
Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems


Abstract
:1. Introduction
2. Conformational Behavior of Ethane and Its Analogs in Nanotubes
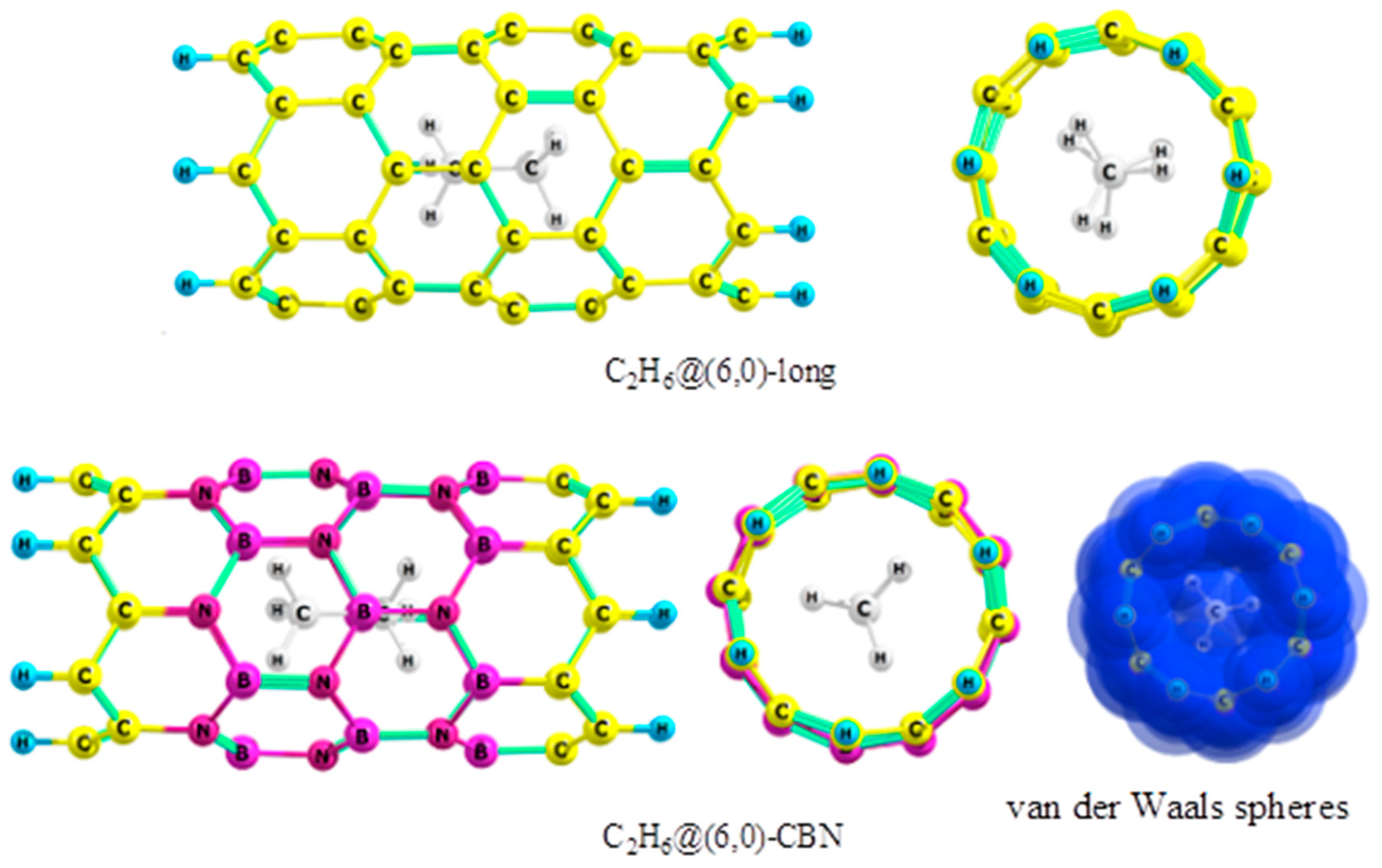
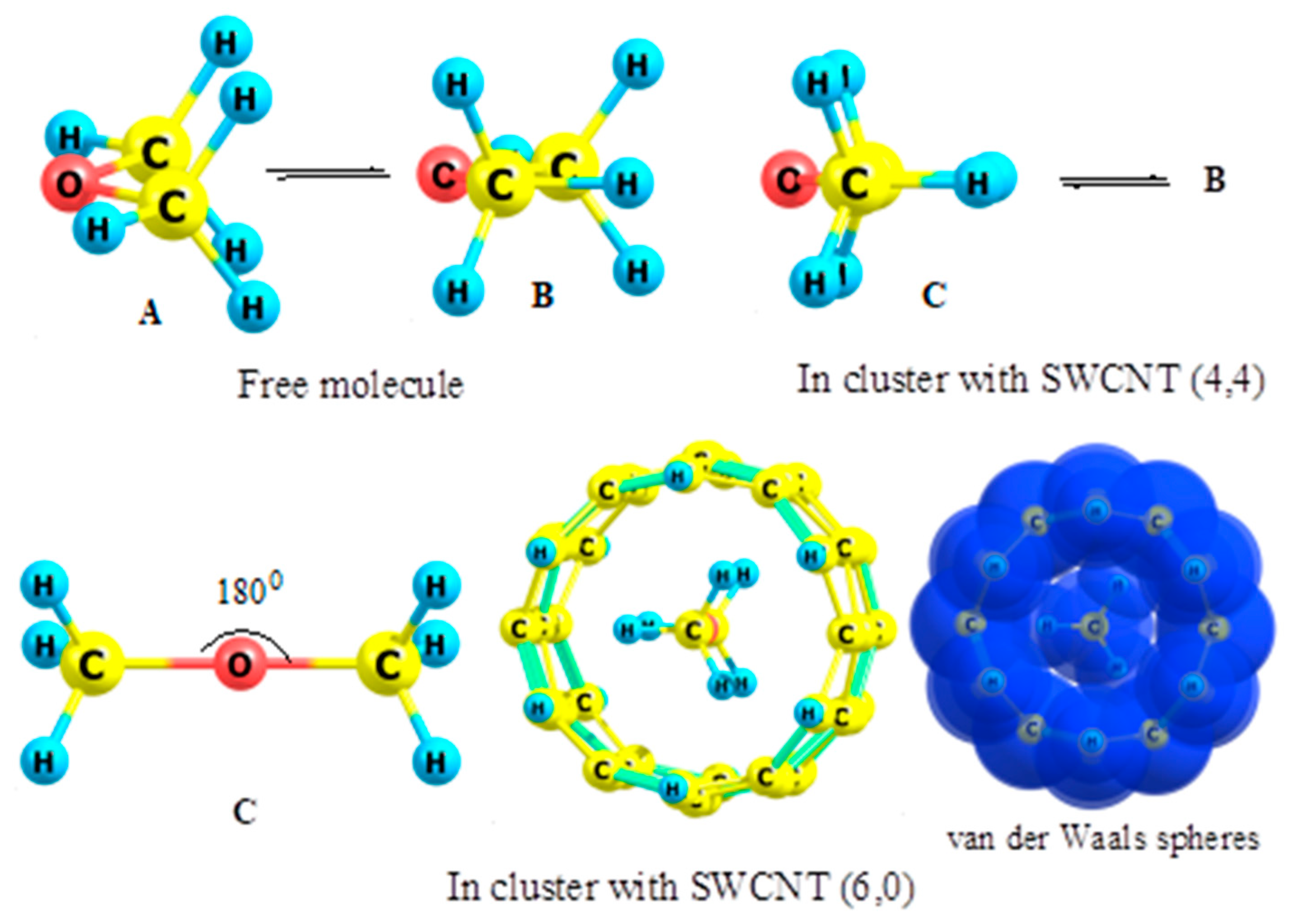
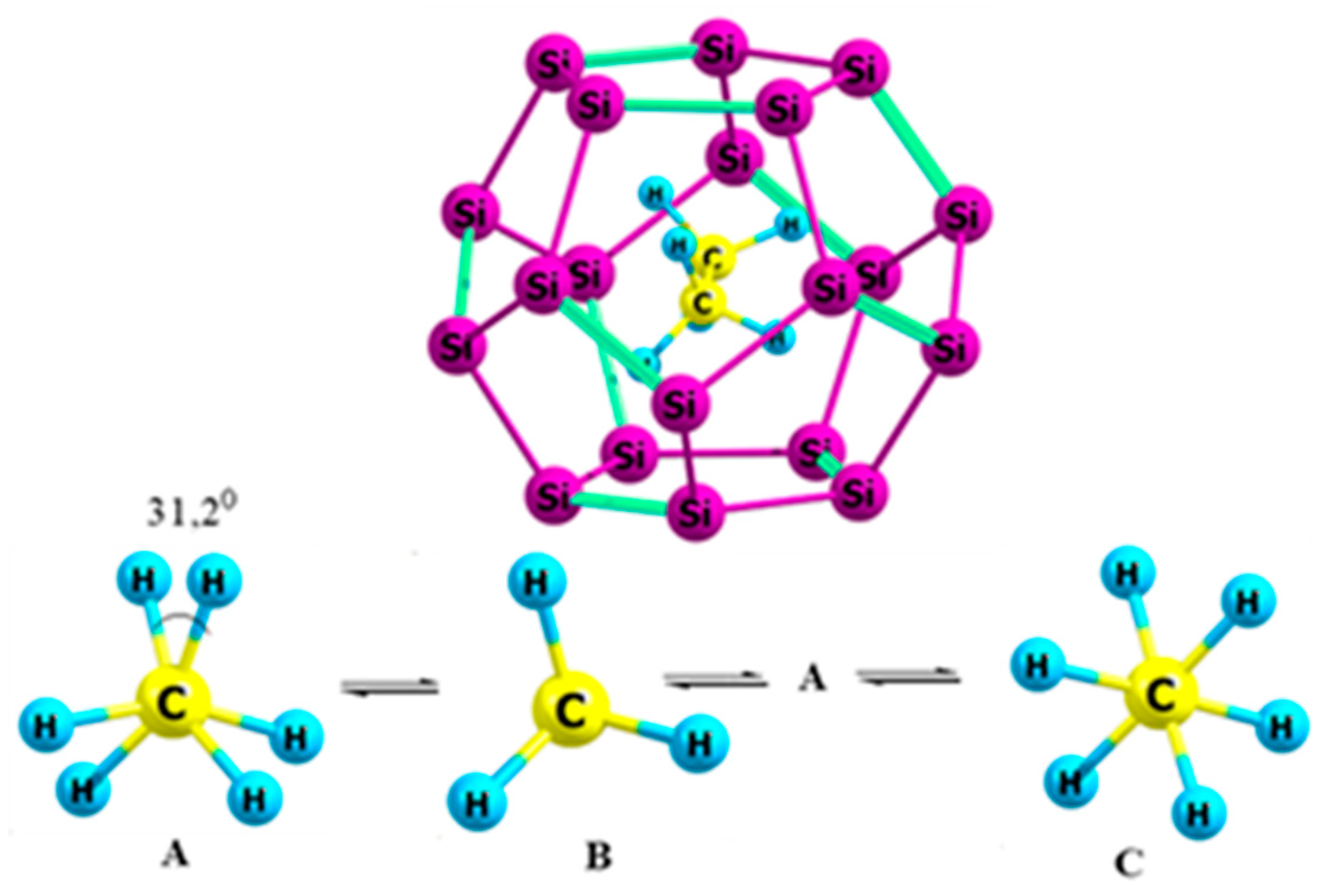
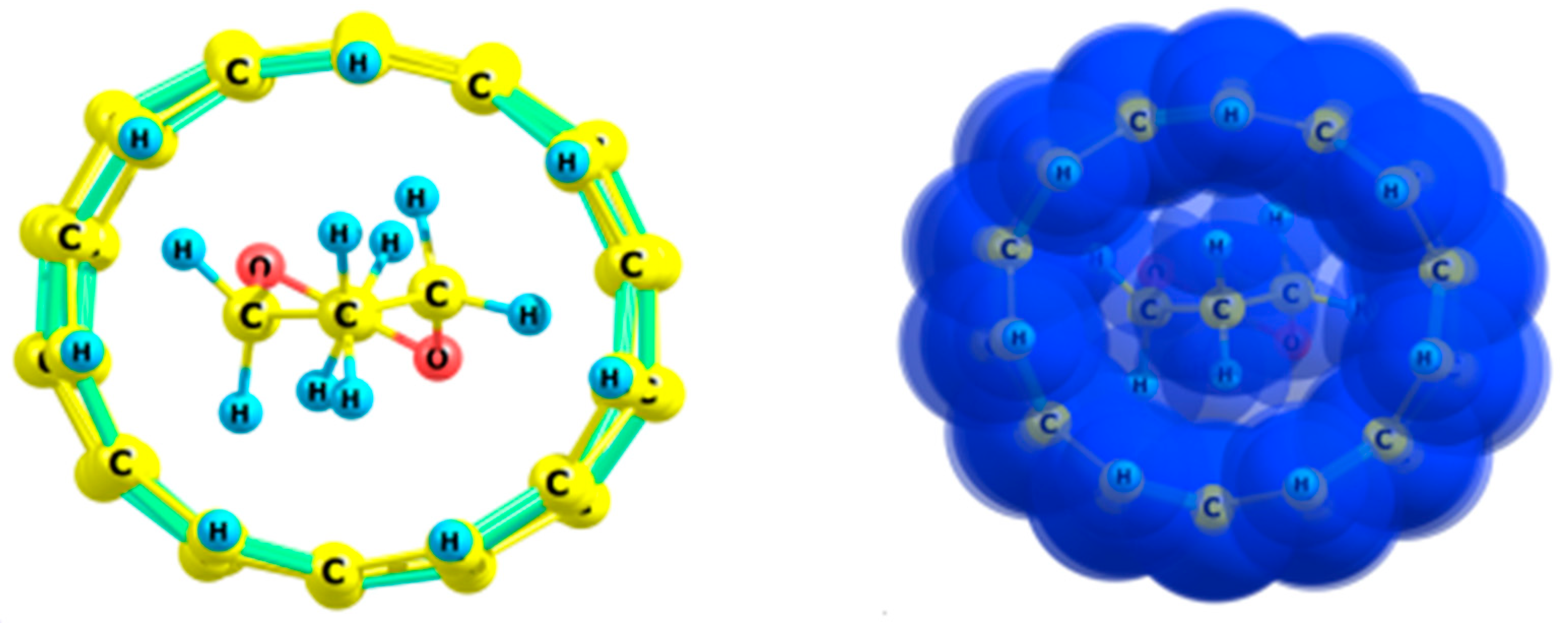
2.1. Ethane
2.2. Propane
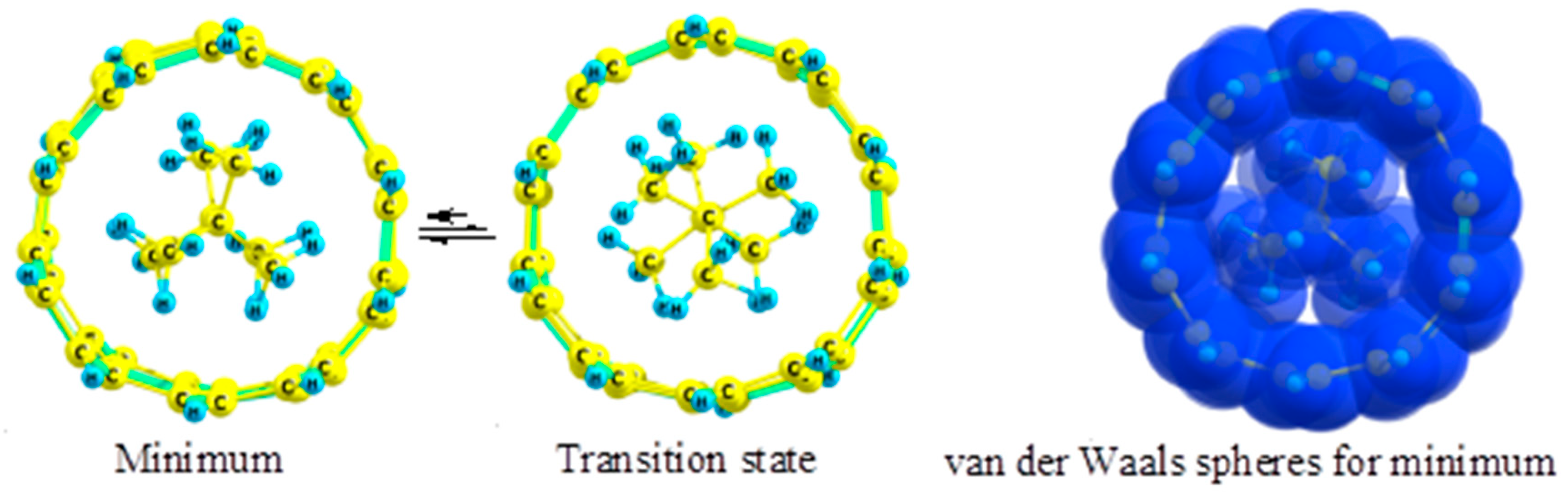
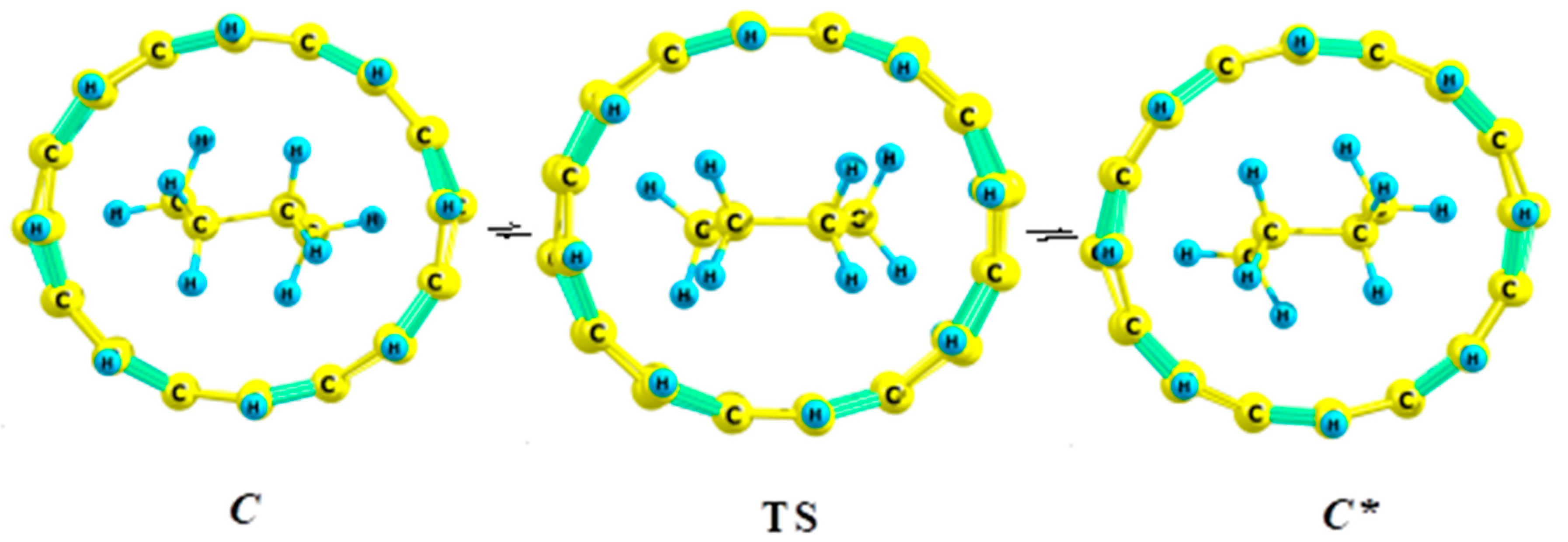
2.3. 2,2,3,3-Tetramethylbutane
2.4. 2,2-Dimethylpropane
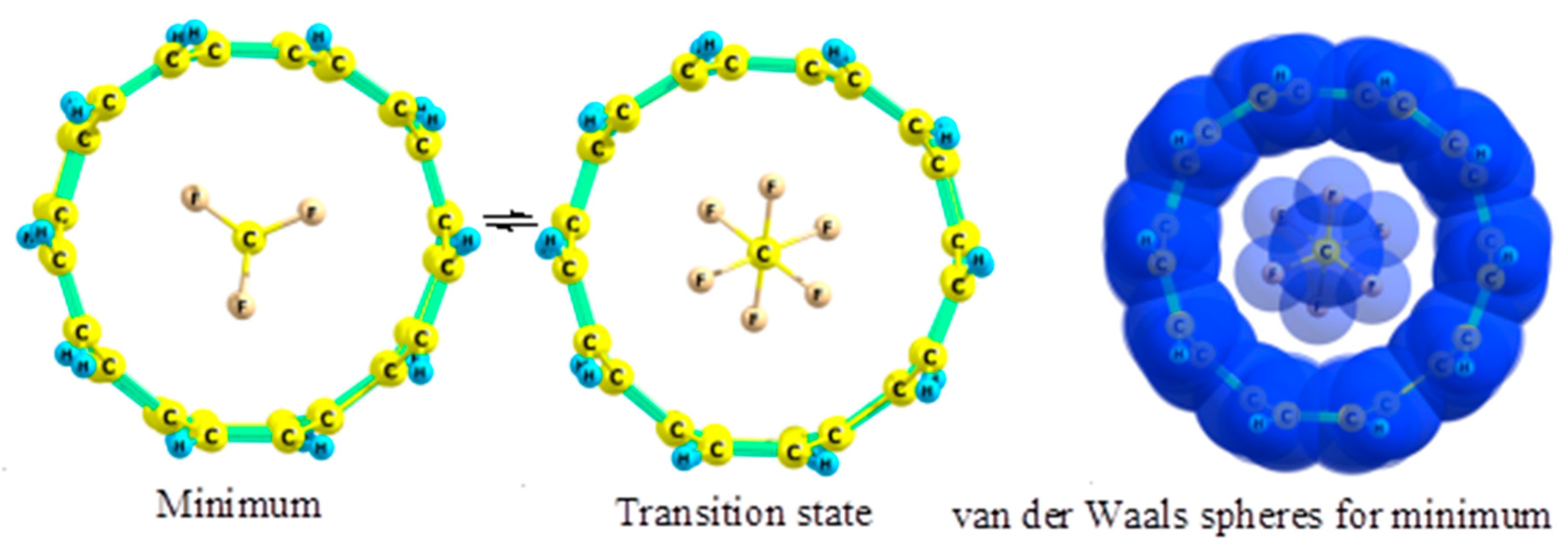
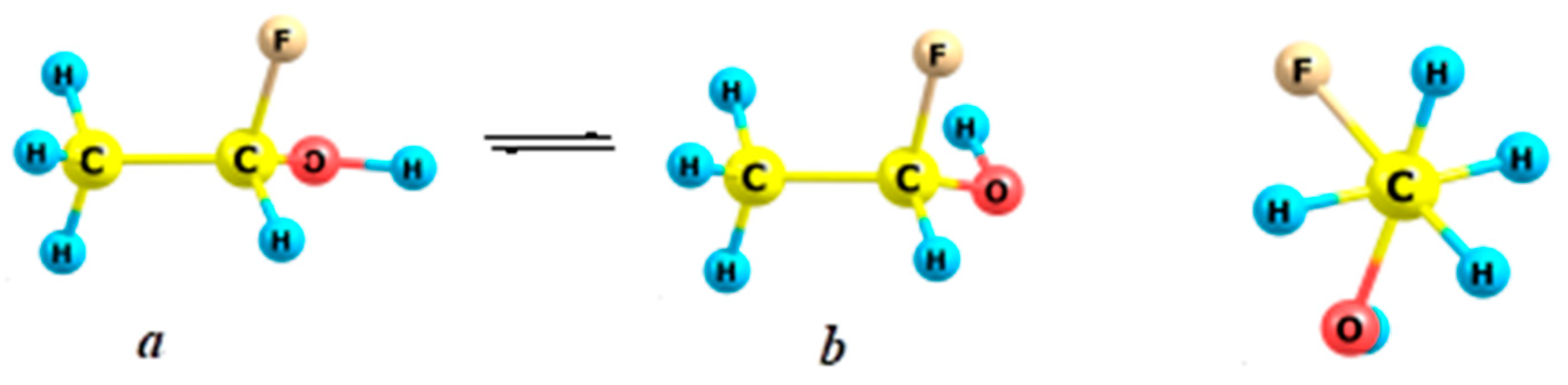
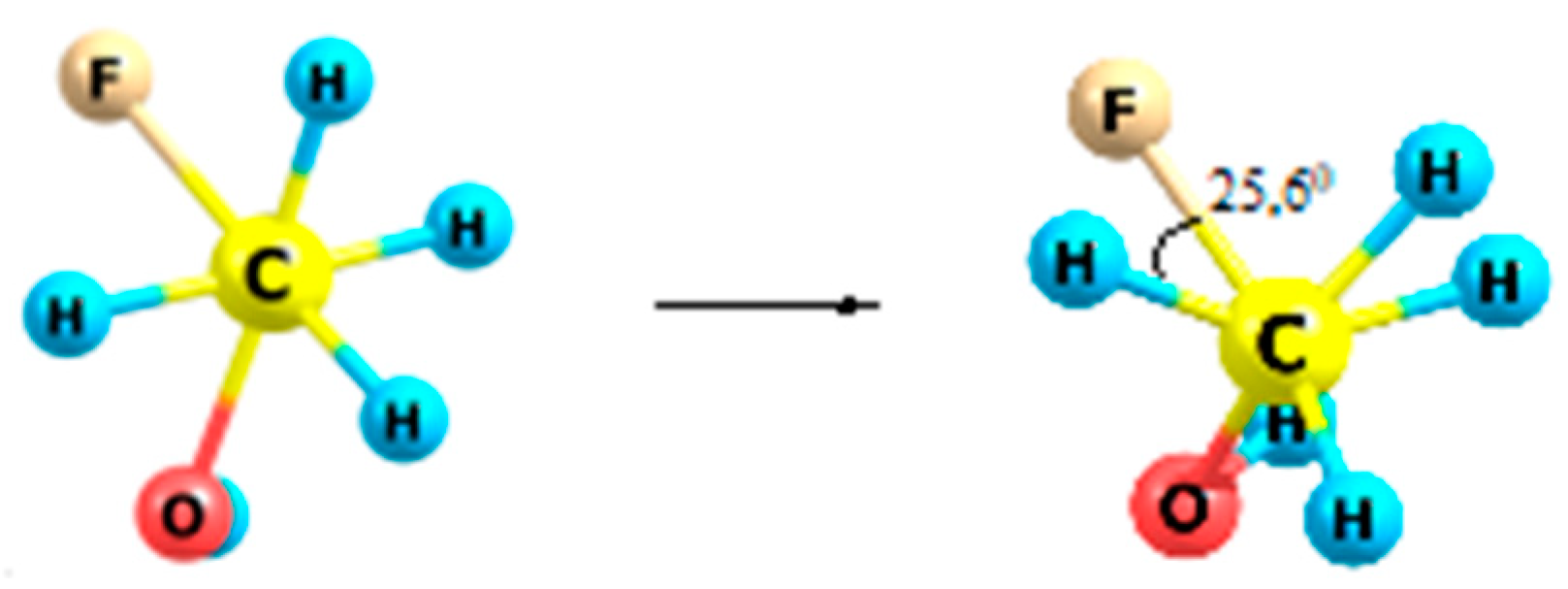
2.5. Fluoroethanes
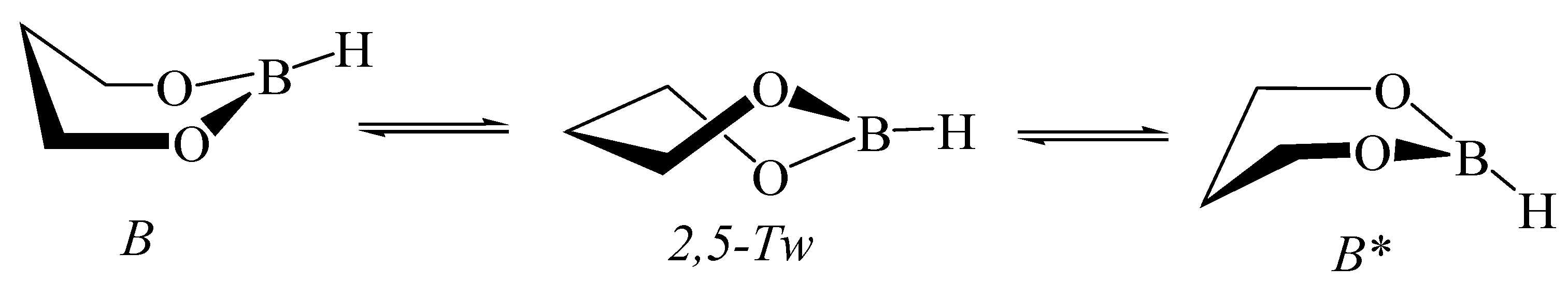
2.6. Ammonia Borane
2.7. Disilane and Digermane
3. Conformational Behavior of Other Acyclic Molecules in Nanotubes
3.1. Hydroxyborane
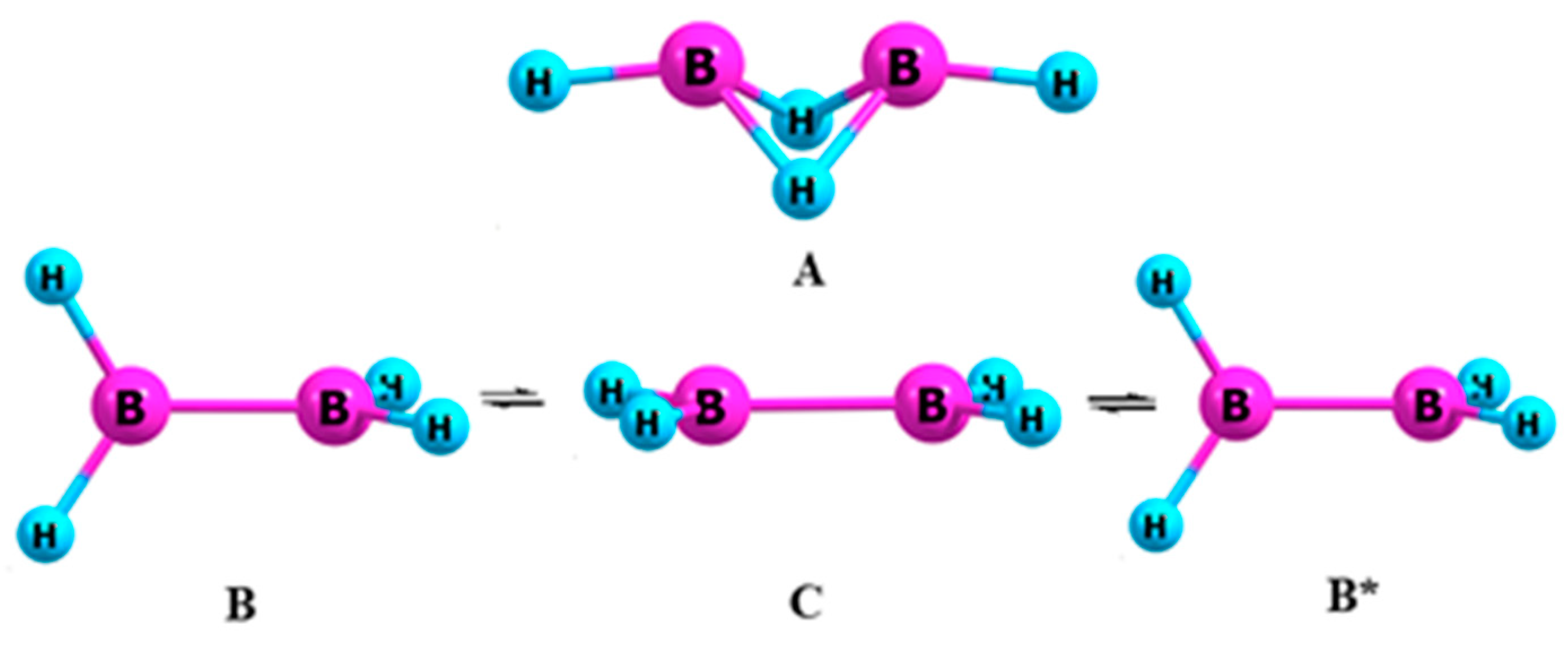
3.2. Diborane (4)
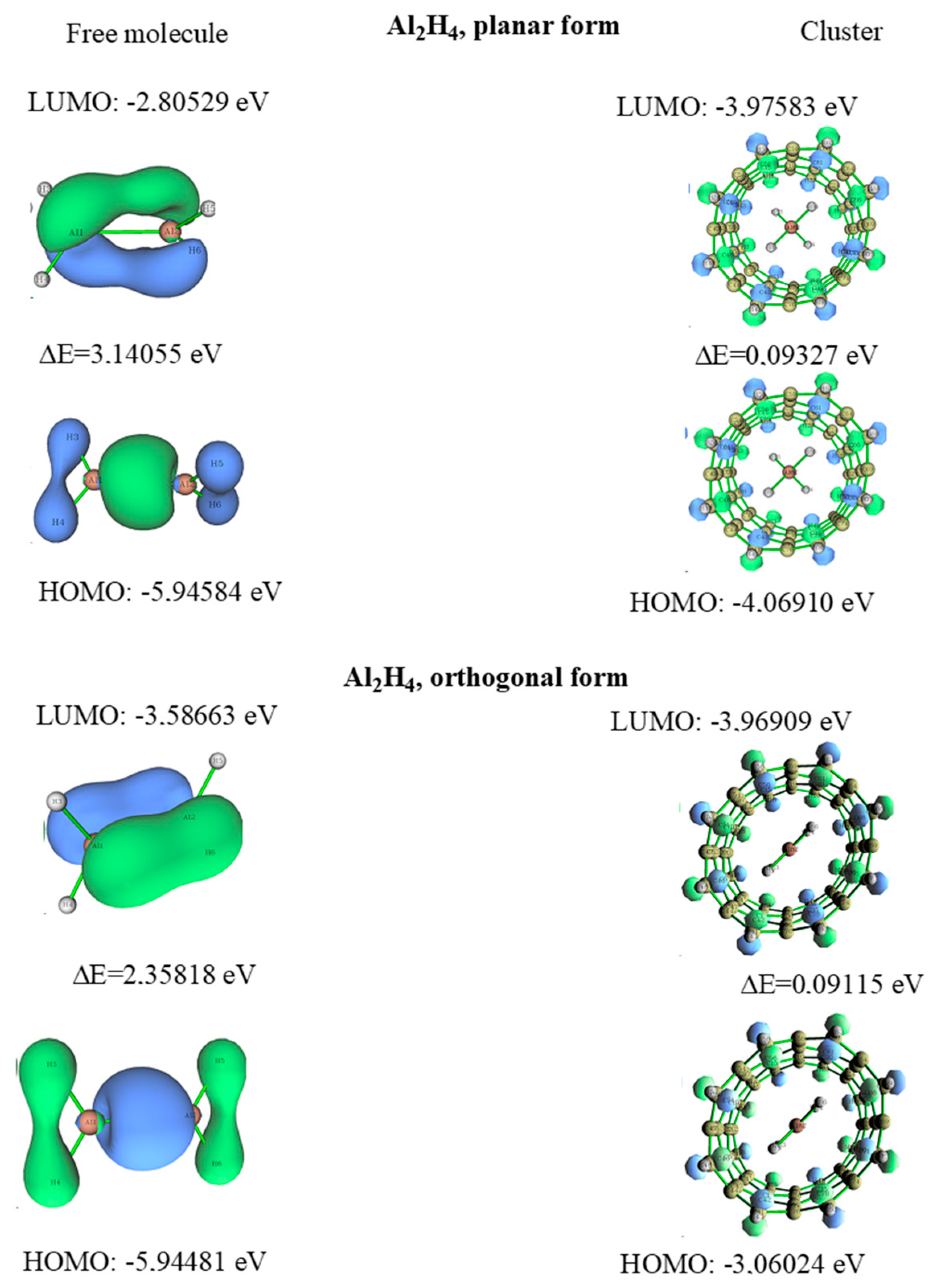
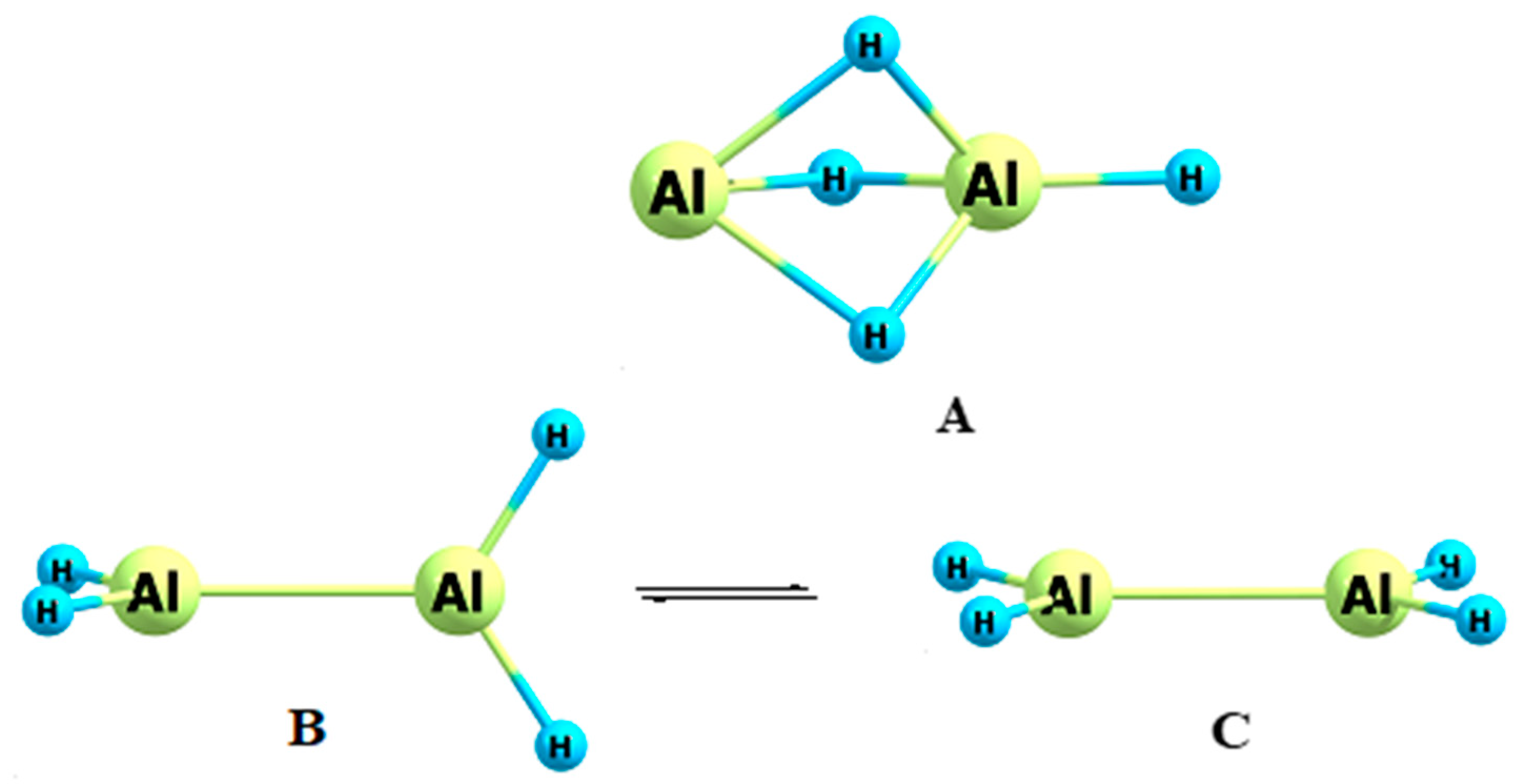
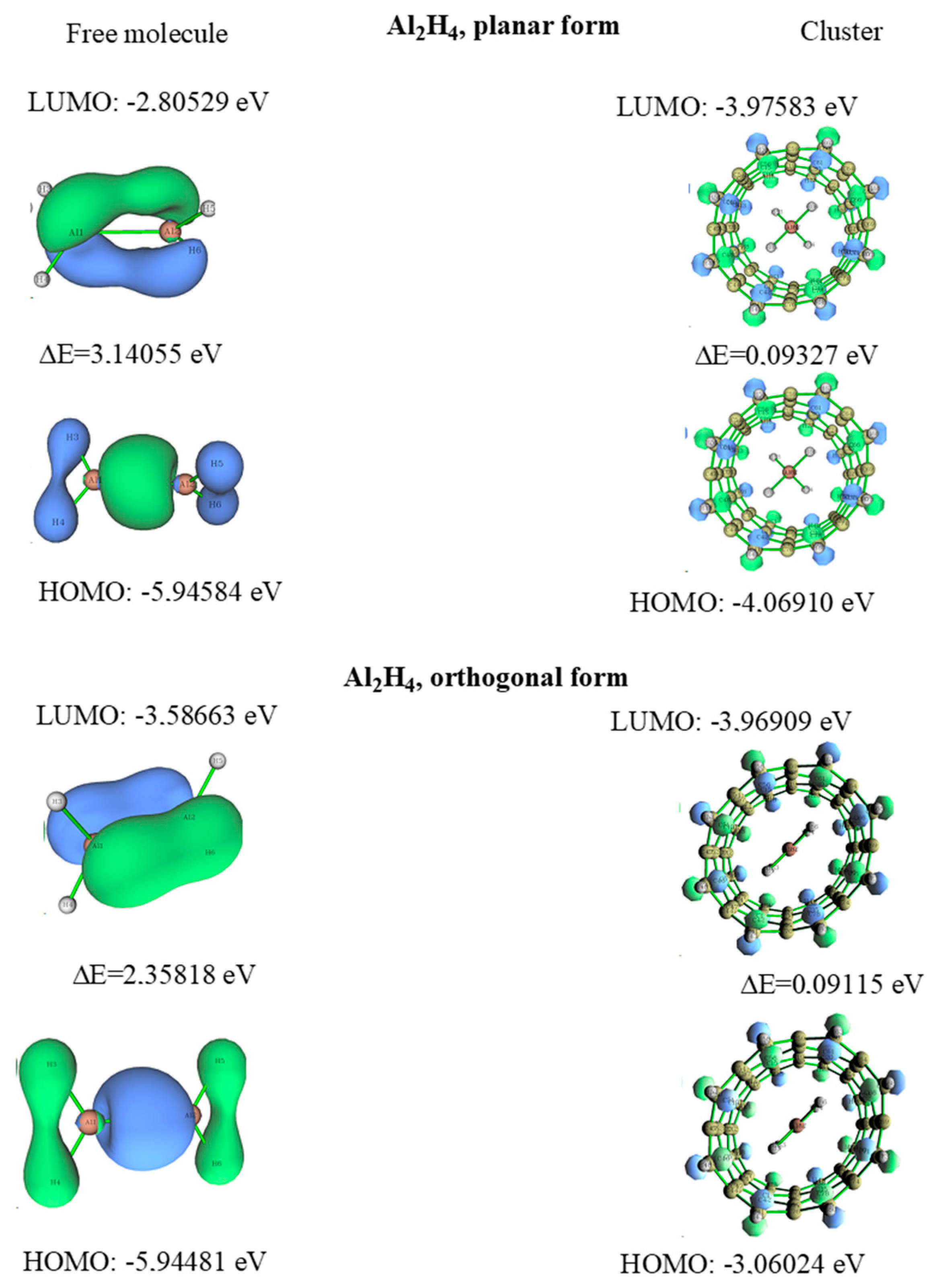
3.3. Dialane (4)
3.4. Hydrazine
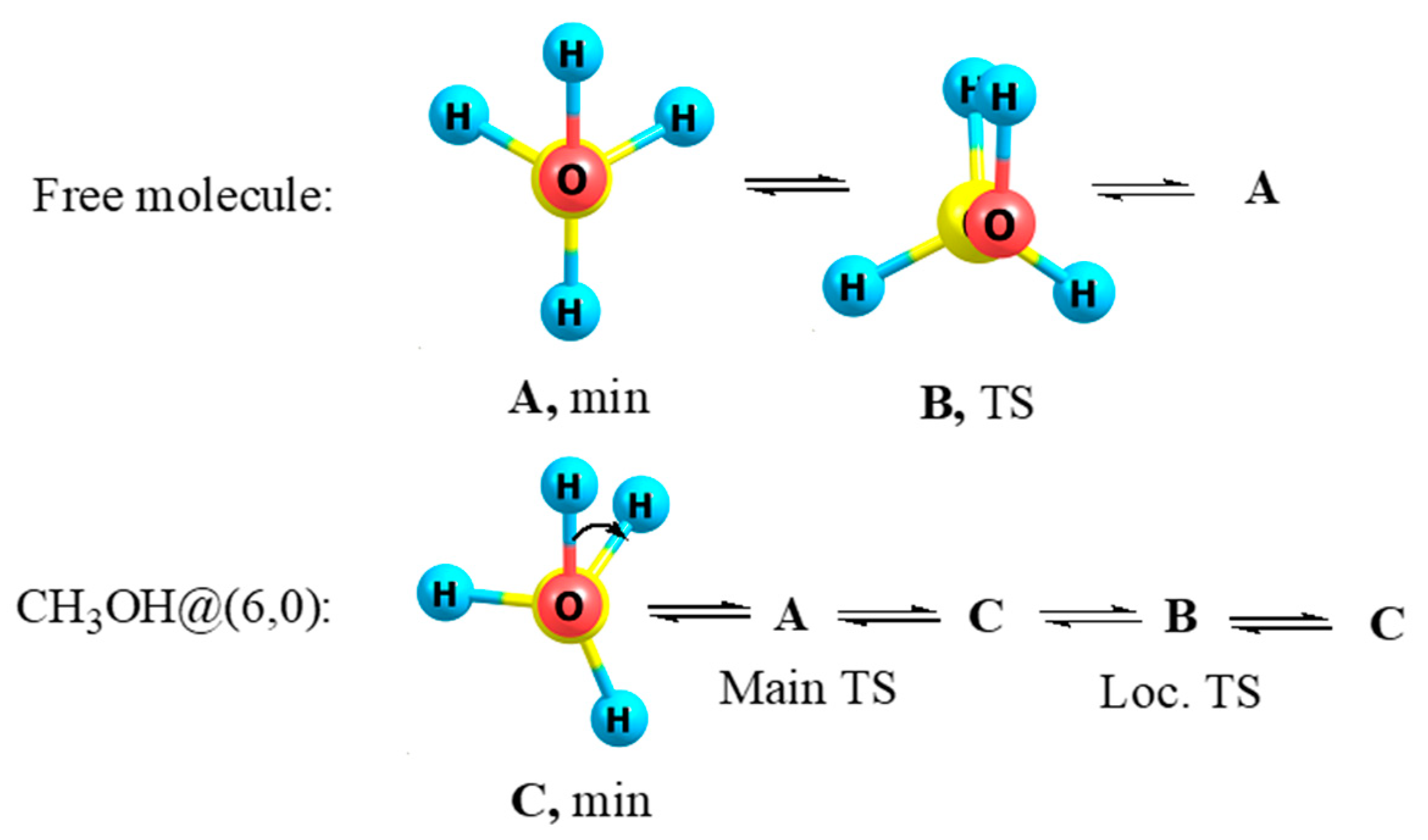
3.5. Methanol and Methanethiol
3.6. Dimethyl Ether
4. Conformational Properties of Simple Molecules in Fullerenes
4.1. Ethane and Its Analogs
4.2. Methanethiol
5. Conformational Behavior of Saturated Cyclic Molecules inside Nanotubes and Fullerenes
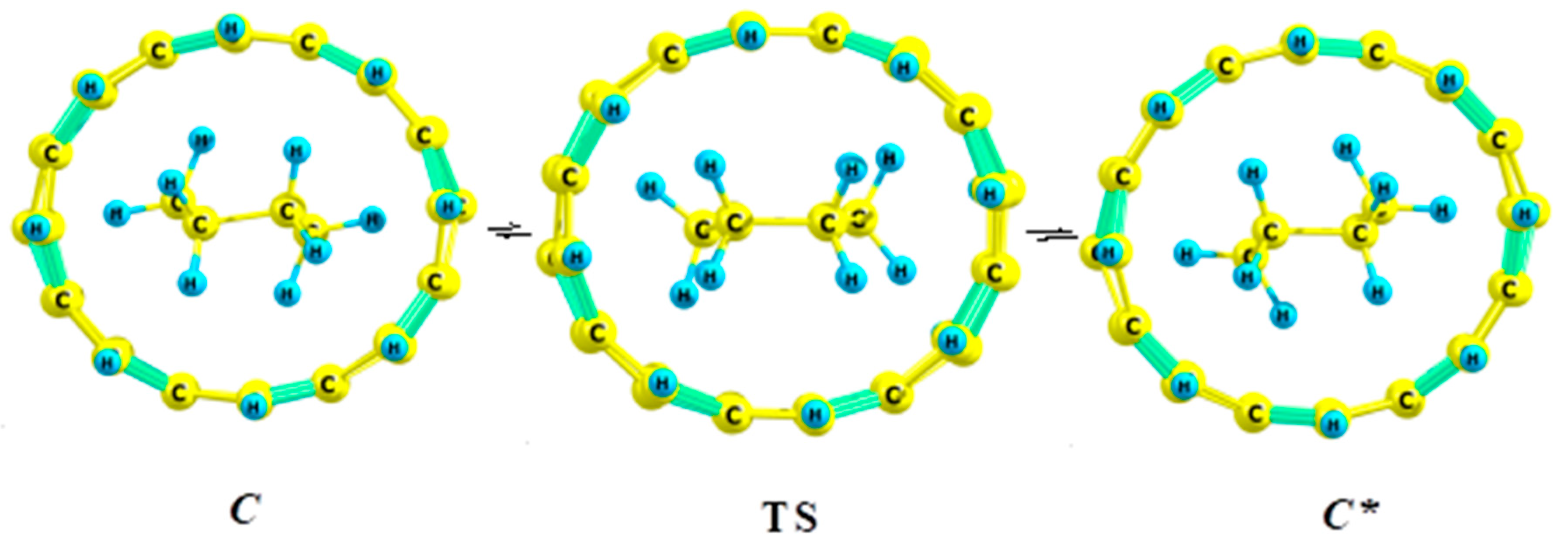
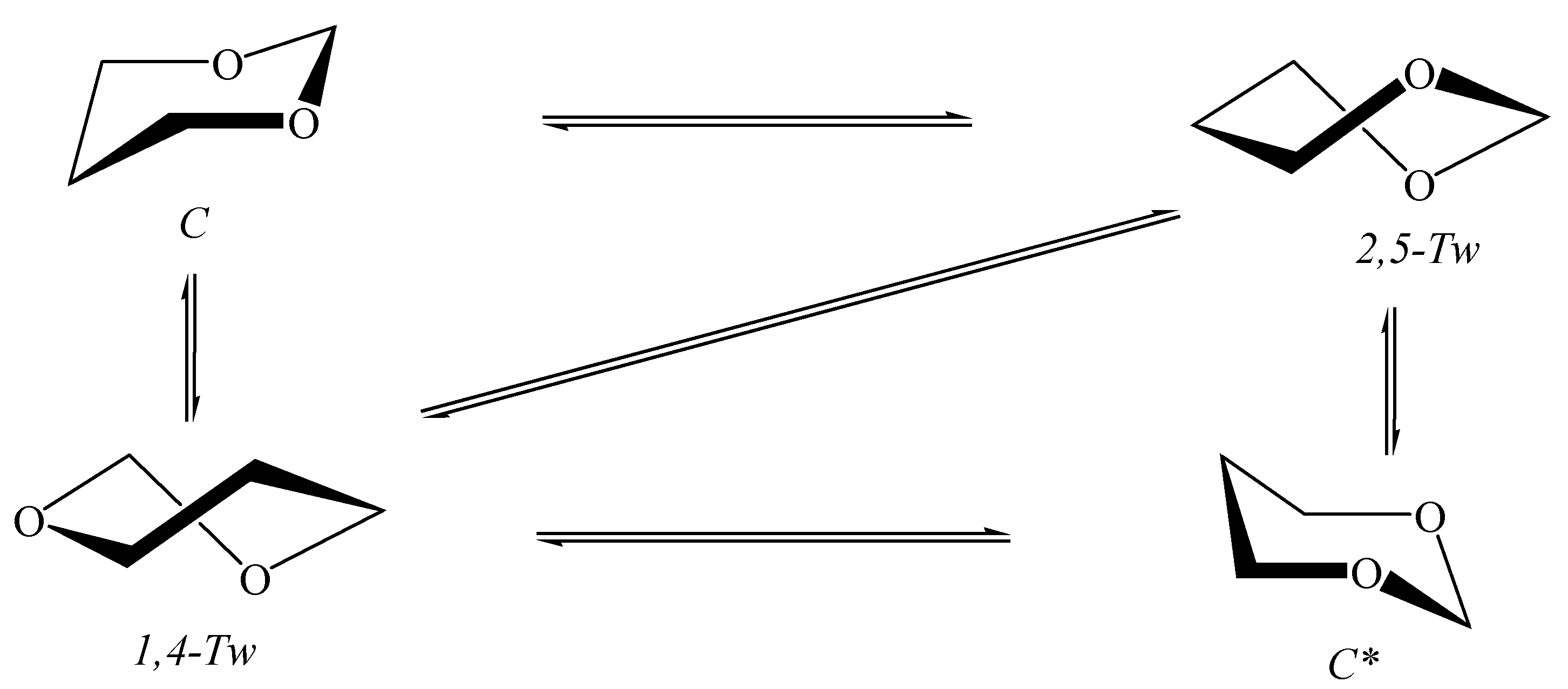
5.1. Cyclohexane in Nanotubes
5.2. 1,3-Dioxane in Nanotubes
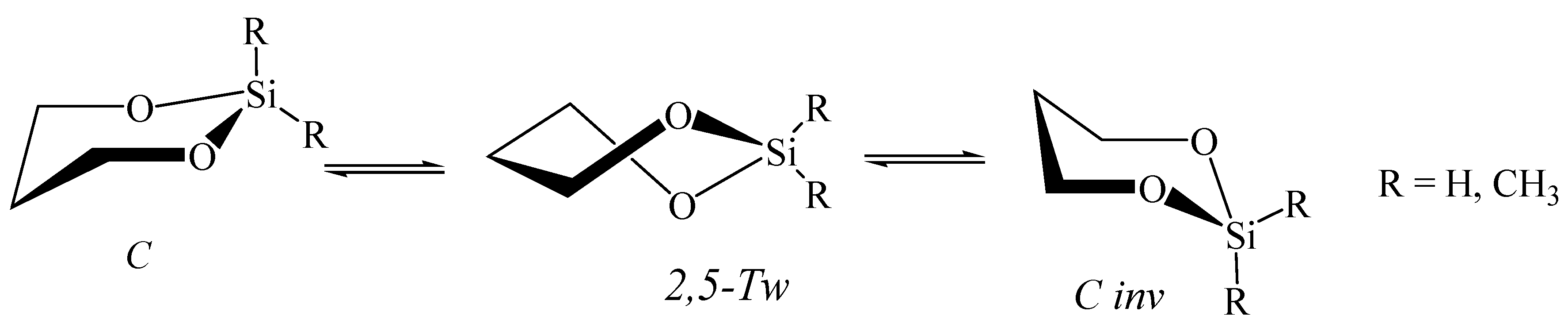
5.3. 1,3-Dioxa-2-Silacyclohexane in Nanotubes
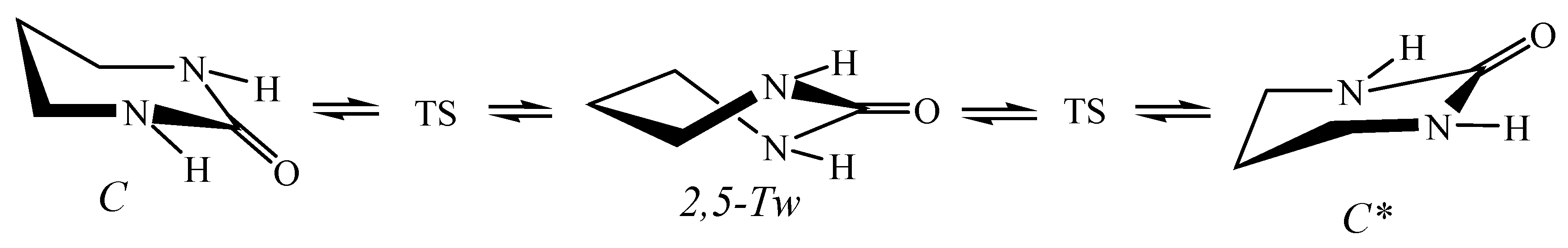
5.4. Hexahydropyrimidin-2-One in Nanotubes
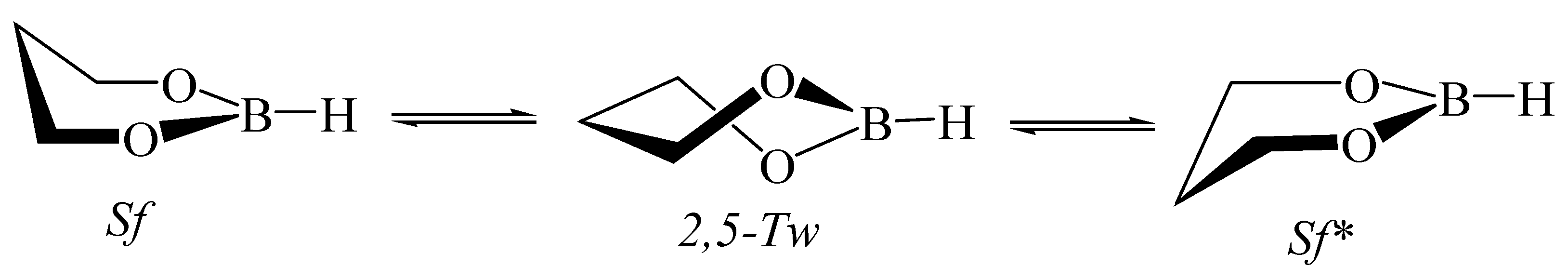
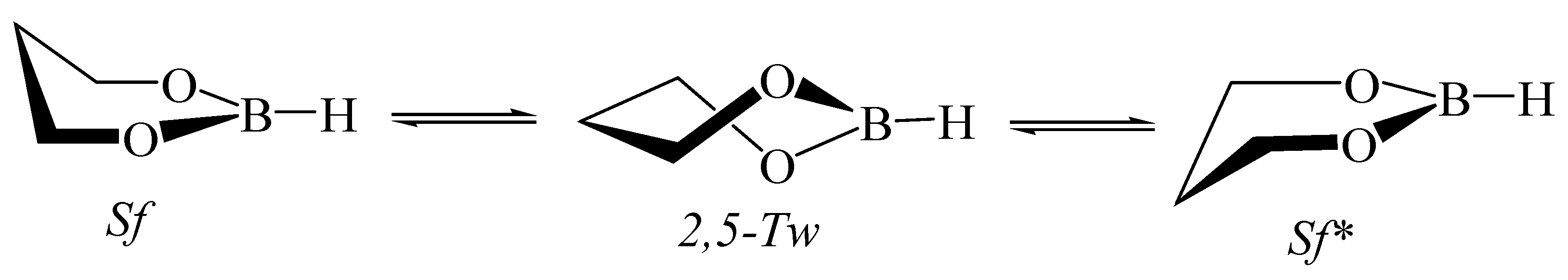
5.5. 1,3,2-Dioxaborinane in Fullerenes
6. Nitrogen Pyramidal Inversion inside Nanotubes and Fullerenes
6.1. Ammonia and Trimethylamine in Nanotubes
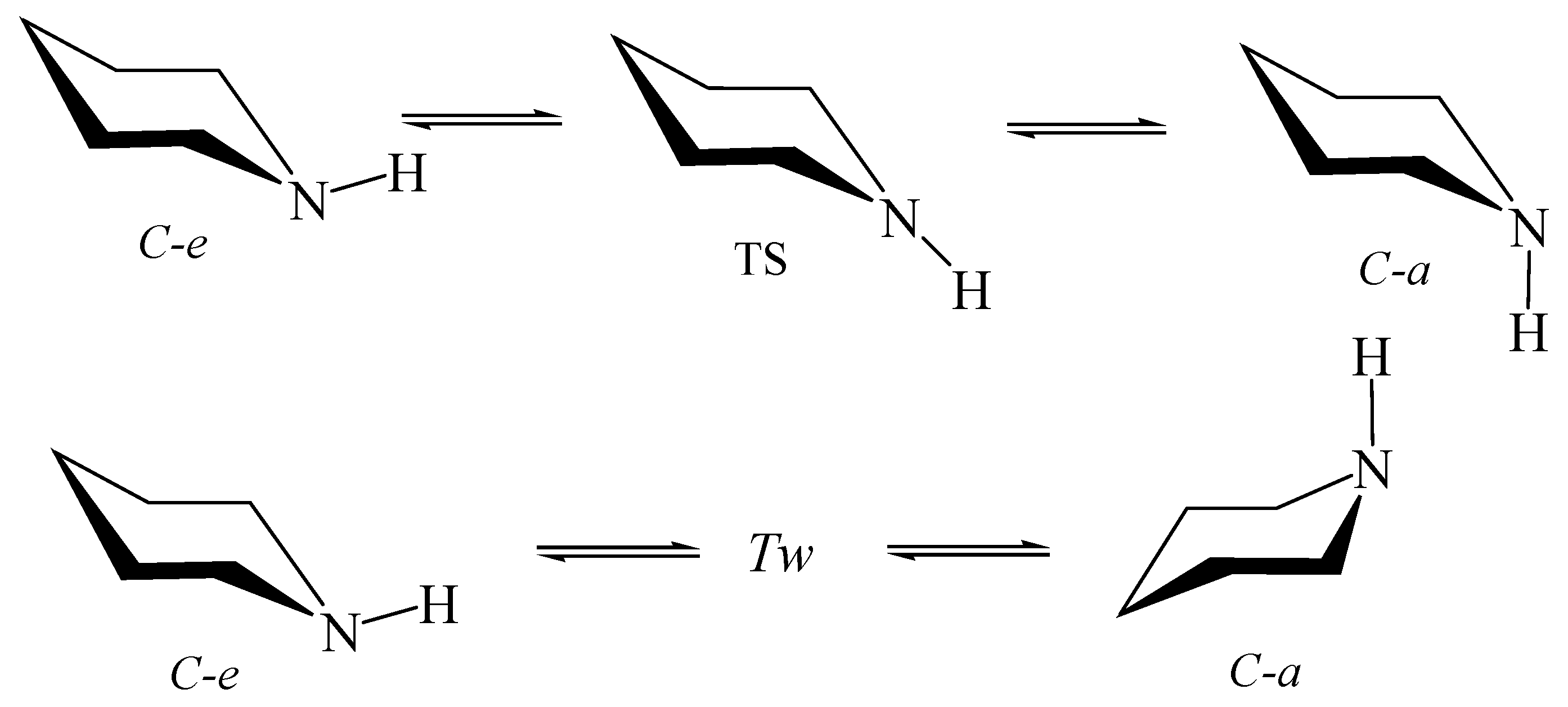
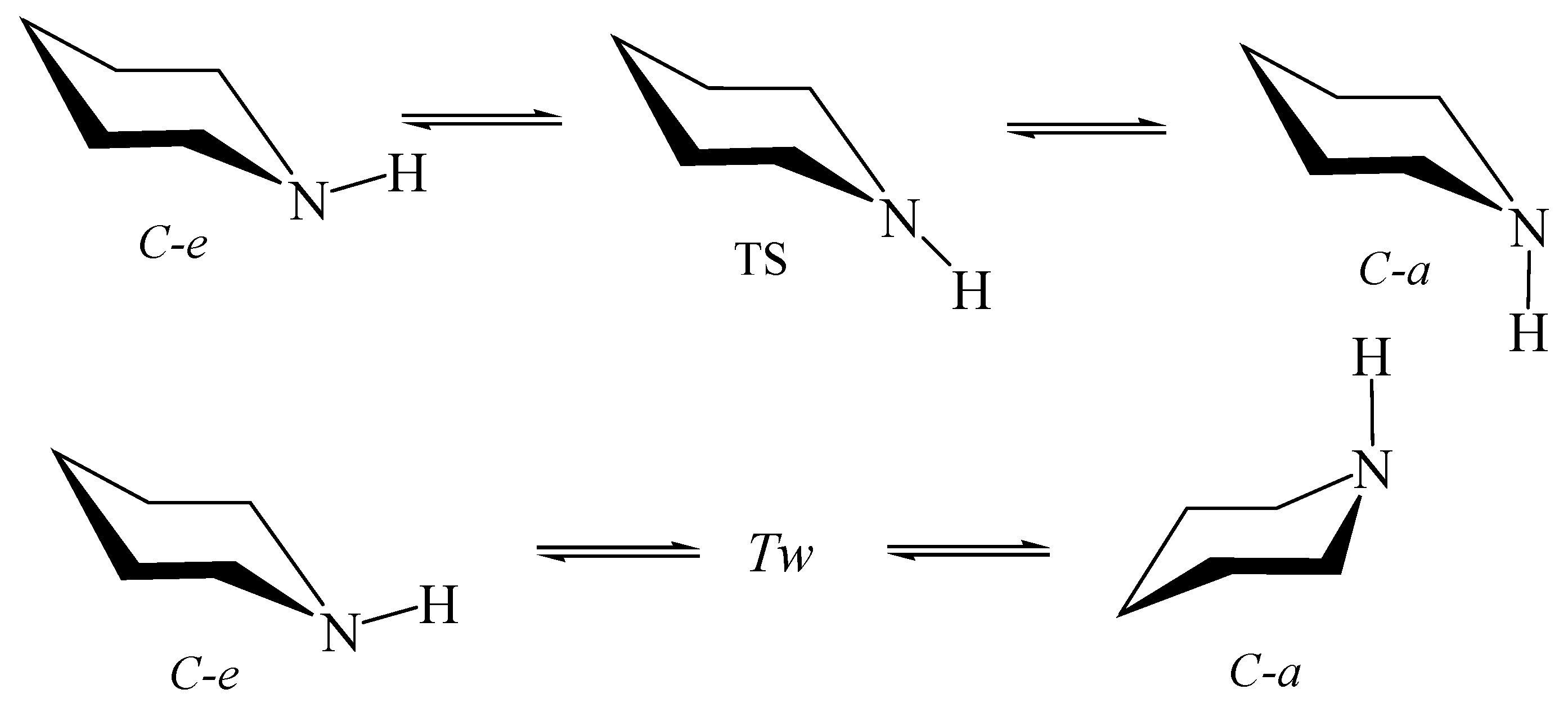
6.2. Piperidine inside Nanotube
6.3. Perhydro-1,3,2-Dioxazine inside Nanotubes
6.4. Ammonia and Trimethylamine in Fullerenes
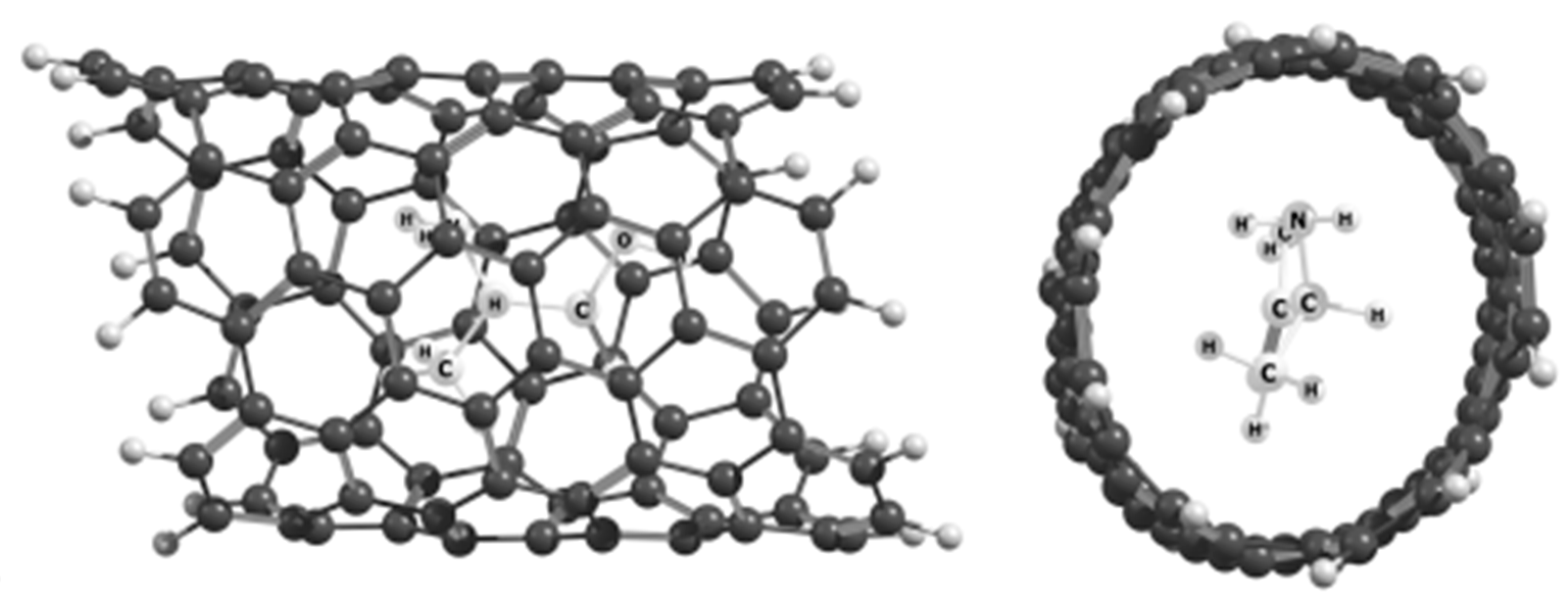
7. Recognition of the R- and S-Isomers by Chiral Nanotubes
7.1. R- and S-Isomers of 1-Fluoroethanol Inside SWCNTs (4,4) and (4,1)
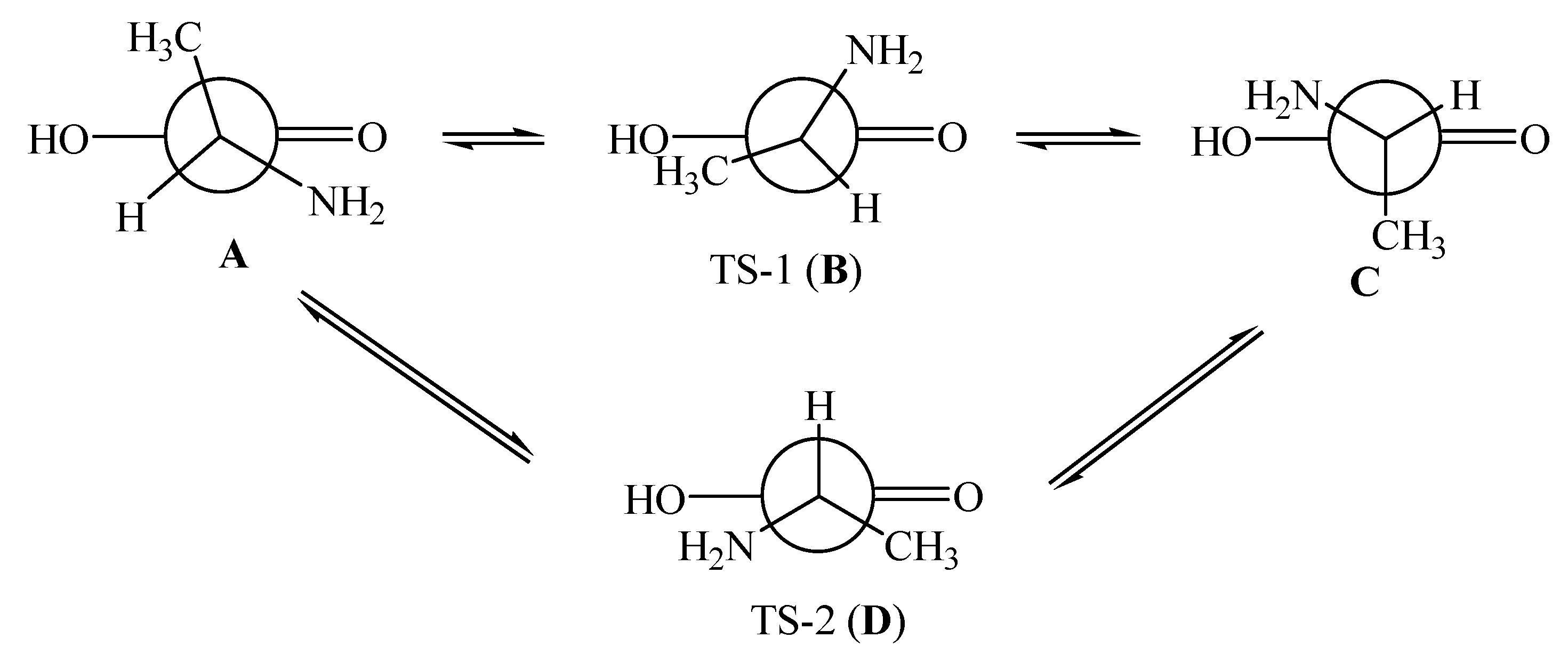
7.2. R- and S-Isomers of α-Alanine inside SWCNTs (n,m)
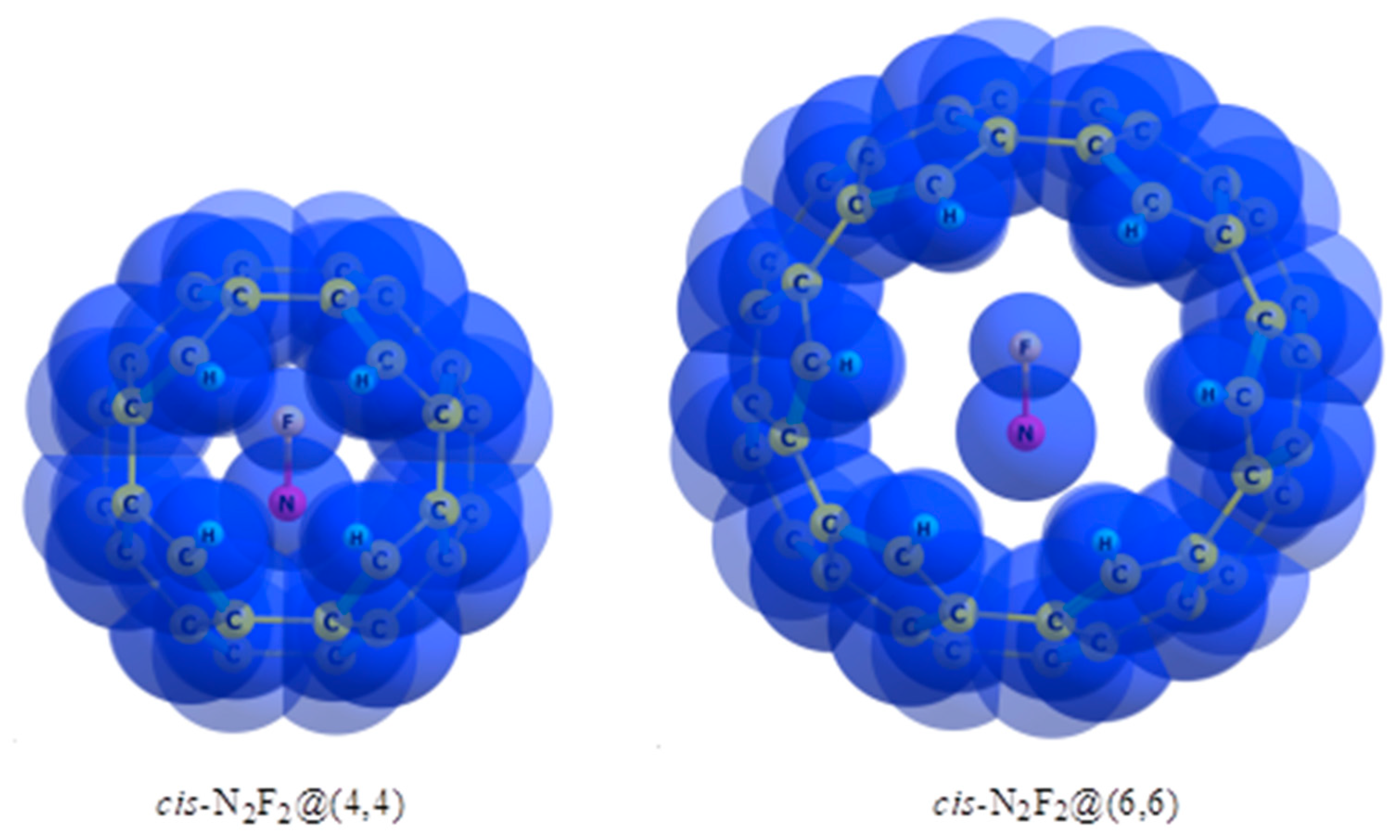
8. Relative Stability of Cis and Trans Isomers inside Nanotubes. The “Trans-Effect”
9. Conclusions
- The preferred conformation of ethane and its analogs inside nanotubes of small diameter is not the staggered, but the eclipsed form. The conformational behavior of ethane-like molecules inside fullerenes is less clear and depends on the size and chemical composition of the nanoobject.
- Hydroxyborane and diborane in endohedral clusters of nanotubes are characterized by reducing the barriers of internal rotation about the B-O and B-B bonds; in the case of dialane, the planar form—the transition state for the free molecule—becomes the minimum on the potential energy surface. A similar change in the nature of the preferred conformation is observed for hydrazine, methanol, methanethiol and dimethyl ether, together with cyclic molecules: cyclohexane, 1,3-dioxa-2-silacyclohexane and hexahydropirimidine-2-one inside nanotubes. In the case of 1,3-dioxane inside SWCNT and 1,3,2-dioxaborinane inside fullerene C60 conformational equilibrium is shifted to forms that can never be realized as a ground state for neither free molecule, nor for these species in any solvent.
- The barrier to pyramidal inversion of amines in endocomplexes varies depending on the nature of the amine and the size of the nanocavity.
- Chiral nanotubes are able to recognize molecules of enantiomers owing to increased affinity of P-SWCNT for the S-isomer and M-SWCNT for the R-form.
- SWCNTs of small diameter are able to drastically change the relative stability of cis and trans isomers in their cavity.
Funding
Conflicts of Interest
Abbreviations
| SWCNTs | single-walled carbon nanotubes |
| PES | potential energy surface |
| TS | transition state |
| DFT | density functional theory |
| HF | Hartree–Fock |
| PBE | Perdew–Burke–Ernzerhof |
| O | Mulliken bond order |
| HOMO | high occupied molecular orbital |
| LUMO | low unoccupied molecular orbital |
| C | conformation of chair form |
| Tw | conformation of twist form |
References
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Iijima, S.; Ichihashi, T. Single-shell carbon nanotubes of 1-nm diameter. Nature 1993, 363, 603–605. [Google Scholar] [CrossRef]
- Kroto, H.W.; Heath, J.R.; Obrien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Y.; Wei, F. Synthesis and Properties of Ultralong Carbon Nanotubes. In Nanotube Superfiber Materials; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 4; pp. 87–136. [Google Scholar] [CrossRef]
- Nanot, S.; Thompson, N.A.; Kim, J.-H.; Wang, X.; Rice, W.D.; Hároz, E.H.; Ganesan, Y.; Pint, C.L.; Kono, J. Single-Walled Carbon Nanotubes. In Springer Handbook of Nanomaterials; Springer Handbooks; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–146. [Google Scholar]
- Tanaka, K.; Iijima, S. Carbon Nanotubes and Graphene, 2nd ed.Elsevier: Amsterdam, The Netherlands, 2014; 458p. [Google Scholar]
- Nasrollahzadeh, M.; Issaabadi, Z.; Sajjadi, M.; Sajadi, S.M.; Atarod, M. Types of nanostructures. Interface Sci. Technol. 2019, 28, 29–80. [Google Scholar] [CrossRef]
- Nimibofa, A.; Newton, E.A.; Cyprain, A.Y.; Donbebe, W. Fullerenes: Synthesis and applications. J. Mater. Sci. Res. 2018, 7, 22–36. [Google Scholar] [CrossRef]
- Manzetti, S.; Gabriel, J.-C.P. Methods for dispersing carbon nanotubes for nanotechnology applications: Liquid nanocrystals, suspensions, polyelectrolytes, colloids, and organization control. Int. Nano Lett. 2019, 9, 31–49. [Google Scholar] [CrossRef] [Green Version]
- Takakura, A.; Beppu, K.; Nishihara, T.; Fukui, A.; Kozeki, T.; Namazu, T.; Miyauchi, Y.; Itami, K. Strength of carbon nanotubes depends on their chemical structures. Nat. Commun. 2019, 10, 3040. [Google Scholar] [CrossRef]
- Abdalla, S.; Al-Marzouki, F.; Al-Ghamdi, A.A.; Abdel-Daiem, A. Different technical applications of carbon nanotubes. Nanoscale Res. Lett. 2015, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, A.; Amadi, E.V.; Chen, Y.; Papadopoulos, C. Carbon nanotube assembly and integration for applications. Nanoscale Res. Lett. 2019, 14, 220. [Google Scholar] [CrossRef]
- Koo, J.H.; Song, J.-K.; Kim, D.-H. Solution-processed thin films of semiconducting carbon nanotubes and their application to soft electronics. Nanotechnology 2019, 30, 132001. [Google Scholar] [CrossRef]
- Akiyama, T. Development of fullerene thin-film assemblies and fullerene-diamine adducts towards practical nanocarbon-based electronic materials. Bull. Chem. Soc. Jpn. 2019, 92, 1181–1199. [Google Scholar] [CrossRef]
- He, H.; Pham-Huy, L.A.; Dramou, P.; Xiao, D.; Zuo, P.; Pham-Huy, C. Carbon nanotubes: Applications in pharmacy and medicine. BioMed Res. Int. 2013, 2013, 578290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sireesha, M.; Babu, V.J.; Kiran, A.S.K.; Ramakrishna, S. A review on carbon nanotubes in biosensor devices and their applications in medicine. Nanocomposites 2018, 4, 36–57. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, M. Biodegradation of carbon nanotubes by macrophages. Front. Mater. 2019, 6, 225–239. [Google Scholar] [CrossRef]
- Castro, E.; Hernandez, G.A.; Zalava, G.; Echegoyen, L. Fullerenes in biology and medicine. J. Mater. Chem. B 2017, 5, 6523–6535. [Google Scholar] [CrossRef] [PubMed]
- Panwar, N.; Soehartono, A.M.; Chan, K.K.; Zeng, S.; Xu, G.; Qu, J.; Coquet, P.; Yong, K.-T.; Chen, X. Nanocarbons for biology and medicine: Sensing, imaging, and drug delivery. Chem. Rev. 2019, 119, 9559–9656. [Google Scholar] [CrossRef]
- Khamitova, K.K.; Kayupov, B.A.; Yegemova, S.S.; Gabdullin, M.T.; Abdullin, K.A.; Ismailov, D.V.; Kerimbekov, D.C. The use of fullerenes as a biologically active molecule. Int. J. Nanotechnol. 2019, 16, 100–108. [Google Scholar] [CrossRef]
- Özkan, E.; Bilge, M.; Bilge, D.; Alver, Ö.; Parlak, C.; Şenyel, M.; Ramasami, P. Sensor application of doped C60 fullerenes in detection of 1-(3-trifluoromethylphenyl)piperazine as an alternative to ecstasy. Main Group Metal. Chem. 2019, 42, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Bourassa, D.J.; Kerna, N.A. Pristine. Nanocarbonbased fullerene-like material. Toxicity and biocompatibility (Part 2 in the series: Will Nanocarbon Onion-Like Fullerenes Play a Decisive Role in the Future of Molecular Medicine?). Determ. Nanomed. Nanotechnol. 2019, 1, DNN.000504.2019. [Google Scholar]
- Rahmati, M.; Mozafari, M. Biological response to carbon-family nanomaterials: Interactions at the nano-bio interface. Front. Bioeng. Biotechnol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Yasuno, T.; Ohe, T.; Ikeda, H.; Takahashi, K.; Nakamura, S.; Mashino, T. Synthesis and antitumor activity of novel pyridinium fullerene derivatives. Int. J. Nanomed. 2019, 14, 6325–6327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.; Leiva, E. Enhanced heavy metal removal from acid mine drainage wastewater using double-oxidized multiwalled carbon nanotubes. Molecules 2019, 25, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, J.R.; O’Brien, S.C.; Zhang, Q.; Liu, Y.; Curi, R.F.; Kroto, H.W.; Tittel, F.K.; Smalley, R.E. Lanthanum complexes of spheroidal carbon shells. J. Am. Chem. Soc. 1985, 107, 7779–7780. [Google Scholar] [CrossRef]
- Khlobystov, A.N.; Britz, D.A.; Briggs, G.A.D. Molecules in carbon nanotubes. Acc. Chem. Res. 2005, 38, 901–909. [Google Scholar] [CrossRef]
- Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chemistry of carbon nanotubes. Chem. Rev. 2006, 106, 1105–1136. [Google Scholar] [CrossRef]
- Britz, D.A.; Khlobystov, A.N. Noncovalent interactions of molecules with single walled carbon nanotubes. Chem. Soc. Rev. 2006, 35, 637–659. [Google Scholar] [CrossRef]
- Zambrano, H.A.; Walther, J.H.; Koumoutsakos, P.; Sbalzarini, I.F. Thermophoretic motion of water nanodroplets confined inside carbon nanotubes. Nano Lett. 2009, 9, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pan, X.; Shen, W.; Ren, P.; Han, X.; Bao, X. NMR study of preferential endohedral adsorption of methanol in multiwalled carbon nanotubes. J. Phys. Chem. C 2012, 116, 7803–7809. [Google Scholar] [CrossRef]
- Sozykin, S.A.; Beskachko, V.P. Structure of endohedral complexes of carbon nanotubes encapsulated with lithium and sodium. Mol. Phys. 2013, 111, 930–938. [Google Scholar] [CrossRef]
- Munusamy, E.; Wheeler, S.E. Endohedral and exohedral complexes of substituted benzenes with carbon nanotubes and graphene. J. Chem. Phys. 2013, 139, 094703. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef] [PubMed]
- Manzetti, S. Molecular and crystal assembly inside the carbon nanotube: Encapsulation and manufacturing approaches. Adv. Manuf. 2013, 1, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Tishchenko, O.; Truhlar, D.G. Atom-cage charge transfer in endohedral metallofullerenes: Trapping atoms within a sphere-like ridge of avoided crossings. J. Phys. Chem. Lett. 2013, 4, 422–425. [Google Scholar] [CrossRef]
- Cerón, M.R.; Li, F.-F.; Echegoyen, L.A. Endohedral fullerenes: The importance of electronic, size and shape complementarity between the carbon cages and the corresponding encapsulated clusters. J. Phys. Org. Chem. 2014, 27, 258–264. [Google Scholar] [CrossRef]
- Min, K.; Farimani, A.B.; Aluru, N.R. Mechanically modulated electronic properties of water-filled fullerenes. MRS Commun. 2015, 5, 305–310. [Google Scholar] [CrossRef]
- Umran, N.M.; Kumar, R. Effect on encapsulation (Au and Tl) molecule in fullerene (C60) on electronic and magnetic properties. Quant. Matter 2015, 4, 492–496. [Google Scholar] [CrossRef]
- Dargouthi, S.; Boughdiri, S.; Tangour, B. Stabilizing of the transitory species (TiO2)2 by encapsulation into carbon nanotubes. Acta Chim. Slov. 2015, 62, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Nikolaenko, T.Yu.; Kryachko, E.S. Formation of dimers of light noble atoms under encapsulation within fullerene’s voids. Nanoscale Res. Lett. 2015, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Junghans, K.; Rosenkranz, M.; Popov, A.A. Sc3CH@C80: Selective 13C enrichment of the central carbon atom. Chem. Commun. 2016, 52, 6561–6564. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, R.L.; Chamberlain, T.W.; Baldoni, M.; Lebedeva, M.A.; Davies, E.S.; Besley, E.; Khlobystov, A.N. Direct measurement of electron transfer in nanoscale host–guest systems: Metallocenes in carbon nanotubes. Chem. Eur. J. A 2016, 22, 13540–13549. [Google Scholar] [CrossRef] [Green Version]
- Popov, A.A. Synthesis, and molecular structures of endohedral fullerenes. In Endohedral Fullerenes: Electron Transfer and Spin; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 1–34. [Google Scholar] [CrossRef] [Green Version]
- Kalugina, Y.N.; Roy, P.-N. Potential energy and dipole moment surfaces for HF@C60: Prediction of spectral and electric response properties. J. Chem. Phys. 2017, 147, 244303. [Google Scholar] [CrossRef]
- Zhang, R.; Murata, M.; Wakamiya, A.; Shimoaka, T.; Hasegawa, T.; Murata, Y. Isolation of the simplest hydrated acid. Sci. Adv. 2017, 3, e1602833. [Google Scholar] [CrossRef]
- Jin, P.; Yang, L.; Liu, C. Computational prediction of the endohedral metalloborofullerenes Tin@B40 (n=1, 2). Theor. Chem. Acc. 2017, 136, 56. [Google Scholar] [CrossRef]
- Calatayud, D.G.; Ge, H.; Kuganathan, N.; Mirabello, V.; Jacobs, R.M.J.; Rees, N.H.; Stoppiello, C.T.; Khlobystov, A.N.; Tyrrell, R.M.; Da Como, E.; et al. Encapsulation of cadmium selenide nanocrystals in biocompatible nanotubes: DFT calculations, X-ray diffraction investigations, and confocal fluorescence imaging. Chem. Open 2018, 7, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Poudel, Y.R.; Wenzhi Li, W. Synthesis, properties, and applications of carbon nanotubes filled with foreign materials: A review. Mater. Today Phys. 2018, 7, 7–34. [Google Scholar] [CrossRef]
- Krylov, D.S.; Liu, F.; Brandenburg, A.; Spree, L.; Bon, V.; Kaskel, S.; Wolter, A.U.B.; Büchner, B.; Avdoshenko, S.M.; Popov, A.A. Magnetization relaxation in the single-ion magnet DySc2N@C80: Quantum tunneling, magnetic dilution, and unconventional temperature dependence. Phys. Chem. Chem. Phys. 2018, 20, 11656–11672. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, G.P. Thermochemistry of complex formation of endofullerene Li+@C60 with the triflate ion. Russ. J. Gen. Chem. 2018, 88, 2335–2338. [Google Scholar] [CrossRef]
- Stasyuk, A.J.; Solà, M.; Voityuk, A.A. Reliable charge assessment on encapsulated fragment for endohedral systems. Sci. Rep. 2018, 8, 2882. [Google Scholar] [CrossRef]
- Xi, C.; Yang, L.; Liu, C.; You, P.; Li, L.; Jin, P. Lanthanide metals in the boron cages: Computational prediction of M@Bn (M = Eu, Gd; n = 38, 40). Int. J. Quant. Chem. 2018, 118, e25576. [Google Scholar] [CrossRef]
- De Munari, S.; Sandoval, S.; Pach, E.; Ballesteros, B.; Tobias, G.; Daniel, C.; Anthony, D.C.; Davis, B.G. In vivo behaviour of glyco-NaI@SWCNT ‘nanobottles’. Inorg. Chim. Acta 2019, 495, 118933. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Encapsulation of cadmium telluride nanocrystals within single walled carbon nanotubes. Inorg. Chim. Acta 2019, 488, 246–254. [Google Scholar] [CrossRef]
- Fujii, S.; Cho, H.; Hashikawa, Y.; Nishino, T.; Murata, Y.; Kiguchi, M. Tuneable single-molecule electronic conductance of C60 by encapsulation. Phys. Chem. Chem. Phys. 2019, 21, 12606–12610. [Google Scholar] [CrossRef]
- Yang, W.; Velkos, G.; Liu, F.; Sudarkova, S.M.; Wang, Y.; Zhuang, J.; Zhang, H.; Li, X.; Zhang, X.; Büchner, B.; et al. Single molecule magnetism with strong magnetic anisotropy and enhanced Dy∙∙∙Dy coupling in three isomers of Dy-oxide clusterfullerene Dy2O@C82. Adv. Sci. 2019, 6, 1901352. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Li, Y.; Magagula, S.; Chen, Z. Exohedral functionalization of endohedral metallofullerenes: Interplay between inside and outside. Coord. Chem. Rev. 2019, 388, 406–439. [Google Scholar] [CrossRef]
- Ye, L.; Liao, M.; Sun, H.; Yang, Y.; Tang, C.; Zhao, Y.; Wang, L.; Xu, Y.; Zhang, L.; Wang, B.; et al. Stabilizing lithium into cross-stacked nanotube sheets with an ultra-high specific capacity for lithium oxygen batteries. Angew. Chem. Int. Ed. 2019, 58, 2437–2442. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Li, M.-Y.; Xiong, Y.-M.; Rahmani, S.; Yuan, K.; Zhao, R.-S.; Ehara, M.; Nagase, S.; Zhao, X. Pivotal role of nonmetal atoms in the stabilities, geometries, electronic structures, and isoelectronic chemistry of Sc3X@C80 (X = C, N, and O). J. Comput. Chem. 2019, 40, 2730–2738. [Google Scholar] [CrossRef]
- Bloodworth, S.; Sitinova, G.; Alom, S.; Vidal, S.; Bacanu, G.R.; Elliott, S.J.; Light, M.E.; Herniman, J.M.; Langley, G.J.; Levitt, M.H.; et al. First synthesis and characterization of CH4@C60. Angew. Chem. Int. Ed. 2019, 58, 5038–5043. [Google Scholar] [CrossRef] [Green Version]
- Sheikhi, M.; Shahab, S.; Alnajjar, R.; Ahmadianarog, M. Theoretical model for surface forces between cytosine and CNT(6,6-6) nanotube: Geometry optimization, molecular structure, intermolecular hydrogen bond, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP, and HOMO–LUMO investigation. Russ. J. Phys. Chem. A 2019, 93, 2429–2443. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, C. Endohedral and exohedral complexes of 1-benzene with carbon nanotubes and high-density assembly of multiple benzenes inside of a carbon nanotube. Int. J. Quant. Chem. 2019, 119, e25936. [Google Scholar] [CrossRef]
- Wang, J.T.-W.; Klippstein, R.; Martincic, M.; Pach, E.; Feldman, R.; Šefl, M.; Michel, Y.; Asker, D.; Sosabowski, J.K.; Kalbac, M.; et al. Neutron activated 153Sm sealed in carbon nanocapsules for in vivo imaging and tumor radiotherapy. ACS Nano 2020, 14, 129–141. [Google Scholar] [CrossRef]
- Wei, T.; Martin, O.; Chen, M.; Yang, S.; Hauke, F.; Hirsch, A. Covalent inter-carbon-allotrope architectures consisting of the endohedral fullerene Sc3N@C80 and single-walled carbon nanotubes. Angew. Chem. Int. Ed. 2019, 58, 8058–8062. [Google Scholar] [CrossRef]
- Dappe, Y.J. Encapsulation of organic molecules in carbon nanotubes: Role of the van der Waals interactions. J. Phys. D Appl. Phys. 2014, 47, 083001. [Google Scholar] [CrossRef]
- Gtari, W.F.; Tangour, B. Interaction of HF, HBr, HCl and HI molecules with carbon nanotubes. Acta Chim. Slov. 2018, 65, 289–295. [Google Scholar] [CrossRef]
- Halls, M.D.; Schlegel, H.B. Chemistry inside carbon nanotubes: the Menshutkin SN2 reaction. J. Phys. Chem. B 2002, 106, 1921–1925. [Google Scholar] [CrossRef] [Green Version]
- Halls, M.D.; Raghavachari, K. Carbon nanotube inner phase chemistry: The Cl− exchange SN2 reaction. Nano Lett. 2005, 5, 1861–1866. [Google Scholar] [CrossRef]
- Botos, A.; Biskupek, J.; Chamberlain, T.W.; Rance, G.A.; Stoppiello, C.T.; Sloan, J.; Liu, Z.; Suenaga, K.; Kaiser, U.; Khlobystov, A.N. Carbon nanotubes as electrically active nanoreactors for multi-step inorganic synthesis: Sequential transformations of molecules to nanoclusters and nanoclusters to nanoribbons. J. Am. Chem. Soc. 2016, 138, 8175–8183. [Google Scholar] [CrossRef]
- Miners, S.A.; Rance, G.A.; Khlobystov, A.N. Chemical reactions confined within carbon nanotubes. Chem. Soc. Rev. 2016, 45, 4727–4746. [Google Scholar] [CrossRef]
- Astle, M.A.; Rance, G.A.; Fay, M.W.; Notman, S.; Sambrook, M.R.; Khlobystov, A.N. Synthesis of hydroxylated group IV metal oxides inside hollow graphitised carbon nanofibers: Nano-sponges and nanoreactors for enhanced decontamination of organophosphates. J. Mater. Chem. A 2018, 6, 20444–20453. [Google Scholar] [CrossRef]
- Astle, M.A.; Rance, G.A.; Loughlin, H.J.; Peters, T.D.; Khlobystov, A.N. Molybdenum dioxide in carbon nanoreactors as a catalytic nanosponge for the efficient desulfurization of liquid fuels. Adv. Funct. Mater. 2019, 29, 1808092. [Google Scholar] [CrossRef]
- Fay, M.W.; Baldoni, M.; Besley, E.; Khlobystov, A.N.; Rance, G.A. Steric and electronic control of 1,3-dipolar cycloaddition reactions in carbon nanotube nanoreactors. J. Phys. Chem. C 2019, 123, 6294–6302. [Google Scholar] [CrossRef]
- Agasti, N.; Astle, M.A.; Rance, G.A.; Fernandes, J.A.; Dupont, J.; Khlobystov, A.N. Cerium oxide nanoparticles inside carbon nanoreactors for selective allylic oxidation of cyclohexene. Nano Lett. 2020, 20, 1161–1171. [Google Scholar] [CrossRef]
- Rance, G.A.; Miners, S.A.; Chamberlain, T.W.; Khlobystov, A.N. The effect of carbon nanotubes on chiral chemical reactions. Chem. Phys. Lett. 2013, 557, 10–14. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 5–91. [Google Scholar]
- Mennucci, B. Solvation Models for Molecular Properties: Continuum versus Discrete Approaches. In Solvation Effects on Molecules and Biomolecules. Computational Methods and Applications; Springer Science & Business Media: Berlin/Heidelberg, Geramny, 2010; pp. 1–21. [Google Scholar]
- Eilmes, A. Solvatochromic probe in molecular solvents: Implicit versus explicit solvent model. Theor. Chem. Acc. 2014, 133, 1538. [Google Scholar] [CrossRef] [Green Version]
- Gaalswyk, K.; Rowley, C.N. An explicit-solvent conformation search method using open software. Peer J. 2016, 4, e2088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; Spoel, D. Comparison of implicit and explicit solvent models for the calculation of solvation free energy in organic solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef]
- Turner, M.A.P.; Turner, R.J.; Horbury, M.D.; Hine, N.D.M.; Stavros, V.G. Examining solvent effects on the ultrafast dynamics of catechol. J. Chem. Phys. 2019, 151, 084305. [Google Scholar] [CrossRef] [Green Version]
- Varghese, J.J.; Mushrif, S.H. Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: A review. React. Chem. Eng. 2019, 4, 165–206. [Google Scholar] [CrossRef]
- Sattasathuchana, T.; Xu, P.; Gordon, M.S. An Accurate quantum-based approach to explicit solvent effects: Interfacing the general effective fragment potential method with ab initio electronic structure theory. J. Phys. Chem. A 2019, 123, 8460–8475. [Google Scholar] [CrossRef]
- Avramov, P.V.; Kudin, K.N.; Scuseria, G.E. Single wall carbon nanotubes density of states: Comparison of experiment and theory. Chem. Phys. Lett. 2003, 370, 597–601. [Google Scholar] [CrossRef]
- Koner, A.; Kumar, C.; Sathyamurthy, N. Heat capacity of endohedral fullerenes Rg@C60 (Rg = He, Ne, Ar and Kr). Mol. Phys. 2018, 116, 2728–2735. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Sabirov, D.S. Anisotropy of polarizability of fullerene higher adducts for assessing the efficiency of their use in organic solar cells. J. Phys. Chem. C 2013, 117, 9148–9153. [Google Scholar] [CrossRef]
- Sabirov, D.S. From endohedral complexes to endohedral fullerene covalent derivatives: A density functional theory prognosis of chemical transformation of water endofullerene H2O@C60 upon its compression. J. Phys. Chem. C 2013, 117, 1178–1182. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Tukhbatullina, A.A.; Sabirov, D.S. Dipole polarizability, structure, and stability of [2+2]-linked fullerene nanostructures (C60)n (n ≤ 7). Physica E 2017, 86, 237–242. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Khatymov, R.V.; Sabirov, D.S.; Yuldashev, A.V. On the upper bound of the thermodynamic stability of fullerenes from small to giant. Physica E 2018, 101, 265–272. [Google Scholar] [CrossRef]
- Laikov, D.N. Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem. Phys. Lett. 1997, 281, 151–156. [Google Scholar] [CrossRef]
- Ramachandran, C.N.; Fazio, D.D.; Sathyamurthy, N.; Aquilanti, V. Guest species trapped inside carbon nanotubes. Chem. Phys. Lett. 2009, 473, 146–150. [Google Scholar] [CrossRef]
- Internal Rotation in Molecules; Orville-Thomas, W.J. (Ed.) Wiley-Interscience: London, UK; New York, NY, USA, 1974; p. 606. [Google Scholar]
- Pophristic, V.; Goodman, L. Exchange repulsion increases internal rotation floppiness. J. Chem. Phys. 2001, 115, 5132–5136. [Google Scholar] [CrossRef]
- Kundu, T.; Pradhan, B.; Singh, B.P. Origin of methyl torsional potential barrier—An overview. J. Chem. Sci. 2002, 114, 623–638. [Google Scholar] [CrossRef] [Green Version]
- Bickelhaupt, F.M.; Baerends, E.J. The case for steric repulsion causing the staggered conformation of ethane. Angew. Chem. Int. Ed. 2003, 42, 4183–4188. [Google Scholar] [CrossRef]
- Rico, J.F.; López, R.; Ema, I.; Ramirez, G. Density and binding forces: Rotational barrier of ethane. J. Chem. Phys. 2003, 119, 12251–12256. [Google Scholar] [CrossRef]
- Sadlej-Sosnowska, N. Energy barriers to internal rotation: Hyperconjugation and electrostatic description. J. Phys. Chem. A 2003, 107, 8671–8676. [Google Scholar] [CrossRef]
- Song, L.; Lin, Y.; Zhang, Q.; Mo, Y. Steric strain versus hyperconjugative stabilization in ethane congeners. J. Phys. Chem. A 2005, 109, 2310–2316. [Google Scholar] [CrossRef]
- Liu, S.B. Steric effect: A quantitative description from density functional theory. J. Chem. Phys. 2007, 126, 244103. [Google Scholar] [CrossRef]
- Mo, Y.; Gao, J. Theoretical analysis of the rotational barrier of ethane. Acc. Chem. Res. 2007, 40, 113–119. [Google Scholar] [CrossRef]
- Liu, S.B.; Govind, N. Toward understanding the nature of internal rotation barriers with a new energy partition scheme: Ethane and n-butane. J. Phys. Chem. A 2008, 112, 6690–6699. [Google Scholar] [CrossRef]
- Liu, S.B.; Govind, N.; Pedersen, L.G. Exploring the origin of the internal rotational barrier for molecules with one rotatable dihedral angle. J. Chem. Phys. 2008, 129, 094104. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y. Rotational barriers in alkanes. WIREs Comput. Mol. Sci. 2011, 1, 164–171. [Google Scholar] [CrossRef]
- Esquivel, R.O.; Liu, S.B.; Angulo, J.C.; Dehesa, J.S.; Antolin, J.; Molina-Espiritu, M. Fisher information and steric effect: Study of the internal rotation barrier of ethane. J. Phys. Chem. A 2011, 115, 4406–4415. [Google Scholar] [CrossRef]
- Quijano-Quiñones, R.F.; Quesadas-Rojas, M.; Cuevas, G.; Mena-Rejón, G.J. The Rotational barrier in ethane: A molecular orbital study. Molecules 2012, 17, 4661–4671. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.B. Origin and nature of bond rotation barriers: A unified view. J. Phys. Chem. A 2013, 117, 962–965. [Google Scholar] [CrossRef]
- Liu, S.B.; Schauer, C.K. Origin of molecular conformational stability: Perspectives from molecular orbital interactions and density functional reactivity theory. J. Chem. Phys. 2015, 142, 054107. [Google Scholar] [CrossRef]
- Baranac-Stojanović, M. Theoretical analysis of the rotational barrier in ethane: Cause and consequences. Struct. Chem. 2015, 26, 989–996. [Google Scholar] [CrossRef]
- Priebe, A.; Pucci, A.; Akemann, W.; Grabhorn, H.; Otto, A. Staggered ethane changes to eclipsed conformation upon adsorption. J. Raman Spect. 2006, 37, 1398–1402. [Google Scholar] [CrossRef]
- Wanjari, P.P.; Sangwai, A.V.; Ashbaugh, H.S. Confinement induced conformational changes in n-alkanes sequestered within a narrow carbon nanotube. Phys. Chem. Chem. Phys. 2012, 14, 2702–2709. [Google Scholar] [CrossRef]
- Velpuri, S.V.V.; Gade, H.M.; Wanjari, P.P. Encapsulation driven conformational changes in n-alkanes inside a hydrogen-bonded supramolecular cavitand assembly. Chem. Phys. 2019, 521, 100–107. [Google Scholar] [CrossRef]
- Gade, H.M.; Velpuri, S.V.V.; Wanjari, P.P. Conformational rearrangements in n-alkanes encapsulated within capsular self-assembly of capped carbon nanotubes. Chem. Phys. 2019, 517, 198–207. [Google Scholar] [CrossRef]
- Bates, S.P.; van Well, W.J.M.; van Santen, R.A.; Smit, B. Location and conformation of n-alkanes in zeolites: An analysis of configurational-bias Monte Carlo calculations. J. Phys. Chem. 1996, 100, 17573–17581. [Google Scholar] [CrossRef] [Green Version]
- Gorgoll, R.M.; Yücelen, E.; Kumamoto, A.; Shibata, N.; Harano, K.; Nakamura, E. Electron microscopic observation of selective excitation of conformational change of a single organic molecule. J. Am. Chem. Soc. 2015, 137, 3474–3477. [Google Scholar] [CrossRef]
- Harano, K.; Takenaga, S.; Okada, S.; Niimi, Y.; Yoshikai, N.; Isobe, H.; Suenaga, K.; Kataura, H.; Koshino, M.; Nakamura, E. Conformational analysis of single perfluoroalkyl chains by single-molecule real-time transmission electron microscopic imaging. J. Am. Chem. Soc. 2014, 136, 466–473. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W. Adsorption of linear ethane molecules in single walled carbon nanotube arrays by molecular simulation. Phys. Chem. Chem. Phys. 2002, 4, 3048–3054. [Google Scholar] [CrossRef]
- Jakobtorweihen, S.; Keil, F.G. Adsorption of alkanes, alkenes and their mixtures in single-walled carbon nanotubes and bundles. Mol. Simul. 2009, 35, 90–99. [Google Scholar] [CrossRef]
- Cruz, F.J.A.L.; Müller, E.A. Behavior of ethylene and ethane within single-walled carbon nanotubes. 1: Adsorption and equilibrium properties. Adsorption 2009, 15, 1–12. [Google Scholar] [CrossRef]
- Cruz, F.J.A.L.; Müller, E.A. Behavior of ethylene and ethane within single-walled carbon nanotubes, 2: Dynamical properties. Adsorption 2009, 15, 13–22. [Google Scholar] [CrossRef]
- Albesa, A.G.; Matías Rafti, M.; Rawat, D.S.; Vicente, J.L.; Migone, A.D. Ethane/ethylene adsorption on carbon nanotubes: Temperature and size effects on separation capacity. Langmuir 2012, 28, 1824–1832. [Google Scholar] [CrossRef]
- Tian, X.; Wang, Z.; Yang, Z.; Xiu, P.; Zhou, B. Adsorptive separation of ethylene/ethane mixtures using carbon nanotubes: A molecular dynamics study. J. Phys. D Appl. Phys. 2013, 46, 395302. [Google Scholar] [CrossRef]
- Boudry, J. van der Waals interactions and decrease of the rotational barrier of methyl-sized rotators: A theoretical study. J. Am. Chem. Soc. 2006, 128, 11088–11093. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical evaluation of conformational preference of ethane molecule encapsulated in a nanotube. Russ. J. Org. Chem. 2013, 49, 313–314. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of ethane molecule encapsulated in a nanotube. Russ. J. Org. Chem. 2013, 49, 1231–1235. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Influence of the nanotube type on the conformational behavior of encapsulated ethane molecule. Russ. J. Gen. Chem. 2013, 83, 2334–2336. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational properties of ethane and its analogs in nanotubes. Russ. J. Gen. Chem. 2019, 89, 1271–1278. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, C. Doped ways of boron and nitrogen doped carbon nanotubes: A theoretical investigation. J. Mol. Struct. (THEOCHEM) 2010, 955, 84–90. [Google Scholar] [CrossRef]
- Rimola, A.; Sodure, M. Gas-phase and microsolvated glycine interacting with boron nitride nanotubes. A B3LYP-D2* periodic study. Inorganics 2014, 2, 334–350. [Google Scholar] [CrossRef] [Green Version]
- Tenne, R.; Enyashkin, A.N. Inorganic fullerene-like nanoparticles and inorganic nanotubes. Inorganics 2014, 2, 649–651. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical evaluation of conformational preference of the propane molecule in nanotubes. Russ. J. Gen. Chem. 2013, 83, 1165–1166. [Google Scholar] [CrossRef]
- Bartell, L.S.; Boates, T.L. Structures of the strained molecules hexamethylethane and 1,1,2,2-tetramethylethane by gas-phase electron diffraction. J. Mol. Struct. 1976, 32, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, V.V. Theoretical evaluation of conformational preference of the 2,2,3,3-tetramethylbutane molecule in nanotubes. Russ. J. Gen. Chem. 2013, 83, 1455–1456. [Google Scholar] [CrossRef]
- Hembree, W.I.; Boudry, J. Three-dimensional mapping of microenvironmental control of methyl rotational barriers. J. Phys. Chem. B 2011, 115, 8575–8580. [Google Scholar] [CrossRef]
- Weiss, S.; Leroi, G.E. Infrared spectra and internal rotation in propane, isobutane and neopentane. Spectrochim. Acta. A 1969, 25, 1759–1766. [Google Scholar] [CrossRef]
- Holm, U.; Kerl, K. Anomalous behaviour of the mean dipole polarizability α of neopentane C(CH3)4 in the temperature range between 250 K and 360 K. Z. Naturforsch. A 1991, 46, 983–988. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational preferences of 2,2-dimethylpropane in nanotubes. Russ. J. Gen. Chem. 2014, 84, 433–438. [Google Scholar] [CrossRef]
- Chen, S.S.; Rodgers, A.C.; Chao, J.; Wilhoit, R.C.; Zwolinski, B.J. Ideal gas thermodynamic properties of six fluoroethanes. J. Phys. Chem. Ref. Data 1975, 4, 441–456. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational analysis of fluoroethane in nanotubes. Russ. J. Org. Chem. 2018, 54, 644–651. [Google Scholar] [CrossRef]
- Brier, P.N.; Higgins, S. Neutron inelastic scattering measurements on 1,1,1-trifluoroethane. J. Mol. Phys. 1970, 19, 645–657. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of 1,1,1-trifluoroethane in nanotubes. Russ. J. Org. Chem. 2014, 50, 1534–1539. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical evaluation of conformational preference of the hexafluoroethane molecule in nanotubes. Russ. J. Gen. Chem. 2013, 83, 1623–1625. [Google Scholar] [CrossRef]
- Thorne, L.R.; Suenram, R.D.; Lovas, F.J. Microwave spectrum, torsional barrier, and structure of BH3NH3. Chem. Phys. 1983, 78, 167–171. [Google Scholar] [CrossRef]
- Parafiniuk, M.; Mitoraj, M.P. On the origin of internal rotation in ammonia borane. J. Mol. Model. 2014, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Demirci, U.B. Ammonia borane, a material with exceptional properties for chemical hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 9978–10013. [Google Scholar] [CrossRef]
- Akbayrak, S.; Özkar, S. Ammonia borane as hydrogen storage materials. Int. J. Hydrogen Energy 2018, 43, 18592–18606. [Google Scholar] [CrossRef]
- Li, L.; Yao, X.; Sun, C.; Du, A.; Cheng, L.; Zhu, Z.; Yu, C.; Zou, J.; Smith, S.C.; Wang, P.; et al. Lithium-catalyzed dehydrogenation of ammonia borane within mesoporous carbon framework for chemical hydrogen storage. Adv. Funct. Mater. 2009, 19, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Wahab, M.A.; Zhao, H.; Yao, X.D. Nano-confined ammonia borane for chemical hydrogen storage. Front. Chem. Sci. Eng. 2012, 6, 27–33. [Google Scholar] [CrossRef]
- Zhang, L.; Xia, G.; Ge, Y.; Wang, C.; Guo, Z.; Li, X.; Yu, X. Ammonia borane confined by nitrogen-containing carbon nanotubes: Enhanced dehydrogenation properties originating from synergetic catalysis and nanoconfinement. J. Mater. Chem. A 2015, 3, 20494–20499. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Ammonia borane in nanotubes: The preference of eclipsed conformation. Russ. J. Inorg. Chem. 2018, 63, 1069–1075. [Google Scholar] [CrossRef]
- Urban, J.; Schireiner, P.R.; Vacec, G.; von RaguéSchleyer, P.; Huang, J.Q.; Leszczynski, J. Molecular structures, vibrational spectra, and rotational barriers of C2H6, Si2H6, SiGeH6, and Ge2H6—experiment and theory in harmony. Chem. Phys. Lett. 1997, 264, 441–448. [Google Scholar] [CrossRef]
- Puzzarini, C. Accurate structure and torsional barrier height of disilane. Phys. Chem. Chem. Phys. 2003, 5, 26–30. [Google Scholar] [CrossRef]
- Pophristic, V.; Goodman, L.; Wu, C.T. Disilane internal rotation. J. Phys. Chem. A 2001, 105, 7454–7459. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Nanotube effect on conformation of encapsulated disilane molecule. Russ. J. Gen. Chem. 2015, 85, 1989–1991. [Google Scholar] [CrossRef]
- Durig, J.R.; Church, J.S. Vibrational spectra of crystalline disilane and disilane-d6, barrier to internal rotation and some normal coordinate calculations on H3SiSiH3, H3SiNCO, and H3SiNCS. J. Chem. Phys. 1980, 73, 4784–4797. [Google Scholar] [CrossRef]
- Dows, D.A.; Hexter, R.M. Infrared spectra of gaseous and solid digermane. J. Chem. Phys. 1956, 24, 1029–1033. [Google Scholar] [CrossRef]
- Isobe, C.; Cho, H.-C.; Crowell, J.E. The photo-induced reaction of digermane with the Si(100)(2 × 1):D surface. Surf. Sci. 1993, 295, 117–132. [Google Scholar] [CrossRef]
- Hart, J.; Hazbun, R.; Eldridge, D.; Hickey, R.; Femando, N.; Adam, T.; Zollner, S.; Kolodzey, J. Tetrasilane and digermane for the ultra-high vacuum chemical vapor deposition of SiGe alloys. Thin Solid Film 2016, 604, 23–27. [Google Scholar] [CrossRef]
- Lin, D.-S.; Huang, K.-H.; Pi, T.-W.; Wu, R.-T. Coverage-dependent thermal reactions of digermane on Si(100)-(2×1). Phys. Rev. B 1996, 54, 16958–16964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatmann, J.-M.; Aubin, J.; Barnes, J.-P. A benchmark of germane and digermane for the low temperature growth of intrinsic and heavily in-situ boron-doped SiGe. ECS Trans. 2016, 75, 281–294. [Google Scholar] [CrossRef]
- Hart, J.; Adam, T.; Kim, Y.; Huang, Y.-Ch.; Reznicer, A.; Hazbun, R.; Gupta, J.; Kolodzey, J. Temperature varying photoconductivity of GeSn alloys grown by chemical vapor deposition with Sn concentrations from 4% to 11%. J. Appl. Phys. 2016, 119, 093105. [Google Scholar] [CrossRef]
- Lu, G.; Crowell, J.E. The adsorption and thermal decomposition of digermane on Ge(111). J. Chem. Phys. 1993, 98, 3415–3421. [Google Scholar] [CrossRef]
- Aubin, J.; Hartmann, J.M.; Buer, M.; Moffatt, S. Very low temperature epitaxy of Ge and Ge rich SiGe alloy with Ge2H6 in a reduced pressure—Chemical vapor deposition tool. J. Cryst. Growth 2016, 445, 65–72. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Bochkor, S.A. Relative stability of digermane conformers in nanotubes. Russ. J. Gen. Chem. 2020, 90, 93–98. [Google Scholar] [CrossRef]
- Gropen, O.; Johansen, R. Barrier of internal rotation and π-bonding in hydroxyborane, H2BOH, studied by ab initio calculations. J. Mol. Struct. 1975, 25, 161–167. [Google Scholar] [CrossRef]
- Lanthier, G.F.; Graham, W.A.G. Dimethylboric anhydride: A convenient preparation and full characterization. Can. J. Chem. 1969, 47, 569–575. [Google Scholar] [CrossRef]
- Finocchiaro, P.; Gust, D.; Mislow, K. Conformational dynamics of alkoxydiarylboranes. J. Am. Chem. Soc. 1973, 95, 7029–7036. [Google Scholar] [CrossRef]
- Brown, N.M.D.; Davidson, F.; Wilson, J.W. Dimesitylboryl compounds. VI. 13C Dynamic nuclear magnetic resonance studies. J. Organometal. Chem. 1981, 210, 1–8. [Google Scholar] [CrossRef]
- Stampf, E.J.; Odom, J.D.; Saari, S.V.; Kim, Y.H.; Bergana, M.M.; During, J.R. Dimethylmethoxyborane: Vibrational assignment, conformational stability, ab initio calculations and barriers to internal rotation. J. Mol. Struct. 1990, 239, 113–137. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformation of hydroxyborane encapsulated within nanotubes. Russ. J. Gen. Chem. 2014, 84, 157–159. [Google Scholar] [CrossRef]
- Vincent, M.A.; Schaefer, H.F. Diborane(4) (B2H4): The boron hydride analog of ethylene. J. Am. Chem. Soc. 1981, 103, 5677–5680. [Google Scholar] [CrossRef]
- Lein, M.; Szabó, A.; Kovács, A.; Frenking, G. Energy decomposition analysis of the chemical bond in main group and transition metal compound. Faraday Discuss. 2003, 124, 365–378. [Google Scholar] [CrossRef]
- Osorio, E.; Olson, J.K.; Tiznado, W.; Boldyrev, A.I. Analysis of why boron avoids sp2 hybridization and classical structures in the BnHn+2 series. Chem. Eur. J. 2012, 18, 9677–9681. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. The influence of carbon nanotubes on the relative stability of diborane molecular forms. Russ. J. Gen. Chem. 2016, 86, 231–240. [Google Scholar] [CrossRef]
- Lammertsma, K.; Güner, O.F.; Drewes, R.M.; Reed, A.E.; Schleyer, P.R. Remarkable structures of dialane(4), Al2H4. Inorg. Chem. 1989, 28, 313–317. [Google Scholar] [CrossRef]
- Wang, X.; Andrews, L.; Tam, S.; DeRose, M.E.; Fajardo, M.E. Infrared spectra of aluminum hydrides in solid hydrogen: Al2H4 and Al2H6. J. Am. Chem. Soc. 2003, 125, 9218–9228. [Google Scholar] [CrossRef]
- Lazarev, V.V.; Kuznetsov, V.V. Influence of nanotubes on the relative stability of orthogonal form of dialane. Russ. J. Gen. Chem. 2019, 89, 1792–1799. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Skancke, A. Thermochemistry, energy comparisons, and conformational analysis of hydrazine, triazane, and triaminoammonia. J. Am. Chem. Soc. 1993, 115, 7465–7471. [Google Scholar] [CrossRef]
- Kobychev, V.B.; Vitkovskaya, N.M.; Pavlova, N.V.; Schmidt, E.Yu.; Trofimov, B.A. Theoretical analysis and experimental study of the spatial structure and isomerism of acetone azine and its cyclization to 3,5,5-trimethyl-4,5-dihydro-1H-pyrazole. J. Struct. Chem. 2004, 45, 748–755. [Google Scholar] [CrossRef]
- Song, L.; Liu, M.; Wu, W.; Zhang, Q.; Mo, Y. Origins of rotational barriers in hydrogen peroxide and hydrazin. J. Chem. Theory Comput. 2005, 1, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Łodyga, W.; Makarewicz, J. Torsion-wagging tunneling and vibrational states in hydrazine determined from its ab initio potential energy surface. J. Chem. Phys. 2012, 136, 174301. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.V. Hydrazine: Structural features and conformational preference in nanotubes. Russ. J. Gen. Chem. 2016, 86, 2000–2007. [Google Scholar] [CrossRef]
- Lees, R.M.; Mohammadi, M.A. Millimetre wave spectrum of methyl mercaptan. Can. J. Phys. 1980, 58, 1640–1648. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of methanol in a nanotube. Russ. J. Org. Chem. 2014, 50, 765–766. [Google Scholar] [CrossRef]
- Tuazon, E.C.; Fateley, W.G. Internal rotation in (CH3)2X molecules of C2υ symmetry–barrier to internal rotation in dimethyl ether. J. Chem. Phys. 1971, 54, 4450–4454. [Google Scholar] [CrossRef]
- Endres, C.P.; Drouin, B.J.; Pearson, J.C.; Müller, H.S.P.; Lewen, F.; Schlemmer, S.; Giesen, T.F. Dimethyl ether: Laboratory spectra up to 2.1 THz. Torsion-rotational spectra within the vibrational ground state. Astron. Astrophys. 2009, 504, 635–640. [Google Scholar] [CrossRef]
- Kimura, K.; Kubo, M. Structures of dimethyl ether and methyl alcohol. J. Chem. Phys. 1959, 30, 151–158. [Google Scholar] [CrossRef]
- Blukis, U.; Kasai, P.H.; Myers, R.J. Microwave spectra and structure of dimethyl ether. J. Chem. Phys. 1963, 38, 2753–2760. [Google Scholar] [CrossRef]
- Goodman, L.; Pophristic, V. Where does the dimethyl ether internal rotation barrier come from? Chem. Phys. Lett. 1996, 259, 287–295. [Google Scholar] [CrossRef]
- Pophristic, V.; Goodman, L.; Guchhait, N. Role of lone-pairs in internal rotation barriers. J. Phys. Chem. A 1997, 101, 4290–4297. [Google Scholar] [CrossRef]
- Pophristic, V.; Goodman, L. Influence of protonation on internal rotation of dimethyl ether. J. Phys. Chem. A 2000, 104, 3231–3238. [Google Scholar] [CrossRef]
- Jimenez-Fabian; Jalbout, A.F. The origin of the rotational barrier in dimethyl ether and dimethyl sulfide. A theoretical study. J. Theor. Comput. Chem. 2007, 6, 421–434. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Dimethyl ether in nanotubes: Structural variations and conformational preferences. Russ. J. Gen. Chem. 2016, 86, 1835–1840. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Fullerene Si20: Influence on the conformational behavior of encapsulated ethane molecule. Russ. J. Gen. Chem. 2016, 86, 1444–1446. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical evaluation of conformational preference of the ethane molecule in fullerene C60. Russ. J. Gen. Chem. 2013, 83, 1163–1164. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational preference of hexafluoroethane molecule encapsulated in fullerenes. Russ. J. Org. Chem. 2014, 50, 456–457. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. The effect of chemical composition of the fullerene on the conformational preference of the encapsulated hexafluoroethane molecule. Russ. J. Gen. Chem. 2016, 86, 1108–1114. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational preference of the hexachloroethane molecule in fullerene C80. Russ. J. Gen. Chem. 2013, 83, 1790–1791. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformation of 2,2-dimethylpropane encapsulated in fullerenes. Russ. J. Org. Chem. 2014, 50, 921–922. [Google Scholar] [CrossRef]
- Shakirova, E.I.; Kuznetsov, V.V. Effect of chemical composition of fullerenes on the structure and internal rotation barrier of encapsulated ammonia borane molecule. Russ. J. Gen. Chem. 2019, 89, 2229–2234. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Bochkor, S.A. The influence of chemical composition of fullerenes on the structural features and conformational preference of encapsulated disilane molecule. Russ. J. Inorg. Chem. 2018, 63, 917–922. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of methanethiol in fullerenes. Russ. J. Org. Chem. 2014, 50, 1073–1074. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Cyclohexane in nanotubes: Direct chair–chair interconversion. Russ. J. Gen. Chem. 2017, 87, 2558–2562. [Google Scholar] [CrossRef]
- Ram, V.J.; Sethi, A.; Nath, M.; Pratar, R. The Chemistry of Heterocycles. Chemistry of Six- to Eight-Membered N, O, S, P and Se Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 340–392. [Google Scholar] [CrossRef]
- Franchini, S.; Sorbi, C.; Linciano, P.; Camevale, G.; Tait, A.; Ronsisvalle, S.; Buccioni, M.; Del Bello, F.; Cilia, A.; Pironal, L.; et al. 1,3-Dioxane as a scaffold for potent and selective 5-HT1AR agonist with in-vivo anxiolytic, anti-depressant and anti-nociceptive activity. Eur. J. Med. Chem. 2019, 176, 310–325. [Google Scholar] [CrossRef]
- Janssens, J.; Risseeuw, M.D.P.; Eycken, J.V.; Calenbergh, S.V. Regioselective ring opening of 1,3-dioxane-type acetals in carbohydrates. Eur. J. Org. Chem. 2018, 2018, 6405–6431. [Google Scholar] [CrossRef]
- Cooksey, J.; Gunn, A.; Philip, J.; Kocienski, P.J.; Kuhl, A.; Uppal, S.; Christopher, J.A.; Bell, R. The nucleophilic addition of α-metallated 1,3-dioxanes to planar chiral cationic η3-allylmolybdenum complexes. Synthesis of (2E,5S,6R,7E)-6-methyl-8-phenylocta-2,7-dienoic acid methyl ester, a key component of the cryptophycins. Org. Biomol. Chem. 2004, 2, 1719–1731. [Google Scholar] [CrossRef]
- Sinz, C.J.; Rychnovsky, S.D. 4-Acetoxy- and 4-cyano-1,3-dioxanes in synthesis. Top. Curr. Chem. 2001, 216, 50–93. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Reactions of 1,3-dioxacycloalkanes and their 2-arsena, 2-bora, 2-germa, 2-sila, and 2-thia analogs with nitriles. Russ. Chem. Bull. 2005, 54, 1543–1551. [Google Scholar] [CrossRef]
- Asare-Nkansah, S.; Wünsch, B. Double intramolecular transacetalization of polyhydroxy acetals: Synthesis of conformationally-restricted 1,3-dioxanes with axially-oriented phenyl moiety. Molecules 2016, 21, 1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, V.V. Computer simulation of conformational transformations of 1,3-dioxanes and their 2-sila and 2-bora analogs. Russ. J. Org. Chem. 2014, 50, 1227–1246. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational analysis of 1,3-dioxane in nanotubes. Russ. J. Org. Chem. 2016, 52, 1688–1693. [Google Scholar] [CrossRef]
- Raskildina, G.Z.; Spirikhin, L.V.; Zlotskij, S.S.; Kuznetsov, V.V. Conformational analysis of 5-ethyl-5-hydroxymethyl-2,2-dimethyl-1,3-dioxan. Russ. J. Org. Chem. 2019, 55, 502–507. [Google Scholar] [CrossRef]
- Khazhiev, S.Y.; Khusainov, M.A.; Khalikov, R.A.; Kataev, V.A.; Tyumkina, T.V.; Mesheryakova, E.S.; Khalilov, L.M.; Kuznetsov, V.V. Structure and conformational analysis of 5,5-bis(bromomethyl)-2,2-diphenyl-1,3-dioxane. Russ. J. Org. Chem. 2020, 56, 1–6. [Google Scholar] [CrossRef]
- Shainyan, B.A.; Kleinpeter, E. Silacyclohexanes and silaheterocyclohexanes — why are they so different from other heterocyclohexanes? Tetrahedron 2013, 69, 5927–5936. [Google Scholar] [CrossRef]
- Schultz, G.; Gergö, E.; Kolonits, M.; Hargittai, I. Molecular structure of 2,2-dimethyl-1,3-dioxa-2-silacyclohexane from gas-phase electron diffraction. J. Mol. Struct. 1993, 295, 143–146. [Google Scholar] [CrossRef]
- Bochkor, S.A.; Kuznetsov, V.V. Unusual conformational isomerization of oxygen-containing silacyclohexanes. Russ. J. Org. Chem. 2010, 46, 945–946. [Google Scholar] [CrossRef]
- Bochkor, S.A.; Kuznetsov, V.V. Comparative conformational analysis of 2,2-dimethyl and 2,2,5-trimethyl-1,3-dioxanes and their 2-heteroanalogs with silicon and germanium atoms. Russ. J. Gen. Chem. 2016, 86, 321–325. [Google Scholar] [CrossRef]
- Bochkor, S.A.; Kuznetsov, V.V. 1,3-Dioxa-2-silacyclohexane in nanotubes: Conformational transformations and structural features. Russ. J. Gen. Chem. 2016, 86, 1300–1305. [Google Scholar] [CrossRef]
- Knabe, J.; Büch, H.P.; Biwersi, J. Zyklische Harnstoffe, 1. Mitt.: Racemate und Enantiomere von Hexahydropyrimidin-2-onen: Synthese, Konfiguration und sedativ-hypnotische Wirkung. Arch. Pharm. 1993, 326, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Shutalev, A.D.; Ignatova, L.A. α-Amido(thioamido)alkylation of dithiocarbamic, O-ethyldithiocarbonic, and arylsulfinic acids by 4-hydroxy(alkoxy)hexahydropyrimidine-2-thiones(ones). Chem. Heterocycl. Compd. 1991, 27, 187–194. [Google Scholar] [CrossRef]
- Fesenko, A.A.; Solovyov, P.A.; Shutalev, A.D. A novel convenient synthesis of 5-acyl-1,2-dihydropyrimidin-2-ones via 4-trichloromethyl-1,2,3,4-tetrahydropyrimidin-2-ones. Tetrahedron 2010, 66, 940–946. [Google Scholar] [CrossRef]
- Tamazyan, R.; Ayvazyan, A.; Martirosyan, V.; Avagyan, K.; Martirosyan, A. 1-Benzyl-6-phenylimino-5-(pyrrol-2-ylidene)hexahydropyrimidine-2,4-dione. Acta Cryst. E 2008, 64, o483. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of hexahydropyrimidin-2-one and its ammonium and oxonium ions. Chem. Heterocycl. Compd. 2011, 47, 651–655. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Hexahydropyrimidin-2-one in nanotubes: Structural changes and conformational preferences. Russ. J. Gen. Chem. 2017, 87, 1461–1465. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Garipova, R.R.; Kinzyabaeva, Z.S. Fullerene–1,4-dioxane adducts: A DFT study of the structural features and molecular properties. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 154–159. [Google Scholar] [CrossRef]
- Yus, M.; González-Gómez, J.C.; Foubelo, F. Diastereoselective allylation of carbonyl compounds and imines: Application to the synthesis of natural products. Chem. Rev. 2013, 113, 5595–5698. [Google Scholar] [CrossRef] [Green Version]
- Partyka, D.V. Transmetalation of unsaturated carbon nucleophiles from boron-containing species to the mid to late d-block metals of relevance to catalytic C−X coupling reactions (X = C, F, N, O, Pb, S, Se, Te). Chem. Rev. 2011, 111, 1529–1595. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, M.; Hartmann, E.; Mewald, M. Activation of the Si–B interelement bond: Mechanism, catalysis, and synthesis. Chem. Rev. 2013, 113, 402–441. [Google Scholar] [CrossRef] [PubMed]
- Brusilovskii, Y.E.; Kuznetsov, V.V. Reactions of cyclic boric acids esters with paraformaldehyde. Russ. J. Gen. Chem. 2011, 81, 542–544. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational behavior of 1,3,2-dioxaborinane molecule encapsulated in fullerenes. Russ. J. Gen. Chem. 2015, 85, 198–199. [Google Scholar] [CrossRef]
- Reimers, J.R.; McKemmish, L.K.; McKenzie, R.H.; Hush, N.S. Bond angle variations in XH3 [X = N, P, As, Sb, Bi]: The critical role of Rydberg orbitals exposed using a diabatic state model. Phys. Chem. Chem. Phys. 2015, 17, 24618–24640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-Q.; Song, P.; Ma, F.-C. Accurate equilibrium inversion barrier of ammonia by extrapolation to the one-electron basis set limit. Chin. Phys. B 2014, 23, 023301. [Google Scholar] [CrossRef]
- Sharma, R.; Goerigk, L. The INV24 test set: How well do quantum-chemical methods describe inversion and racemization barriers? Can. J. Chem. 2016, 94, 1133–1143. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Simulation of pyramidal inversion of nitrogen in tetrahydro-1,3-oxazines in polar medium. J. Struct. Chem. 2018, 59, 1374–1380. [Google Scholar] [CrossRef]
- Holleman-Wiberg. Inorganic Chemistry; ACADEMIC PRESS: San Diego, CA, USA, 2001; pp. 616–617. [Google Scholar]
- Kuznetsov, V.V. Theoretical evaluation of inversion barrier of trimethylamine in nanotubes. Russ. J. Gen. Chem. 2013, 83, 1453–1454. [Google Scholar] [CrossRef]
- Shinsaku, F. Group-theoretical framework for characterizing the ring flipping and N-inversion of piperidine derivatives. Extended pseudo-point groups and subsymmetry-itemized enumeration. Bull. Chem. Soc. Jpn. 1999, 72, 1759–1768. [Google Scholar] [CrossRef]
- Anet, F.L.A.; Yavari, I. Nitrogen inversion in piperidine. J. Am. Chem. Soc. 1977, 99, 2794–2796. [Google Scholar] [CrossRef]
- Lambert, J.B.; Featherman, S.I. Conformational analysis of pentamethylene heterocycles. Chem. Rev. 1975, 75, 611–626. [Google Scholar] [CrossRef]
- Carballiera, L.; Pérez-Juste, I. Influence of calculation level and effect of methylation on axial/equatorial equilibria in piperidines. J. Comput. Chem. 1998, 19, 961–976. [Google Scholar] [CrossRef]
- Blackburn, I.D.; Katritzky, A.R.; Takeuchi, Y. Conformation of piperidine and of derivatives with additional ring hetero atoms. Acc. Chem. Res. 1975, 8, 300–306. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical estimation of conformational preference of piperidine molecule encapsulated in a nanotube. Russ. J. Org. Chem. 2014, 50, 143–144. [Google Scholar] [CrossRef]
- Rudchenko, V.F.; Shevchenko, V.I.; Kostyanovskii, R.G. 2-Dimethylcarbamoyl-1,3,2-dioxazolidine, 2-dimethylcarbamoyl- and 2H-perhydro-1,3,2-dioxazines. Russ. Chem. Bull. 1985, 34, 1543. [Google Scholar] [CrossRef]
- Rudchehko, V.F.; Shevschenko, V.I.; Kostyanovskii, R.G. Geminal systems 33. Reactions of 1,1-dialkoxyureas with electrophiles and nucleophiles. Synthesis of cyclic 1,1-dialkoxyureas and N,N-dialkoxyamines. Russ. Chem. Bull. 1987, 36, 1436–1440. [Google Scholar] [CrossRef]
- Kostyanovskii, R.G.; Rudchehko, V.F.; Shtamburg, V.G.; Chervin, I.I.; Nasibov, S.S. Asymmetrical nonbridgehead nitrogen-XXVI1: Synthesis, configurational stability, and resolution of N,N-dialkoxyamines into antipodes. Tetrahedron 1981, 37, 4245–4254. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational transformations of of perhydro-1,3,2-dioxazine. Russ. J. Gen. Chem. 2012, 82, 783–784. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Conformational preference of perhydro-1,3,2-dioxazine inside nanotubes. Russ. J. Gen. Chem. 2014, 84, 525–530. [Google Scholar] [CrossRef]
- Kuznetsova, M.V.; Kuznetsov, V.V. Theoretical estimation of the barrier to pyramidal inversion of ammonia and trimethylamine encapsulated in fullerenes. Russ. J. Org. Chem. 2013, 49, 1845–1847. [Google Scholar] [CrossRef]
- Nitti, A.; Pacini, A.; Pasini, D. Chiral nanotubes. Nanomaterials 2017, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, J.; Lin, X.; Tang, Y.; Liu, Y.; Wang, T.; Zhu, H. The electronic properties of chiral carbon nanotubes. Comput. Mater. Sci. 2017, 129, 290–294. [Google Scholar] [CrossRef]
- Wei, X.; Tanaka, T.; Akizuki, N.; Miyauchi, Y.; Matsuda, K.; Ohfuchi, M.; Kataura, H. Single-chirality separation and optical properties of (5,4) single-wall carbon nanotubes. J. Phys. Chem. C 2016, 120, 10705–10710. [Google Scholar] [CrossRef]
- De Faria, C.G.; Grassi, M.; Carvalho, A.C.M. Conformational analysis and electronic structure of chiral carbon and carbon nitride nanotubes. Mater. Res. 2011, 14, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Hemasa, A.L.; Naumovski, N.; Maher, W.A.; Ghanem, A. Application of carbon nanotubes in chiral and achiral separations of pharmaceuticals, biologics and chemicals. Nanomaterials 2017, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Kameta, N.; Masuda, M.; Shimizu, T. Soft nanotubes acting as confinement effecters and chirality inducers for achiral polythiophenes. Chem. Commun. 2016, 52, 1346–1349. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Recognition of the R- and S-isomers of 1-fluoroethanol by a chiral nanotube. Russ. J. Gen. Chem. 2015, 85, 2813–2815. [Google Scholar] [CrossRef]
- Lathan, W.A.; Radon, L.; Hehre, W.J.; Pople, J.A. Molecular orbital theory of the electronic structure of organic compounds. XVIII. Conformations and stabilities of trisubstituted methanes. J. Am. Chem. Soc. 1973, 95, 699–703. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Recognition of R- and S-isomers of α-alanine by chiral nanotubes. Russ. J. Gen. Chem. 2018, 88, 930–934. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.A.V.; Ribeiro da Silva, M.D.M.C.; Santos, A.F.L.O.M.; Roux, M.V.; Foces-Foces, C.; Notario, R.; Guzmán-Mejıa, R.; Juaristi, E. Experimental and computational thermochemical study of α-alanine (DL) and β-alanine. J. Phys. Chem. B 2010, 114, 16471–16480. [Google Scholar] [CrossRef] [PubMed]
- Craig, N.C.; Piper, L.G.; Wheeler, V.L. Thermodynamics of cis-trans isomerizations. II. 1-Chloro-2-fluoroethylenes, 1,2-difluorocyclopropanes, and related molecules. J. Phys. Chem. 1971, 75, 1453–1460. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry, 4th ed.; John Wiley & Sons Ltd: New York, NY, USA, 1980; p. 434. [Google Scholar]
- Jana, J. Relative stabilities of two difluorodiazene isomers: Density functional and molecular orbital studies. Rep. Theor. Chem. 2012, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.; Ghosh, A.; Chattopadhyay, S.; Ghosh, P.; Chaudhuri, R.K. Revisiting the ‘cis-effect’ in 1,2-difluoro derivatives of ethylene and diazene using ab initio multireference methods. J. Mol. Phys. 2014, 112, 3206–3224. [Google Scholar] [CrossRef]
- Mourão, Z.S.; Melo, A. Energy decomposition analysis of cis and trans isomers of 1,2-dihaloethylenes and 2-butene. J. Mol. Struct. Teochem. 2010, 946, 7–12. [Google Scholar] [CrossRef]
- Teixeira, F.; Melo, A.; Cordeiro, M.N.D.S. Exploring rare chemical phenomena using fractional nuclear charges: The cis-effect in N2F2. Int. J. Quantum Chem. 2018, 118, e25662. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical evaluation of relative stability of diazadifluoride isomers in nanotubes. Russ. J. Gen. Chem. 2013, 83, 140–142. [Google Scholar] [CrossRef]
- Kuznetsov, V.V. Theoretical estimation of the stability of cis- and trans-difluoroethylene in nanotubes. Russ. J. Org. Chem. 2013, 49, 765–767. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form | ΔG≠298 (kcal/mol) | rC–C (Å) | OC–C | Charge |
|---|---|---|---|---|
| C2H6 | ||||
| Staggered Eclipsed | 0 2.5 | 1.531 1.544 | 1.02 1.00 | 0 0 |
| C2H6@(4,4)-closed | ||||
| Staggered Eclipsed | 0.6 0 | 1.495 1.499 | 0.80 0.78 | −0.54 −0.57 |
| C2H6@(6,0)-short | ||||
| Staggered Eclipsed | 2.0 0 | 1.431 1.429 | 0.51 0.53 | 0.72 0.73 |
| C2H6@(6,0)-long | ||||
| Staggered Eclipsed | 1.7 0 | 1.434 1.432 | 0.49 0.50 | 0.40 0.48 |
| C2H6@(6,0)-CBN | ||||
| Staggered Eclipsed | 0.9 0 | 1.449 1.449 | 0.66 0.66 | −1.16 −1.22 |
| Form | ΔG≠298 (kcal/mol) | rC–C (Å) | OC–C | Charge | Reference |
|---|---|---|---|---|---|
| C2H5F | |||||
| Staggered Eclipsed | 0 3.1 (exp. 3.31) | 1.514 1.530 | 1.03 1.01 | 0 0 | [94,139,140] |
| C2H5F@(4,4) | |||||
| Staggered Eclipsed | 0.8 0 | 1.487 1.485 | 0.68 0.73 | −0.19 −0.16 | [140] |
| C2H5F@(5,5) | |||||
| Staggered Eclipsed | 0 2.4 | 1.508 1.522 | 0.96 0.97 | −0.41 −0.40 | [140] |
| CH3−CF3 | |||||
| Staggered Eclipsed | 0 2.7 (exp. 3.17) | 1.507 1.521 | 1.01 1.01 | 0 0 | [139,141,142] |
| CH3−CF3@(4,4) | |||||
| Staggered Eclipsed | 1.2 0 | 1.505 1.518 | 0.94 0.92 | −0.22 −0.20 | [142] |
| CH3−CF3@(5,5) | |||||
| Staggered Eclipsed | 0 2.0 | 1.501 1.513 | 0.99 0.98 | −0.27 −0.26 | [142] |
| CF3−CF3 | |||||
| Staggered Eclipsed | 0 4.5 (exp. 4.30) | 1.565 1.600 | 0.88 0.88 | 0 0 | [139,143] |
| CF3−CF3@(5,5) | |||||
| Staggered Eclipsed | 2.3 0 | 1.562 1.587 | 0.88 0.86 | −0.15 −0.16 | [143] |
| CF3−CF3@(6,6) | |||||
| Staggered Eclipsed | 0 4.2 | 1.539 1.572 | 0.89 0.87 | −0.13 −0.13 | [143] |
| Cluster; ΔG≠298 (kcal/mol) 1 | rC–C (Å) 1 | O 1 | Charge | Reference |
|---|---|---|---|---|
| C2H6@C60; 3.3 (2,5) | 1.465 (1.531) | 1.12 (1.00) | −0.47 | [196] |
| C2F6@C60; 15.1 (4,6) | 1.374 (1.565) | 0.86 (0.88) | 0.20 | [197,198] |
| C2F6@C12B24N24; 6.5 | 1.405 | 0.83 | 0.46 | [198] |
| C2F6@B36N24; 8.2 | 1.464 | 0.86 | 0.48 | [198] |
| C2F6@C80; 7.2 | 1.477 | 0.89 | −0.27 | [197,198] |
| C2F6@C14B33N33; 10.0 | 1.493 | 0.90 | 0.15 | [198] |
| C2F6@B47N33; 7.6 | 1.507 | 0.92 | 0.15 | [198] |
| C2Cl6@C80; 43.5 (13,8) | 1.431 (1.590) | 0.98 (1.06) | −0.22 | [199] |
| C5H12@C60; 10.7 (3,8) | 1.366 (1.539) | 0.86 (1.00) | 2.07 | [200] |
| C5H12@C80; 8.3 | 1.459 | 0.86 | −1.27 | [200] |
| H3B←NH3@C60; 4.4 (2,0) | 1.522 (1.652) | 0.43 (0.65) | −0.71 | [201] |
| H3B←NH3@C12B24N24; 1.5 | 1.544 | 0.54 | −0.79 | [201] |
| H3B←NH3@B36N24; 2.3 | 1.558 | 0.48 | −0.22 | [201] |
| H3B←NH3@C70; 2.1 | 1.595 | 0.62 | −0.90 | [201] |
| H3B←NH3@B41N29; 1.8 | 1.610 | 0.72 | −0.11 | [201] |
| H3B←NH3@C80; 1.8 | 1.601 | 0.58 | −0.64 | [201] |
| H3B←NH3@C14B33N33; 1.1 | 1.605 | 0.57 | −0.11 | [201] |
| H3B←NH3@B47N33; 1.6 | 1.618 | 0.64 | −0.06 | [201] |
| Si2H6@Si60; 1.0 (1,3) | 2.351 (3.355) | 0.97 (1.00) | −0.12 | [202] |
| Si2H6@C80; 2.4 | 2.184 | 1.52 | −1.16 | [202] |
| Si2H6@B47N33; 1.6 | 2.252 | 1.08 | 0.01 | [202] |
| Object | ΔG2980 (ΔG298≠), kcal/mol | Electric Charge |
|---|---|---|
| (5,5) | 0 | 0 |
| P,M(5,1) 1 | 5.9 | 0 |
| P,M(5,2) 1 | 16.1 | 0 |
| S-C3H7NO2 (A) | 0 | 0 |
| S-C3H7NO2 (B) 2 | (3.1) | 0 |
| S-C3H7NO2 (D) 2 | (2.7) | 0 |
| R,S-C3H7NO2@(5,5) 3 | 13.9 | −0.69 |
| S-C3H7NO2@M(5,1) 3 | 12.3 | −0.69 |
| S-C3H7NO2@P(5,1) 3 | 12.1 | −0.67 |
| R-C3H7NO2@M(5,1) 3 | 12.1 | −0.67 |
| R-C3H7NO2@P(5,1) 3 | 12.3 | −0.69 |
| S-C3H7NO2@M(5,2) 3 | 4.6 | −0.64 |
| S-C3H7NO2@P(5,2) | 0 | −0.67 |
| R-C3H7NO2@M(5,2) 3 | 0.1 | −0.67 |
| R-C3H7NO2@P(5,2) 3 | 4.7 | −0.65 |
| Object | ΔG2980, kcal/mol | OX=X | Charge |
|---|---|---|---|
| CHF=CHF, cis- trans- | 0 0.6 | 1.78 1.77 | 0 0 |
| FN=NF, cis- trans- | 0 3.4 | 1.76 1.66 | 0 0 |
| CHF=CHF@(4,4), cis- trans- | 26.2 0 | 1.32 1.44 | 0.46 0.52 |
| CHF=CHF@(6,6), cis- trans- | 0.9 0 | 1.72 1.73 | −0.16 −0.17 |
| FN=NF@(4,4), cis- trans- | 27.4 0 | 1.46 1.73 | −0.36 −0.36 |
| FN=NF@(6,6), cis- trans- | 0 3.8 | 1.71 1.69 | −0.03 −0.004 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, V. Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems. Molecules 2020, 25, 2437. https://doi.org/10.3390/molecules25102437
Kuznetsov V. Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems. Molecules. 2020; 25(10):2437. https://doi.org/10.3390/molecules25102437
Chicago/Turabian StyleKuznetsov, Valerij. 2020. "Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems" Molecules 25, no. 10: 2437. https://doi.org/10.3390/molecules25102437
APA StyleKuznetsov, V. (2020). Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems. Molecules, 25(10), 2437. https://doi.org/10.3390/molecules25102437