3. Materials and Methods
3.1. General Methods and Reagents
Mass spectra were recorded with a Thermoquest Automass Multi system (ThermoQuest, CE, Instruments, Milan, Italy), a GCMS-2020 (Shimadzu, Tokyo, Japan) both operated in EI mode (70 eV) and a Thermo Q-Exactive HPLC/MS/MS (Thermo Scientific, Waltham, MA, USA) with ESI(+) ionization. Elemental analyses were obtained with a Fisons EA 1110 CHNS elemental analyzer (Fisons Instruments, Milan, Italy). The melting points were determined with a Boetius micromelting point apparatus (Franz Küstner Nachf. K. G., Dresden, Germany). The
1H-NMR spectra were recorded with a Bruker Avance 3 Ascend 500 system (Bruker BioSpin Corp., Karslruhe, Germany) operated at 500 MHz, and the
13C-NMR spectra were obtained at 125 MHz and
31P-NMR 202 MHz in CDCl
3 or DMSO-
d6 at 298 K. The “in situ” reduction of the nitroxides was achieved by addition of five equivalents of hydrazobenzene ((PhNH)
2/radical). The
O-acetyl derivative of compound
22 for NMR measurement was prepared as described previously [
49]. The EPR (electron paramagnetic resonance) spectra were recorded on MiniScope MS 200 (Magnettech GMBH, Berlin, Germany) instrument in CHCl
3 solution, and the concentrations were 1.0 × 10
−4 M. All radicals gave a 3-line spectra characteristic of monoradicals, a
N = 14.4–15.6 G, radical concentration was > 98% in each case and referred for TEMPO (1-oxyl-2,2,6,6-tetramethylpiperidine. The IR spectra were obtained using a Bruker Alpha FT-IR instrument (Bruker Optics, Ettlingen, Germany) with ATR support on a diamond plate. Spectrophotometric measurements were performed on a Specord 40 UV/VIS Spectrophotometer (Specord, Jena, Germany) at 732 nm in a 1 × 1 cm quartz cuvette. Hydrogenation was performed with an H-Cube Mini Plus, ThalesNano, Budapest, Hungary) instrument with a 10%Pd/C cartridge at 5 bar hydrogen pressure, 35 °C, and a flow rate of 1 mL/min. Flash column chromatography was performed on a Kieselgel 60 (0.040–0.063 mm) column (Merck, Darmstadt, Germany). Qualitative TLC was performed on commercially available plates (20 cm × 20 cm × 0.02 cm) coated with Merck Kieselgel GF
254. Compounds
1a [
19],
1b [
20],
1c [
21],
3 [
22],
5a [
33],
5b [
23],
6 [
25],
9a [
34],
9b [
20],
9c [
21],
15 [
40],
18 [
46], TEMPO [
23] and TEMPOL [
23] were synthesized as previously described. The reagents LiHMDS, Trolox
®,
m-CPBA, diethylphosphite, triphenyl-phosphine, triethylphosphite, DEAD, FeCl
3, MnO
2, NaH
, NaN
3, DDQ, POCl
3, ABTS, Dess–Martin periodinane, benzaldehyde, 2-thiophencarbaldehyde, undecanal, 3-pyridinecarbaldehyde, NiCl
2, diethyl chlorophosphate, BuLi, DMSO-
d6, CDCl
3, hydrazobenzene were purchased from Sigma Aldrich (St. Louis, MO, USA) and hexane, DCM, CHCl
3, methanol (MeOH), methyliodide (MeI), ethyl acetatate (EtOAc), toluene, benzene, THF, MgSO
4, FeSO
4 .7H
2O, NaCl, Na
2HPO
4, KH
2PO
4 from Molar Chemicals (Halásztelek, Hungary).
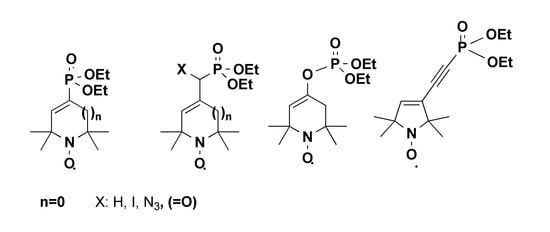
3.2. General Procedure for Arbusov Reactions (2a–c, 4)
In a well-ventilated hood, a mixture of compound 1a or 1b or 1c or 3 (10.0 mmol) and triethylphosphite (2.5 g, 15.0 mmol, or 5.0 g, 30.0 mmol, for compound 3) was stirred in an open vessel at 120 °C in an oil bath. The ethylbromide byproduct was allowed to escape. The reaction mixture was monitored by TLC, and after consumption of the starting material (~2 h), the mixture was allowed to cool spontaneously with stirring. After cooling, the resulting mixture was purified by flash column chromatography to give the allylic phosphonates.
3.2.1. Diethyl ((1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl)phosphonate Radical (2a)
Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to produce an orange oil (1.88 g, 65%); TLC (CHCl3/Et2O, 2:1): Rf = 0.33. 31P-NMR (CDCl3 + (PhNH)2) δ:26.9. 13C-NMR (CDCl3 + (PhNH)2) δ: 16.6 (d, J = 6.0 Hz, 2C), 24.2 (2C), 24.3 (d, J = 143.0 Hz, 1C), 25.8 (2C), 62.2 (d, J = 6.6 Hz, 2C), 68.2 (d, J = 1.1 Hz, 1C) 71.6 (d, J = 9 Hz, 1C), 132.6 (d, J = 8.0 Hz, 1C), 134.0 (d, J = 8.0 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.32 (s, 6H), 1.36 (s, 6H), 1.39 (t, J = 6.9 Hz, 6H), 2.56 (d, J = 22 Hz, 2H), 4.19 (quint, J = 1.2 Hz, 4H), 5.86 (s, 1H). IR: 2976, 2931, 1650 cm−1. MS (EI): m/z (%): 290 (M+, 13) 260 (70), 245 (15), 138 (22), 122 (100).
3.2.2. Diethyl ((4-bromo-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl)phosphonate Radical (2b)
Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to afford an orange oil (2.83 g, 77%); TLC (CHCl3/Et2O, 2:1): Rf =0.48. 31P-NMR (CDCl3 + (PhNH)2) δ:26.9. 13C-NMR (CDCl3 + (PhNH)2) δ: 16.5 (d, J = 6.0 Hz, 2C), 24.1 (d, J = 143.0 Hz, 1C), 24.2 (2C), 25.8 (2C), 62.1 (d, J = 6.7 Hz, 2C), 68.0 (d, J = 2.1 Hz, 1C) 71.4 (d, J = 8.8 Hz, 1C), 132.6 (d, J = 8.1 Hz, 1C), 133.9 (d, J = 11.1 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.35 (s, 6H), 1.37 s (6H), 1.45 (bs, 6H)., 2.58 (d, J = 21.5 Hz, 2H), 4.25 (bs, 4H). IR: 2979, 2932, 1644 cm−1. MS (EI): m/z (%): 370/368 (M+, 44), 340/338 (4/4), 259(35), 121 (100).
3.2.3. Diethyl ((1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl)phosphonate Radical (2c)
Obtained by method A: Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to afford a red oil (2.46 g, 81%); TLC (CHCl3/Et2O, 2:1): Rf = 0.35. 31P-NMR (CDCl3 + (PhNH)2) δ: 27.1. 13C-NMR (CDCl3 + (PhNH)2) δ: 16.5 (d, J = 6.1 Hz, 2C),25.0 (1C), 26.3 (bs, 1C), 34.3 (d, J = 38.1 Hz, 2C), 44.0 (d, J = 2.3 Hz, 1C), 57.7 (1C), 59.0 (d, J = 2.3 Hz, 1C), 61.9 (d, J = 6.8 Hz, 2C), 122.5 (d, J = 11.0 Hz, 1C), 134.1 (d, J = 12.0 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.28 (s, 6H), 1.32 (s, 6H), 1.38 (t, J = 7 Hz, 6H), 2.29 (d, J = 3.5 Hz, 2H), 2.55 (d, J = 21.5 Hz, 2H), 4.13–4.20 (m, 4H), 5,43 (d, J = 5.3 Hz, 1H). IR: 2977, 2932, 1645 cm−1. MS (EI): m/z (%): 304 (M+, 27) 274 (100), 259 (27), 152 (16), 81 (60).
3.2.4. Diethyl ((1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl)phosphonate Radical (2c)
Obtained by method B: To a stirred suspension of NaH (240 mg, 10.0 mmol) in toluene (10 mL), a solution of tetraethyl methylenediphosphonate (2.88 mg, 10.0 mmol) in toluene (10 mL) was added dropwise at 0 °C under N2. After 30 min, a solution of compound 5b (1.7 g, 10.0 mmol) in toluene (10 mL) was added dropwise at 0 °C. The mixture was refluxed for 3 hours. After cooling, the solvent was evaporated, and the residue was partitioned between water (30 mL) and EtOAc (50 mL). The organic phase was separated, dried (MgSO4), filtered, and evaporated, and the residue was purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give a red oil (1.77 g, 58%), TLC (CHCl3/Et2O 2:1): Rf = 0.35. IR: 2977, 2932, 1645 cm−1, and all other spectral data were identical to those of one of the compounds obtained with method A.
3.2.5. Tetraethyl ((1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3,4-diyl)bis(methylene)) bisphosphonate Radical (4)
Purified by flash column chromatography (eluent: CHCl3/Et2O, 2:1) to give a brownish powder (3.1 g, 70%); mp 85–87 °C; TLC (CHCl3/MeOH 29:1): Rf = 0.33. 31P-NMR (DMSO-d6 + (PhNH)2) δ:27.4. 13C-NMR ((DMSO-d6 + (PhNH)2) δ: 16.7 (4C), 23.6 (d, J = 133.0 Hz, 2C), 24.7 (4C), 61.7 (4C), 69.4 (2C), 132.7 (2C). 1H-NMR (DMSO-d6 + (PhNH)2) δ: 1.11 (s, 12H), 1.23 (t, J = 6.8 Hz, 12H), 2.92 (d, J = 20.0 Hz, 4H), 4.01 (quint, J = 6.5 Hz, 8H). IR: 2982, 2933, 2920 cm−1. MS (EI): m/z (%): 440 (M+, 10), 410 (38), 395 (28), 273 (77), 152 (8), 135 (100)
3.3. Diethyl (1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)phosphate Radical (7)
In a well-ventilated hood, a mixture of compound 6 (2.49 g, 10.0 mmol) and triethylphosphite (2.5 g, 15.0 mmol) was stirred in an open vessel at 60 °C in an oil bath. The ethylbromide byproduct was allowed to escape. The reaction mixture was monitored by TLC, and after ~ 2 h, the temperature was increased to 100 °C for ~ 1 h. The mixture was allowed to cool spontaneously with stirring. After cooling, the resulted mixture was purified by flash column chromatography to give the Perkow product, which was purified by flash column chromatography (hexane/EtOAc, 1:1) to give a red oil (1.05 g, 34%); TLC (CHCl3/Et2O, 2:1): Rf = 0.50. 31P-NMR (CDCl3 + (PhNH)2) δ: −6.2. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.2 (d, J = 6.5 Hz, 2C), 25.3 (2C), 26.7 (2C), 42.1 (d, J = 3.8 Hz, 1C), 58.4 (1C), 59.1 (1C), 64.3 (d, J = 6.1 Hz, 2C), 118.0 (d, J = 5.4 Hz, 1C), 142.3 (d, J = 8.8 Hz, 1C). 1H-NMR (DMSO-d6 + (PhNH)2) δ: 1.30 (s, 6H), 1.35 (s, 6H), 1.43 (t, J = 7.1 Hz, 6H), 2.38 (s, 2H), 4.23 (quint, J = 7.1 Hz, 2H), 5.43 (d, J = 1.8 Hz, 1H). IR: 2980, 2935, 2911, 1696 cm−1. MS (EI): m/z (%): 306 (M+, 8), 276(10), 155 (70) 107 (100).
3.4. General Procedure for Pudovik α-hydroxyphosphonate Synthesis from Paramagnetic Aldehydes and Ketones 8a, 8b, 10a–c
To a stirred mixture of compound 5a or 5b or 9a or 9b or 9c and diethyl phosphite (1.38 g, 10.0 mmol), K3PO4 (106 mg, 0.5 mmol) was added, and the stirring continued at room temperature for 1 h. Subsequently, 10% aq. Na2CO3 (50 mL) was added, followed by extraction with EtOAc (2 × 50 mL). The combined organic phases were dried (MgSO4), filtered, and evaporated, and the residue was purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give the α-hydroxy-phosphonate products.
3.4.1. Diethyl (3-hydroxy-1-oxyl-2,2,5,5-tetramethylpyrrolidin-3-yl)phosphonate Radical (8a)
Purified by flash column chromatography (eluent: CHCl3/Et2O, 2:1) to give a yellow powder (2.7 g, 92%); mp 100–103 °C; TLC (CHCl3/MeOH, 56:4): Rf = 0.51. 31P-NMR (DMSO-d6 (PhNH)2) δ: 23.2. 13C-NMR ((DMSO-d6 + (PhNH)2) δ: 17.0 (d, J = 5.2 Hz, 2C), 20.0 (1C), 22.1 (1C), 27.0 (1C), 31.1 (1C), 46.5 (d, J = 4.0 Hz, 1C), 61.9 (d, J = 8.2 Hz, 1C), 62.6 (d, J = 5.6 Hz, 1C), 77.8 (1C), 79.1 (1C). 1H-NMR (DMSO-d6 + (PhNH)2) δ: 1.11–1.25 (m, 18H), 1.85 (d, J = 13.4 Hz, 1H), 2.35 (t, J = 11.9 Hz, 1H), 4.04–4.11 (m, 4H). IR: 3258, 2982, 2938, 2910 cm−1. MS (EI): m/z (%): 294 (M+, 12), 264(2), 249 (5) 180 (100), 138 (78).
3.4.2. Diethyl (4-hydroxy-1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)phosphonate Radical (8b)
Purified by flash column chromatography (eluent: CHCl3/Et2O, 2:1) to give red crystals (2.77 g, 90%); mp 115–117 °C; TLC (CHCl3/MeOH, 56:4): Rf = 0.53. 31P-NMR (CDCl3 (PhNH)2) δ: 24.4. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.6 (d, J = 5.1 Hz, 2C), 21.0 (4 C), 33.3 (2C), 43.1 (2C), 57.9 (d, J = 14.5 Hz, 1C), 63.1 (d, J = 7.5 Hz), 71.3 (1C), 72.6 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.28 (s, 6H), 1.40 (t, J = 7 Hz, 6H), 1.48 (s, 6H), 2.02 (d, J = 4.01 Hz, 4H), 3.11 (bs, 1H), 4.23 (quint, J = 7.1 Hz, 4H), 4.69 (bs, 1H). IR: 3198, 2993, 2973, 2929 cm−1. MS (EI): m/z (%): 308 (M+, 13), 259(10), 222 (38), 194 (16), 156 (18), 138 (100).
3.4.3. Diethyl (hydroxy(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl)phosphonate Radical (10a)
Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give an orange oil (2.61 g, 85%); TLC (CHCl3/MeOH, 58:2): Rf = 0.33. 31P-NMR (CDCl3 (PhNH)2) δ: 21.8. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.5 (t, J = 5.1 Hz, 2C), 24.5. (1C), 24.9 (1C), 25.4 (1C), 25.5 (1C), 63.1 (d, J = 185.0 Hz, 1C), 63.9 (d, J = 164.0 Hz, 2C), 68.0 (1C), 71.2 (d, J = 9.4 Hz, 1C), 135.1 (d, J = 6.2 Hz, 1C), 140.3 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.34–1.42 (m, 18H), 4.26 (q, J = 7.0 Hz, 4H), 4.35 (d, J = 10.8 Hz, 1H), 6.13 (s, 1H). IR: 3286, 2977, 2931, 1645 cm−1. MS (EI): m/z (%): 306 (M+, 7), 276 (9), 154 (26), 138 (100).
3.4.4. Diethyl (hydroxyl(4-bromo-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl)phosphonate Radical (10b)
Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give an orange powder (2.98 g, 78%); mp 107–109 °C; TLC (CHCl3/MeOH, 58:2): Rf = 0.34. 31P-NMR (CDCl3 (PhNH)2) δ: 20.7. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.5 (d, J = 5.7 Hz, 2C), 23.7 (1C), 24.5 (1C) 24.9 (1C), 25.1 (1C), 63.0 (d, J = 7.2 Hz, 1C) 63.6 (d, J = 7.2 Hz, 1C), 67.5 (d, J = 162.1 Hz, 1C), 70.8 (1C), 71.5 (1C), 127.1 (d, J = 12.6 Hz, 1C), 137.5 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.33–1.47 (m, 18H), 4.18-4.30 (m, 4H), 4.94 (d, J = 16.7 Hz, 1H). IR: 3263, 2980, 2934, 2908, 1631 cm−1. MS (EI): m/z (%): 386/384 (M+, 16/16), 356/354 (4/4), 275 (38), 138 (100).
3.4.5. Diethyl (hydroxy(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl)-phosphonate Radical (10c)
Purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give a red oil (2.56 g, 80%); TLC (CHCl3/MeOH, 58:2): Rf = 0.38. 31P-NMR (CDCl3 (PhNH)2) δ: 22.0. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.5 (d, J = 5.7 Hz, 2C), 39.8. (1 C), 57.7 (1C), 59.8 (1C), 62.8 (d, J = 7.4 Hz, 1C) 63.1 (d, J = 7 Hz, 1C), 71.3 (d, J = 158.1 Hz, 1C), 127.4 (d, J = 4.3 Hz, 1C), 132.8 (d, J = 11.5 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.27 (s, 6H), 1.33 (s, 6H), 1.34 (s, 6H), 1.38 (t, J = 7 Hz, 3H), 2.30 (dq, J1= 2.5 Hz, J2=9.9 Hz, 2H), 4.18–4.27 (m, 4H), 4.38 (d, J = 10 Hz, 1H), 5.67 (d, J = 4.6 Hz, 1H). IR: 3290, 2978, 2933, 1649 cm−1. MS (EI): m/z (%): 320 (M+, 5), 290 (7), 272 (8), 182 (10), 152 (100).
3.5. Diethyl (1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)phosphonate Radical (11)
To a solution of compound 8b (1.54 g, 5.0 mmol) in anhydrous pyridine (10 mL), POCl3 (1.0 mL, 10.6 mmol) was added dropwise at 0 °C, and the mixture was allowed to remain at r.t for 48 h. The mixture was poured onto 100 g crushed ice, extracted with CH2Cl2 (3 × 15 mL), and the combined organic phase was washed with aq. 1N HCl (2 × 40 mL). The organic phase was dried (MgSO4), filtered, and evaporated, and the residue was purified by flash column chromatography (eluent: hexane/EtOAc, 2:1) to give a red powder (420 mg, 29%); mp 53–55 °C; TLC (CHCl3/Et2O, 2:1): Rf = 0.44. 31P-NMR (CDCl3 (PhNH)2) δ: 19.3. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.4 (d, J = 6.1 Hz, 2C), 24.6 (2C), 25.5 (2C), 39.1. (d, J = 7.3 Hz, 1C), 57.3 (d, J = 4.9 Hz, 1C), 60.5 (d, J = 17.8 Hz, 1C), 61.7 (d, J = 5.4 Hz, 1C), 120.5 (d, J = 182.6 Hz, 1C), 149.3 (d, J = 7.6 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.27 (s, 6H), 1.38–1.41 (m, 12H), 2.31 (d, J = 6.1 Hz, 2H), 4.10-4.2 (m, 4H), 6.6 (d, J = 21.5 Hz, 1H). IR: 2979, 2932, 2903, 1658 cm−1. MS (EI): m/z (%): 320 (M+, 5), 290 (7), 272 (8), 182 (10), 152 (100).
3.6. Diethyl (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carbonyl)phosphonate Radical (12)
To a stirred solution of compound 10a (1.53 g, 5.0 mmol) in anhydr. CH2Cl2 (DCM) (10 mL), Dess–Martin periodinane (6.36 g, 15.0 mmol, 3 eq.) was added in 3 portions at 0 °C over a period of 10 min. The stirring was continued for 1 h at r.t. The resulting mixture was filtered on a sintered glass funnel. The filtrate was diluted with DCM (25 mL) and washed with 10% aq NaHCO3 solution (25 mL) and 10% aq Na2S2O3 (25 mL). The organic phase was separated, dried (MgSO4), filtered, and evaporated, and the residue was purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give a red powder (950 mg, 62%); mp 35–37 °C; TLC (CHCl3/Et2O, 2:1): Rf = 0.56, 31P-NMR (CDCl3 (PhNH)2) δ: −2.9. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.4 (d, J = 5.7 Hz, 2C), 24.3. (2 C), 24.7 (2C) 63.9 (d, J = 7.3 Hz, 2C), 69.0 (1C), 70.3 (d, J = 10.8 Hz, 1C), 143.2 (d, J = 64.0 Hz, 1C), 155.2 (1C), 196 (d, J = 174.0 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.42-1.46 (m, 18H), 4.28 (quint, J = 7.24 Hz, 4H). IR: 3067, 2976, 2931, 1637, 1601 cm−1. MS (EI): m/z (%): 304 (M+, 4), 274 (6), 246 (3), 137 (49), 109 (100).
3.7. Diethyl (azido (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl)phosphonate Radical (13)
To a stirred suspension of Ph3P (3.14 g, 12 mmol) in anhydrous DCM (10 mL), a solution of DEAD 5.2 mL (2.09 g, 12.0 mmol in 40% toluene) diluted anhydr. DCM (5 mL) was added dropwise at −78 °C under N2. A 1.85 M solution of HN3 (6.7 mL, 12.5 mmol) in benzene was added dropwise, and the stirring was continued for 5 min at 0 °C followed by dropwise addition of a solution of compound 10a (3.06 g, 10.0 mmol) in anhydr. DCM (10 mL). After the addition was completed, the mixture was held for 30 min at 0 °C, and stirring was continued for 24 h at r.t. The resulted mixture was filtered on a sintered glass funnel, and solvents were evaporated. The residue was purified by flash column chromatography (eluent: hexane/EtOAc, 2:1) to give a yellow powder (1.97 g, 60%); mp 50–52 °C; TLC (CHCl3/Et2O, 2:1): Rf = 0.60, 31P-NMR (CDCl3 (PhNH)2) δ: 15.6. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.4 (t, J = 5.7 Hz, 2C), 19.7 (1 C), 22.4 (1C) 25.0 (1C), 30.9 (1C), 61.7 (d, J = 5.6 Hz, 1C), 62.0 (d, J = 5.6 Hz, 1C), 66.1 (1C), 66.3 (1C), 66.5 (d, J = 6.4 Hz, 1c), 114.1 (d, J = 191 Hz, 1C), 167.0 (d, J = 5.4 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.38–1.43 (m, 18H), 4.12–4.28 (m, 4H), 4.98 (s, 1H), 5.81 (d, J = 13.1 Hz). IR: 2981, 2935, 2096, 1635 cm−1. HRMS (ESI) m/z [M+H]+ calc. for C13H25N4O4P: 332.1613; found: 332.1609.
3.8. Diethyl ((1-oxl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)iodomethyl)phosphonate Radical (14)
To a stirred suspension of compound 10a (3.06 g, 10.0 mmol) and Ph3P (3.14 g, 12.0 mmol) in benzene (20 mL), a solution of DEAD (2.09 g, 12.0 mmol in 40% toluene) diluted with benzene (5 mL) was added dropwise at 0 °C under N2. After 10 min to complete the addition, a solution of CH3I (0.7 mL, 12.0 mmol) in benzene (5 mL) was added dropwise. After the addition was completed, the mixture was held for 30 min at 0 °C, and stirring was continued for 24 h at r.t. The solvent was evaporated, and the residue was partitioned between water (20 mL) and EtOAc (50 mL). The organic phase was separated, dried (MgSO4), filtered, and evaporated, and the crude was purified by flash column chromatography (eluent: hexane/EtOAc, 1:1) to give a yellow semi-solid (2.0 g, 48%); TLC (CHCl3/Et2O, 2:1): Rf = 0.40, IR: 3040, 2990 1528. HRMS (ESI) m/z [M]+ calc. for C13H25INO4P: 417.0566; found: 417.1311; [M-HI]+ calc. for C13H24NO4P: 289.1443; found 289.1434.
3.9. 3-Bromo-1-methoxy-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole (16)
To a stirred solution of 15 (1.1 g, 5.0 mmol) and FeSO4·7H2O (6.9 g, 25.0 mmol) in DMSO (30 mL) at 0 °C, 30% aq H2O2 (5 mL) was added dropwise over 2 h. The reaction was monitored by TLC. Upon consumption of the starting material, distilled H2O (50 mL) was added, and the aqueous solution was extracted with Et2O (3 × 30 mL). The combined organic phases were dried (MgSO4), filtered, and evaporated, and the crude product was purified by flash column chromatography (hexane–Et2O, 2:1) to give a colorless oil (700 mg, 60%); TLC (hexane/Et2O, 9:1): Rf = 0.42. 13C-NMR (CDCl3) δ: 22.3 (2C) 28.6 (2C), 65.0 (1C) 68.9 (1C), 71.7 (1C), 125.6 (1C), 134.0 (1C). 1H-NMR (CDCl3) δ: 1.27 (s, 6H), 1.29 (s, 6H), 3.69 (s, 3H), 5.69 (s, 1H). IR: 2921, 2852, 1642. MS (EI): m/z (%): 235/233 (M+, 3/3), 220/218 (33/33), 139 (100), 108 (25).
3.10. Diethyl (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)phosphonate Radical (17)
To a stirred solution of compound 16 (470 mg, 2.0 mmol) in anhydrous THF (10 mL), n-BuLi solution in hexane (0.8 mL, 2.0 mmol, 2.5 M) diluted with anhydr. THF (10 mL) was added dropwise at −78 °C under N2.. After the addition was completed, the mixture was continuously stirred for 1 h at −78 °C. A solution of diethylchlorophosphate (345 mg, 2.0 mmol) in anhydr. THF (10 mL) was added dropwise. After stirring at this temperature for 30 min, the reaction mixture was allowed to warm to r.t. with continuous stirring for 2 h. A sat. aq. NH4Cl solution (5 mL) was added, the mixture was extracted with CH2Cl2 (2 × 10 mL), and the combined organic phase was dried (MgSO4), filtered and evaporated. The crude residue (480 mg, 1.65 mmol) was dissolved in anhydr. DCM (10 mL), and 3-chloroperbenzoic acid (~60%, 1.18 g, 4.1 mmol, 2.5 eq) was added in 2–3 portions at 0 °C over a period of 10 min. Stirring was continued for an additional 1 h at ambient temperature. The solution was washed with 10% aq. Na2CO3 solution (2 × 20 mL), and the organic phase was separated, dried (MgSO4), filtered and evaporated. The residue was purified by flash column (eluent: hexane/EtOAc, 1:1) to give a yellow powder (140 mg, 50%); mp 60–62 °C; TLC (CHCl3/MeOH, 2:1): Rf = 0.50. 31P-NMR (CDCl3 (PhNH)2) δ: 14.6. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.3 (d, J = 6.3 Hz, 2C), 25.0 (2C), 25.3 (2C), 61.9 (d, J = 5.6 Hz, 2C), 68.7 (d, J = 15.6 Hz, 1C), 71.3 (d, J = 15.6 Hz, 1C), 133.8 (d, J = 4.0 Hz, 1C), 150.6 (d, J = 8.1 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.33 (s, 6H), 1.40 (t, J = 7.5 Hz, 6H), 1.44 (s, 6H) 4.13–4.21 (m, 4H), 6.57 (d, J = 13.5 Hz, 1H). IR: 3079, 2977, 2931, 2866, 1609 cm−1. MS (EI): m/z (%): 276 (M+, 15), 246 (65), 231 (100), 203 (5), 175 (44), 107 (78).
3.11. Diethyl ((1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)ethynyl)phosphonate Radical (19)
To a stirred solution of compound 18 (492 mg, 3.0 mmol) in anhydr. THF (10 mL), LiHMDS (3.0 mL 3.0 mmol, 1 M THF solution) was added dropwise at −78 °C under N2.. After the addition was completed, the mixture was stirred for 1 h at −78 °C. A solution of diethylchlorophosphate (517 mg, 3.0 mmol) in anhydr. THF (10 mL) was added dropwise, and the temperature was allowed to warm to r.t. spontaneously with stirring for 2 h. The reaction mixture was quenched with sat. NH4Cl solution (5 mL). The mixture was diluted with EtOAc (20 mL), the organic phase was separated, the aq. phase was extracted with EtOAc (10 mL), and the combined phases were dried (MgSO4), filtered and evaporated. The residue was subjected to flash column chromatography purification (eluent: hexane/EtOAc, 1:1) to offer a yellow solid (470 mg, 52%); mp 50–52 °C; TLC (CHCl3/Et2O, 2:1): Rf = 0.43. 31P-NMR (CDCl3 (PhNH)2) δ: –6.4. 13C-NMR ((CDCl3 + (PhNH)2) δ: 16.1 (d, J = 6.9 Hz, 2C), 24.9 (2C), 25.2 (2C), 63.2 (d, J = 5.5 Hz, 2C), 69.2 (1C), 71.3 (1C), 81.7 (d, J = 297.8 Hz, 1C), 93.8 (d, J = 52.7 Hz, 1C), 125.6 (d, J = 3.6 Hz, 1C), 146.2 (d, J = 3.0 Hz, 1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.32 (s, 6H), 1.38 (s, 6H) 1.45 (t, J = 7.1 Hz, 6H), 4.22–4.28 (m, 4H), 6.22 (d, J = 0.7 Hz, 1H). IR: 3073, 2976, 2931, 2908, 2866, 2171, 1612 cm−1. MS (EI): m/z (%): 300 (M+, 14), 285 (33), 270 (20), 241 (7), 132 (100), 117 (52).
3.12. General Procedure for HWE Olefination with 2a Nitroxide Phosphonate: Compounds 20a–d
To a stirred suspension of oil-free NaH (120 mg, 5.0 mmol) in anhydr. toluene (10 mL), a solution of compound 2a (1.45 g, 5.0 mmol) in anhydr. toluene (5 mL) was added dropwise at 0 °C under N2. After 30 min, a solution of the appropriate aldehyde (5.0 mmol) in anhydr. toluene (10 mL) was added dropwise at 0 °C. The mixture was refluxed for 3 h and allowed to stand overnight at r.t. The solvent was evaporated, and the residue was partitioned between sat. aq. NH4Cl solution (25 mL) and EtOAc (50 mL). The organic phase was separated, dried (MgSO4), filtered, and evaporated, and the crude product was purified by flash column chromatography to yield the olefinated nitroxides.
3.12.1. (E)-3-(Dodec-1-en-1-yl)-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole Radical (20a)
Purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give a brown oil (950 mg, 62%); TLC (hexane/Et2O, 5:1): Rf = 0.56. 13C-NMR ((CDCl3 + (PhNH)2) δ: 24.9 (2C), 25.0 (2C) 25.7 (1C), 29.0 (1C), 29.1 (1C), 29.2 (1C), 29.3 (1C)29.4 (1C), 29.5 (1C), 33.3 (1C), 33.8 (1C), 65.4 (1C), 67.4 (1C), 114.2 (1C), 130.8 (1C), 131.25 (1C), 139.1 (1C). 139.2 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.33–1.37 (m, 33H), 2.13 (d, J = 6.1 Hz, 2H) 5.02 (d, J =10.0 Hz, 1H), 5.08 (d, J = 17 Hz, 1H), 5.88–5.95 (m, 1H). IR: 3075, 2975, 2924, 2853, 1640 cm−1. MS (EI): m/z (%): 306 (M+, 2), 281 (7), 207 (28), 149 (25), 55 (100).
3.12.2. (E)-1-Oxyl-3-styryl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole Radical (20b)
Purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give a to give an orange powder; mp 67–70 °C (730 mg, 60%); TLC (hexane/Et2O, 2:1): Rf = 0.53. 13C-NMR ((CDCl3 + (PhNH)2) δ: 25.4 (2C), 26.0 (2C), 67.6 (1C), 70.3 (1C), 122.4 (1C), 126.4 (2C), 127.7 (1C), 128.8 (2C), 129.9 (1C), 131.9 (1C), 137.4 (1C), 142.7 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.45 (s, 6H), 1.56 (s, 6H) 5.86 (s, 1H), 6.7 (d, J = 16.5 Hz, 1H), 7.36-7.47 (m, 3H). 3H are overlapped with peaks of diphenyl hydrazine. IR: 3023, 2972, 2927, 2865, 1634, 1596 cm−1. MS (EI): m/z (%): 242 (M+, 12), 227 (22), 212 (100), 197 (71), 91 (28).
3.12.3. (E)-1-Oxyl-3-(2-(pyridin-3-yl)vinyl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole Radical (20c)
Purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give an orange powder; mp 90–93 °C (680 mg, 56%); TLC (CHCl3/Et2O, 2:1): Rf = 0.33. 13C-NMR ((CDCl3 + (PhNH)2) δ: 25.2 (2C), 25.7 (2C), 67.5 (1C), 70.0 (1C), 123.5 (1C), 124.5 (1C), 126.0 (1C), 132.5 (1C), 133.0 (1C), 133.3 (1C), 142.4 (1C), 148.4 (1C), 148.5 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.37 (s, 6H), 1.48 (s, 6H) 5.86 (s, 1H), 6.68 (d, J = 16.5 Hz, 1H), 6.82 (d, J = 16.5 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 8.54 (d, J = 4.4 Hz, 1H), 8.71 (s, 1H). 1H is overlapped with diphenyl hydrazine peaks. IR: 3042, 3017, 2974, 2928, 2868, 1633, 1566 cm−1. MS (EI): m/z (%): 243 (M+, 20), 228 (42), 213 (100), 198 (75), 125 (37), 93 (61).
3.12.4. (E)-(1-Oxyl-2,2,5,5-tetramethyl-3-(2-(thiophen-2-yl)vinyl)-2,5-dihydro-1H-pyrrol Radical (20d)
Purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give brown crystals; mp 75–77 °C (635 mg, 51%); TLC (CHCl3/Et2O, 2:1): Rf = 0.5. 13C-NMR ((CDCl3 + (PhNH)2) δ: 25.3 (2C), 25.9 (2C), 67.5 (1C), 70.0 (1C), 122.1 (1C), 124.4 (1C), 125.9 (1C), 127.6 (1C), 132.1 (1C), 142.4 (1C), 143.0 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.40 (s, 6H), 1.51 (s, 6H) 5.81 (s, 1H), 6.52 (d, J = 16.2 Hz, 1H), 7.03 (d, J = 16.2 Hz, 1H), 7.08–7.26 (m, 3H). IR: 3101, 3059, 3037, 2979, 2930, 2862. 1624 cm−1. MS (EI): m/z (%): 248 (M+, 16), 233 (24), 218 (100), 203 (59), 175 (48), 44 (73).
3.13. (R,S)-1-Oxyl-3-phenethyl-2,2,5,5-tetramethylpyrrolidine Radical (21)
A solution of compound 20b (485 mg, 2.0 mmol) in anhydr. EtOH (75 mL) was subjected to continuous flow hydrogenation by a H-Cube Mini Plus apparatus with a 10% Pd/C catalyst cartridge. After consumption of the starting material (monitored by TLC and the content of the receiving flask), the solvent was evaporated, the residue was dissolved in CHCl3 (25 mL), MnO2 (17.4 mg, 0.2 mmol) was added, and the mixture was bubbled with O2 for 30 min., followed by filtration through a Celite pad. After rinsing the pad with CHCl3 (10 mL), the filtrate was evaporated and the crude product was purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give an orange powder; mp 60–62 °C (367 mg, 74%); TLC (hexane/Et2O, 2:1): Rf = 0.35. 13C-NMR ((CDCl3 + (PhNH)2) δ: 17.2 (1C), 26.6 (1C), 27.2 (1C), 29.9 (1C), 32.4(1C), 34.7(1C), 43.0 (1C), 43.1 (1C), 61.4 (1C), 66.5(1C), 125.9 (1C), 128.4 (2C), 128.5 (2C), 142.6 (1C). 1H-NMR (CDCl3 + (PhNH)2) δ: 1.10 (s, 3H), 1.29 (s, 3H), 1.33 (s, 3H), 1.36 (s, 3H), 1.54–1.59 (m, 2H), 1.86–1.89 m (2H), 1.98–2.02 (m 1H), 2.61–2.67 (m, 1H), 2.77–2.82 (m, 1H), 7.42–7.45 (m, 3H). 2H are overlapped with peaks of with diphenyl hydrazine. IR: 3066, 3025, 2965, 2917, 2879, 2857, 1602 cm−1. MS (EI): m/z (%): 246 (M+, 43), 216 (26), 117 (19), 91 (100).
3.14. 6-Diphenyl-2-Oxyl-1,1,3,3-tetramethylisoindoline Radical (22)
To a suspension of oil-free NaH (144 mg, 6.0 mmol) in anhydrous toluene (10 mL), a solution of compound 4 (1.32 g, 3.0 mmol) in anhydrous toluene (10 mL) was added dropwise at 0 °C under N2. After 30 min, a solution of freshly distilled benzaldehyde (848 mg, 8.0 mmol) in toluene (10 mL) was added dropwise at 0 °C. The mixture was refluxed for 3 h. After cooling, sat. aq. NH4Cl solution (5 mL) and Et2O (30 mL) were added to the mixture and stirred for 10 min. The organic phase was separated, dried (MgSO4), filtered, and evaporated. The residue was dissolved in toluene (20 mL), followed by the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 681 mg, 3.0 mmol), and the mixture was refluxed with stirring for 2 h. After cooling, the solvent was evaporated, and the residue was partitioned between 10% aq. Na2CO3 solution (25 mL) and EtOAc (50 mL). The organic phase was separated, dried (MgSO4), filtered, and evaporated, and the crude product was purified by flash column chromatography (eluent: hexane/Et2O, 2:1) to give a beige powder; mp 213–216 °C (500 mg, 48%); TLC (hexane/Et2O, 2:1): Rf = 0.40. 13C-NMR of O-acetyl ((CDCl3 + (PhNH)2) δ: 19.3 (1C), 25.3 (1C), 28.9 (4C), 68.3 (2C), 123.7 (2C), 126.5 (2C) 127.8 (4C), 129.9 (2C), 140.4 (2C), 141.6 (2C), 143.3 (2C), 171.7 (2C). 1H-NMR of O-acetyl (CDCl3 + (PhNH)2) δ: 1.53 (s, 6H), 1.59 (s, 6H) 2.28 (s, 3H), 7.17–7.28 (m, 12H). IR: 3057, 3026, 2979, 2925, 2853, 1601 cm−1. MS (EI): m/z (%): 342 (M+, 1), 312 (100), 297 (21), 141 (10).
3.15. ABTS Scavenging Assay
The measurements were collected on a Specord 40 instrument. ABTS was dissolved in PBS buffer (0.136 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.00176 M KH2PO4) to a 7.0 mM concentration. ABTS radical cations (ABTS•+) were produced by reacting the ABTS stock solution with potassium persulfate at a final concentration of 2.45 mM and allowing the mixture to stand in the dark at room temperature for 16 h before use. For study of the compounds, the ABTS•+ solution was diluted with water to an absorbance of 0.70(±0.02) at 734 nm and equilibrated at 37 °C. Stock solutions of new compounds and Trolox in dimethylsulfoxide (DMSO) were added to the diluted ABTS•+solution in final concentrations of 12.5, 10, 7.5, and 2.5 µM. After addition, the mixtures were incubated for 6 min at 37 °C before measuring their absorbance at 734 nm. All determinations were repeated three times. The percentage inhibition of absorbance at 734 nm is calculated with the usual formula: (A0—Aantioxidant)/A0, where A0 is the absorbance of the diluted ABTS•+ solution. The concentration–response curves of new compounds were compared with the curve of Trolox.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}