
Design, Synthesis, Antitumor Activity and Molecular Docking Study of Novel 5-Deazaalloxazine Analogs


Abstract
:1. Introduction
2. Results and Discussion
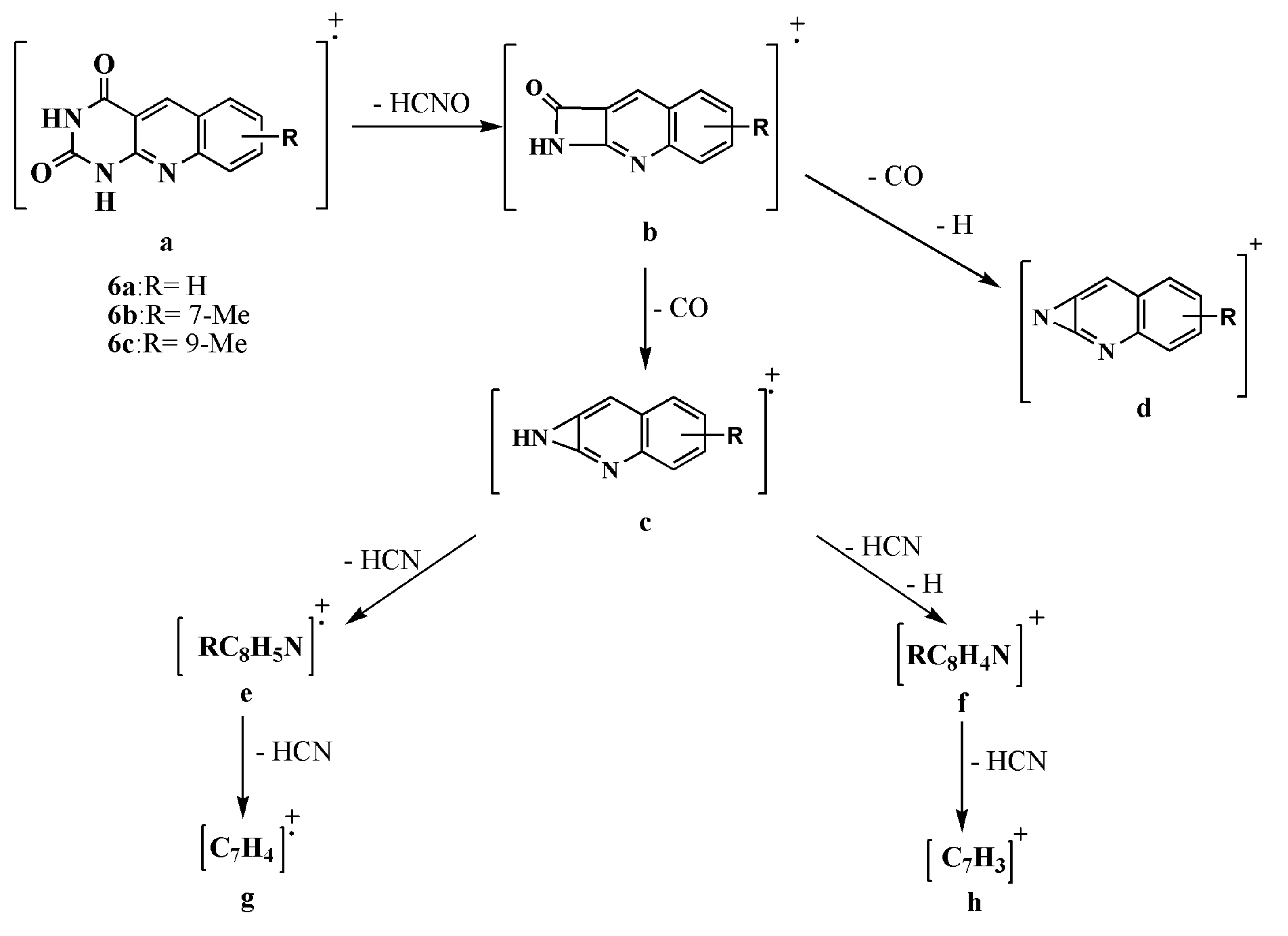
2.1. Synthesis and Structural Elucidation of 5-Deazaalloxazine Derivatives
2.2. Antiproliferative MTT Assay
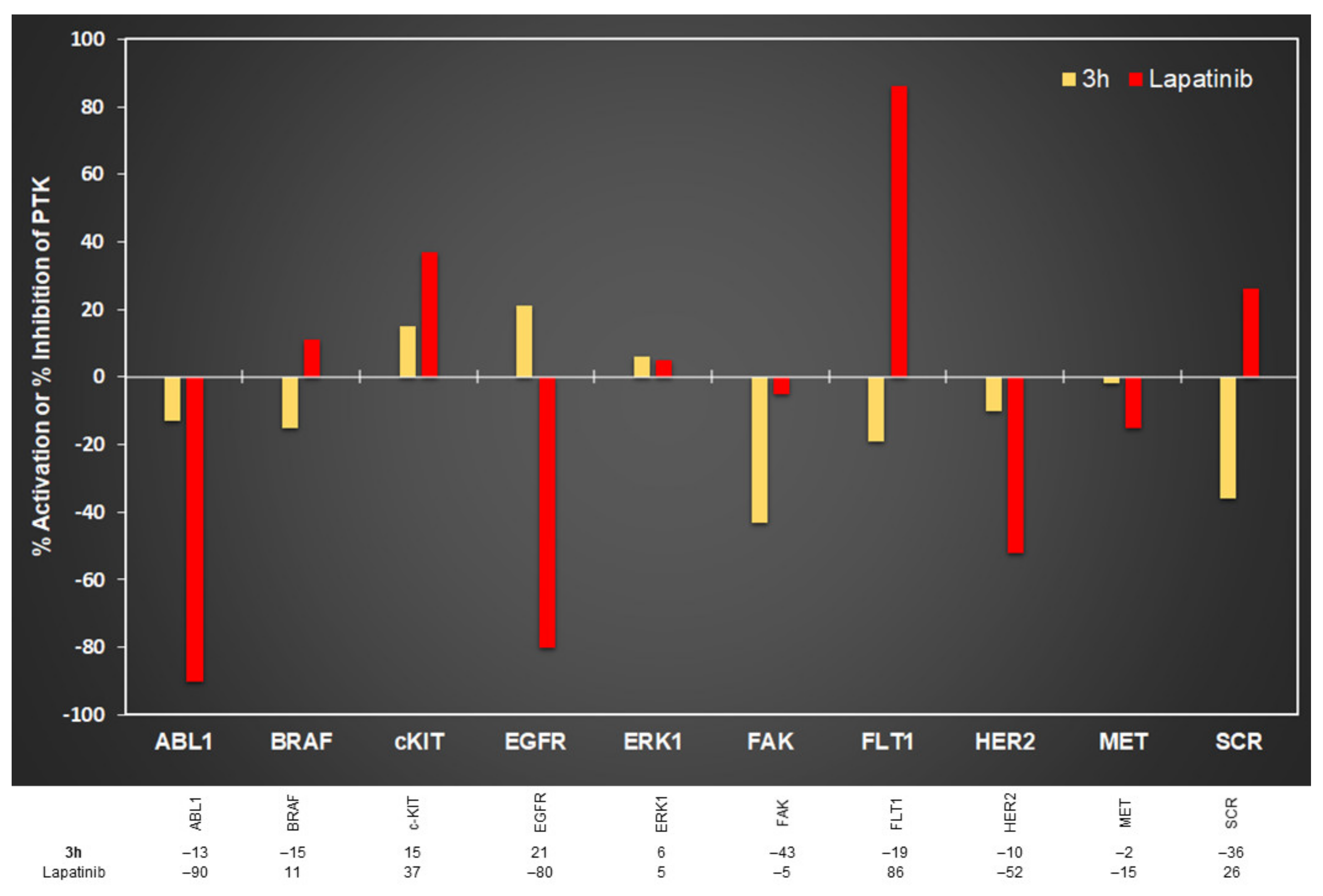
2.3. In Vitro Protein Kinase Assay
2.4. Annexin V PI/FITC Apoptosis Assay
2.5. Structure-Activity Relationship (SAR)
2.6. Molecular Docking Study
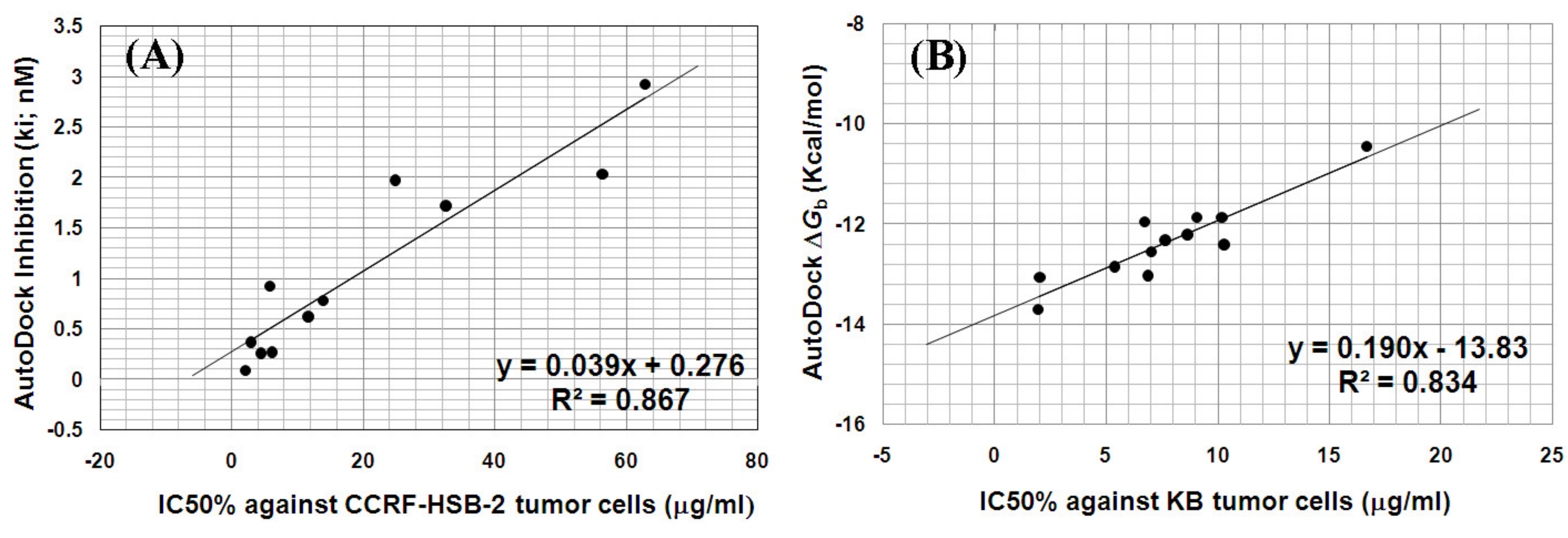
2.6.1. AutoDock Validation
2.6.2. AutoDock Binding Affinities of the Synthesized Compounds into C-Kit Tyrosine Kinase
3. Experimental
3.1. Synthesis and Structural Elucidation of 5-Deazaalloxazine Derivatives
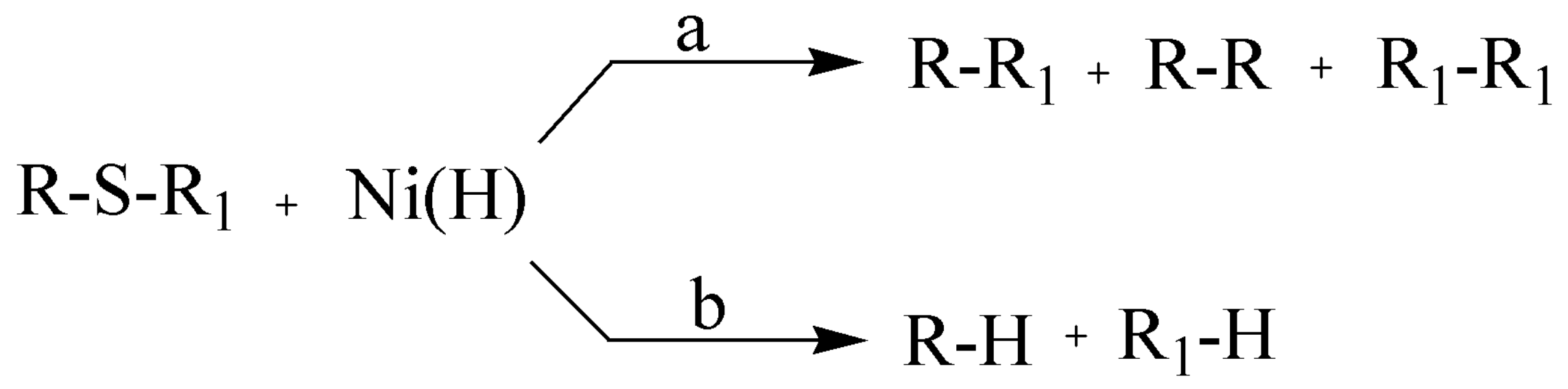
3.1.1. General Procedure for the Preparation of 2-Alkylthiopyrimido[4,5-b] Quinoline-4(3H)-Ones {2-Deoxo-2-Alkylthio-5-Deazaalloxazines} (2a–e)
3.1.2. General Procedure for the Preparation of 2-(Substituted alkyl amino)-pyrimido[4,5-b] quinoline-4(3H)-ones {2-(substituted alkyl amino)-2-deoxo-5-deazaalloxazines} (3a–m)
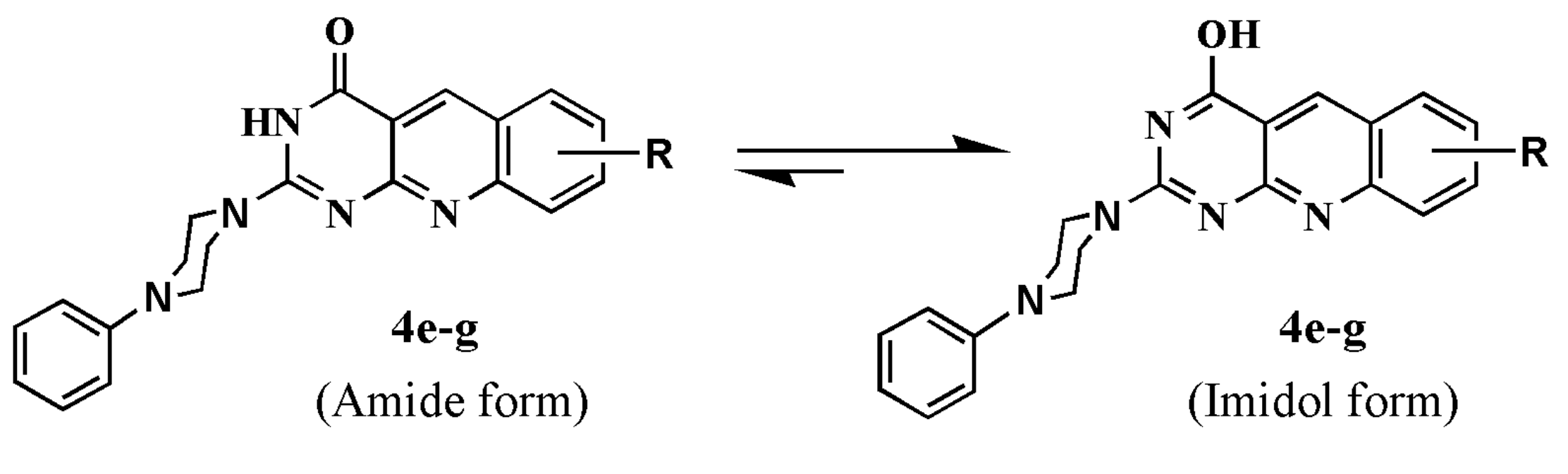
3.1.3. General Procedure for the Preparation of 2-(heterocyclic substituted)pyrimido[4,5-b]quinoline-4(3H)-ones {2-(heterocyclic substituted)-2-deoxo-5-deazaalloxazines} (4a–g)
3.1.4. General Procedure for the Preparation of 2-Aminopyrimido[4,5-b]quinoline-4(3H)-ones {2-amino-2-deoxo-5-deazaalloxazines} (5a,b)
3.1.5. General Procedure for the Preparation of Pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones{2,4-dioxo-5-deazaalloxzines} (6a–c)
3.1.6. General Procedure for the Preparation of 6-(o-tolylamino)pyrimidin-4(3H)-one (7)
3.1.7. General Procedure for the Preparation of 9-(methyl)pyrimido[4,5-b]quinolin-4-ol {2-deoxo-5-deazaalloxazine} (8)
3.2. Antiproliferative MTT Assay
3.2.1. Antiproliferative Activities for Compounds (2b–d, 3a, d, i, j, k, m, 4a, c–g, 5a, b) Against CCRF-HSB and KB Tumor Cell Lines
3.2.2. Antiproliferative Activities for Compounds (2b–e, 3b, c, e, f, g, h, l, 4b, 6a–c, 8) Against MCF-7 and HeLa Tumor Cell Lines
3.3. [γ-32P] ATP Radiometric Protein Kinase Assay Method
3.4. Annexin V PI/FITC Apoptosis Assay Using MCF7 Cells
3.5. Molecular Docking Study
3.5.1. Preparation of Target C-Kit Kinase and Ligands
3.5.2. Calculation of Autogrid Maps for the C-Kit Kinase Embracing the STI-571 Ligand
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Statistics Center. 2020. Available online: https://cancerstatisticscenter.cancer.org/#!/ (accessed on 26 May 2020).
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase–role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Burke, T.R., Jr.; Yao, Z.J.; Liu, D.G.; Voigt, J.; Gao, Y. Phosphoryltyrosyl mimetics in the design of peptide—Based signal transduction inhibitors. Biopolymers 2001, 60, 32–44. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, K.J.; Kim, S.K.; Wood, R.; et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2002, 8, 935–942. [Google Scholar] [PubMed]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer. 2018, 17, 36–48. [Google Scholar] [CrossRef]
- Ali, H.I.; Tomita, K.; Akaho, E.; Kambara, H.; Miura, S.; Hayakawa, H.; Yoneda, F. Antitumor studies. Part 1: Design, synthesis, antitumor activity and AutoDock study of 2-deoxo-2-phenyl-5-deazaflavins and 2-deoxo-2-phenylflavin-5-oxides as a new class of antitumor agents. Bioorgan. Med. Chem. 2007, 15, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, H.I.; Tomita, K.; Akaho, E.; Kunishima, M.; Kawashima, Y.; Yamagishi, T.; Nagamatsu, T. Antitumor studies–Part 2: Structure–activity relationship study for flavin analogs including investigations on their in vitro antitumor assay and docking simulation into protein tyrosine kinase. Eur. J. Med. Chem. 2008, 43, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.I.; Ashida, N.; Nagamatsu, T. Antitumor studies. Part 4: Design, synthesis, antitumor activity and molecular docking study of novel 2-substituted 2-deoxoflavin-5-oxides, 2-deoxoalloxazine-5-oxides and their 5-deaza analogs. Bioorgan. Med. Chem. 2008, 16, 922–940. [Google Scholar] [CrossRef] [Green Version]
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Ghorab, W.M. Design and synthesis of some novel quinoline derivatives as anticancer and radiosensitizing agents targeting VEGFR tyrosine kinase. J. Het. Chem. 2011, 48, 1269–1279. [Google Scholar] [CrossRef]
- Berezovskii, V.M.; Eremenko, T.V. Chemistry of alloxazines and isoalloxazines. Russ. Chem. Rev. 1963, 32, 290–307. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein. J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolfrey, J.R.; Weston, G. The use of computational methods in the discovery and design of kinase inhibitors. Curr. Pharm. Des. 2002, 8, 1527–1545. [Google Scholar] [CrossRef] [PubMed]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2019, 7, 873–898. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.H.; Chiu, C.C.; Lin, C.L.; Chen, T.S.; Jheng, B.R.; Lee, Y.C.; Chen, J.; Chen, B.S. A new era for cancer target therapies: Applying systems biology and computer-aided drug design to cancer therapies. Curr. Pharm. Biotechnol. 2016, 17, 1246–1267. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.; Choi, S. Expediting the design, discovery and development of anticancer drugs using computational approaches. Curr. Med. Chem. 2017, 24, 4753–4778. [Google Scholar] [CrossRef]
- De, B.; Bhandari, K.; Mendonça, F.J.; Scotti, M.T.; Scotti, L. Computational studies in drug design against cancer. Anti-Cancer Agents Med. Chem. (Formerly Curr. Med. Chem. Anti-Cancer Agents) 2019, 19, 587–591. [Google Scholar] [CrossRef]
- Cichero, E.; D’Ursi, P.; Moscatelli, M.; Bruno, O.; Orro, A.; Rotolo, C.; Milanesi, L.; Fossa, P. Homology modeling, docking studies and molecular dynamic simulations using graphical processing unit architecture to probe the type-11 phosphodiesterase catalytic site: A computational approach for the rational design of selective inhibitors. Chem. Boil. Drug Des. 2013, 82, 718–731. [Google Scholar] [CrossRef]
- Franchini, S.; Battisti, U.M.; Prandi, A.; Tait, A.; Borsari, C.; Cichero, E.; Fossa, P.; Cilia, A.; Prezzavento, O.; Ronsisvalle, S.; et al. Scouting new sigma receptor ligands: Synthesis, pharmacological evaluation and molecular modeling of 1, 3-dioxolane-based structures and derivatives. Eur. J. Med. Chem. 2016, 112, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Rusnati, M.; Sala, D.; Orro, A.; Bugatti, A.; Trombetti, G.; Cichero, E.; Urbinati, C.; Somma, M.D.; Millo, E.; Galietta, L.J.V.; et al. Speeding up the identification of cystic fibrosis transmembrane conductance regulator-targeted drugs: An approach based on bioinformatics strategies and surface plasmon resonance. Molecules 2018, 23, 120. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C. Beware of docking. Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Kimachi, T.; Yoneda, F.; Sasaki, T. New synthesis of 5-amino-5-deazaflavin derivatives by direct coupling of 5-deazaflavins and amines. J. Heterocycl. Chem. 1992, 29, 763–765. [Google Scholar] [CrossRef]
- Hall, L.H.; Orchard, B.J.; Tripathy, S.K. The structure and properties of flavins: Molecular orbital study based on totally optimized geometries. II. Molecular orbital structure and electron distribution. Int. J. Quantum C. 1987, 31, 217–242. [Google Scholar] [CrossRef]
- Dow, R.L.; Bechle, B.M.; Chou, T.T.; Goddard, C.; Larson, E.R. Selective inhibition of the tyrosine kinase pp60src by analogs of 5, 10-dihydropyrimido [4, 5-b] quinolin-4 (1H)-one. Bioorg. Med. Chem. Lett. 1995, 5, 1007–1010. [Google Scholar] [CrossRef]
- Ali, H.I.; Ashida, N.; Nagamatsu, T. Antitumor studies. Part 3: Design, synthesis, antitumor activity and molecular docking study of novel 2-methylthio-, 2-amino- and 2-(N-substituted amino)-10-alkyl-2-deoxo-5-deazaflavins. Bioorgan. Med. Chem. 2007, 15, 6336–6352. [Google Scholar] [CrossRef] [Green Version]
- Curran, W.V.; Angier, R.B. Some new syntheses of amino-and alkylaminopyrimidines and pteridines. J. Org. Chem. 1963, 28, 2672–2677. [Google Scholar] [CrossRef]
- Taylor, E.C.; Cheng, C.C. Purine Chemistry. VII. An Improved Synthesis of Hypoxanthine. J. Org. Chem. 1960, 25, 148–149. [Google Scholar] [CrossRef]
- Mozingo, R.; Wolf, D.E.; Harris, T.A.; Folkers, K. Hydrogenolysis of sulfur compounds by Raney nickel catalyst. J. Am. Chem. Soc. 1943, 65, 1013–1016. [Google Scholar] [CrossRef]
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 2000, 28, 283–296. [Google Scholar] [CrossRef]
- Prukata, D.; Sikorski, M. Electron ionization mass spectrometric study of substituted alloxazine-5-oxides and iso-alloxazine-5-oxide. Rapid Commun. Mass Spectrom. 2009, 23, 619–628. [Google Scholar] [CrossRef]
- Miura, S.; Yoshimura, Y.; Endo, M.; Machida, H.; Matsuda, A.; Tanaka, M.; Sasaki, T. Antitumor activity of a novel orally effective nucleoside, 1-(2-deoxy-2-fluoro-4-thio-β-d-arabinofuranosyl) cytosine. Cancer Lett. 1998, 129, 103–110. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ali, H.I.; Nagamatsu, T.; Akaho, E. Structurebased drug design and AutoDock study of potential protein tyrosine kinase inhibitors. Bioinform. 2011, 5, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, W.H.; Gouda, A.M.; Ali, H.E.; Al-Rousan, R.; Samaha, D.; Abdalla, A.N.; Ali, H.I. Structural-based design, synthesis and antitumor activity of novel alloxazine analogues with potential selective kinase inhibition. Eur. J. Med. Chem. 2018, 152, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Dickens, M.P.; Roxburgh, P.; Hock, A.; Mezna, M.; Kellam, B.; Vousden, K.H.; Fischer, P.M. 5-Deazaflavin derivatives as inhibitors of p53 ubiquitination by HDM2. Bioorgan. Med. Chem. 2013, 21, 6868–6877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.M.; Henderson, G.; Black, F.; Sutherland, A.; Ludwig, R.L.; Vousden, K.H.; Robins, D.J. Synthesis of 5-deazaflavin derivatives and their activation of p53 in cells. Bioorgan. Med. Chem. 2007, 15, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hao, L.; Pei, J.; Yin, W. Promotion of apoptosis does not necessarily mean inhibition of remodeling. Hypertension. 2012, 60, e3. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin, V. J. Immunol Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Sepehr, K.S.; Baradaran, B.; Mazandarani, M.; Yousefi, B.; Alitappeh, M.A.; Khori, V. Growth-inhibitory and apoptosis-inducing effects of Punica granatum, L. var. spinosa (apple punice) on fibrosarcoma cell lines. Adv. Pharmaceut. Bull. 2014, 4, 583–590. [Google Scholar]
Sample Availability: Samples of the compounds (2a–e), (3a–m), (4a–g), (5a,b), (6a–c), and (8) are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ions | m/z | Elemental Composition | % Relative Abundance | ||
|---|---|---|---|---|---|
| 6a | 6b | 6c | |||
| a [M+] | 213 | C11H7N3O2 | 83 | ─ | ─ |
| 227 | C12H9N3O2 | ─ | 100 | 39 | |
| b | 170 | C10H6N2O | 35 | ─ | ─ |
| 184 | C11H8N2O | ─ | 34 | ─ | |
| c | 142 | C9H6N2 | 20 | ─ | ─ |
| 156 | C10H8N2 | ─ | 26 | ─ | |
| d | 141 | C9H5N2 | 15 | ─ | ─ |
| 155 | C10H7N2 | ─ | 23 | 36 | |
| e | 115 | C8H5N | 80 | ─ | ─ |
| 129 | C9H7N | ─ | 21 | 50 | |
| f | 114 | C8H4N | 100 | ─ | ─ |
| 128 | C9H6N | ─ | 30 | 100 | |
| g | 88 | C7H4 | 36 | ─ | ─ |
| 102 | C8H6 | ─ | 21 | 32 | |
| h | 87 | C7H3 | 28 | ─ | ─ |
| 101 | C8H5 | ─ | 17 | 44 | |
| Inhibitory Activity Against Tumor Cell Lines [IC50 µM)] | |||||
|---|---|---|---|---|---|
| Compd | CCRF–HSB-2 | KB | Compd | MCF-7 | HeLa |
| 2b | 23.66 | 26.73 | 2b | 1.79 | >100 |
| 2c | 35.56 | 30.44 | 2c | 1.24 | >100 |
| 2d | >100 | >100 | 2d | 24.72 | 3.40 |
| 3a | 47.21 | 35.47 | 2e | >100 | >100 |
| 3d | 14.91 | 6.93 | 3b | 4.40 | 16.67 |
| 3i | 6.71 | 6.35 | 3c | 8.49 | 0.57 |
| 3j | >100 | 24.9 | 3e | 1.10 | >100 |
| 3k | >100 | 11.62 | 3f | 1.74 | >100 |
| 3m | 9.17 | 17.47 | 3g | 0.22 | >100 |
| 4a | 49.58 | 36.74 | 3h | 0.17 | 2.00 |
| 4c | 39.14 | 23.79 | 3l | >100 | 0.13 |
| 4d | 19.41 | 26.01 | 4b | 1.25 | >100 |
| 4e | 69.38 | 28.53 | 6a | 2.17 | >100 |
| 4f | 50.61 | 60.30 | 6b | 29.45 | 1.39 |
| 4g | >100 | 23.28 | 6c | 2.10 | >100 |
| 5a | >100 | 1.46 | 8 | 18.22 | 0.33 |
| 5b | 44.64 | 73.81 | 5-FU | 5.15 | 7.76 |
| Ara-c | 0.14 | 0.26 | |||
| Comp. | ΔGb a (kcal/mol) | Kib | Hydrogen Bonds | RMSD c (Å) | ||
|---|---|---|---|---|---|---|
| Atoms of Comp. | Amino Acids | The Most Relevant Amino Acids | ||||
| 2b | −13.04 | 274.65 pM | N10 | HN of D810 | D810 | 1.31 |
| 2e | −12.60 | 577.10 pM | 3-NH 4-C=O | O=C of I789 HS of C788 | - | 7.28 |
| 3a | −11.87 | 2.00 nM | N1 2-NH Terminal OH | HN of D810 O=C of E640 O=C of D810 | E640 D810 | 0.86 |
| 3d | −13.07 | 261.56 pM | 2-NH 3-NH 4-C=O | OH of T670 O=C of E671 HN of C673 | T670 E671 C673 | 6.41 |
| 3e | −12.35 | 889.03 pM | 3-NH Terminal OH | O=C of D810 O=C of E640 | E640 D810 | 3.07 |
| 3h | −11.07 | 7.68 nM | 3-NH Terminal OH | OH of T670 O=C of E640 | E640 T670 | 6.13 |
| 3i | −13.71 | 88.93 pM | 3-NH 4-C=O 2-NH | O=C of E671 HN of C673 OH of T670 | T670 C673 E671 | 6.82 |
| 3j | −11.96 | 1.72 nM | 2-NH Terminal HO | O=C of D677 HN of N680 | - | 10.33 |
| 3k | −11.64 | 2.92 nM | 4-C=O | HN of C673 | C673 | 7.82 |
| 3m | −12.86 | 373.23 pM | 4-C=O | HN of C673 | C673 | 7.71 |
| 4a | −12.42 | 782.55 pM | 4-C=O | HN of C673 | C673 | 7.44 |
| 4b | −12.41 | 806.85 pM | 7-O 4-C=O | HO of T670 HN of C673 | T670 C673 | 7.88 |
| 4c | −12.56 | 624.08 pM | 4-C=O | HN of C673 | C673 | 7.56 |
| 4d | −12.33 | 917.97 pM | 4-C=O | HN of C673 | C673 | 7.57 |
| 4e | −11.88 | 1.97 nM | 3-NH 4-C=O | O=C of I789 HS of C788 | - | 8.91 |
| 4g | −12.22 | 1.10 nM | 3-NH 4-C=O | O=C of I789 HS of C788 | - | 8.44 |
| 5a | −11.86 | 2.03 nM | 4-C=O 3-NH 2-NH1 2-NH1 | HN of K623 O=C of D810 O=C of D810 O=C of E640 | E640 D810 | 2.46 |
| 5b | −10.45 | 21.92 nM | 4-C=O | HN of C673 | C673 | 8.04 |
| STId | −16.42 | 915.03 fM | HNC=O Ph-NH | HN of D810 OH of T670 | T670 D810 | 0.17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, S.; Samaha, D.; Mohamed, M.S.; Abou Taleb, N.A.; Elsawy, M.A.; Nagamatsu, T.; Ali, H.I. Design, Synthesis, Antitumor Activity and Molecular Docking Study of Novel 5-Deazaalloxazine Analogs. Molecules 2020, 25, 2518. https://doi.org/10.3390/molecules25112518
Mahmoud S, Samaha D, Mohamed MS, Abou Taleb NA, Elsawy MA, Nagamatsu T, Ali HI. Design, Synthesis, Antitumor Activity and Molecular Docking Study of Novel 5-Deazaalloxazine Analogs. Molecules. 2020; 25(11):2518. https://doi.org/10.3390/molecules25112518
Chicago/Turabian StyleMahmoud, Sawsan, Doaa Samaha, Mosaad S. Mohamed, Nageh A. Abou Taleb, Mohamed A. Elsawy, Tomohisa Nagamatsu, and Hamed I. Ali. 2020. "Design, Synthesis, Antitumor Activity and Molecular Docking Study of Novel 5-Deazaalloxazine Analogs" Molecules 25, no. 11: 2518. https://doi.org/10.3390/molecules25112518
APA StyleMahmoud, S., Samaha, D., Mohamed, M. S., Abou Taleb, N. A., Elsawy, M. A., Nagamatsu, T., & Ali, H. I. (2020). Design, Synthesis, Antitumor Activity and Molecular Docking Study of Novel 5-Deazaalloxazine Analogs. Molecules, 25(11), 2518. https://doi.org/10.3390/molecules25112518