Two-Phase Dibromocyclopropanation of Unsaturated Alcohols Using Flow Chemistry

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Representative Procedure, Synthesis of (2,2-Dibromo-3,3-Dimethylcyclopropyl)Methanol (1)
3.3. Synthesis of 5-(2,2-Dibromo-3,3-Dimethylcyclopropyl)-3-Methyl-1-Penten-3-Ol (2)
3.4. Synthesis of 5-(2,2-Dibromo-3,3-Dimethylcyclopropyl)-3-Methylpentan-1-Ol (4)
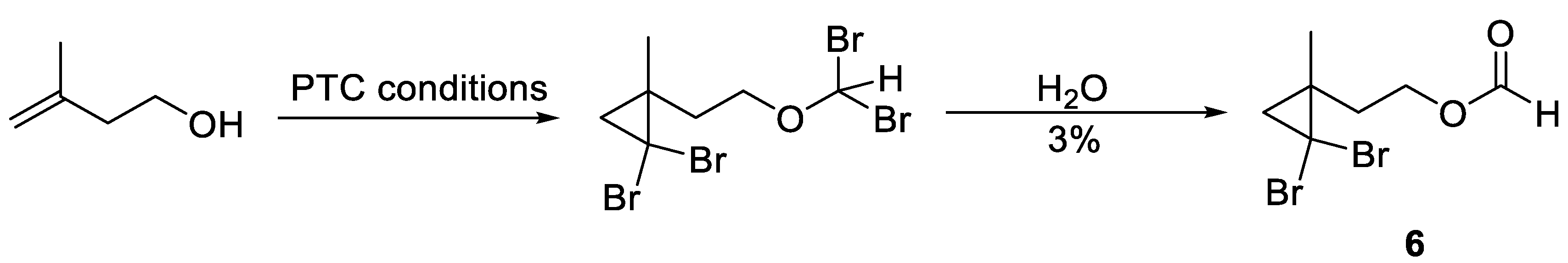
3.5. Synthesis of 2-(2,2-Dibromo-1-Methylcyclopropyl)Ethan-1-Ol (5) and 2-(2,2-Dibromo-1-Methylcyclopropyl)Ethyl Formate (6)
3.6. Synthesis of 4-(2,2-Dibromo-3,3-Dimethylcyclopropyl)Butan-2-Ol (7)
3.7. Synthesis of (2,2-Dibromo-1,3-Dimethylcyclopropyl)Methanol (9)
3.8. Synthesis of 1-(2,2-Dibromo-1-Methylcyclopropyl)Ethanol (10)
3.9. Synthesis of (2,2-Dibromo-3-Methyl-3-Phenylcyclopropyl)Methanol (16)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Skattebøl, L. The synthesis of allenes from 1,1-dihalocyclopropane derivatives and alkyllithium. Acta Chem. Scand. 1963, 17, 1683–1693. [Google Scholar] [CrossRef]
- Kleveland, K.; Skattebøl, L. Chemistry of gem-dihalocyclopropanes. IX. Stereospecific synthesis of conjugated meso-diallenes. Acta Chem. Scand. Ser. B 1975, B29, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Skattebøl, L. Novel route to cumulenes. Addition of dihalocarbenes to 2,5-dimethyl-2,3,4-hexatriene. Tetrahedron Lett. 1965, 6, 2175–2179. [Google Scholar] [CrossRef]
- Holm, K.H.; Mohamed, E.A.; Skattebøl, L. Chemistry of dihalocyclopropanes. XXV. The influence of halogen substituents on the vinylcyclopropylidene-cyclopentenylidene rearrangement. Acta Chem. Scand. 1993, 47, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Holm, K.H.; Skattebøl, L. The vinylcyclopropylidene - cyclopentadiene rearrangement. Tetrahedron Lett. 1977, 18, 2347–2350. [Google Scholar] [CrossRef]
- Arct, J.; Skattebøl, L.; Stenstrøm, Y. Chemistry of gem-dihalocyclopropanes. XIX. Formation of 3-oxabicyclo[3.1.0]hexane derivatives by intramolecular insertion of cyclopropylidenes. Acta Chem. Scand. Ser. B 1983, B37, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Skattebøl, L.; Stenstrøm, Y.; Stjerna, M.B. Chemistry of gem-dihalocyclopropanes. XXIII. Reaction of some gem-dibromocyclopropyl acetals with methyllithium. Acta Chem. Scand. Ser. B 1988, B42, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Fedorynski, M. Syntheses of gem-dihalocyclopropanes and their use in organic synthesis. Chem. Rev. 2003, 103, 1099–1132. [Google Scholar] [CrossRef]
- Banwell, M.G.; Beck, D.A.S.; Stanislawski, P.C.; Sydnes, M.O.; Taylor, R.M. Pyrroles and gem-dihalocyclopropanes as building blocks for alkaloid synthesis. Curr. Org. Chem. 2005, 9, 1589–1600. [Google Scholar] [CrossRef]
- Banwell, M.G.; Lehmann, A.L.; Menon, R.S.; Willis, A.C. New methods for the synthesis of certain alkaloids and terpenoids. Pure Appl. Chem. 2011, 83, 411–423. [Google Scholar] [CrossRef]
- Doering, W.V.E.; Hoffmann, A.K. The addition of dichlorocarbene to olefins. J. Am. Chem. Soc. 1954, 76, 6162–6165. [Google Scholar] [CrossRef]
- Ma̧kosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662. [Google Scholar] [CrossRef]
- Makosza, M.; Fedorynski, M. Phase transfer catalysis in dichlorocarbene chemistry: Basic principles and specific features. Russ. Chem. Bull. 2011, 60, 2141–2146. [Google Scholar] [CrossRef]
- Kleveland, K.; Skattebøl, L.; Sydnes, L.K. Chemistry of gem-dihalocyclopropanes. XI. Reactions of dihalocarbenes with allylic alcohols. Acta Chem. Scand. Ser. B 1977, B31, 463–468. [Google Scholar] [CrossRef]
- Bolesov, I.G.; Solov’eva, V.A.; Baird, M.S. Functionalized α-bromocyclopropylmagnesium bromides: Generation and some reactions. Russ. J. Org. Chem. 2013, 49, 1580–1593. [Google Scholar] [CrossRef]
- Piers, E.; Morton, H.E.; Nagakura, I.; Thies, R.W. Seven-membered ring annulation and spiro-annulation processes via preparation and thermal rearrangement of β-(2-vinylcyclopropyl) α,β-unsaturated ketones. Can. J. Chem. 1983, 61, 1226–1238. [Google Scholar] [CrossRef]
- Skattebøl, L.; Aziz Abskharoun, G.; Greibrokk, T. Chemistry of gem-dihalocyclopropanes VIII A convenient preparation of gem-dibromocyclopropanes. Tetrahedron Lett. 1973, 14, 1367–1370. [Google Scholar] [CrossRef]
- Sirovski, F.; Gorokhova, M.; Ruban, S. Phase-transfer catalysis: Kinetics and mechanism of dichlorocyclopropane formation in liquid/liquid and solid/liquid systems. J. Mol. Catal. A Chem. 2003, 197, 213–222. [Google Scholar] [CrossRef]
- Starks, C.M. Interfacial area generation in two-phase systems and its effect on kinetics of phase transfer catalyzed reactions. Tetrahedron 1999, 55, 6261–6274. [Google Scholar] [CrossRef]
- Dummann, G.; Quittmann, U.; Groschel, L.; Agar, D.W.; Worz, O.; Morgenschweis, K. The capillary-microreactor: A new reactor concept for the intensification of heat and mass transfer in liquid-liquid reactions. Catal. Today 2003, 79–80, 433–439. [Google Scholar] [CrossRef]
- Jas, G.; Kirschning, A. Continuous flow techniques in organic synthesis. Chem. Eur. J. 2003, 9, 5708–5723. [Google Scholar] [CrossRef]
- Geyer, K.; Codee, J.D.C.; Seeberger, P.H. Microreactors as tools for synthetic chemists - the chemists’ round-bottomed flask of the 21st century? Chem. Eur. J. 2006, 12, 8434–8442. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P. Continuous flow reactors, a tool for the modern synthetic chemist. Eur. J. Org. Chem. 2008, 2008, 1655–1671. [Google Scholar] [CrossRef]
- Valera, F.E.; Quaranta, M.; Moran, A.; Blacker, J.; Armstrong, A.; Cabral, J.T.; Blackmond, D.G. The Flow’s the Thing···Or Is It? Assessing the Merits of Homogeneous Reactions in Flask and Flow. Angew. Chem. Int. Ed. 2010, 49, 2478–2485. [Google Scholar] [CrossRef]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef]
- Burns, J.R.; Ramshaw, C. The intensification of rapid reactions in multiphase systems using slug flow in capillaries. Lab Chip 2001, 1, 10–15. [Google Scholar] [CrossRef]
- Kashid, M.N.; Gerlach, I.; Goetz, S.; Franzke, J.; Acker, J.F.; Platte, F.; Agar, D.W.; Turek, S. Internal Circulation within the Liquid Slugs of a Liquid-Liquid Slug-Flow Capillary Microreactor. Ind. Eng. Chem. Res. 2005, 44, 5003–5010. [Google Scholar] [CrossRef]
- Zhang, L.; Geng, M.; Teng, P.; Zhao, D.; Lu, X.; Li, J.-X. Ultrasound-promoted intramolecular direct arylation in a capillary flow microreactor. Ultrason. Sonochem. 2012, 19, 250–256. [Google Scholar] [CrossRef]
- Sinkovec, E.; Krajnc, M. Phase Transfer Catalyzed Wittig Reaction in the Microtube Reactor under Liquid-Liquid Slug-Flow Pattern. Org. Process Res. Dev. 2011, 15, 817–823. [Google Scholar] [CrossRef]
- Østby, R.B.; Stenstrøm, Y.H.; Didriksen, T. The use of flow chemistry for two-phase dibromocyclopropanation of alkenes. J. Flow Chem. 2015, 5, 69–73. [Google Scholar] [CrossRef] [Green Version]
- von Keutz, T.; Cantillo, D.; Kappe, C.O. Enhanced mixing of biphasic liquid-liquid systems for the synthesis of gem-dihalocyclopropanes using packed bed reactors. J. Flow Chem. 2019, 9, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Richey, H.G., Jr.; Moses, L.M. Configurational stability of a cyclopropyl Grignard reagent containing a metalated 2-hydroxymethyl group. J. Org. Chem. 1983, 48, 4013–4017. [Google Scholar] [CrossRef]
- Sydnes, L.K.; Skare, S. Reactions of 2,2-dibromocyclopropyl carboxylic acids with methyllithium. Can. J. Chem. 1984, 62, 2073–2078. [Google Scholar] [CrossRef]
- Holm, K.H.; Skattebøl, L. Chemistry of gem-dihalocyclopropanes. XX. The effect of methyl and phenyl substituents on the vinylcyclopropylidene-cyclopentenylidene rearrangement. Acta Chem. Scand. Ser. B 1984, B38, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Nishii, Y.; Matsumura, H.; Muroya, Y.; Tsuchiya, T.; Tanabe, Y. Novel synthetic pyrethroids containing a halocyclopropane structure. Biosci. Biotechnol. Biochem. 1995, 59, 1355–1357. [Google Scholar] [CrossRef]
- Molchanov, A.P.; Kostikov, R.R. Reaction of dichlorocarbene with allyl and cinnamyl alcohols. Zh. Org. Khim. 1987, 23, 69–71. [Google Scholar]
- Skell, P.S.; Garner, A.Y. Reactions of bivalent carbon compounds. Reactivities in olefin-dibromocarbene reactions. J. Am. Chem. Soc. 1956, 78, 5430–5433. [Google Scholar] [CrossRef]
- Jonczyk, A.; Fedorynski, M. Dihalocarbenes or Equivalents. In Houben-Weyl-Methods of Organic Chemistry: Cyclopropanes: A. Synthesis; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1997; Volume E 17a, pp. 589–734. [Google Scholar]
- Kobayashi, Y.; Yoshida, S.; Nakayama, Y. Total synthesis of korormicin. Eur. J. Org. Chem. 2001, 2001, 1873–1881. [Google Scholar] [CrossRef]
- Yadav, J.S.; Yadav, N.N.; Reddy, B.V.S. Studies directed towards the total synthesis of polyether antibiotic ionomycin: Asymmetric synthesis of fragments C(24)-C(32) and C(1)-C(23). Tetrahedron 2015, 71, 7539–7549. [Google Scholar] [CrossRef]
- Kumar, H.M.S.; Reddy, B.V.S.; Reddy, E.J.; Yadav, J.S. Iodine-catalyzed mild and efficient tetrahydropyranylation/depyranylation of alcohols. Chem. Lett. 1999, 857–858. [Google Scholar] [CrossRef]
- Jung, Y.H.; Kim, J.D. One-pot synthesis of cinnamyl amines with various protecting groups from cinnamyl ethers. Arch. Pharmacal Res. 2001, 24, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, U.; Knauer, L.; Strohmann, C. The Reactivity of Benzyl Lithium Species is Regulated by Intermediate Structures. Angew. Chem. Int. Ed. 2017, 56, 6232–6235. [Google Scholar] [CrossRef] [PubMed]
- Wijtmans, M.; Maussang, D.; Sirci, F.; Scholten, D.J.; Canals, M.; Mujic-Delic, A.; Chong, M.; Chatalic, K.L.S.; Custers, H.; Janssen, E.; et al. Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur. J. Med. Chem. 2012, 51, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.H.; Perkins, R.J.; Smith, J.A.; Moeller, K.D. Solvolysis, Electrochemistry, and Development of Synthetic Building Blocks from Sawdust. J. Org. Chem. 2015, 80, 11953–11962. [Google Scholar] [CrossRef] [PubMed]
- Sheshenev, A.E.; Baird, M.S.; Bolesov, I.G. Synthesis of 1-bromo-substituted analogs of cis-deltamethrinic and cis-permethrinic acids. Russ. J. Org. Chem. 2005, 41, 1601–1609. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | CHBr3 (eq) b | Product | Yield (%) | Litt. Yield (%) c | Reference | |
|---|---|---|---|---|---|---|---|
| 1 | 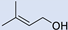 | 1.5 |  | (1) | 70 d | 36 | [32] |
| 2 |  | 2 | 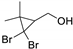 | (1) | 74 d | 36 | [32] |
| 3 |  | 2.5 | 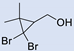 | (1) | 78 d | 36 | [32] |
| 4 | 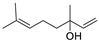 | 2 |  | (2) | 89 d | 93 | [14] |
| 5 |  | 2 | 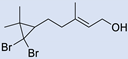 | (3) | -e | - | [14] |
| 6 |  | 2.5f |  | (4) | 57 g | - | - |
| 7 | 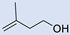 | 2 | 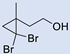 | (5) | 47 g | 58 | [33] |
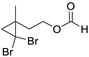 | (6) | 3 g | - | - | |||
| 8 | 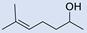 | 2 | 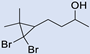 | (7) | 77 g | - | - |
| 9 | 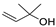 | 2 | 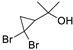 | (8) | - h | 2 | [34] |
| 10 | 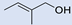 | 2 | 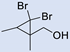 | (9) | 62 d,i | 45 | [14] |
| 11 | 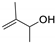 | 2 |  | (10) | 49 g,j | 62k | [14] |
| 12 |  | 2 |  | (11)l | - m | - | - |
| 13 |  | 2 | 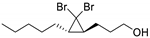 | (12)l | - m | - | - |
| 14 | 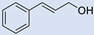 | 2 |  | (13) | - m | - | - |
| 15 |  | 2 |  | (14) | - m | - | - |
| 16 | 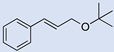 | 2 |  | (15) | - m | - | - |
| 17 |  | 2 |  | (16) | 41 g | - | - |
| 18 | 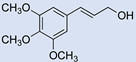 | 2 | 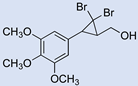 | (17) | - m | - | - |
| 19 | 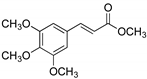 | 2 | 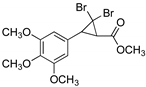 | (18) | - m | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Østby, R.B.; Didriksen, T.; Antonsen, S.G.; Nicolaisen, S.S.; Stenstrøm, Y. Two-Phase Dibromocyclopropanation of Unsaturated Alcohols Using Flow Chemistry. Molecules 2020, 25, 2364. https://doi.org/10.3390/molecules25102364
Østby RB, Didriksen T, Antonsen SG, Nicolaisen SS, Stenstrøm Y. Two-Phase Dibromocyclopropanation of Unsaturated Alcohols Using Flow Chemistry. Molecules. 2020; 25(10):2364. https://doi.org/10.3390/molecules25102364
Chicago/Turabian StyleØstby, Runa Berg, Terje Didriksen, Simen Gjelseth Antonsen, Steinar Sollien Nicolaisen, and Yngve Stenstrøm. 2020. "Two-Phase Dibromocyclopropanation of Unsaturated Alcohols Using Flow Chemistry" Molecules 25, no. 10: 2364. https://doi.org/10.3390/molecules25102364