
Resolving DNA Damage: Epigenetic Regulation of DNA Repair



Abstract
:1. Introduction
2. Types of DNA Damage and DNA Damage Response
2.1. Sources of DNA Damage
2.2. DNA Repair Is a Multi-Step Process
2.3. DDR Initiation Requires Recruitment of Sensor Proteins to Damaged Chromatin
3. Chromatin Regulation during DNA Repair
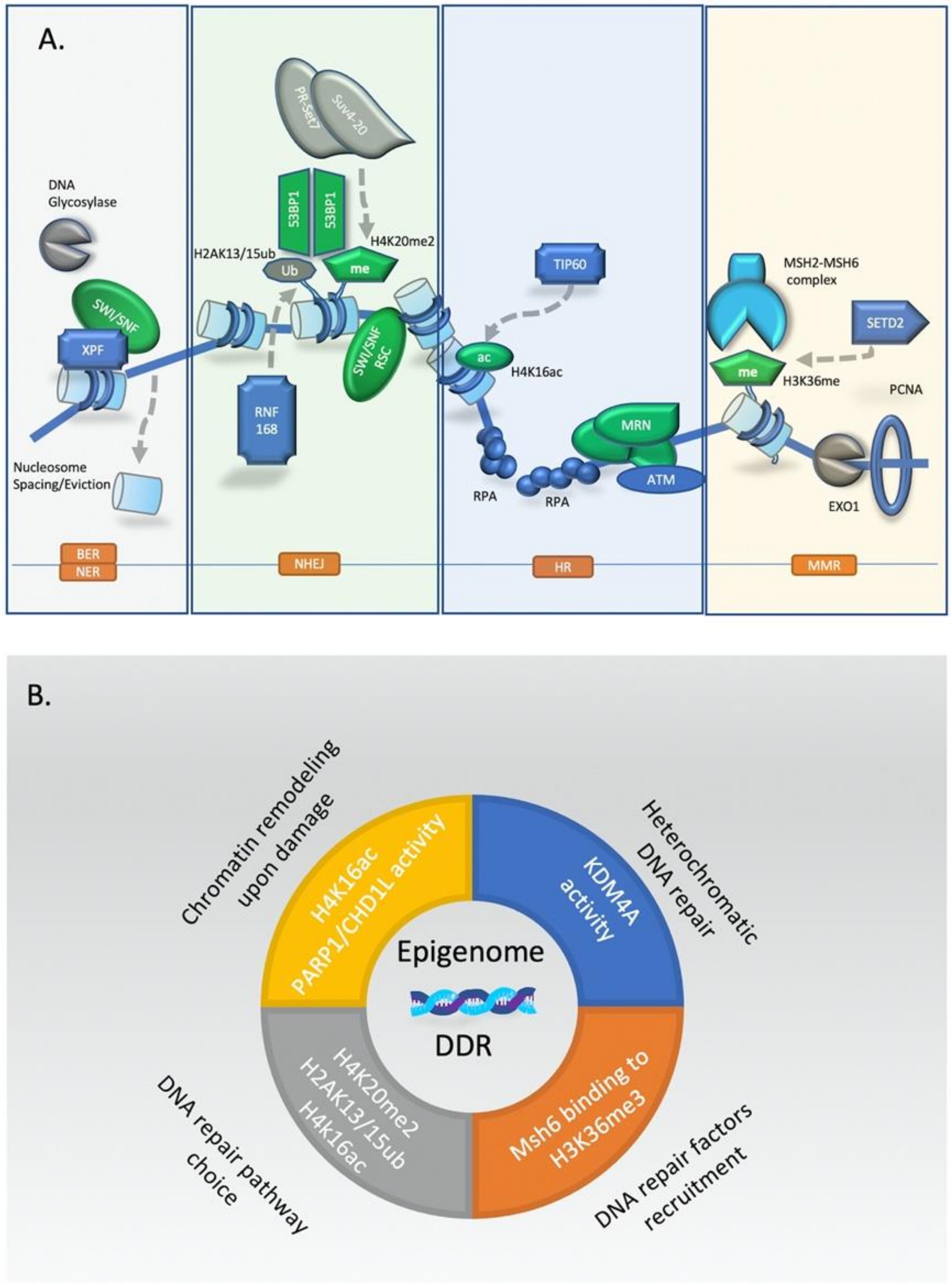
3.1. Epigenetic Driven Chromatin Remodeling Is One of the Earliest Responses to DNA Damage
3.2. Regulation of Chromatin During Repair of DSBs in Heterochromatin
3.3. Epigenetic Factors and DNA Repair Choice between HR and NHEJ
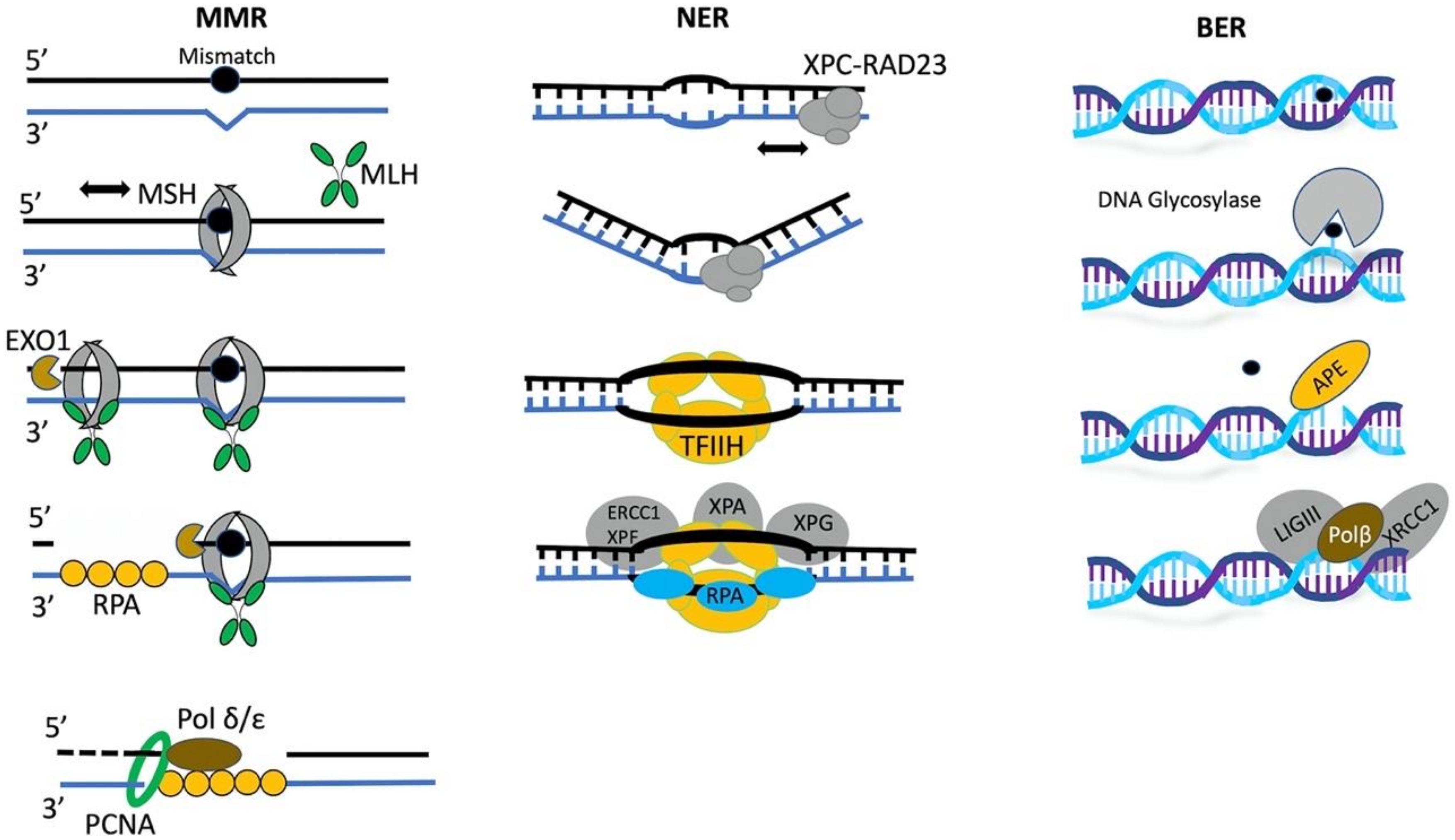
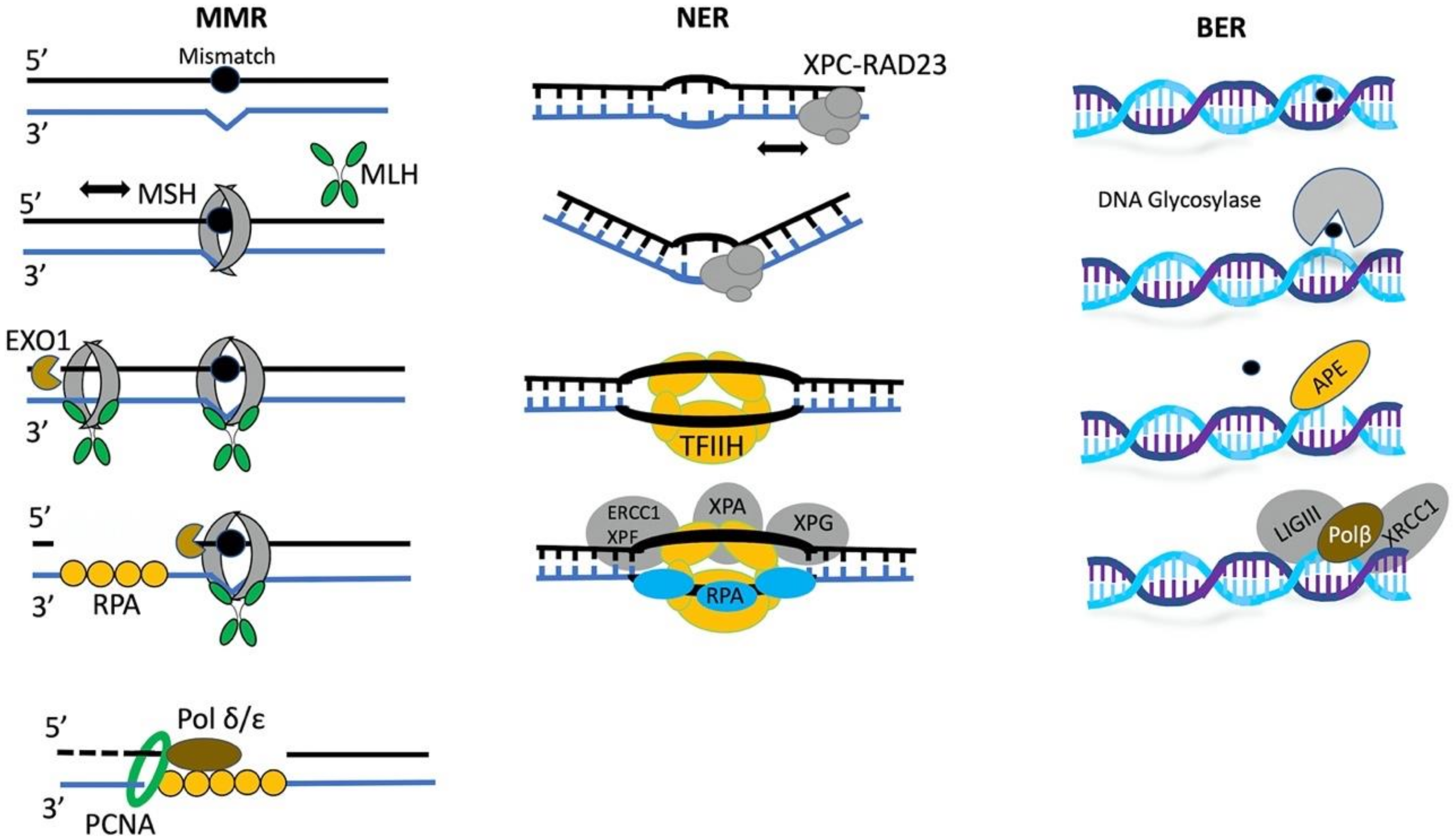
3.4. Epigenetic Regulation of Mismatch Repair
3.5. The Role of Chromatin Remodelers in BER and NER
4. Epigenetic Regulation of DNA Repair Factors Though Transcription
4.1. Epigenetic Regulation of DNA Repair Component Expression
4.2. Epi-miRs on the Epigenetic Regulation of DDR Components Transcription
5. Nucleosome-Independent Regulation of DDR by Epigenetic Factors
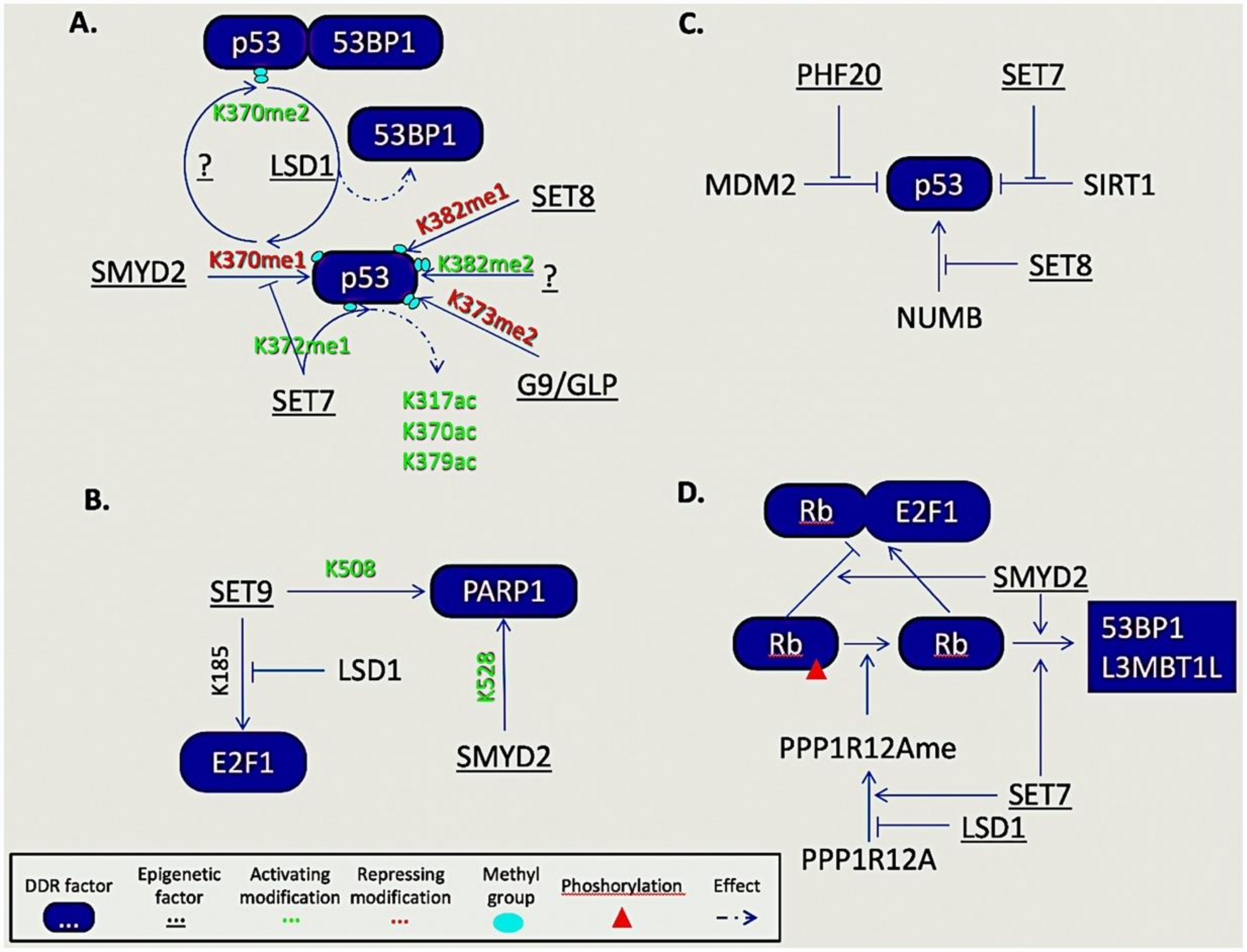
5.1. Non-Histone Lysine Methylation
5.2. Non-Histone Arginine Methylation
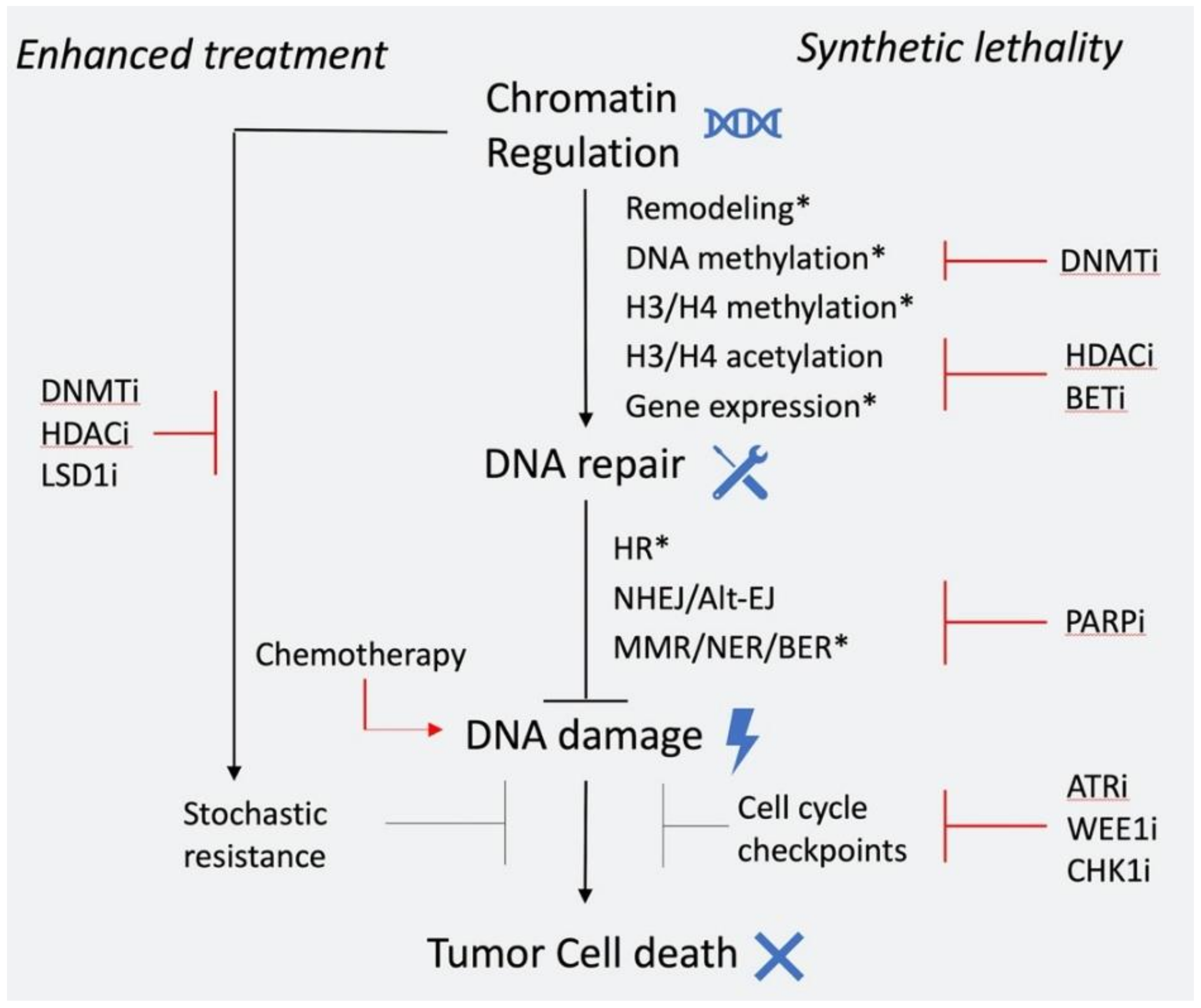
6. Epigenetic Chromatin Regulation and DNA Repair: Synthetic Lethal Interactions and Clinical Applications
6.1. Epigenetic Inhibitors in Combination with Chemotherapy/Radiotherapy
6.2. Epigenetic Inhibitors in Combination with Drugs that Target DNA Repair Components
6.3. Targeting DNA Repair in Tumors with Epigenetic Alterations
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershkovitz, M.; Riggs, A.D. Metaphase chromosome analysis by ligation-mediated PCR: Heritable chromatin structure and a comparison of active and inactive X chromosomes. Proc. Natl. Acad. Sci. USA 1995, 92, 2379–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R. Epigenetics and memigenetics. Cell. Mol. Life. Sci. 2014, 71, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Peterson, C.L. ATP-dependent chromatin remodeling. Curr. Top. Dev. Biol. 2005, 65, 115–148. [Google Scholar]
- Henikoff, S.; Smith, M.M. Histone variants and epigenetics. Cold. Spring. Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Gonzalez-Ramirez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Patel, D.R.; Weiss, R.S. A tough row to hoe: When replication forks encounter DNA damage. Biochem. Soc. Trans. 2018, 46, 1643–1651. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Huang, Y.; Li, L. DNA crosslinking damage and cancer—A tale of friend and foe. Transl. Cancer Res. 2013, 2, 144–154. [Google Scholar]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Diffley, J.F. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef] [Green Version]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordelet, H.; Dubrana, K. Keep moving and stay in a good shape to find your homologous recombination partner. Curr. Genet. 2019, 65, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef]
- Uematsu, N.; Weterings, E.; Yano, K.; Morotomi-Yano, K.; Jakob, B.; Taucher-Scholz, G.; Mari, P.O.; van Gent, D.C.; Chen, B.P.; Chen, D.J. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 2007, 177, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003, 5, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem. 2010, 285, 1097–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000, 407, 703–710. [Google Scholar] [CrossRef]
- Dip, R.; Camenisch, U.; Naegeli, H. Mechanisms of DNA damage recognition and strand discrimination in human nucleotide excision repair. DNA Repair. 2004, 3, 1409–1423. [Google Scholar] [CrossRef]
- Buterin, T.; Meyer, C.; Giese, B.; Naegeli, H. DNA quality control by conformational readout on the undamaged strand of the double helix. Chem. Biol. 2005, 12, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Min, J.H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef]
- Crenshaw, C.M.; Wade, J.E.; Arthanari, H.; Frueh, D.; Lane, B.F.; Nunez, M.E. Hidden in plain sight: Subtle effects of the 8-oxoguanine lesion on the structure, dynamics, and thermodynamics of a 15-base pair oligodeoxynucleotide duplex. Biochemistry 2011, 50, 8463–8477. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Smerdon, M.J.; Tlsty, T.D.; Lieberman, M.W. Distribution of ultraviolet-induced DNA repair synthesis in nuclease sensitive and resistant regions of human chromatin. Biochemistry 1978, 17, 2377–2386. [Google Scholar] [CrossRef]
- Jaberaboansari, A.; Nelson, G.B.; Roti Roti, J.L.; Wheeler, K.T. Postirradiation alterations of neuronal chromatin structure. Radiat. Res. 1988, 114, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Sidik, K.; Smerdon, M.J. Nucleosome rearrangement in human cells following short patch repair of DNA damaged by bleomycin. Biochemistry 1990, 29, 7501–7511. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Arnould, C.; Legube, G. The Secret Life of Chromosome Loops upon DNA Double-Strand Break. J. Mol. Biol. 2020, 432, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Hittelman, W.N.; Pollard, M. Visualization of chromatin events associated with repair of ultraviolet light-induced damage by premature chromosome condensation. Carcinogenesis 1984, 5, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. Pretreatment with UV light renders the chromatin in human fibroblasts more susceptible to the DNA-damaging agents bleomycin, gamma radiation and 8-methoxypsoralen. Carcinogenesis 1989, 10, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Strickfaden, H.; McDonald, D.; Kruhlak, M.J.; Haince, J.F.; Th’ng, J.P.; Rouleau, M.; Ishibashi, T.; Corry, G.N.; Ausio, J.; Underhill, D.A.; et al. Poly(ADP-ribosyl)ation-dependent Transient Chromatin Decondensation and Histone Displacement following Laser Microirradiation. J. Biol. Chem. 2016, 291, 1789–1802. [Google Scholar] [CrossRef] [Green Version]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Muller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell. Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell. 2016, 27, 3791–3799. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Sellou, H.; Chapuis, C.; Huet, S.; Timinszky, G. CHD3 and CHD4 recruitment and chromatin remodeling activity at DNA breaks is promoted by early poly(ADP-ribose)-dependent chromatin relaxation. Nucleic. Acids Res. 2018, 46, 6087–6098. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, G.; Wiegant, W.W.; Marteijn, J.A.; Luijsterburg, M.S.; Sroczynski, N.; Costelloe, T.; Romeijn, R.J.; Pastink, A.; Mailand, N.; Vermeulen, W.; et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell. Sci. 2013, 126, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell. 2016, 61, 547–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q.; Harmon, K.E.; Megee, P.C.; Grant, P.A.; Smith, M.M.; Christman, M.F. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Erler, J.; Langowski, J. Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J. 2017, 112, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gursoy-Yuzugullu, O.; Parasuram, R.; Price, B.D. The tale of a tail: Histone H4 acetylation and the repair of DNA breaks. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160284. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenares, S.U.; Swenson, J.M.; Langley, S.A.; Kennedy, C.; Costes, S.V.; Karpen, G.H. Drosophila Histone Demethylase KDM4A Has Enzymatic and Non-enzymatic Roles in Controlling Heterochromatin Integrity. Dev. Cell 2017, 42, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Van Attikum, H.; Fritsch, O.; Gasser, S.M. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007, 26, 4113–4125. [Google Scholar] [CrossRef] [Green Version]
- Lademann, C.A.; Renkawitz, J.; Pfander, B.; Jentsch, S. The INO80 Complex Removes H2A.Z to Promote Presynaptic Filament Formation during Homologous Recombination. Cell Rep. 2017, 19, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Murr, R.; Loizou, J.; Yang, Y.-G.; Cuenin, C.; Li, H.; Wang, Z.-Q.; Herceg, Z. Histone acetylation by Trrap–Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature 2005, 8, 91–99. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.-P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.L.; Portoso, M.; Mata, J.; Bähler, J.; Allshire, R.C.; Kouzarides, T. Methylation of Histone H4 Lysine 20 Controls Recruitment of Crb2 to Sites of DNA Damage. Cell 2004, 119, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forné, I.; Reverón-Gómez, N.; Foster, B.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzon, C.; Spektor, T.; Kong, X.; Congdon, L.M.; Wu, S.; Schotta, G.; Yokomori, K.; Rice, J.C. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014, 8, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, J.; Li, G.-M.; Longley, M.; Modrich, P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268, 1909–1912. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P.L. Endonucleolytic Function of MutLα in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-M. Decoding the histone code: Role of H3K36me3 in mismatch repair and implications for cancer susceptibility and therapy. Cancer Res. 2013, 73, 6379–6383. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Gu, L.; Li, G.-M. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J. Boil. Chem. 2018, 293, 7811–7823. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Terui, R.; Nagao, K.; Kawasoe, Y.; Taki, K.; Higashi, T.L.; Tanaka, S.; Nakagawa, T.; Obuse, C.; Masukata, H.; Takahashi, T. Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genome Res. 2018, 32, 806–821. [Google Scholar] [CrossRef] [Green Version]
- Hause, R.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Brooks, S.C.; Adhikary, S.; Rubinson, E.H.; Eichman, B.F. Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta BBA Proteins Proteom. 2013, 1834, 247–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA Repair in Mammalian Cells. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The FEN-1 family of structure-specific nucleases in eukaryotic dna replication, recombination and repair. BioEssays 1997, 19, 233–240. [Google Scholar] [CrossRef]
- Li, C.; Delaney, S. Challenges for base excision repair enzymes: Acquiring access to damaged DNA in chromatin. Pept. Cancer Derived Enzyme Product. 2019, 45, 27–57. [Google Scholar] [CrossRef]
- Menoni, H.; Gasparutto, D.; Hamiche, A.; Cadet, J.; Dimitrov, S.; Bouvet, P.; Angelov, D. ATP-Dependent Chromatin Remodeling Is Required for Base Excision Repair in Conventional but Not in Variant H2A.Bbd Nucleosomes. Mol. Cell. Boil. 2007, 27, 5949–5956. [Google Scholar] [CrossRef] [Green Version]
- Menoni, H.; Shukla, M.S.; Gerson, V.; Dimitrov, S.; Angelov, D. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res. 2011, 40, 692–700. [Google Scholar] [CrossRef]
- Nakanishi, S.; Prasad, R.; Wilson, S.H.; Smerdon, M.J. Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Res. 2007, 35, 4313–4321. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Boil. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Gaillard, H.; Fitzgerald, D.J.; Smith, C.L.; Peterson, C.L.; Richmond, T.J.; Thoma, F. Chromatin Remodeling Activities Act on UV-damaged Nucleosomes and Modulate DNA Damage Accessibility to Photolyase. J. Boil. Chem. 2003, 278, 17655–17663. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Sancar, A. The SWI/SNF Chromatin-Remodeling Factor Stimulates Repair by Human Excision Nuclease in the Mononucleosome Core Particle. Mol. Cell. Boil. 2002, 22, 6779–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Wang, Q.-E.; Ray, A.; Wani, G.; Han, C.; Milum, K.; Wani, A.A. Modulation of Nucleotide Excision Repair by Mammalian SWI/SNF Chromatin-remodeling Complex. J. Boil. Chem. 2009, 284, 30424–30432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothandapani, A.; Gopalakrishnan, K.; Kahali, B.; Reisman, D.; Patrick, S.M. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp. Cell Res. 2012, 318, 1973–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banin, S.; Moyal, L.; Shieh, S.-Y.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced Phosphorylation of p53 by ATM in Response to DNA Damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Shieh, S.-Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genome Res. 2000, 14, 289–300. [Google Scholar]
- Adimoolam, S.; Ford, J.M. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. USA 2002, 99, 12985–12990. [Google Scholar] [CrossRef] [Green Version]
- Myung, K.; Braastad, C.; He, D.M.; Hendrickson, E.A. KARP-1 is induced by DNA damage in a p53- and ataxia telangiectasia mutated-dependent fashion. Proc. Natl. Acad. Sci. USA 1998, 95, 7664–7669. [Google Scholar] [CrossRef] [Green Version]
- Poletto, M.; Legrand, A.; Fletcher, S.C.; Dianov, G.L. p53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Scherer, S.J.; Maier, S.M.; Seifert, M.; Hanselmann, R.G.; Zang, K.D.; Muller-Hermelink, H.K.; Angel, P.; Welter, C.; Schartl, M. p53 and c-Jun functionally synergize in the regulation of the DNA repair gene hMSH2 in response to UV. J. Biol. Chem. 2000, 275, 37469–37473. [Google Scholar] [CrossRef] [Green Version]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genome Res. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [Green Version]
- Hartman, A.-R.; Ford, J.M. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat. Genet. 2002, 32, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.B.; E Quinn, J.; Harkin, D.P. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habraken, Y.; Piette, J. NF-κB activation by double-strand breaks. Biochem. Pharmacol. 2006, 72, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Lavon, I.; Fuchs, D.; Zrihan, D.; Efroni, G.; Zelikovitch, B.; Fellig, Y.; Siegal, T. Novel Mechanism whereby Nuclear Factor B Mediates DNA Damage Repair through Regulation of O6-Methylguanine-DNA-Methyltransferase. Cancer Res. 2007, 67, 8952–8959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Jiang, S.W.; Thangaraju, M.; Wu, G.; Couch, F.J. Induction of the BRCA2 promoter by nuclear factor-kappa B. J. Biol. Chem. 2000, 275, 35548–35556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yang, J.; Zhu, F.; Xu, F. Response of REV3 promoter to N-methyl-N′-nitro-N-nitrosoguanidine. Mutat. Res. Mol. Mech. Mutagen. 2004, 550, 49–58. [Google Scholar] [CrossRef]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019, 234, 14951–14965. [Google Scholar] [CrossRef]
- Boldogh, I.; Ramana, C.V.; Chen, Z.; Biswas, T.; Hazra, T.K.; Grösch, S.; Grombacher, T.; Mitra, S.; Kaina, B. Regulation of expression of the DNA repair gene O6-methylguanine-DNA methyltransferase via protein kinase C-mediated signaling. Cancer Res. 1998, 58, 3950–3956. [Google Scholar]
- Chang, H.W.; Lai, Y.C.; Cheng, C.Y.; Ho, J.L.; Ding, S.T.; Liu, Y.C. UV inducibility of rat proliferating cell nuclear antigen gene promoter. J. Cell. Biochem. 1999, 73, 423–432. [Google Scholar] [CrossRef]
- Das, A.; Hazra, T.K.; Boldogh, I.; Mitra, S.; Bhakat, K.K. Induction of the Human Oxidized Base-specific DNA Glycosylase NEIL1 by Reactive Oxygen Species. J. Boil. Chem. 2005, 280, 35272–35280. [Google Scholar] [CrossRef] [Green Version]
- Humbert, O.; Achour, I.; Lautier, D.; Laurent, G.; Salles, B. hMSH2 expression is driven by AP1-dependent regulation through phorbol-ester exposure. Nucleic Acids Res. 2003, 31, 5627–5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, C.-K.; Kim, M.-H.; Cho, H.-J.; Chang, I.-Y.; Chung, M.-H.; You, H.J. Oncogenic H-Ras Up-Regulates Expression of ERCC1 to Protect Cells from Platinum-Based Anticancer Agents. Cancer Res. 2004, 64, 4849–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakura, Y.; Sugano, K.; Konishi, F.; Ichikawa, A.; Maekawa, M.; Shitoh, K.; Igarashi, S.; Kotake, K.; Koyama, Y.; Nagai, H. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology 2001, 121, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.F. Pathology of Endometrial Carcinoma. Adv. Exp. Med. Biol. 2016, 943, 75–96. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lu, Y.P.; Tseng, R.C.; Lin, R.K.; Chang, J.W.; Chen, J.T.; Shih, C.M.; Chen, C.Y. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J. Clin. Investig. 2003, 111, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Demokan, S.; Suoglu, Y.; Demir, D.; Gozeler, M.; Dalay, N. Microsatellite instability and methylation of the DNA mismatch repair genes in head and neck cancer. Ann. Oncol. 2006, 17, 995–999. [Google Scholar] [CrossRef]
- Furihata, M.; Takeuchi, T.; Ohtsuki, Y.; Terao, N.; Kuwahara, M.; Shuin, T. Genetic analysis of hMLH1 in transitional cell carcinoma of the urinary tract: Promoter methylation or mutation. J. Urol. 2001, 165, 1760–1764. [Google Scholar] [CrossRef]
- Liu, Q.; Thoms, J.A.; Nunez, A.C.; Huang, Y.; Knezevic, K.; Packham, D.; Poulos, R.C.; Williams, R.L.; Beck, D.; Hawkins, N.; et al. Disruption of a −35 kb Enhancer Impairs CTCF Binding and MLH1 Expression in Colorectal Cells. Clin. Cancer Res. 2018, 24, 4602–4611. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Alsop, K.; Etemadmoghadam, D.; Hondow, H.; Mikeska, T.; Dobrovic, A.; DeFazio, A.; Smyth, G.K.; Levine, U.A.; Mitchell, G.; et al. Nonequivalent Gene Expression and Copy Number Alterations in High-Grade Serous Ovarian Cancers with BRCA1 and BRCA2 Mutations. Clin. Cancer Res. 2013, 19, 3474–3484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5, 17869. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Y.; Shao, C.-J.; Chen, F.-R.; Kwan, A.-L.; Chen, Z.-P. Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas. Int. J. Cancer 2009, 126, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Karagiannis, D.; Avgeris, M.; Polyzos, A.; Kokkalis, A.; Kanaki, Z.; Kousidou, E.; Tzetis, M.; Kanavakis, E.; Stravodimos, K.; et al. The lysine-specific methyltransferase KMT 2C/MLL 3 regulates DNA repair components in cancer. EMBO Rep. 2019, 20, e46821. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.; Varambally, S.; Cao, Q.; Chinnaiyan, A.M.; Ferguson, D.O.; Merajver, S.D.; Kleer, C.G. The Polycomb Group Protein EZH2 Impairs DNA Repair in Breast Epithelial Cells. Neoplasia 2005, 7, 1011–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.; Farh, K.K.-H.; Burge, C.B.; Bartel, B. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Dai, E.; Yu, X.; Zhang, Y.; Meng, F.; Wang, S.; Liu, X.; Liu, D.; Wang, J.; Li, X.; Jiang, W. EpimiR: A database of curated mutual regulation between miRNAs and epigenetic modifications. Database 2014, 2014, bau023. [Google Scholar] [CrossRef] [Green Version]
- Reale†, E.; Taverna†, D.; Cantini, L.; Martignetti, L.; Osella, M.; De Pittà, C.; Virga, F.; Orso, F.; Caselle‡, M. Investigating the epi-miRNome: Identification of epi-miRNAs using transfection experiments. Epigenomics 2019, 11, 1581–1599. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics 2013, 5, 671–683. [Google Scholar] [CrossRef]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [Green Version]
- Ugalde, A.P.; Ramsay, A.J.; De La Rosa, J.; Varela, I.; Mariño, G.; Cadiñanos, J.; Lu, J.; Freije, J.M.P.; Lopez-Otin, C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011, 30, 2219–2232. [Google Scholar] [CrossRef]
- Lyu, G.; Guan, Y.; Zhang, C.; Zong, L.; Sun, L.; Huang, X.; Huang, L.; Zhang, L.; Tian, X.L.; Zhou, Z.; et al. TGF-beta signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018, 9, 2560. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Zemp, F.; Kolb, B.E.; Kovalchuk, O. Sex-specific radiation-induced microRNAome responses in the hippocampus, cerebellum and frontal cortex in a mouse model. Mutat. Res. Toxicol. Environ. Mutagen. 2011, 722, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Spaety, M.E.; Gries, A.; Badie, A.; Venkatasamy, A.; Romain, B.; Orvain, C.; Yanagihara, K.; Okamoto, K.; Jung, A.C.; Mellitzer, G.; et al. HDAC4 Levels Control Sensibility toward Cisplatin in Gastric Cancer via the p53-p73/BIK Pathway. Cancers 2019, 11, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duru, N.; Gernapudi, R.; Zhang, Y.; Yao, Y.; Lo, P.K.; Wolfson, B.; Zhou, Q. NRF2/miR-140 signaling confers radioprotection to human lung fibroblasts. Cancer Lett. 2015, 369, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Liu, J.; Zhu, Y.; Yuan, X.; Gao, J.; Cheng, J.; Yan, X. Diesel exhaust particles induce toxicity to beta cells by suppressing miR-140–5p. Int. J. Clin. Exp. Pathol. 2019, 12, 2858–2866. [Google Scholar]
- Mueller, A.C.; Sun, D.; Dutta, A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene 2012, 32, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Biggar, K.K.; Li, S.S.-C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Boil. 2014, 16, 5–17. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Huang, J.; Berger, S. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 2008, 18, 152–158. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 Function by SET8-Mediated Methylation at Lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Dorsey, J.; Chuikov, S.; Perez-Burgos, L.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Sengupta, R.; Espejo, A.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurash, J.K.; Lei, H.; Shen, Q.; Marston, W.L.; Granda, B.W.; Fan, H.; Wall, D.; Li, E.; Gaudet, F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 2008, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Kontaki, H.; Talianidis, I. Lysine Methylation Regulates E2F1-Induced Cell Death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R.; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the Retinoblastoma Tumor Suppressor by SMYD2. J. Boil. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 Methylation by SMYD2 Enhances Cell Cycle Progression through an Increase of RB1 Phosphorylation. Neoplasia 2012, 14, 476-IN8. [Google Scholar] [CrossRef] [Green Version]
- Thandapani, P.; Couturier, A.; Yu, Z.; Li, X.; Couture, J.-F.; Li, S.; Masson, J.-Y.; Richard, S. Lysine methylation of FEN1 by SET7 is essential for its cellular response to replicative stress. Oncotarget 2017, 8, 64918–64931. [Google Scholar] [CrossRef] [Green Version]
- Hahm, J.Y.; Kim, J.Y.; Park, J.W.; Kang, J.Y.; Kim, K.B.; Kim, S.R.; Cho, H.; Seo, S.B. Methylation of UHRF1 by SET7 is essential for DNA double-strand break repair. Nucleic Acids Res. 2019, 47, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassner, I.; Andersson, A.; Fey, M.; Tomas, M.; Ferrando-May, E.; Hottiger, M.O. SET7/9-dependent methylation of ARTD1 at K508 stimulates poly-ADP-ribose formation after oxidative stress. Open Biol. 2013, 3, 120173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, L.; Kang, D.; Suzuki, T.; Masuda, A.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia 2014, 16, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Galka, M.; Mori, E.; Liu, X.; Lin, Y.F.; Wei, R.; Pittock, P.; Voss, C.; Dhami, G.; Li, X.; et al. A method for systematic mapping of protein lysine methylation identifies functions for HP1beta in DNA damage response. Mol. Cell 2013, 50, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhou, J.; Liu, X.; Lu, D.; Shen, C.; Du, Y.; Wei, F.Z.; Song, B.; Lu, X.; Yu, Y.; et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc. Natl. Acad. Sci. USA 2013, 110, 5516–5521. [Google Scholar] [CrossRef] [Green Version]
- Karakaidos, P.; Verigos, J.; Magklara, A. LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers 2019, 11, 1821. [Google Scholar] [CrossRef] [Green Version]
- Alsulami, M.; Munawar, N.; Dillon, E.; Oliviero, G.; Wynne, K.; Alsolami, M.; Moss, C.; O’Gaora, P.; O’Meara, F.; Cotter, D.; et al. SETD1A Methyltransferase Is Physically and Functionally Linked to the DNA Damage Repair Protein RAD18. Mol. Cell Proteom. 2019, 18, 1428–1436. [Google Scholar] [CrossRef]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Hoshii, T.; Cifani, P.; Feng, Z.; Huang, C.H.; Koche, R.; Chen, C.W.; Delaney, C.D.; Lowe, S.W.; Kentsis, A.; Armstrong, S.A. A Non-catalytic Function of SETD1A Regulates Cyclin K and the DNA Damage Response. Cell 2018, 172, 1007–1021. [Google Scholar] [CrossRef] [Green Version]
- Dery, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.Y. A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol. Cell Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, F.M.; Dery, U.; Masson, J.Y.; Richard, S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 2005, 19, 671–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hebert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Chen, T.; Hebert, J.; Li, E.; Richard, S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell Biol. 2009, 29, 2982–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, F.M.; Rhie, A.; Richard, S.; Doherty, A.J. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 2005, 4, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.M.; Wang, B.; Xia, Z.; Morales, J.C.; Lu, X.; Donehower, L.A.; Bochar, D.A.; Elledge, S.J.; Carpenter, P.B. 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle 2005, 4, 1854–1861. [Google Scholar] [CrossRef] [Green Version]
- Guendel, I.; Carpio, L.; Pedati, C.; Schwartz, A.; Teal, C.; Kashanchi, F.; Kehn-Hall, K. Methylation of the tumor suppressor protein, BRCA1, influences its transcriptional cofactor function. PLoS ONE 2010, 5, e11379. [Google Scholar] [CrossRef] [Green Version]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Hassa, P.O.; Gehrig, P.; Covic, M.; Hubscher, U.; Hottiger, M.O. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J. 2007, 21, 26–34. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Steinacher, R.; Focke, F.; Gehrig, P.; Covic, M.; Hassa, P.O.; Schar, P.; Hubscher, U.; et al. Arginine methylation regulates DNA polymerase beta. Mol. Cell 2006, 22, 51–62. [Google Scholar] [CrossRef]
- Hu, D.; Gur, M.; Zhou, Z.; Gamper, A.; Hung, M.C.; Fujita, N.; Lan, L.; Bahar, I.; Wan, Y. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 2015, 6, 8419. [Google Scholar] [CrossRef] [Green Version]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef]
- Scoumanne, A.; Zhang, J.; Chen, X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009, 37, 4965–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zheng, L.; Xu, H.; Dai, H.; Zhou, M.; Pascua, M.R.; Chen, Q.M.; Shen, B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 2010, 6, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Li, X.; Frank, G.; Shen, B. Cell cycle-dependent and DNA damage-inducible nuclear localization of FEN-1 nuclease is consistent with its dual functions in DNA replication and repair. J. Biol. Chem. 2001, 276, 4901–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Zhou, M.; Chai, Q.; Parrish, J.; Xue, D.; Patrick, S.M.; Turchi, J.J.; Yannone, S.M.; Chen, D.; Shen, B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005, 6, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesche, J.; Kuhn, S.; Kessler, B.M.; Salton, M.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell Mol. Life Sci. 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Qiu, T.; Zhou, L.; Zhu, W.; Wang, T.; Wang, J.; Shu, Y.; Liu, P. Effects of treatment with histone deacetylase inhibitors in solid tumors: A review based on 30 clinical trials. Future Oncol. 2013, 9, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Morel, D.; Almouzni, G.; Soria, J.C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017, 28, 254–269. [Google Scholar] [CrossRef]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell Mol. Life Sci. 2020, 77, 677–703. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, M.; Gallardo-Rincon, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J.L.; Arrieta, O.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; de la Cruz-Hernandez, E.; Camargo, M.F.; et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 2007, 18, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Zheng, Y.; Wang, J.Z.; Lu, X.X.; Wang, Z.; Chen, L.B.; Guan, X.X.; Tong, J.D. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics 2014, 9, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/KDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [Green Version]
- Glasspool, R.M.; Brown, R.; Gore, M.E.; Rustin, G.J.; McNeish, I.A.; Wilson, R.H.; Pledge, S.; Paul, J.; Mackean, M.; Hall, G.D.; et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer. 2014, 110, 1923–1929. [Google Scholar] [CrossRef]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camphausen, K.; Tofilon, P.J. Inhibition of histone deacetylation: A strategy for tumor radiosensitization. J. Clin. Oncol. 2007, 25, 4051–4056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Carr, T.; Dimtchev, A.; Zaer, N.; Dritschilo, A.; Jung, M. Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat. Res. 2007, 168, 115–124. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Mei, H.; Yin, Z.; Geng, Y.; Zhang, T.; Wu, G.; Lin, Z. The BET bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett. 2017, 391, 141–151. [Google Scholar] [CrossRef]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Yu, C.H.; Zhang, Y.; Yu, J.; Li, J.; Zhang, W.; Zhang, B.; Li, Y.; Guo, N. EZH2 silencing with RNAi enhances irradiation-induced inhibition of human lung cancer growth in vitro and in vivo. Oncol. Lett. 2012, 4, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, Y.; Tan, L.; Fu, X. The pivotal role of DNA methylation in the radio-sensitivity of tumor radiotherapy. Cancer Med. 2018, 7, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Wilson, A.J.; Saskowski, J.; Wass, E.; Khabele, D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 2014, 133, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [Green Version]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell. 2018, 33, 401–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell. 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Kim, G.W.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020, 34, 3461–3484. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.A.; Liu, X.Y.; Sturm, D.; Korshunov, A.; Jones, D.T.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.H.; Kapur, P.; Joseph, R.W.; Serie, D.J.; Eckel-Passow, J.E.; Tong, P.; Wang, J.; Castle, E.P.; Stanton, M.L.; Cheville, J.C.; et al. Loss of histone H3 lysine 36 trimethylation is associated with an increased risk of renal cell carcinoma-specific death. Mod. Pathol. 2016, 29, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, Y.; Jia, J.; Fang, Y.; Tang, Y.; Wu, H.; Fang, D. H3K36me3, message from chromatin to DNA damage repair. Cell Biosci. 2020, 10, 9. [Google Scholar] [CrossRef]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.C.; Zalmas, L.P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell. 2015, 28, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, M.; Saito, M.; Min, A.K.T.; Ujiie, D.; Saito, K.; Sato, T.; Kikuchi, T.; Okayama, H.; Fujita, S.; Endo, H.; et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci. Rep. 2019, 9, 6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih Ie, M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR Protein | Methylated Residue | Epigenetic Enzyme | Effect | |
|---|---|---|---|---|
| LYSINE METHYLATION | TP53 | K370me1 | SMYD2 | repression |
| K373me2 | G9/GLP | repression | ||
| K382me1 | SET8 | repression | ||
| K370me2-->me1 | LSD1 | repression | ||
| K370me2 | activation | |||
| K372me1 | SET7 | activation (promotes p53 acetylation) | ||
| K382me2 | activation | |||
| E2F1 | K185 | SET9 LSD1 | suppresses E2F1 accumulation | |
| NUMB | K158 K136 | SET8 | p53 degradation | |
| Rb | SMYD2 | enhances Rb phosphorylation | ||
| K810 | SET7 | docking site for 53BP1 | ||
| K860 | SMYD2 | docking site for L3MBTL1 | ||
| SITR1 | SET7 | prevents SIRT1-p53 interaction, p53 activation | ||
| PPP1R12A | K442me1 | SET7 | dephosphorylates Rb | |
| K442me1-->K442 | LSD1 | unable to dephosphorylate Rb | ||
| PCNA | K248 | SET8 | activation | |
| MDC1 | K45 | JMJD1C | activation | |
| FEN1 | K377 | SET7 | activation | |
| UHRF1 | K385me1 | SET7 | PCNA polyubiquitination | |
| K385me-->K385 | LSD1 | prevents UHRF1-PCNA interaction | ||
| PARP1 | K508 K528 | SET7 SMYD2 | enhances PARP1 at damage sites | |
| DNA-PKcs | K1150me3 | activation | ||
| K2746me2 | ||||
| K3248me2 | ||||
| KU80 | K7me3 | activation | ||
| SUV39H1 | K105 K123 | SET7 | heterochromatin relaxation | |
| ARGININE METHYLATION | MRE11 | PRMT1 | activation | |
| 53BP1 | R1406 | PRMT1 | activation (enhances binding at damage sites) | |
| R1413 | ||||
| BRCA1 | PRMT1 | activation | ||
| Polβ | R83 | PRMT1 PRMT6 | activation | |
| R137 | ||||
| R154 | ||||
| KLF4 | R374 | PRMT5 | KLF4 accumulation | |
| R376 | ||||
| R377 | ||||
| TP53 | PRMT5 | activation | ||
| FEN1 | R192 | PRMT5 | activation |
| Inhibitor Type | Representative Drugs | Target | Status | Cancer Type |
|---|---|---|---|---|
| HDAC | Vorinostat | All HDACs | FDA approved | T-cell Lymphoma |
| Romidepsin | HDAC1-3 | FDA approved | T-cell Lymphoma | |
| Belinostat | All HDACs | FDA approved | T-cell Lymphoma | |
| Panobinostat | All HDACs | FDA approved | Refractory multiple myeloma | |
| BET | OTX015/MK-8628 | BRD2/3/4 | phase 1b | NUT midline carcinoma |
| I-BET762 | BRD2/3/5 | phase 1/2 | NUT midline carcinoma & hematological cancers | |
| DNMT | 5-azacitidine | DNMTs | FDA appoved | AML, MDS |
| Decitabine | DNMTs | FDA appoved | AML, MDS | |
| HDM | tranylcypromine | LSD1 | phase 1 | AML |
| HMT | tazemetostat | EZH2 | phase 1/2 | B-cell Lymphoma |
| Pinometostat | DOT1L | phase 1 | MLL-r Leukemia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules 2020, 25, 2496. https://doi.org/10.3390/molecules25112496
Karakaidos P, Karagiannis D, Rampias T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules. 2020; 25(11):2496. https://doi.org/10.3390/molecules25112496
Chicago/Turabian StyleKarakaidos, Panagiotis, Dimitris Karagiannis, and Theodoros Rampias. 2020. "Resolving DNA Damage: Epigenetic Regulation of DNA Repair" Molecules 25, no. 11: 2496. https://doi.org/10.3390/molecules25112496
APA StyleKarakaidos, P., Karagiannis, D., & Rampias, T. (2020). Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules, 25(11), 2496. https://doi.org/10.3390/molecules25112496