
Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer

 and
and
Abstract
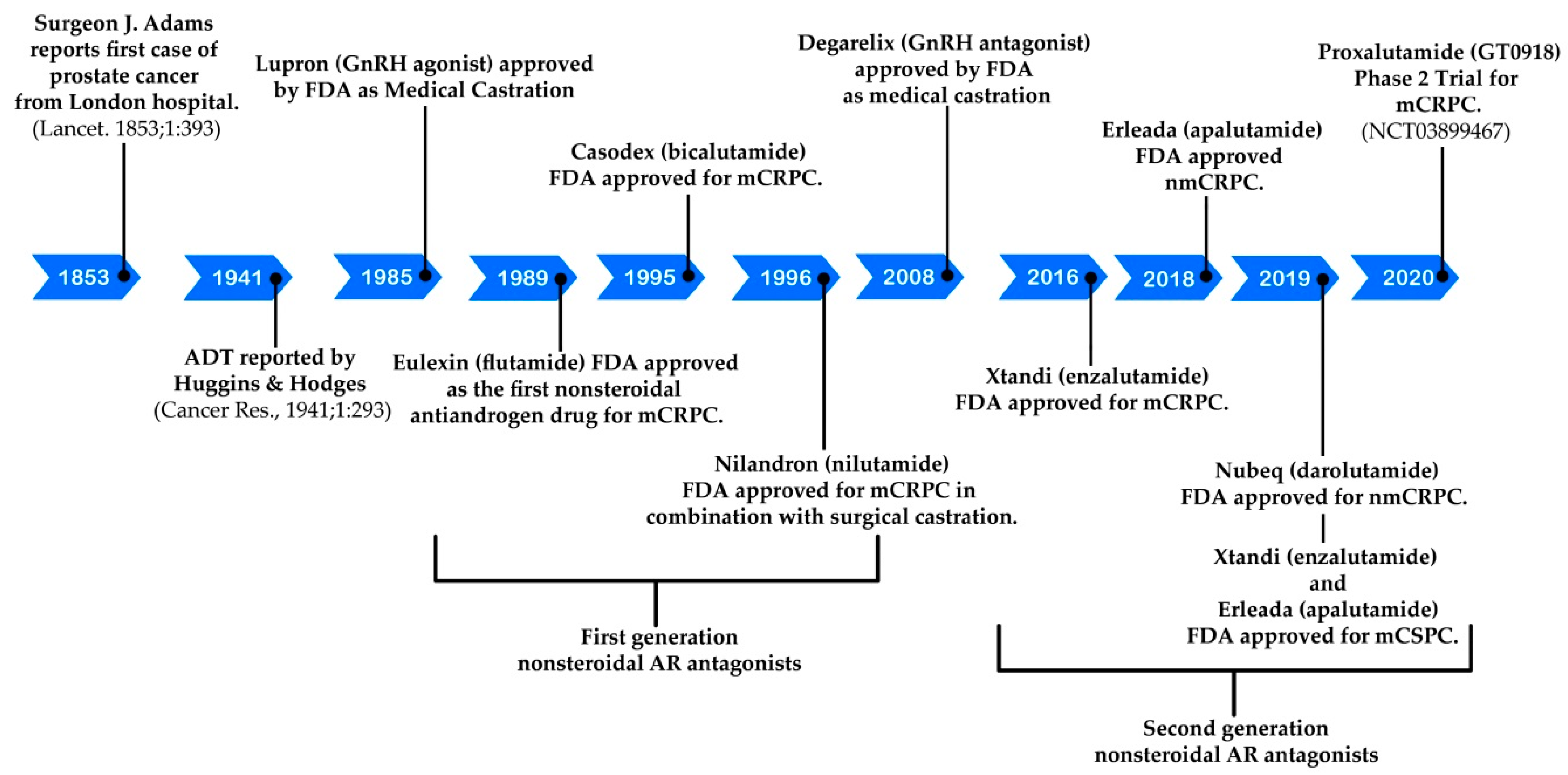
:1. Introduction
1.1. Prostate Cancer
1.2. Hormonal Therapeutics
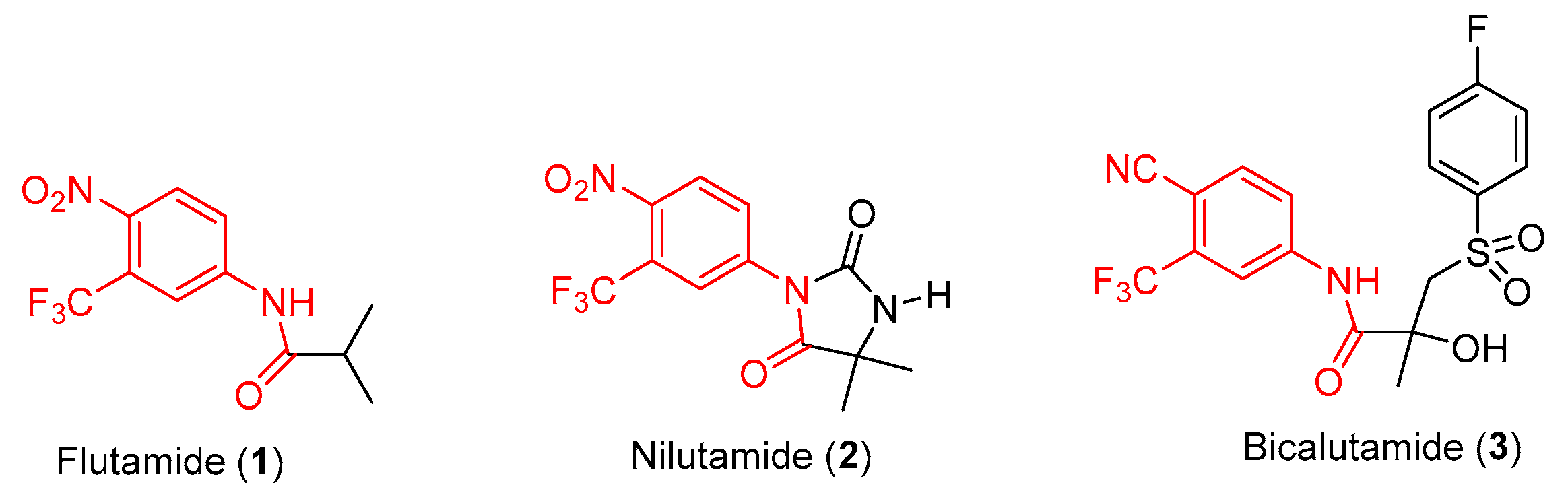
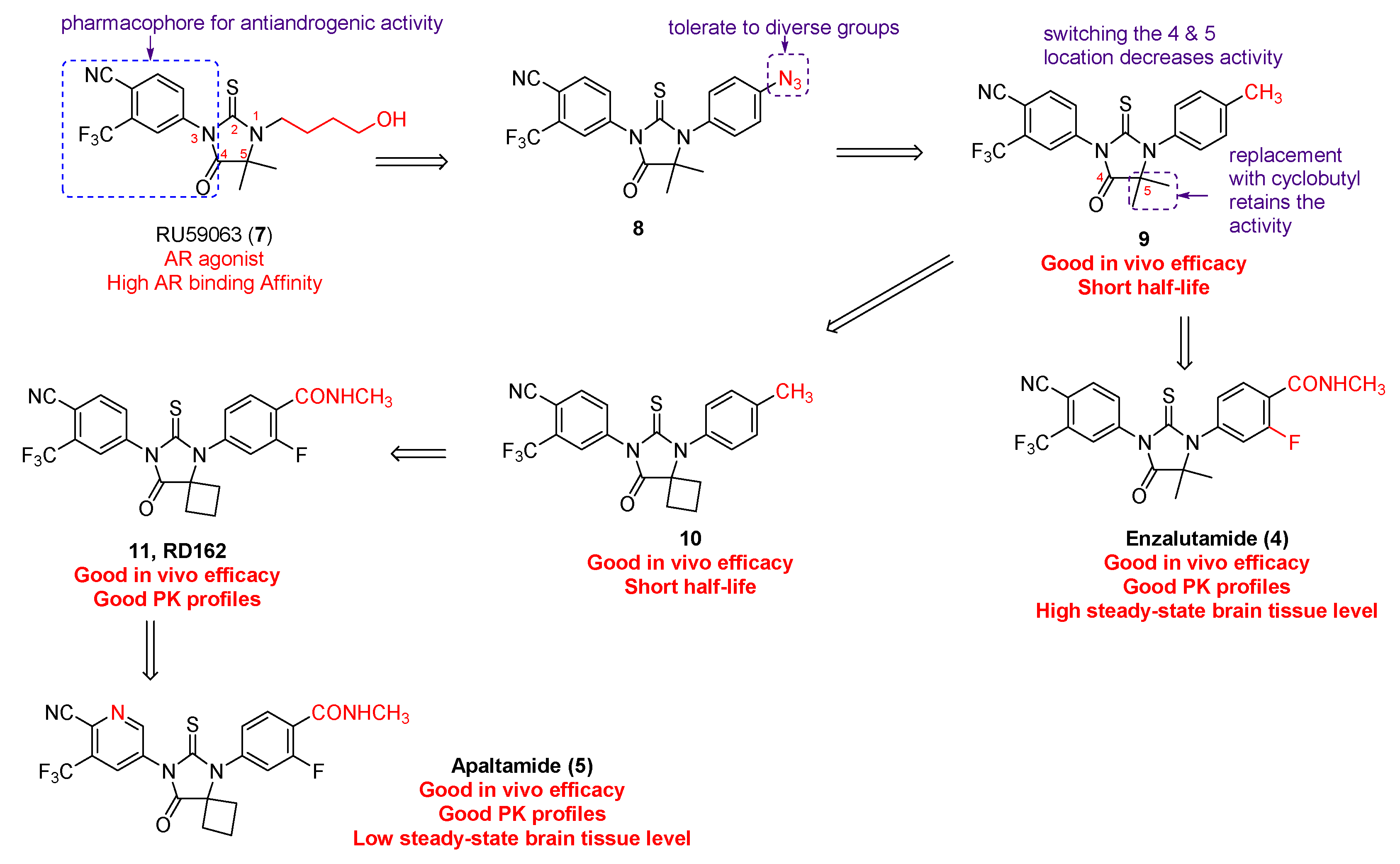
2. Discovery and Preclinical Studies
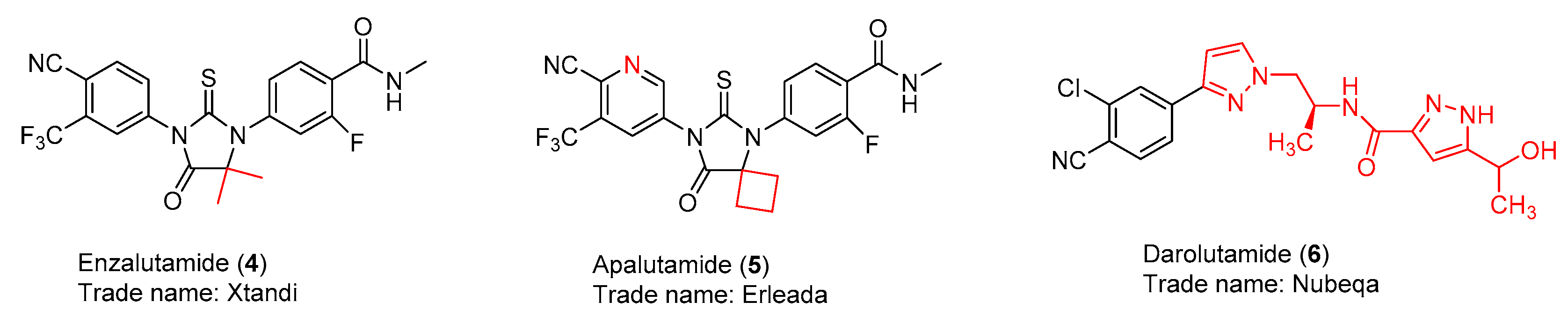
2.1. Enzalutamide (4) and Apalutamide (5)
2.2. Darolutamide (6)
3. Syntheses
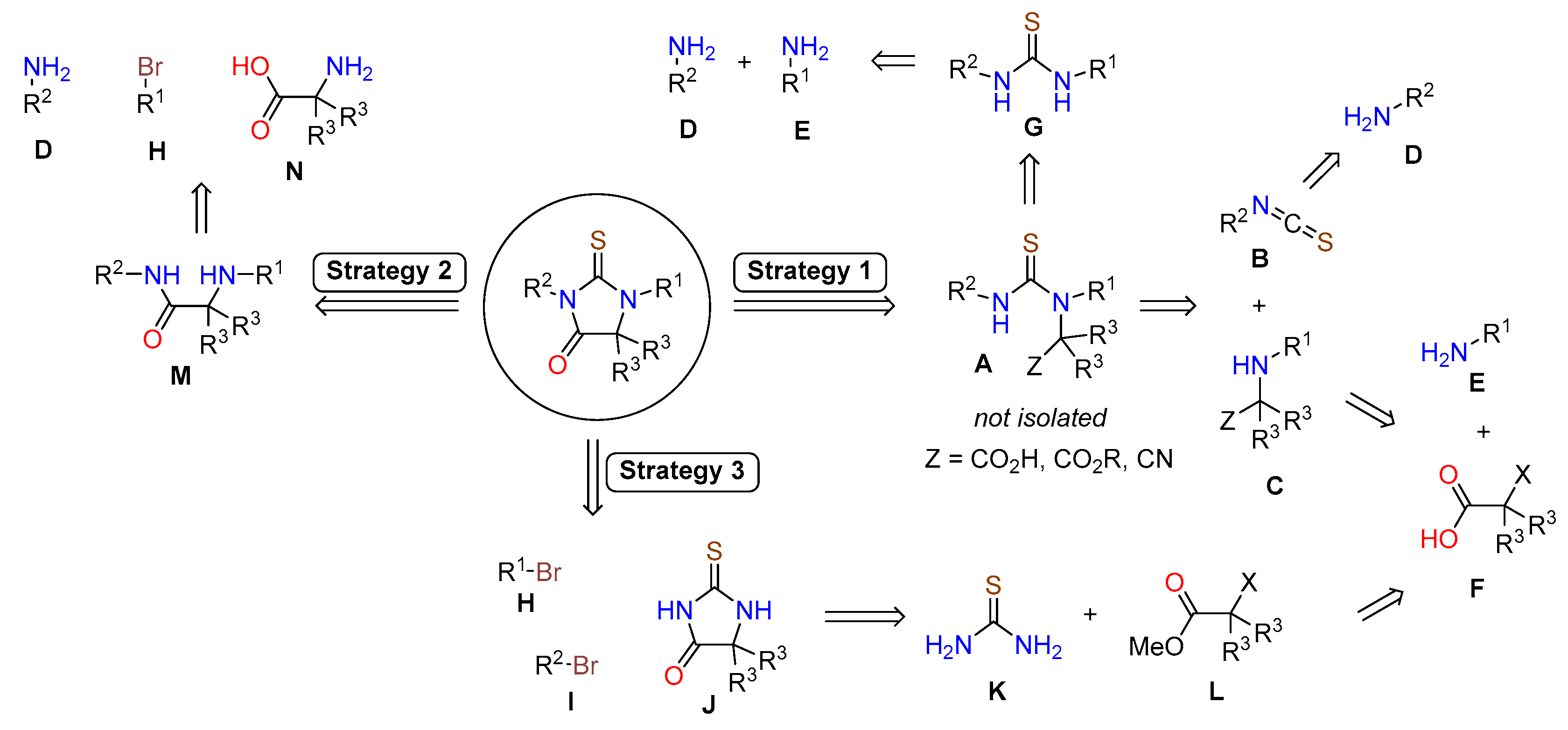
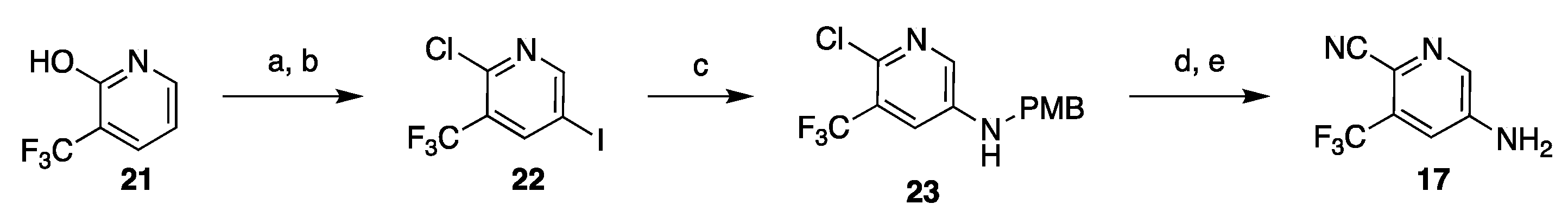
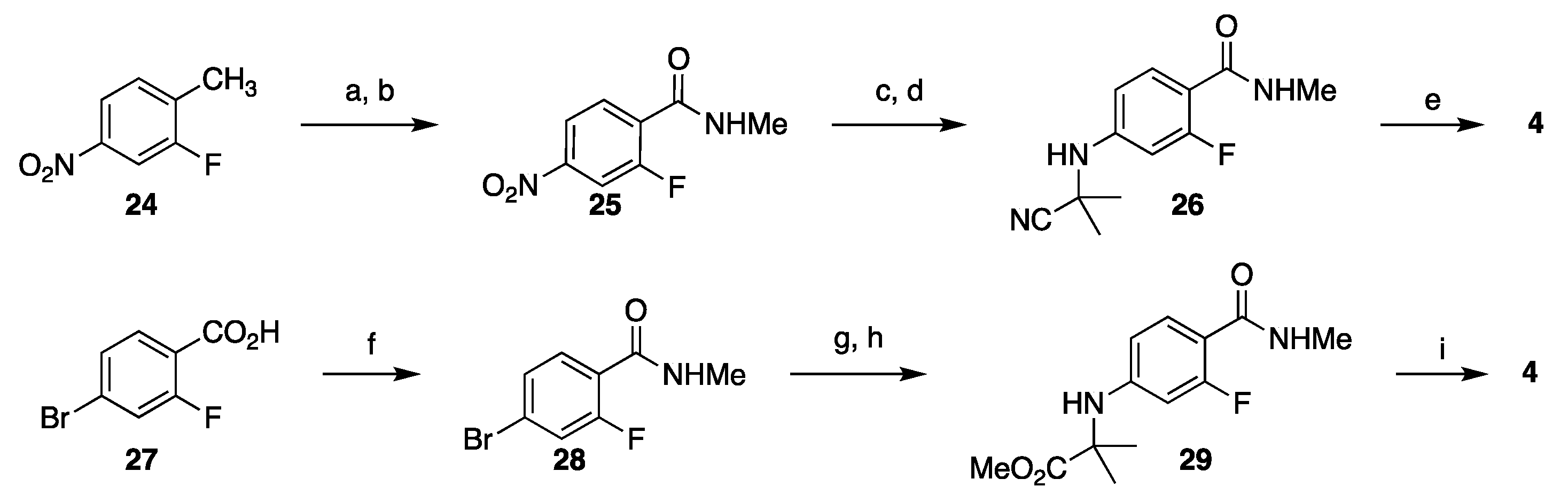
3.1. Synthetic Approaches to Enzalutamide (4) and Apalutamide (5)
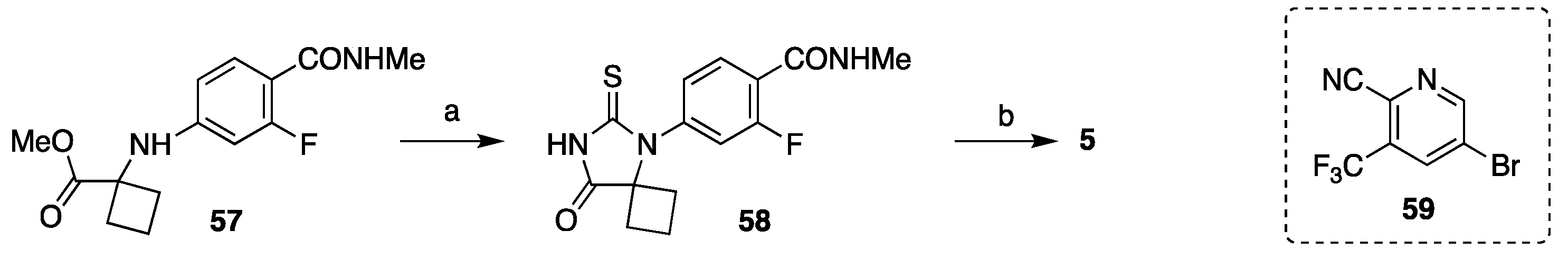
3.1.1. Strategy 1: Cyclization of Isothiocyanate
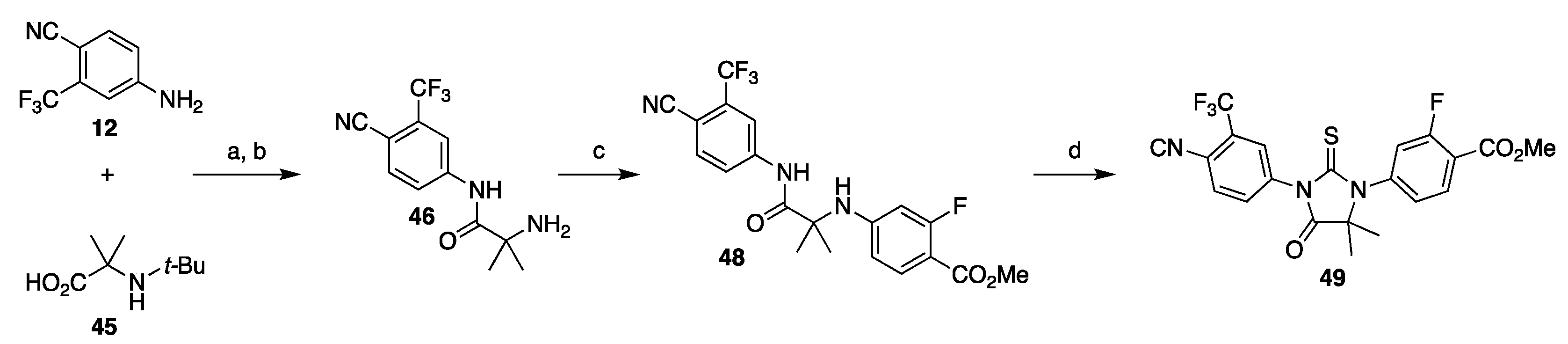
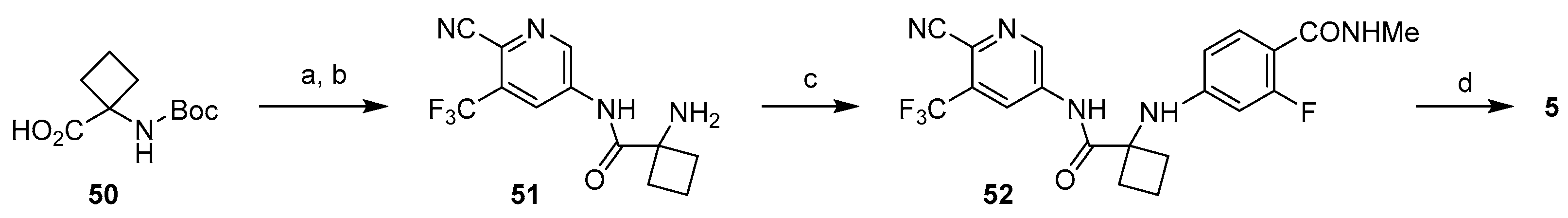
3.1.2. Strategy 2: Late Stage Cyclization of Amino Amide
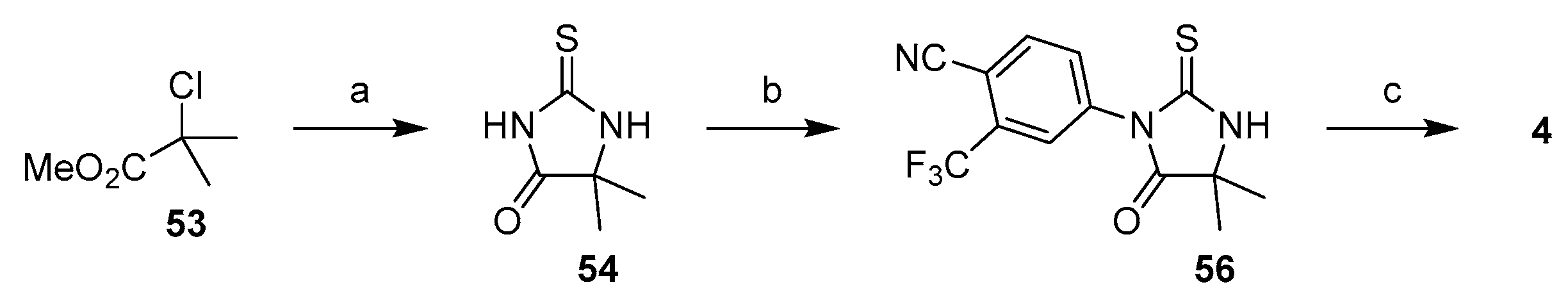
3.1.3. Strategy 3: Functionalization of Thiohydantoin
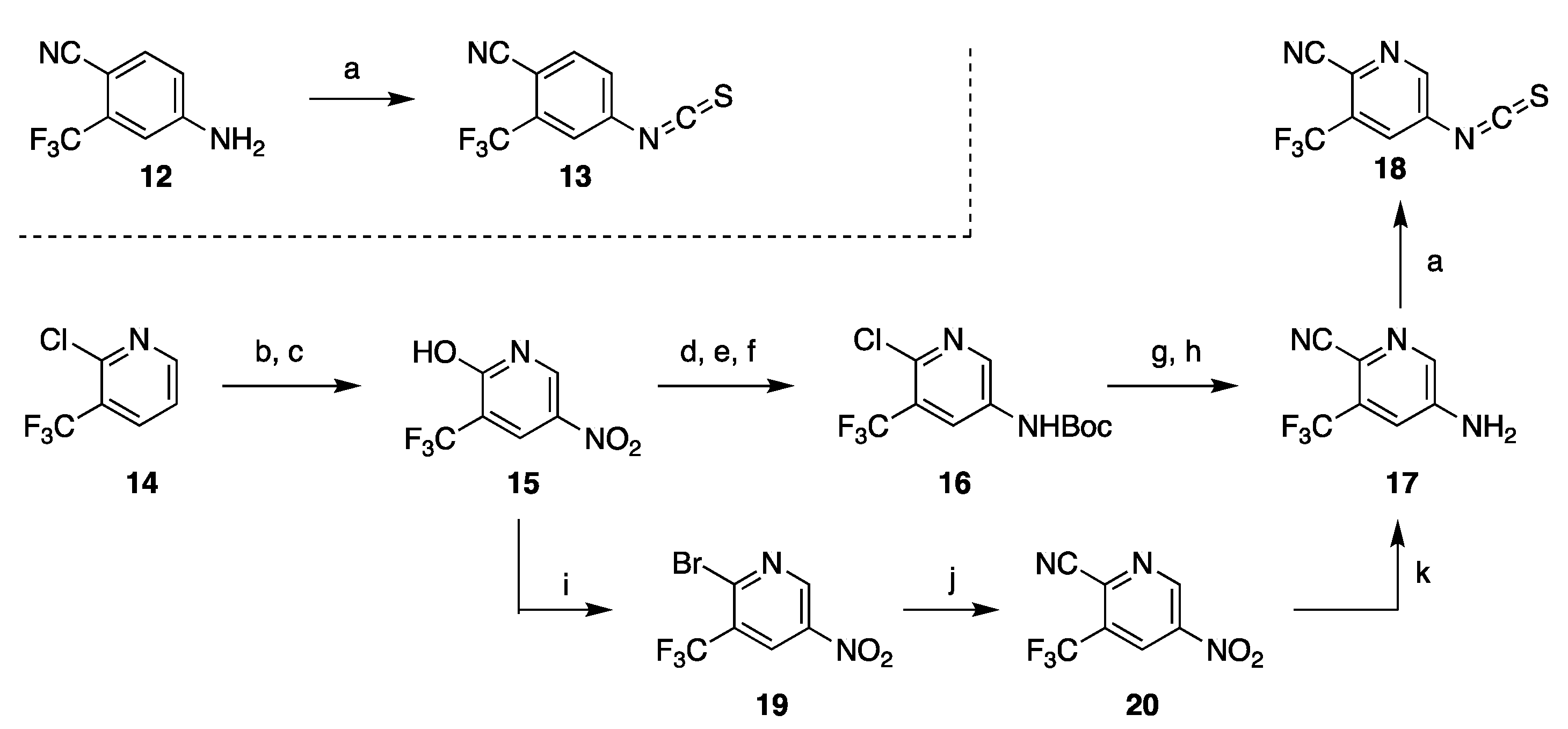
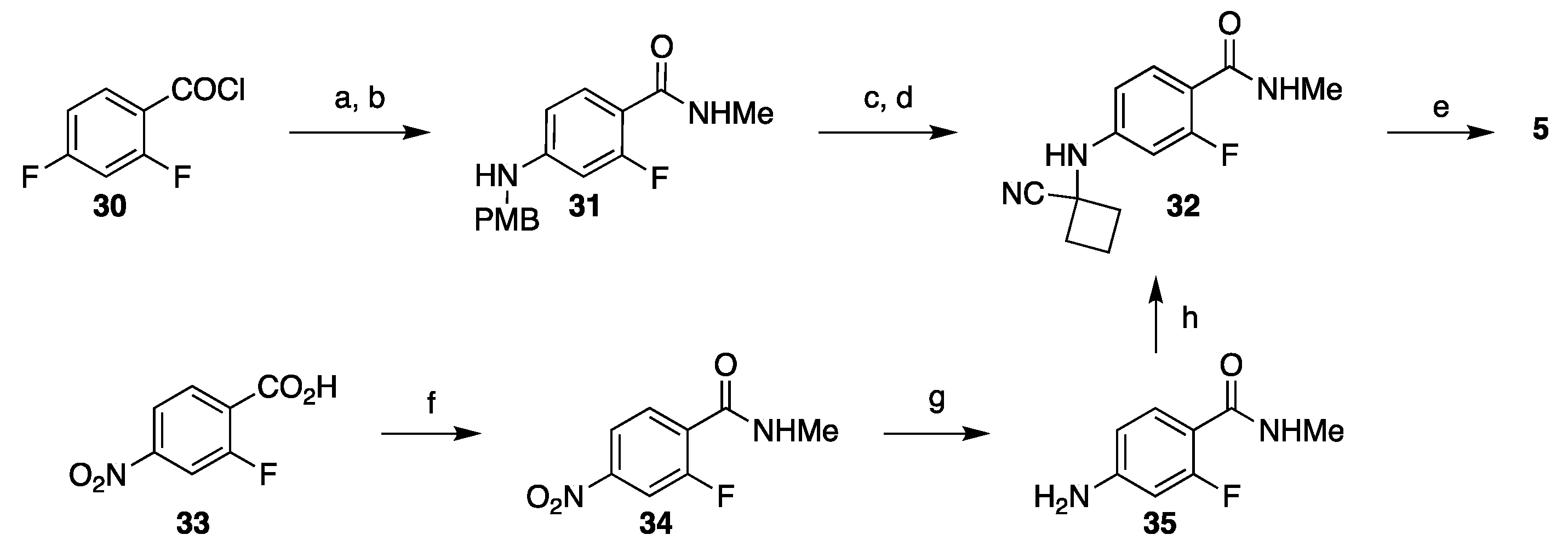
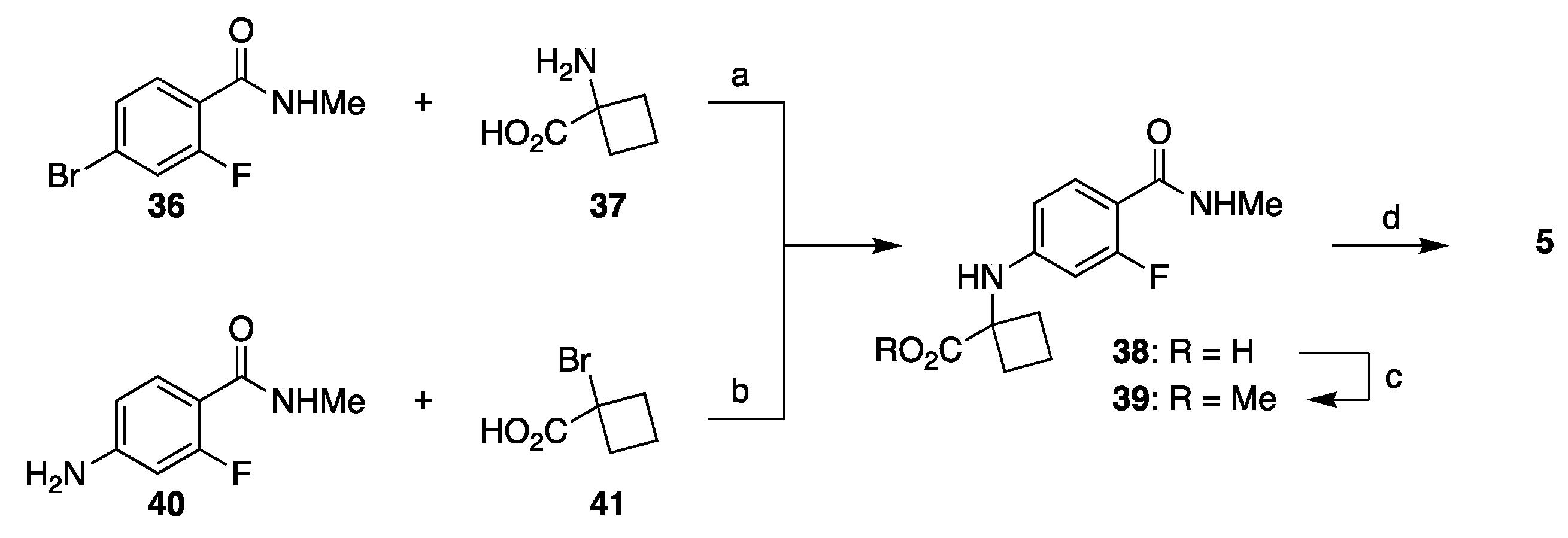
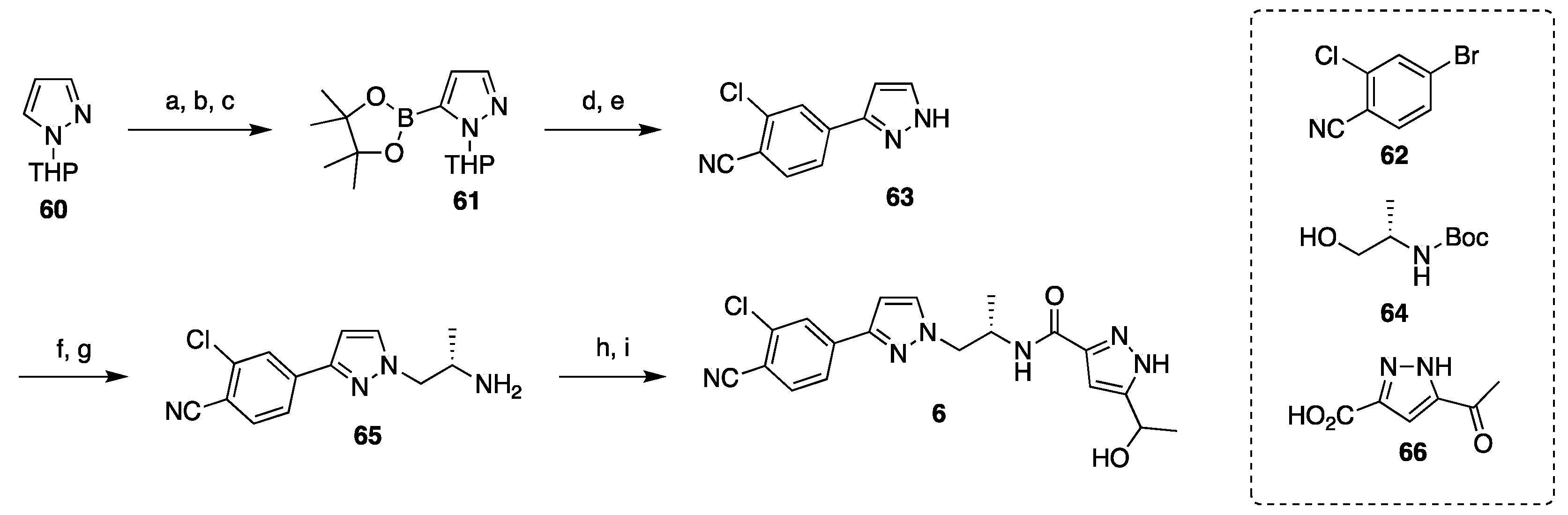
3.2. Synthesis of Darolutamide
4. Clinical Studies and Use of Second Generation Nonsteroidal AR Antagonists
4.1. Enzalutamide
4.1.1. Enzalutamide for mCRPC
4.1.2. Enzalutamide for nmCRPC
4.1.3. Enzalutamide for mCSPC
4.2. Apalutamide
4.2.1. Apalutamide for nmCRPC
4.2.2. Apalutamide for mCSPC
4.2.3. Indirect Comparison with Enzalutamide
4.3. Darolutamide
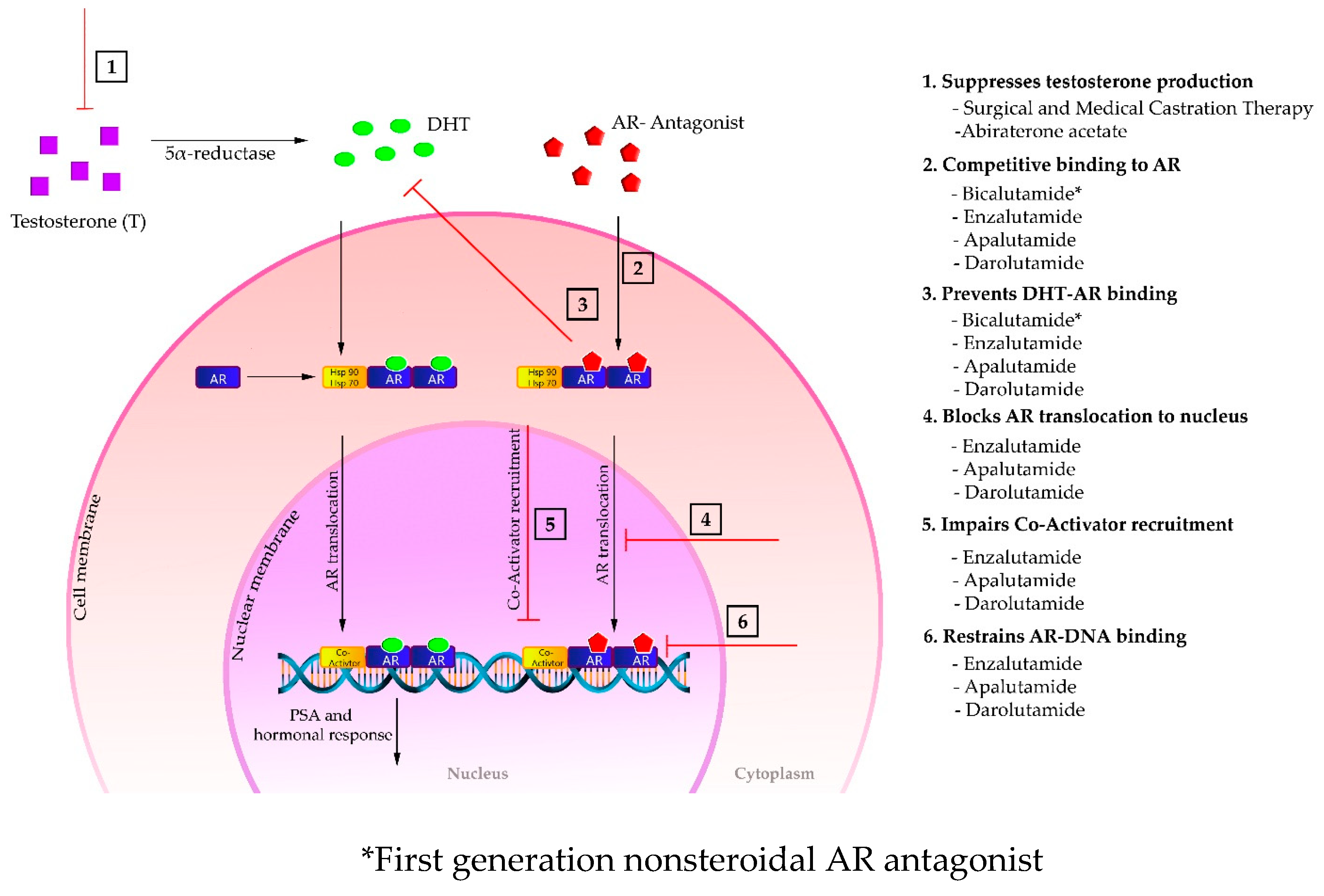
5. Mechanism of Action of the Second-Generation AR Antagonists
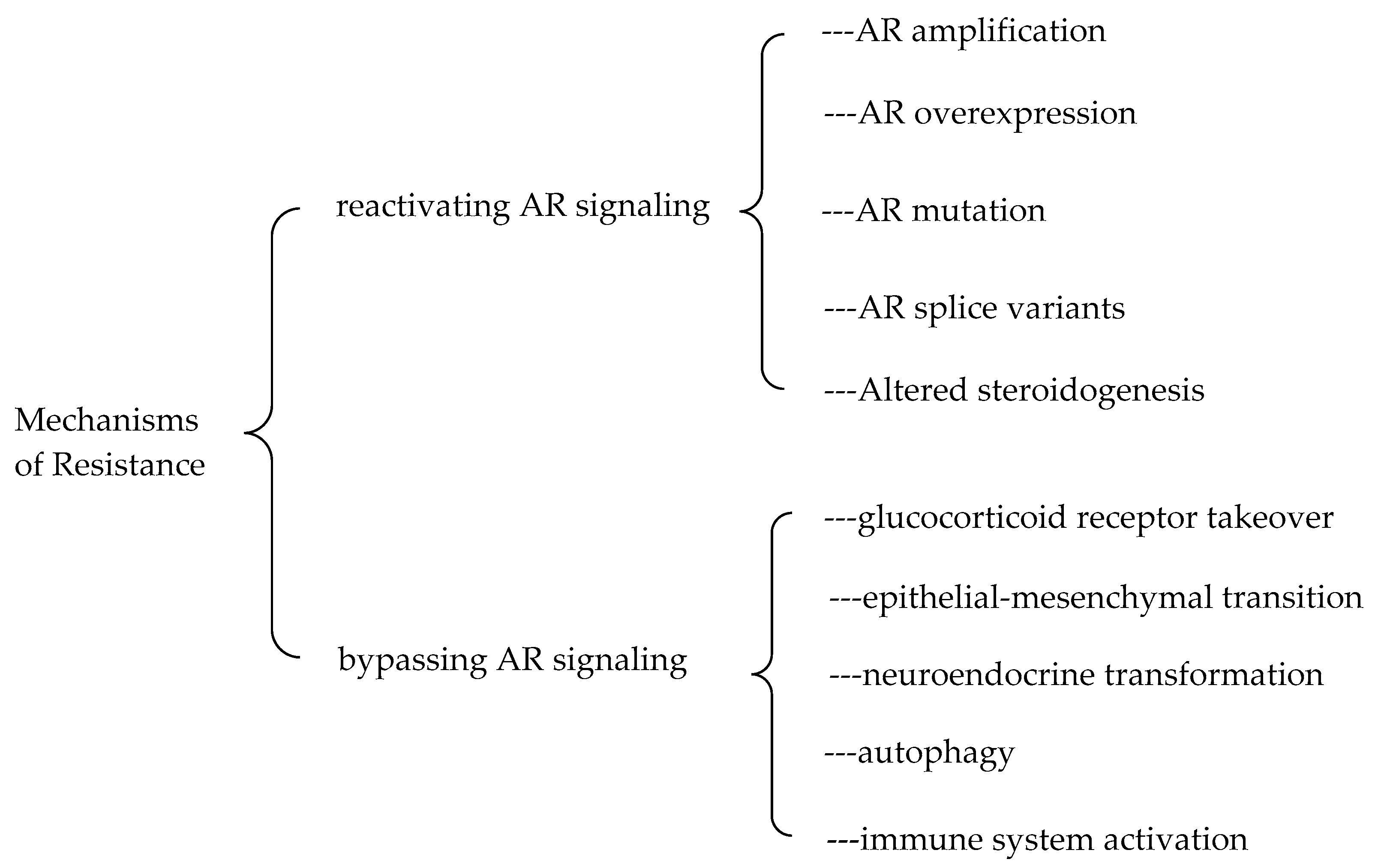
6. Strategies to Overcome the Resistance to the Second-Generation AR Antagonists
6.1. Mechanisms of the Resistance to the Second-Generation AR Antagonists
6.2. Strategies to Overcome the Resistance to the Second-Generation AR Antagonists
6.2.1. Combination Therapy
6.2.2. Target AR with Other Strategies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller Kimberly, D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2014, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshan, N.G.R.D.; Rettig, M.; Jung, M.E. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med. Res. Rev. 2018, 39, 910–960. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Blackard, C.E. The Veterans’ Administration Cooperative Urological Research Group studies of carcinoma of the prostate: A review. Cancer Chemother. Rep. 1975, 59, 225–227. [Google Scholar] [PubMed]
- Redding, T.W.; Schally, A.V. Inhibition of prostate tumor growth in two rat models by chronic administration of D-Trp6 analog of luteinizing hormone-releasing hormone. Proc. Natl. Acad. Sci. USA 1981, 78, 6509–6512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobi, G.H.; Wenderoth, U.K. Gonadotropin-releasing hormone analogs for prostate cancer: Untoward side effects of high-dose regimens acquire a therapeutical dimension. Eur. Urol. 1982, 8, 129–134. [Google Scholar] [CrossRef]
- Walker, K.; Turkes, A.; Turkes, A.; Zwink, R.; Beacock, C.; Buck, A.; Peeling, W.; Griffiths, K. Treatment of patients with advanced cancer of the prostate using a slow-release (depot) formulation of the lhrh agonist ici 118630 (zoladex®). J. Endocrinol. 1984. [Google Scholar] [CrossRef]
- The Leuprolide Study Group Leuprolide versus Diethylstilbestrol for Metastatic Prostate Cancer. N. Engl. J. Med. 1984, 311, 1281–1286. [CrossRef]
- Schröder, F.H.; Crawford, E.; Axcrona, K.; Payne, H.; Keane, T. Androgen deprivation therapy: Past, present and future. BJU Int. 2012, 109, 1–12. [Google Scholar] [CrossRef]
- Anderson, J. The role of antiandrogen monotherapy in the treatment of prostate cancer. BJU Int. 2003, 91, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, P.F.; Sharifi, R.; Block, N.L.; Soloway, M.S.; Venner, P.M.; Patterson, A.L.; Sarosdy, M.F.; Vogelzang, N.J.; Schellenger, J.J.; Kolvenbag, G.J. Clinical benefits of bicalutamide compared with flutamide in combined androgen blockade for patients with advanced prostatic carcinoma: Final report of a double-blind, randomized, multicenter trial. Casodex Combination Study Group. Urology 1997, 50, 330–336. [Google Scholar] [CrossRef]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2003, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, N.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. Yearb. Paediatr. Endocrinol. 2019, 380, 1235–1346. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.E.; Ouk, S.; Yoo, N.; Sawyers, C.L.; Chen, C.; Tran, C.; Wongvipat, J. Structure−Activity Relationship for Thiohydantoin Androgen Receptor Antagonists for Castration-Resistant Prostate Cancer (CRPC). J. Med. Chem. 2010, 53, 2779–2796. [Google Scholar] [CrossRef] [Green Version]
- Teutsch, G.; Goubet, F.; Battmann, T.; Bonfils, A.; Bouchoux, F.; Cerede, E.; Gofflo, D.; Gaillard-Kelly, M.; Philibert, D. Non-steroidal antiandrogens: Synthesis and biological profile of high-affinity ligands for the androgen receptor. J. Steroid Biochem. Mol. Biol. 1994, 48, 111–119. [Google Scholar] [CrossRef]
- Van Dort, M.E.; Robins, D.M.; Wayburn, B. Design, synthesis, and pharmacological characterization of 4-[4,4-dimethyl-3-(4-hydroxybutyl)-5-oxo-2-thioxo-1-imidazolidinyl]-2-iodobenzonitrile as a high-affinity nonsteroidal androgen receptor ligand. J. Med. Chem. 2000, 43, 3344–3347. [Google Scholar] [CrossRef]
- Scher, H.I.; Sawyers, C.L. Biology of Progressive, Castration-Resistant Prostate Cancer: Directed Therapies Targeting the Androgen-Receptor Signaling Axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef]
- Tucker, H.; Crook, J.W.; Chesterson, G.J. Nonsteroidal antiandrogens. Synthesis and structure-activity relationships of 3-substituted derivatives of 2-hydroxypropionanilides. J. Med. Chem. 1988, 31, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Foster, W.; Car, B.D.; Shi, H.; Levesque, P.C.; Obermeier, M.T.; Gan, J.; Arezzo, J.C.; Powlin, S.S.; Dinchuk, J.E.; Balog, A.; et al. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate 2010, 71, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moilanen, A.-M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykänen, P.S.; Törmäkangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huges, D.L. Review of synthetic routes and crystalline forms of the antiandrogen oncology drugs enzalutamide, apalutamide, and darolutamide. Org. Process Res. Dev. 2020, 24, 347–362. [Google Scholar] [CrossRef]
- Sawyers, C.L.; Jung, M.E.; Chen, C.D.; Ouk, S.; Welsbie, D.; Tran, C.; Wongvipat, J.; Yoo, D. Diarylhydantoin Compounds. PCT Int. Patent Appl. WO2006124118, 23 November 2006. [Google Scholar]
- Sawyers, C.L.; Jung, M.E.; Chen, C.D.; Ouk, S.; Welsbie, D.; Tran, C.; Wongvipat, J.; Yoo, D. Diarylhydantoin Compounds. U.S. Patent 7709517B2, 4 May 2010. [Google Scholar]
- Jung, M.E.; Yoo, D.; Sawyers, C.L.; Tran, C.; Wongvipat, J. Diarylhydantoin Compounds. U.S. Patent 8110594B2, 7 February 2012. [Google Scholar]
- Chivukula, K.R.; Karuturi, V.V.R.; Benda, S.; Anke, R.; Gajula, D.; Moturu, V.R.K.M.; Indukuri, V.S.; Gorantla, S.R.A.; Chava, S. Process for the Preparation of Enzalutamide. U.S. Patent 10131636B2, 20 November 2018. [Google Scholar]
- Suzuki, Y.; Nakagawa, S.; Kitamura, T. Process for Producing Enzalutamide Crystal Form. Int. Patent Appl. WO 2016/194813, 27 May 2016. [Google Scholar]
- Song, L.J.; Wang, Y.; Lu, X.F.; Li, Z.Y. Synthesis of androgen receptor antagonist MDV3100. Fine Chem. Intermed. 2012, 42, 34. [Google Scholar]
- Fu, Q.; Yue, L.; Lin, Q.; Liao, X.; Zhao, M.; Qin, Y. Method for Synthesizing Enzalutamide. Chinese Patent Appl. CN 104844520, 5 September 2017. [Google Scholar]
- Jung, M.E.; Sawyers, C.L.; Ouk, S.; Tran, C.; Wongvipat, J. Androgen Receptor Modulator for the Treatment of Prostate Cancer and Androgen Receptor-Associated Diseases. Int. Patent Appl. WO2007126765A2, 8 November 2007. [Google Scholar]
- Jung, M.E.; Sawyers, C.L.; Ouk, S.; Tran, C.; Wongvipat, J. Androgen Receptor Modulator for the Treatment of Prostate Cancer and Androgen Receptor-Associated Diseases. U.S. Patent 8445507B2, 21 May 2013. [Google Scholar]
- Ouerfelli, O.; Dilhas, A.; Yang, G.; Zhao, H. Synthesis of Thiohydantoins. Int. Patent Appl. WO2008/119015A2, 2 October 2008. [Google Scholar]
- Ouerfelli, O.; Dilhas, A.; Yang, G.; Zhao, H. Synthesis of Thiohydantoins. U.S. Patent Appl. 2010/0190991A1, 29 July 2010. [Google Scholar]
- Pang, X.; Wang, Y.; Chen, Y. Design, synthesis, and biological evaluation of deuterated apalutamide with improved pharmacokinetic profiles. Bioorg. Med. Chem. Lett. 2017, 27, 2803–2806. [Google Scholar] [CrossRef]
- Chen, Y.; Gong, Y. Imidazole Diketone Compound and Use Thereof. Int. Patent Appl. WO2014/190895, 4 December 2014. [Google Scholar]
- Muthusamy, A.R.; Kanniah, S.L.; Arote, N.D.; Bhagwat, O.V.; Sonar, J.K.; Poundkar, V.B.; Wagh, Y.D. Solid State Forms of Apalutamide. Int. Patent Appl. WO 2018/112001, 21 June 2018. [Google Scholar]
- Muthusamy, A.R.; Kanniah, S.L.; Arote, N.D.; Bhagwat, O.V.; Sonar, J.K.; Poundkar, V.B.; Wagh, Y.D. Solid State Forms of Apalutamide. U.S. Patent Appl. 2019/0322640A1, 24 October 2019. [Google Scholar]
- Bodhuri, P.; Ceccarelli, A.P.; Emmett, M.R.; Karadeolian, A.; Souza, F.E.S.; Weeratunga, G.; Gorin, B. Processes for the Preparation of Apalutamide and Intermediates Thereof. U.S. Patent Appl. 2019/ 0276424, 12 September 2019. [Google Scholar]
- Haim, C.B.; Horvath, A.; Weerts, E.; Albaneze-Walker, J. Processes for the Preparation of a Diarylthiohydantoin Compound. U.S. Patent Appl. 2019/0135775A9, 9 May 2019. [Google Scholar]
- Haim, C.B.; Horvath, A.; Weerts, E.; Albaneze-Walker, J. Processes for the Preparation of a Diarylthiohydantoin Compound. U.S. Patent Appl. 2018/0002309A1, 4 January 2018. [Google Scholar]
- Meng, X.; Shao, L.; Huang, G.; Wu, K.; Tian, P.; Xu, C.; Jiang, Z.; Xiong, L. Preparation Method of Enzalutamide of Formula (VIII). Chinese Patent Appl. CN 109651256, 4 April 2019. [Google Scholar]
- Haim, C.B.; Horvath, A.; Edmond, J.E. Processes for the Preparation of a Diarylthiohydantoin Compound. U.S. Patent Appl. 2016/0176845A1, 23 June 2016. [Google Scholar]
- Koyama, M.; Kawakami, T.; Okazoe, T.; Nozaki, K. Cyanide-Free One-Pot Synthesis of Methacrylic Esters from Acetone. Chem. - A Eur. J. 2019, 25, 10913–10917. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y. Method for Synthesizing Apalutamide and Intermediate Thereof. Chinese Patent Appl. CN 108383749, 10 August 2018. [Google Scholar]
- Laitinen, I.; Karjalainen, O. Process for the Preparation of Androgen Receptor Antagonists and Intermediates Thereof. U.S. Patent 10189789B2, 29 January 2016. [Google Scholar]
- Wohlfahrt, G.; Törmäkangas, O.; Salo, H.; Höglung, L.; Karjalainen, A.; Knuuttila, P.; Holm, P. Androgen Receptor Modulating Compounds. Int. Patent Appl. WO 2011/051540A1, 5 May 2011. [Google Scholar]
- Törmäkangas, O.; Wohlfahrt, G.; Salo, H.; Ramasubramanian, R.D.; Patra, P.K.; Martin, A.E.; Heikkinen, T.; Vesalainen, A.; Moilanen, A.; Karjalainen, A. Androgen Receptor Modulating Carboxamides. Int. Patent Appl. WO 2012/143599A1, 26 October 2012. [Google Scholar]
- Wohlfahrt, G.; Törmäkangas, O.; Salo, H.; Höglung, L.; Karjalainen, A.; Koivikko, P.; Holm, P.; Rasku, S.; Vesalainen, A. Androgen Receptor Modulating Compounds. U.S. Patent 9657003 B2, 23 May 2017. [Google Scholar]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.-E.; Efstathiou, E.; Rathkopf, D.E.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mülders, P.; Hadaschik, B.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Scher, H.I.; Solo, K.; Valant, J.; Todd, M.B.; Mehra, M. Prevalence of Prostate Cancer Clinical States and Mortality in the United States: Estimates Using a Dynamic Progression Model. PLoS ONE 2015, 10, e0139440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Morris, M.J.; Fox, J.J.; Danila, D.C.; Slovin, S.F.; Hager, J.H.; Rix, P.J.; Maneval, E.C.; Chen, I.; Gönen, M.; et al. Phase I Study of ARN-509, a Novel Antiandrogen, in the Treatment of Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2013, 31, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Small, E.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.; Graff, J.; Olmos, D.; Mainwaring, P.; Lee, J.; Uemura, H.; et al. Apalutamide and overall survival in non-metastatic castration-resistant prostate cancer. Ann. Oncol. 2019, 30, 1813–1820. [Google Scholar] [CrossRef] [Green Version]
- Saad, F.; Cella, D.; Basch, E.; Hadaschik, B.; Mainwaring, P.N.; Oudard, S.; Graff, J.N.; McQuarrie, K.; Li, S.; Hudgens, S.; et al. Effect of apalutamide on health-related quality of life in patients with non-metastatic castration-resistant prostate cancer: An analysis of the SPARTAN randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1404–1416. [Google Scholar] [CrossRef]
- Kwon Daniel, H.-M.; Friedlander, T.; Kwon Daniel, H.-M.; Friedlander, T. A TITAN step forward: Apalutamide for metastatic castration-sensitive prostate cancer. Ann. Transl. Med. 2019, 7 (Suppl. 8), S364. [Google Scholar]
- Chowdhury, S.; Oudard, S.; Uemura, H.; Joniau, S.; Pilon, D.; Ladouceur, M.; Behl, A.S.; Liu, J.; Dearden, L.; Sermon, J.; et al. Matching-Adjusted Indirect Comparison of the Efficacy of Apalutamide and Enzalutamide with ADT in the Treatment of Non-Metastatic Castration-Resistant Prostate Cancer. Adv. Ther. 2019, 37, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Oudard, S.; Uemura, H.; Joniau, S.; Pilon, D.; Lefebvre, P.; McQuarrie, K.; Liu, J.; Dearden, L.; Sermon, J.; et al. Matching-Adjusted Indirect Comparison of Health-Related Quality of Life and Adverse Events of Apalutamide Versus Enzalutamide in Non-Metastatic Castration-Resistant Prostate Cancer. Adv. Ther. 2019, 37, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Fizazi, K.; Gillessen, S.; Heidenreich, A.; Perez-Lopez, R.; Oyen, W.; Shore, N.; Smith, M.; Sweeney, C.; Tombal, B.; et al. Managing Nonmetastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 75, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Massard, C.; Bono, P.; Jones, R.; Kataja, V.; James, N.; A Garcia, J.; Protheroe, A.; Tammela, T.L.; Elliott, T.; et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): An open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014, 15, 975–985. [Google Scholar] [CrossRef]
- Shore, N.D.; Tammela, T.L.; Massard, C.; Bono, P.; Aspegren, J.; Mustonen, M.; Fizazi, K. Safety and Antitumour Activity of ODM-201 (BAY-1841788) in Chemotherapy-naïve and CYP17 Inhibitor-naïve Patients: Follow-up from the ARADES and ARAFOR Trials. Eur. Urol. Focus 2018, 4, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, M.; Zichi, C.; Buttigliero, C.; Vignani, F.; Scagliotti, G.V.; Di Maio, M. Enzalutamide-resistant castration-resistant prostate cancer: Challenges and solutions. OncoTargets Ther. 2018, 11, 7353–7368. [Google Scholar] [CrossRef] [Green Version]
- Prekovic, S.; Broeck, T.V.D.; Linder, S.; Van Royen, M.E.; Houtsmuller, A.B.; Handle, F.; Joniau, S.; Zwart, W.; Claessens, F. Molecular underpinnings of enzalutamide resistance. Endocr.-Relat. Cancer 2018, 25, R545–R557. [Google Scholar] [CrossRef]
- Claessens, F.; Helsen, C.; Prekovic, S.; Broeck, T.V.D.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef]
- Centenera, M.; Selth, L.A.; Ebrahimie, E.; Butler, L.M.; Tilley, W.D. New Opportunities for Targeting the Androgen Receptor in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Qiu, Y.; Xu, J. Current opinion and mechanistic interpretation of combination therapy for castration-resistant prostate cancer. Asian J. Androl. 2019, 21, 270–278. [Google Scholar] [CrossRef]
- Borgmann, H.; Lallous, N.; Ozistanbullu, D.; Beraldi, E.; Paul, N.; Dalal, K.; Fazli, L.; Haferkamp, A.; Lejeune, P.; Cherkasov, A.; et al. Moving Towards Precision Urologic Oncology: Targeting Enzalutamide-resistant Prostate Cancer and Mutated Forms of the Androgen Receptor Using the Novel Inhibitor Darolutamide (ODM-201). Eur. Urol. 2018, 73, 4–8. [Google Scholar] [CrossRef]
- Roell, D.; Rösler, T.W.; Hessenkemper, W.; Kraft, F.; Hauschild, M.; Bartsch, S.; Abraham, T.E.; Houtsmuller, A.B.; Matusch, R.; Van Royen, M.E.; et al. Halogen-substituted anthranilic acid derivatives provide a novel chemical platform for androgen receptor antagonists. J. Steroid Biochem. Mol. Biol. 2019, 188, 59–70. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.R.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. 2007, 104, 10418–10423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, S.; He, Y.; Hwang, D.-J.; Thiyagarajan, T.; Houtman, R.; Bocharova, V.; Sumpter, B.G.; Fernandez, E.; Johnson, D.L.; Du, Z.; et al. Orally Bioavailable Androgen Receptor Degrader, Potential Next-Generation Therapeutic for Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today: Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Zaidi, S.; Gandhi, J.; Joshi, G.; Smith, N.L.; Khan, S.A. The anticancer potential of metformin on prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 351–361. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, Z.; Ratnam, M.; Dou, Q.P. The interplay of AMP-activated protein kinase and androgen receptor in prostate cancer cells. J. Cell. Physiol. 2014, 229, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.A.H.; Albrecht, H.; Milne, R.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr.-Relat. Cancer 2016, 23. [Google Scholar] [CrossRef] [Green Version]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
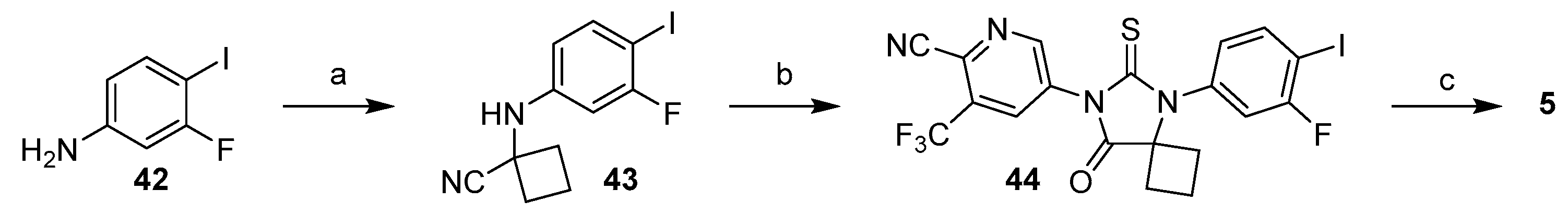{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand Name | Generic Name | Approval Date | Treatments | Category |
|---|---|---|---|---|
| Taxotere | Docetaxel in combination with prednisone | 19 May 2004 | mCRPC | Chemotherapy |
| Jevtana | Cabazitaxel in combination with prednisone | 17 June 2010 | mCRPC after docetaxel | Chemotherapy |
| Xofigo | radium-223 | 15 May 2013 | mCRPC | Radiotherapy |
| Provenge | Sipuleucel-T | 29 April 2010 | Asymptomatic or minimally symptomatic mCRPC | Immunotherapy |
| Zytiga | Abiraterone acetate in combination with prednisone | 28 April 2011 | mCRPC after docetaxel | Hormonal therapy |
| Zytiga | Abiraterone acetate in combination with prednisone | 10 December 2012 | mCRPC before chemotherapy | Hormonal therapy |
| Zytiga | Abiraterone acetate in combination with prednisone | 7 February 2018 | mCSPC | Hormonal therapy |
| Erleada | Apalutamide | 14 February 2018 | nmCRPC | Hormonal therapy |
| Erleada | Apalutamide | 17 September 2019 | mCSPC | Hormonal therapy |
| XTANDI | Enzalutamide | 31 August 2012 | mCRPC after docetaxel | Hormonal therapy |
| XTANDI | Enzalutamide | 13 July 2018 | nmCRPC | Hormonal therapy |
| NUBEQA | Darolutamide | 30 July 2019 | nmCRPC | Hormonal therapy |
| XTANDI | Enzalutamide | 16 December 2019 | mCSPC | Hormonal therapy |
| Brand Name | Generic Name | mCRPC | nmCRPC | mCSPC |
|---|---|---|---|---|
| XTANDI | enzalutamide | yes | yes | yes |
| Erleada | apalutamide | no | yes | yes |
| NUBEQA | darolutamide | no | yes | no |
| Enzalutamide AFFIRM Phase III Trial (NCT00974311) | |||
|---|---|---|---|
| End Points | Enzalutamide (n = 800) | Placebo (n = 399) | Hazard Ratio (95% CI) |
| Primary End Points | |||
| Median OS (mo) | 18.4 | 13.6 | 0.63 |
| Secondary End Points | |||
| Median time to rPFS (mo) | 8.3 | 2.9 | 0.4 |
| Median time to first SRE (mo) | 16.7 | 13.3 | 0.69 |
| Median time to PSA progression (mo) | 8.3 | 3 | 0.25 |
| * PSA response (%) of no. | |||
| decline ≥90% from baseline | 25 (731) | 1 (3300 | |
| decline ≥50% from baseline | 54 (731) | 1 (330) | |
| Serious AEs (%) | 39.9 | 38.8 | |
| Enzalutamide PREVAIL Phase III Trial (NCT01212991) | |||
|---|---|---|---|
| End Points | Enzalutamide (n = 872) | Placebo (n = 845) | Hazard Ratio (95% CI) |
| Primary end point | |||
| Median OS (mo) | 18.4 | 13.6 | 0.63 |
| Median time to rPFS (mo) | 3.9 | 0.19 | |
| Secondary end points | |||
| Median time to first SRE (mo) | 16.7 | 13.3 | 0.69 |
| Median CC initiation time (mo) | 28 | 10.8 | 0.35 |
| Median time to PSA progression (mo) | 11.2 | 2.8 | 0.17 |
| * PSA response (%) of no. | |||
| decline ≥ 90% from baseline | 47 (854) | 1 (777) | |
| decline ≥ 50% from baseline | 78 (854) | 3 (777) | |
| * Serious AEs (%) of no. | 44.1 (871) | 3.5 (844) | |
| Enzalutamide PROSPER Phase III Trial (NCT02003924) | |||
|---|---|---|---|
| End Points | Enzalutamide (n = 933) | Placebo (n = 468) | Hazard Ratio (95% CI) |
| Primary end point | |||
| Median MFS (mo) | 36.6 | 14.7 | 0.29 |
| Median time to rPFS (mo) | 3.9 | 0.19 | |
| Secondary end points | |||
| Median OS (mo) | |||
| Median time to C-FS (mo) | 38.1 | 34 | |
| Median first time use of CC (mo) | 39.7 | 0.38 | |
| Median time to PP (mo) | 18.5 | 18.4 | 0.96 |
| Median time to PSA progression (mo) | 37.2 | 3.9 | 0.07 |
| * PSA response (%) | |||
| decline ≥ 90% from baseline | 55.9 | 0.4 | |
| decline ≥ 50% from baseline | 76.3 | 2.4 | |
| decline to undetectable level | 9.6 | 0 | |
| * Serious AEs (%) of no. | 24.3 (930) | 18.9 (465) | |
| Enzalutamide ARCHES Phase III Trial (NCT02677896) | |||
|---|---|---|---|
| End Points | Enzalutamide + ADT (n = 574) | Placebo + ADT (n = 576) | Hazard Ratio (95% CI) |
| Primary End Point | |||
| ‡ Median time to rPFS based on ICR via PCWG2 (mo) | 19.4 | 0.39 | |
| † Median time to rPFS based on ICR via PAC (mo) | 19.0 | 0.39 | |
| Secondary End Points | |||
| Median OS (mo) | 0.19 | ||
| Time to NAT | 30.2 | 0.28 | |
| Median time to CR (mo) | 13.9 | 0.28 | |
| PSA undetctable rate (%) of no. | 68.1 (511) | 17.6 (506) | |
| * Serious AEs (%) of no. | 18.2 (572) | 19.5 (574) | |
| Apalutamide SPARTAN Phase III Trial (NCT01946204) | |||
|---|---|---|---|
| End Points | Apalutamide (n = 806) | Placebo (n = 401) | Hazard Ratio (95% CI) |
| Primary End Points | |||
| Median MFS (mo) | 40.5 | 16.2 | 0.28 |
| Secondary End Points | |||
| Median time to metastasis (mo) | 40.5 | 16.6 | 0.28 |
| Median time to PFS (mo) | 40.5 | 14.7 | 0.29 |
| Median OS (mo) | 39.0 | ||
| Median time to CC (mo) | |||
| * Serious AEs (%) of no. | 24.8 (803) | 23.1 (398) | |
| Apalutamide TITAN Phase III Trial (NCT02489318) | |||
|---|---|---|---|
| End Points | Apalutamide + ADT (n = 525) | Placebo + ADT (n = 527) | Hazard Ratio (95% CI) |
| Primary End Points | |||
| OS (% alive) | 82..4 | 73.5 | 0.67 |
| rPFS (%) | 68.2 | 47.5 | 0.48 |
| Secondary End Points | |||
| Median time to PSA progression (mo) | 12.9 | 0.26 | |
| * Serious AEs (%) of no. | 19.2 (574) | 20.3 (527) | |
| Darolutamide ARMIS Phase III Trial (NCT02200614) | |||
|---|---|---|---|
| End Points | Darolutamide (n = 955) | Placebo (n = 554) | Hazard Ratio (95% CI) |
| Primary End Point | |||
| Median MFS (mo) | 40.4 | 18.4 | 0.41 |
| Secondary End Points | |||
| Median OS (mo) | 0.71 | ||
| Median first time use of CC (mo) | 38.2 | 0.43 | |
| Median time to PP (mo) | 40.3 | 25.36 | 0.65 |
| Median time to SSE (mo) | 0.43 | ||
| * Serious AEs (%) of no. | 24.8 (954) | 10.5 (554) | |
| Trial Name | AR Antagonist | Primary End Point | Hazard Ratio (95% CI) | Patients |
|---|---|---|---|---|
| AFFIRM | enzalutamide | Median overall survival | 0.63 | mCRPC |
| PROPSER | enzalutamide | Median metastasis-free survival | 0.29 | nmCRPC |
| SPARTAN | apalutamide | Median metastasis-free survival | 0.28 | nmCRPC |
| ARAMIS | darolutamide | Median metastasis-free survival | 0.41 | nmCRPC |
| ARCHES | enzalutamide | Median rPFS | 0.31 | mCSPC |
| TITAN | apalutamide | Median rPFS | 0.48 | mCSPC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajaram, P.; Rivera, A.; Muthima, K.; Olveda, N.; Muchalski, H.; Chen, Q.-H. Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer. Molecules 2020, 25, 2448. https://doi.org/10.3390/molecules25102448
Rajaram P, Rivera A, Muthima K, Olveda N, Muchalski H, Chen Q-H. Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer. Molecules. 2020; 25(10):2448. https://doi.org/10.3390/molecules25102448
Chicago/Turabian StyleRajaram, Pravien, Alyssa Rivera, Kevin Muthima, Nicholas Olveda, Hubert Muchalski, and Qiao-Hong Chen. 2020. "Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer" Molecules 25, no. 10: 2448. https://doi.org/10.3390/molecules25102448
APA StyleRajaram, P., Rivera, A., Muthima, K., Olveda, N., Muchalski, H., & Chen, Q.-H. (2020). Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer. Molecules, 25(10), 2448. https://doi.org/10.3390/molecules25102448