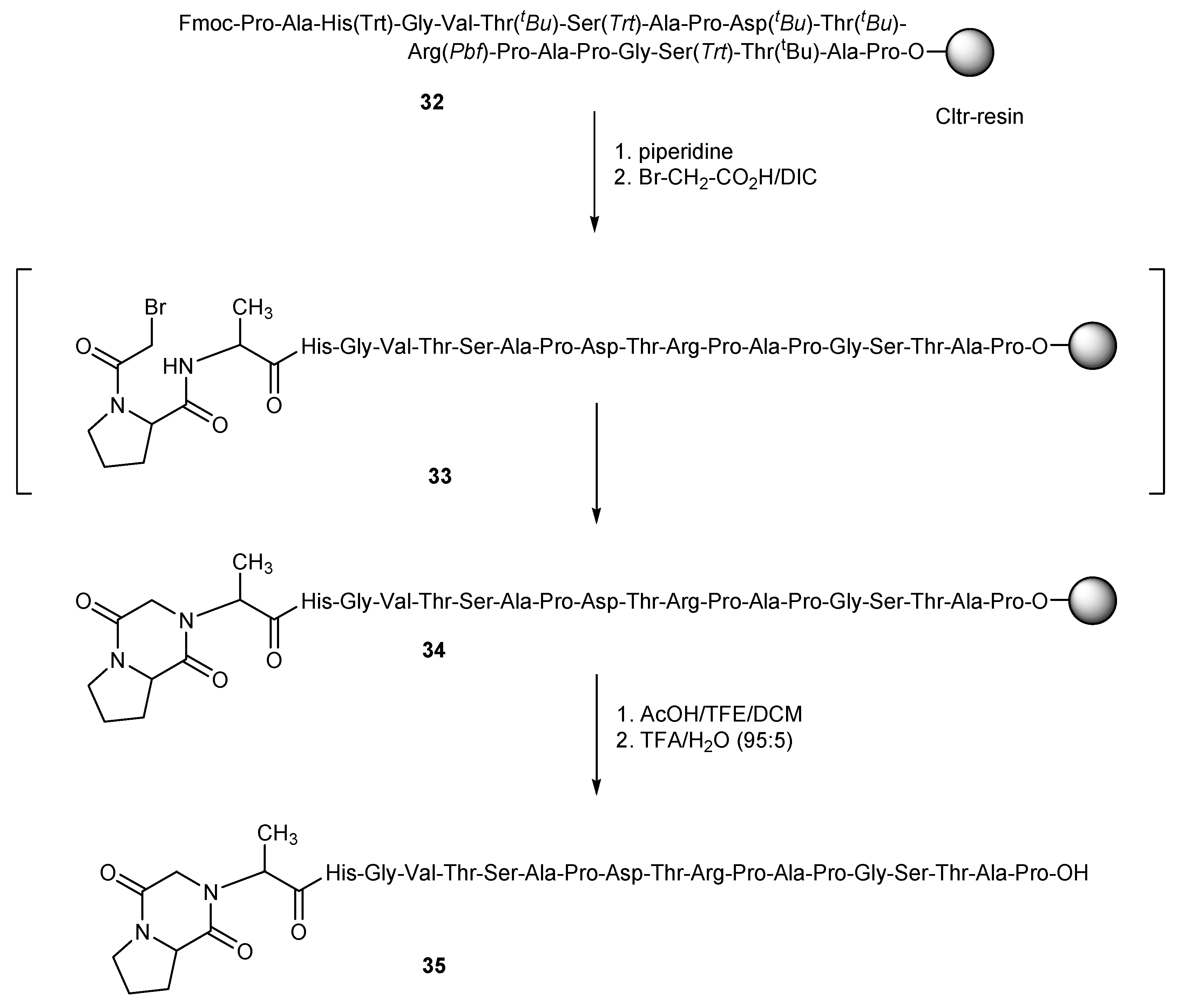
In case of haloacetyl-peptides, which contain various strong nucleophiles, one should be aware of possible side reactions of the nucleophiles with the haloacetyl moiety. This is a well-known problem especially for bromoacetylated-methionine (Met)-peptides [
31,
32,
33]. In this work we found similar instability issues for bromoacetylated-proline (Pro)-peptides. As an example, resin-bound MUC-1
32 was treated with piperidine to liberate the
N-terminus of the peptide sequence and this was reacted with a three-fold molar excess bromoacetic acid and DIC in NMP (
Scheme 6). After cleavage of the obtained peptide from the resin and deprotection with TFA/water (95:5) for 3 h at room temperature, the main product of the synthesis, instead of the expected haloacylated peptide, was a product with a molecular mass [M − 81] ([M − 81 + 2H]: calc.: 963.97; found: 964.09). This corresponds to a peptide with one less HBr, which was attributed to the diketopiperazine peptide derivative
35, obviously prepared by the nucleophilic attack of the
N-terminus group of alanine to the carbon atom that bears the bromine. This nucleophilic attack is possibly favored by the proximity of these atoms, which is due to the presence of proline just before the haloacid in the peptide chain (
Scheme 6).
4. Conclusions
Besides classical step-by-step synthesis, the convergent solid phase peptide synthesis (CSPPS) was used for the preparation of complex and difficult peptides and small proteins.
Peptides where the amide bond is replaced by its CH2–S isostere (thioether containing peptides) are very important peptide mimetics. Thus, we were interested in the synthesis of thioether containing peptides by convergent methods.
Comparing the methods discussed, route C (
Scheme 3) offers significant advantages, over routes A and B, for the preparation of thioether containing peptides, mainly because the condensation of the thiol-peptide fragment
20 with the resin-bound bromoacetyl peptide
12 proceeds faster than: (i) the use of the thioether containing building block
8 in SPPS (
Scheme 1; route A), or (ii) the condensation of the thioether containing peptide
9 with the peptide fragment
10 (
Scheme 1; route A) or (iii) the condensation of the thiol-containing peptide fragment
14 with the peptide fragment
15 (
Scheme 2; route B).
Regarding the synthesis of the thiol-peptide fragment 20, in case that acid sensitive (cleaved under acidic solutions ≤1% TFA) amino acid side chain protecting groups are needed in the peptide sequence, we suggest its synthesis through reductive cleavage from resin 19, instead of its acidic cleavage from resin 18 (by which the acid labile protecting group would be cleaved).
In contrary, routes B and C, which were based on the synthesis of the resin-bound haloacylated peptide 12, have to be avoided when the sequence of 12 contains various strong nucleophiles, which may react with the haloacetyl moiety. This is a well-known problem for bromoacetylated-Met-peptides and we proved that bromoacetylated-Pro-peptides also suffered from instability issues, by their conversion to the corresponding diketopiperazine peptide derivatives. In this case, route A should be preferred, as in this route the thioether formation is performed on the resin-bound esterified haloacid 5, thus, amino acids have not yet been introduced into the peptide sequence, avoiding the nucleophilic attack on the haloacetyl moiety by the nucleophilic groups that are present in the peptide chain of routes B and C.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}