
In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus




Abstract
:
1. Introduction
2. Results and Discussion
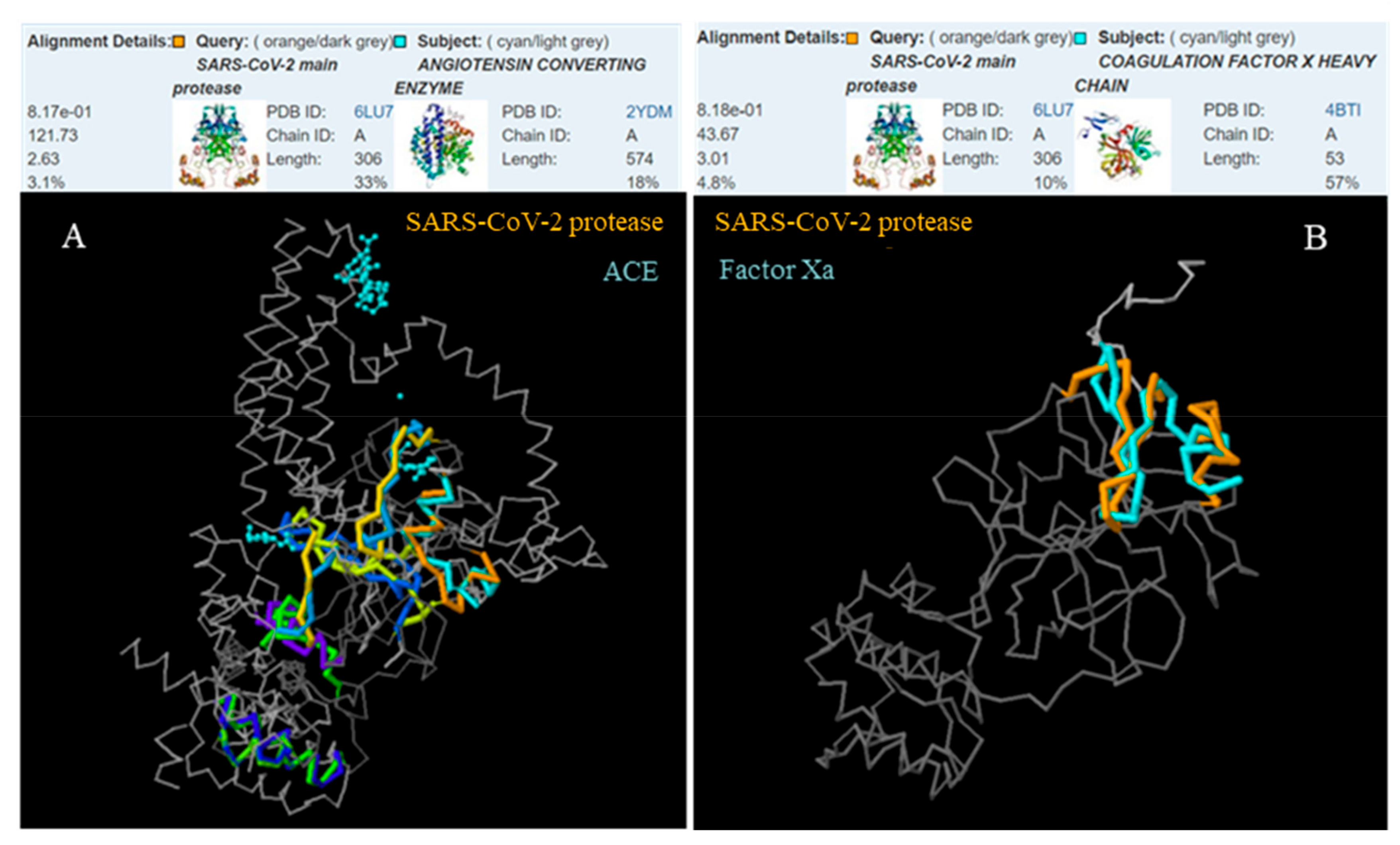
2.1. The 3D Structure Protein Alignment
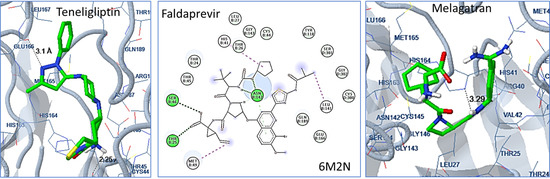
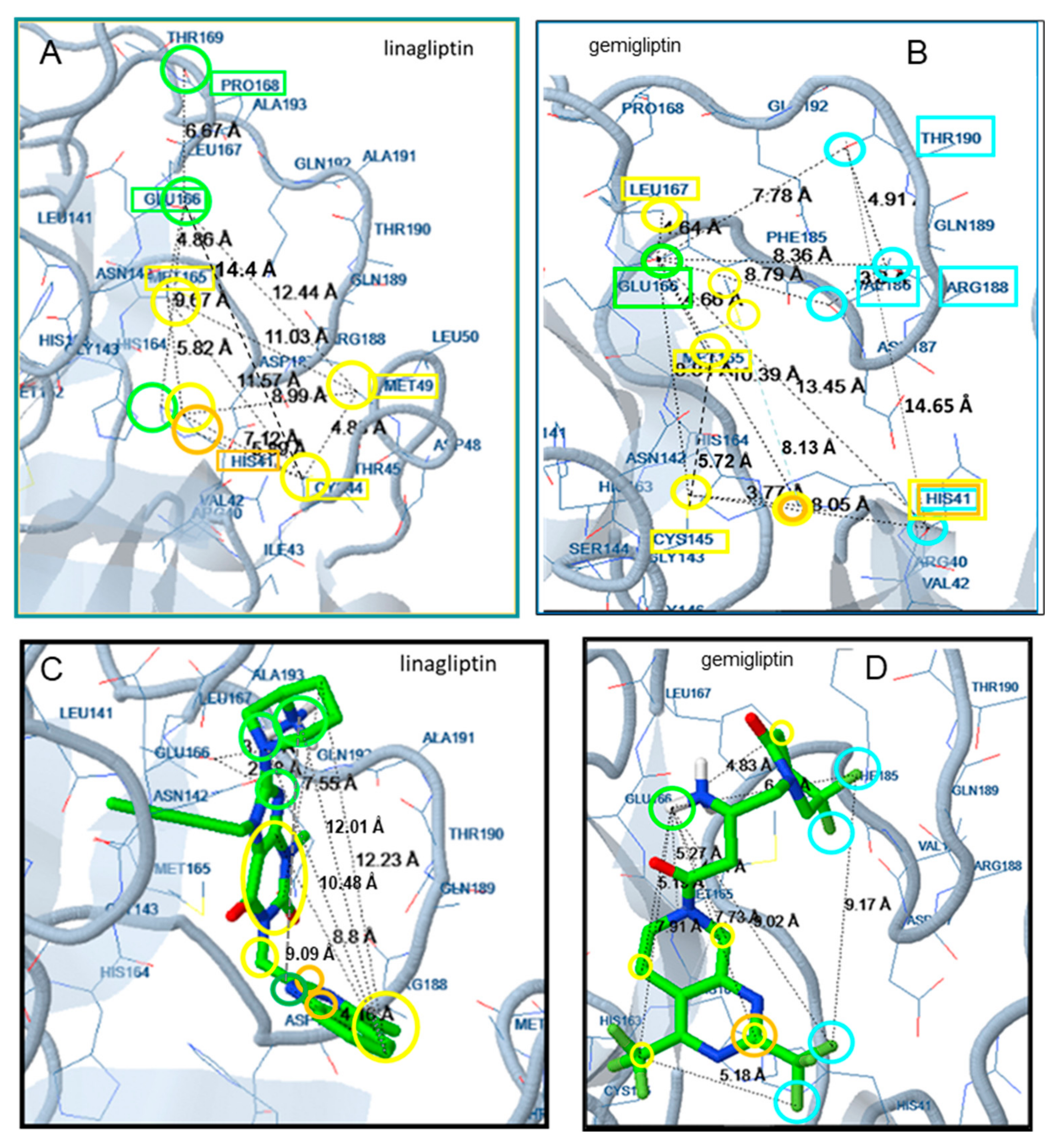
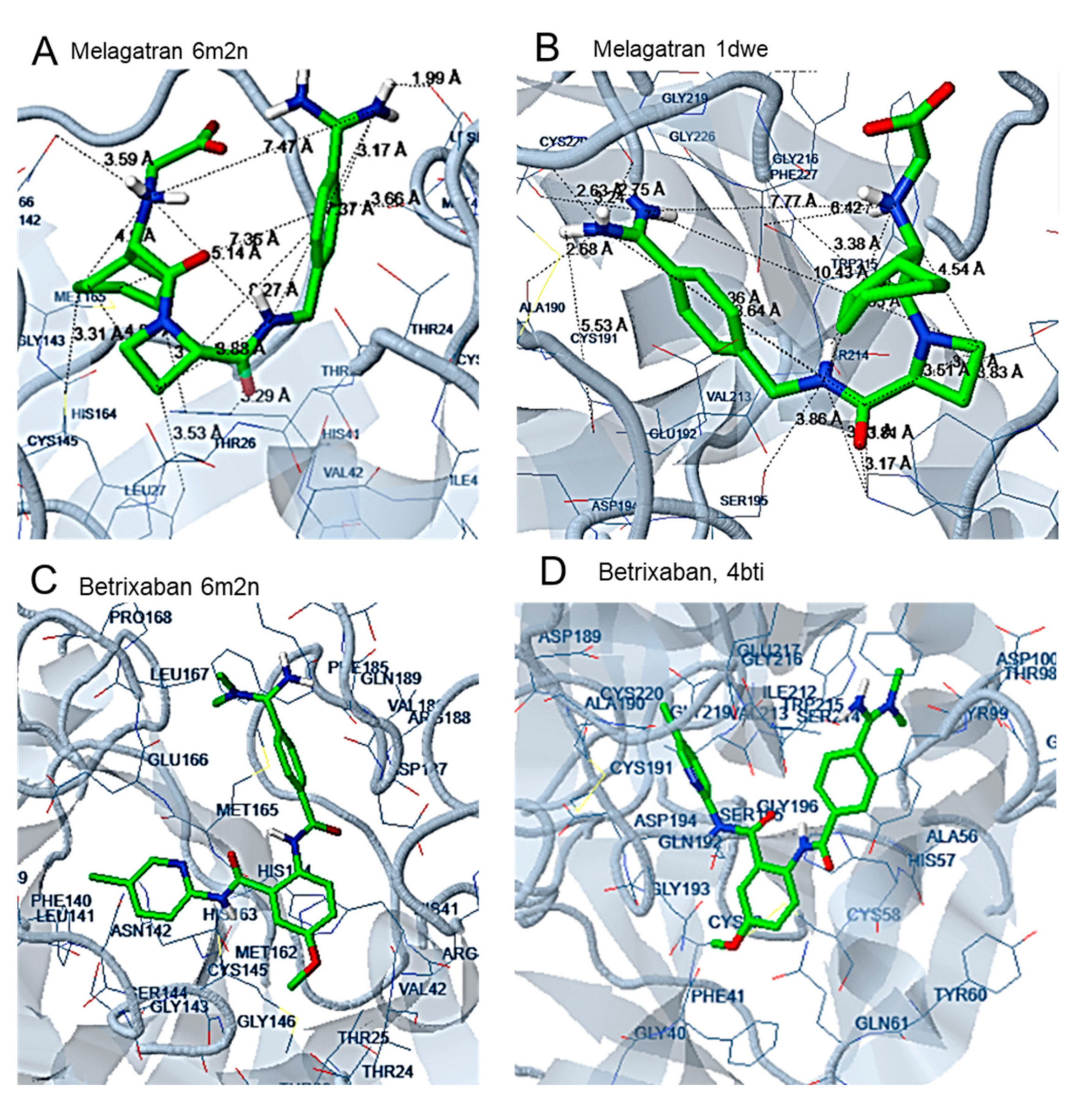
2.2. Docking Analysis
3. Experimental Part
3.1. The 3D Structure Protein Alignment
3.2. Docking Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RDRP | RNA-dependent RNA polymerase |
| DPP-4 | dipeptidyl peptidase-4 |
References
- Coronavirus Disease (COVID-19) Outbreak Situation. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 16 April 2020).
- COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 16 April 2020).
- Chan, J.F.-W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16–20. [Google Scholar] [CrossRef] [Green Version]
- UBC, Apeiron Biologics to Trial Coronavirus Drug Candidate in China. Available online: https://www.clinicaltrialsarena.com/news/ubc-apeiron-biologics-covid-19-trial/ (accessed on 25 March 2020).
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, X.-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 47, 119–121. [Google Scholar] [CrossRef]
- Joshi, T.; Joshi, T.; Sharma, P.; Mathpal, S.; Pundir, H.; Bhatt, V.; Chandra, S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4529–4536. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.M. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 20-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Calligari, P.; Bobone, S.; Ricci, G.; Bocedi, A. Molecular Investigation of SARS–CoV-2 Proteins and Their Interactions with Antiviral Drugs. Viruses 2020, 12, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Sarmah, S.; Lyndem, S.; Singha Roy, A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential Inhibitors of Coronavirus 3-Chymotrypsin-Like Protease (3CLpro): An in silico screening of Alkaloids and Terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [Green Version]
- The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances; World Health Organization: Geneva, Switzerland, 2016; Available online: https://www.who.int/medicines/services/inn/StemBook_2013_Final.pdf (accessed on 26 March 2020).
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R.; Zhang, L.; et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J. Comparative Studies on Retroviral Proteases: Substrate Specificity. Viruses 2010, 2, 147–165. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, S.A.; Thomsen, E.R.; Cieplak, P.; Chudin, E.; Cheltsov, A.V.; Chee, M.S.; Kozlov, I.A.; Strongin, A.Y. New Details of HCV NS3/4A Proteinase Functionality Revealed by a High-Throughput Cleavage Assay. PLoS ONE 2012, 7, e35759. [Google Scholar] [CrossRef] [Green Version]
- Lankas, G.R.; Leiting, B.; Roy, R.S.; Eiermann, G.J.; Beconi, M.G.; Biftu, T.; Chan, C.C.; Edmondson, S.; Feeney, W.P.; He, H.; et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: Potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes 2005, 54, 2988–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.-J.; Tsai, K.-C.; Sun, Y.-K.; Chang, H.-J.; Huang, Y.-J.; Yu, H.-M.; Lin, C.-H.; Mao, S.-S.; Yang, A.-S. Factor Xa active site substrate specificity with substrate phage display and computational molecular modeling. J. Biol. Chem. 2008, 283, 12343–12353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallwitz, M.; Enoksson, M.; Thorpe, M.; Hellman, L. The Extended cleavage specificity of human thrombin. PLoS ONE 2012, 7, e31756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuyer, S.G.; Schwager, S.L.U.; Sturrock, E.D.; Isaac, R.E.; Acharya, K.R. Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci. Rep. 2012, 2, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Akaki, J.; Satou, R.; Takaya, M.; Iwata, H.; Katsurada, A.; Nishiuchi, K.; Ohmura, Y.; Suzuki, F.; Nakamura, Y. The His-Pro-Phe motif of angiotensinogen is a crucial determinant of the substrate specificity of renin. Biol. Chem. 2007, 388, 237–246. [Google Scholar] [CrossRef]
- Di Nisio, M.; Middeldorp, S.; Büller, H. Direct thrombin inhibitors. N. Engl. J. Med. 2005, 353, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Lechtenberg, B.C.; Freund, S.M.; Huntington, J.A. An ensemble view of thrombin allostery. Biol. Chem. 2012, 393, 889–898. [Google Scholar] [CrossRef]
- Ganou, C.A.; Eleftheriou, P.T.; Theodosis-Nobelos, P.; Geronikaki, A.A.; Lialiaris, T.; Rekka, E.A. Docking analysis targeted to the whole enzyme: An application to the prediction of inhibition of PTP1b by thiomorpholine and thiazolyl derivatives. SAR QSAR Environ. Res. 2018, 29, 133–149. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Petrou, A.; Geronikaki, A.; Liaras, K.; Dirnali, S.; Anna, M. Prediction of enzyme inhibition and mode of inhibitory action based on calculation of distances between hydrogen bond donor/acceptor groups of the molecule and docking analysis: An application on the discovery of novel effective PTP1B inhibitors. SAR QSAR Environ. Res. 2015, 26, 557–576. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2019. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Huang, M.; Li, D.; Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis 2020, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCSB PDB Protein Comparison Tool. Available online: http://www.rcsb.org/pdb/workbench/workbench.do (accessed on 12 March 2020).
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of Auto-Dock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Solis, F.J.; Wets, R.J.B. Minimization by Random Search Techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- A Phase 3 Study in Combination with BMS-790052 and BMS-650032 in Japanese Hepatitis C Virus (HCV) Patients. Available online: https://clinicaltrials.gov/ (accessed on 5 April 2020).
- Kumada, H.; Suzuki, Y.; Ikeda, K.; Toyota, J.; Karino, Y.; Chayama, K.; Kawakami, Y.; Ido, A.; Yamamoto, K.; Takaguchi, K.; et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 2014, 59, 2083–2091. [Google Scholar] [CrossRef]
- Huang, F.; Moschetti, V.; Lang, B.; Halabi, A.; Petersen-Sylla, M.; Yong, C.-L.; Elgadi, M. Pharmacokinetics, Safety, and Tolerability of Faldaprevir in Patients with Renal Impairment. Antimicrob. Agents Chemother. 2014, 59, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.L.; Morgan, T.R. Management of Adverse Events during the Treatment of Chronic Hepatitis C Infection. Clin. Liver Dis. (Hoboken) 2012, 1, 54–57. [Google Scholar] [CrossRef]
- Lawitz, E.; Sulkowski, M.; Jacobson, I.; Kraft, W.K.; Maliakkal, B.; Al-Ibrahim, M.; Gordon, S.C.; Kwo, P.; Rockstroh, J.K.; Panorchan, P.; et al. Characterization of Vaniprevir, a Hepatitis C Virus NS3/4A Protease Inhibitor, in Patients With HCV Genotype 1 Infection: Safety, Antiviral Activity, Resistance, and Pharmacokinetics. Antivir. Res. 2013, 99, 214–220. [Google Scholar] [CrossRef]
- FDA Drug Safety Communication: FDA Adds Warnings about Heart Failure Risk to Labels of Type 2 Diabetes Medicines Containing Saxagliptin and Alogliptin. Available online: https://www.fda.gov/ (accessed on 5 April 2020).
- Gu, N.; Park, M.K.; Kim, T.; Bahng, M.; Lim, K.S.; Cho, S.; Yoon, S.H.; Cho, J.; Jang, I.; Yu, K. Multiple-dose pharmacokinetics and pharmacodynamics of evogliptin (DA-1229), a novel dipeptidyl peptidase IV inhibitor, in healthy volunteers. Drug Des. Dev. Ther. 2014, 8, 1709–1721. [Google Scholar] [CrossRef] [Green Version]
- Salvo, F.; Moore, N.; Arnaud, M.; Robinson, P.; Raschi, E.; De Ponti, F.; Bégaud, B.; Pariente, A. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: Systematic review and meta-analysis. BMJ 2016, 353, i2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DPP-4 Inhibitors for Type 2 Diabetes: Drug Safety Communication—May Cause Severe Joint Pain; FDA: Rockville, MD, USA, 2015.
- Onglyza. Available online: https://www.rxlist.com/onglyza-drug.htm (accessed on 5 April 2020).
- American Society of Health-System Pharmacists. Sitagliptin Phosphate Monograph for Professionals. Available online: https://www.drugs.com/ (accessed on 5 April 2020).
- Galvus (PDF). Available online: https://www.ema.europa.eu/ (accessed on 5 April 2020).
- Argatroban Side Effects. Available online: https://www.drugs.com/ (accessed on 5 April 2020).
- Dabigatran Etexilate Mesylate Monograph for Professionals; American Society of Health-System Pharmacists: Bethesda, MD, USA; Available online: https://www.drugs.com/ (accessed on 5 April 2020).
- Apixaban Monograph for Professionals; American Society of Health-System Pharmacists: Bethesda, MD, USA; Available online: https://www.drugs.com/ (accessed on 5 April 2020).
- Betrixaban. Available online: https://www.drugs.com/mtm/betrixaban.html (accessed on 5 April 2020).
- Edoxaban, Oral Tablet. Available online: https://www.healthline.com/health/edoxaban-oral-tablet (accessed on 5 April 2020).
Sample Availability: Samples of the compounds are not available from the authors. |



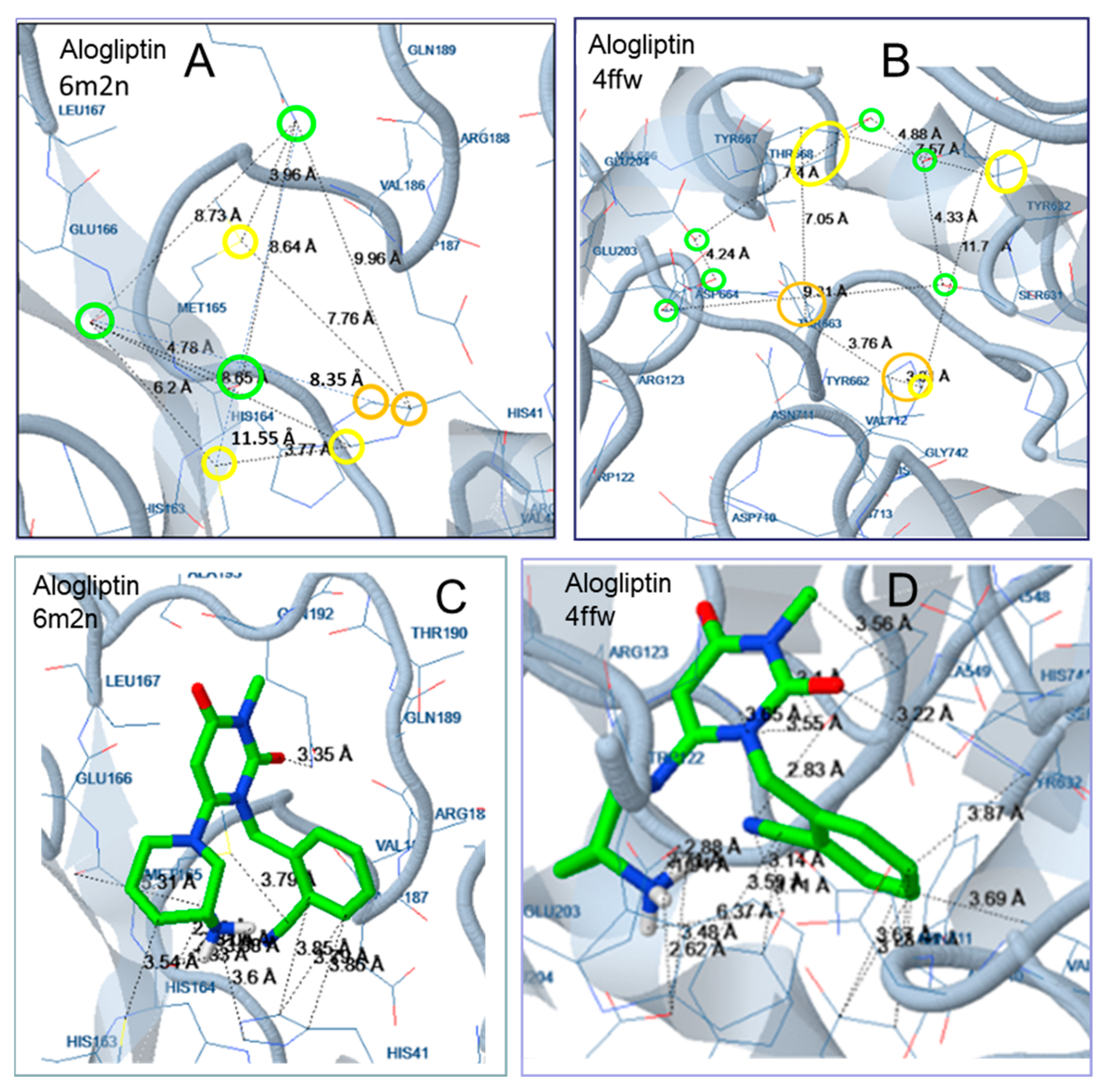
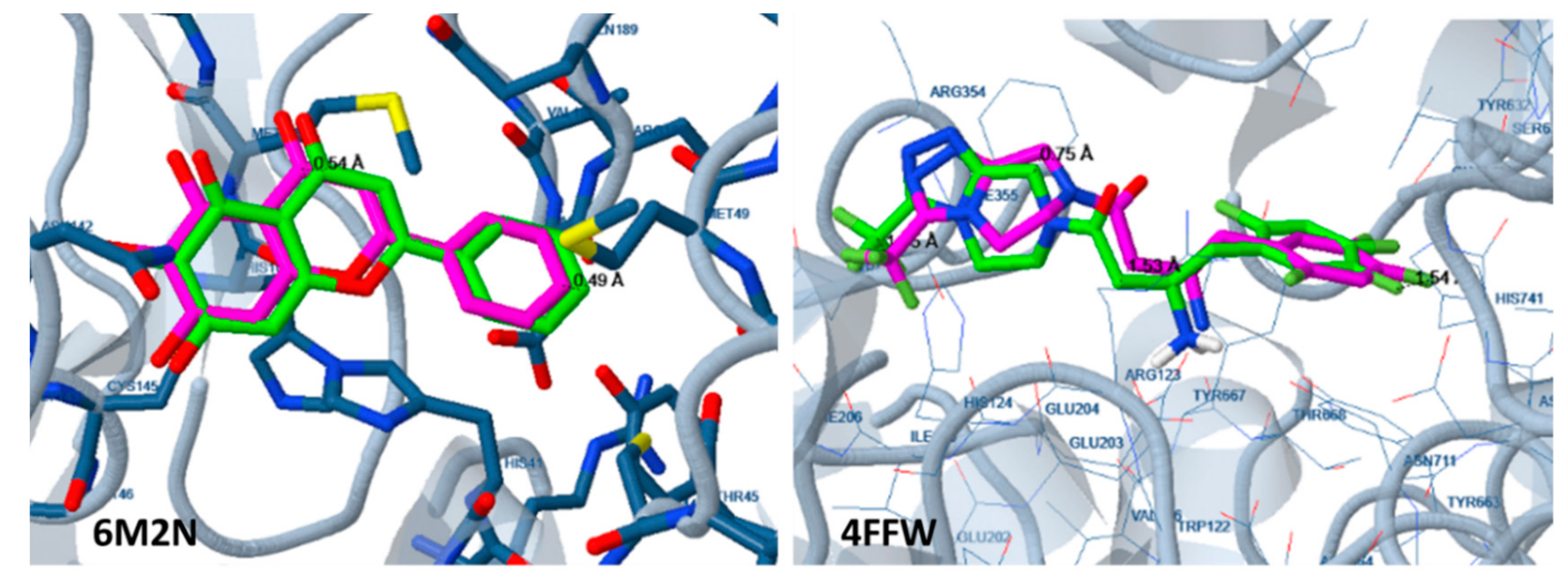

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease | Inhibitors |
|---|---|
| HIV-1 Protease | amprenavir, atazanavir, brecanavir, darunavir, droxinavir, fosamprenavir, indinavir, lasinavir, lopinavir, mozenavir, nelfinavir, palinavir, ritonavir, saquinavir, telinavir, and tipranavir |
| HCV Protease | asunaprevir, boceprevir, ciluprevir, danoprevir, faldaprevir, narlaprevir, neceprevir, simeprevir, sovaprevir, telaprevir, vaniprevir, and vedroprevir |
| Dipeptidyl Peptidase-4 (DPP-4) | alogliptin, anagliptin, bisegliptin, carmegliptin, denagliptin, dutogliptin, evogliptin, gemigliptin, gosogliptin, linagliptin, melogliptin, omarigliptin, saxagliptin, sitagliptin, teneligliptin, trelagliptin, and vildagliptin |
| Renin | aliskiren, ciprokiren, ditekiren, enalkiren, remikiren, terlakiren, and zankiren |
| ACE | alacepril, benazepril, captopril, ceronapril, cilazapril, delapril, enalapril, fosinopril, idrapril, imidapril, indolapril, libenzapril, lisinopril, moexipril, moveltipril, orbutopril, pentopril, perindopril, pivopril, quinapril, ramipril, rentiapril, spirapril, temocapril, trandolapril, utibapril, zabicipril, and zofenopril |
| Thrombin | direct inhibitors: argatroban, inogatran, melagatran and its pro-drug ximelagatran, and dabigatran [30,31] |
| Coagulation Factor Xa | rivaroxaban, apixaban, betrixaban, darexaban, edoxaban, otamixaban, and letaxaban |
| Approved Drug | Lowest Free Binding Energy to the Active Site (kcal mol−1) |
|---|---|
| HIV-1 protease inhibitors | |
| Lopinavir | −8.65 (2nd) |
| Ritonavir | −8.96 (2nd) |
| HCV protease inhibitors | |
| Telaprevir | −9.23 |
| Boceprevir | −9.16 (2nd) |
| DPP-4 inhibitor | |
| Sitagliptin | −8.80 |
| Thrombin inhibitors | |
| Argatroban | −9.03 |
| Dabigatran | −6.57 |
| Factor Xa inhibitor | |
| Rivaroxaban | −7.97 |
| ACE inhibitor | |
| Captopril | −5.17 |
| Renin inhibitor | |
| Aliskiren | −4.66 |
| HCV Protease Inhibitors | Est. Free Binding Energy (kcal/mol) | DPP-4 Inhibitors | Est. Free Binding Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | HCV | HIV-1 | SARS-CoV-2 | DPP-4 | HIV-1 | ||||
| 6LU7 | 6M2N | 2WF8 | 4RVJ | 6LU7 | 6M2N | 2FFW | 4RVJ | ||
| Asunaprevir | −7.77 | −7.52 | Alogliptin | −7.42 | −7.19 | −9.14 | |||
| Danoprevir | −8.05 | −8.00 | −11.53 | −7.16 | Anagliptin | −8.05 | −8.20 | −9.16 | |
| Faldaprevir | −10.92 | −11.15 | −11.27 | −6.02 | Evogliptin | −8.38 | −8.74 | −9.84 | |
| Narlaprevir | −5.70 | −6.11 | −12.38 | −6.90 | Gosogliptin | −8.30 | −8.75 | −9.18 | |
| Sovaprevir | −8.17 | −8.53 | −10.45 | −4.80 | Gemigliptin | −8.99 | −9.95 −9.94 * | −10.18 | −7.86 |
| Vaniprevir | −7.12 | −7.44 | Linagliptin | −9.48 | −8.25 −8.35 * | −9.52 | −2.20 | ||
| 2WF8 initial ligand | −12.42 | Melogliptin | −8.06 | −7.01 | −9.00 | ||||
| 4RVJ initial ligand | −9.86 | Omarigliptin | −8.69 | −7.91 | −10.02 | ||||
| 6M2N initial ligand | −7.45 | Saxagliptin | −8.67 | −8.14 | −8.28 | ||||
| N3 inhibitor | −7.41 | Teneligliptin | −9.58 | −9.16 −9.52 * | −9.87 | −6.55 | |||
| Trelagliptin | −8.92 | −8.41 | −9.30 | ||||||
| Vildagliptin | −8.55 | −8.21 | −8.67 | ||||||
| 4FFW initial ligand (sitagliptin) | −10.78 | ||||||||
| α-Thrombin Inhibitors | Est. Free Binding Energy (kcal/mol) | Factor Xa Inhibitors | Est. Free Binding Energy (kcal/mol) | ||||||
| SARS-CoV-2 | thrombin | SARS-CoV-2 | FXa | ||||||
| 6LU7 | 6M2N | 1DWE | 6LU7 | 6M2N | 4BTI | ||||
| Inogatran | −10.30 (3rd) | −8.32 | −10.39 | Apixaban | −7.53 | −7.11 | |||
| Melagatran | −8.64 | −8.70 | −10.35 | Betrixaban | −9.25 | −8.44 | −8.76 | ||
| 1DWE initial ligand | −9.00 | Edoxaban | −10.51 (2nd) | −9.11 | −11.59 | ||||
| Otamixaban | −7.27 | −7.33 | |||||||
| Inhibitors | CheMBL ID | Phase | Industry | Most Common Side Effects | |
|---|---|---|---|---|---|
| HCV protease inhibitors | Asunaprevir | ChEMBL2105735 | Phase III clinical trials [41] | Bristol-Myers Squibb | Generally well tolerated, increased ALT/AST [42] |
| Danoprevir | ChEMBL2311191 | Approved in China 2018 | Ascletis by Roche | - | |
| Faldaprevir | ChEMBL1241348 | Phase III clinical trials in 2011 | Boehringer-Ingelheim | Gastrointestinal events [43] | |
| Narlaprevir | ChEMBL1255891 | Approved | Schering, ℞-Pharm | Pregnancy lactation, severe neutropenia [44] | |
| Sovaprevir | ChEMBL2105750 | Investigational, received Fast Track status from FDA in 2012 | Achillion Pharmaceuticals | - | |
| Telaprevir | ChEMBL231813 | Approved 2011 | Vertex Pharmaceuticals and Johnson & Johnson | Rash, anemia, leukopenia/neutropenia [44] | |
| Vaniprevir | ChEMBL599872 | Approved Japan 2014 | Merck & Co. | Diarrhea, nausea [45] | |
| DPP-4 inhibitors | Alogliptin | ChEMBL376359 | Approved 2013 | Takeda Pharmaceutical Company | Increased risk of heart failure [46] |
| Anagliptin | - | Approved in Japan 2012 | Sanwa Kagaku Kenkyusho | - | |
| Dutogliptin | - | Phase III | Phenomix Corporation | Not known yet | |
| Evogliptin | ChEMBL1779710 | Approved 2015 | Dong-A ST | Headache, nasopharyngitis, upper respiratory tract infection [47] | |
| Gemigliptin | - | Approved 2011 | LG Life Sciences | Generally well tolerated | |
| Gosogliptin | - | Approved in Russia 2016 | Pfizer | Hypoglycemia [48] | |
| Linagliptin | ChEMBL237500 | Approved 2011 | Eli Lilly and Company and Boehringer Ingelheim | Angioedema, pancreatitis, joint pain [49] | |
| Melogliptin | - | Phase III | Glenmark Pharmaceuticals and Merck KGaA | Not known yet | |
| Omarigliptin | ChEMBL2105762 | Approved in Japan 2012 | Merck & Co. | Generally well tolerated | |
| Saxagliptin | ChEMBL385517 | Approved 2009 | Bristol-Myers Squibb; AstraZeneca | Upper respiratory tract infection, may cause joint pain [49,50] | |
| Sitagliptin | ChEMBL1422 | Approved 2006 | Merck & Co. | Headache, swelling of the legs, upper respiratory tract infection [51] | |
| Teneligliptin | - | Approved in Japan 2012 | Mitsubishi Tanabe Pharma | Generally well tolerated | |
| Trelagliptin | - | Approved in Japan 2015 | Takeda | Generally well tolerated | |
| Vildagliptin | ChEMBL142703 | Approved 2007 | Novartis | Nausea, hypoglycemia, headache, dizziness [52] | |
| α-Thrombin inhibitors | Argatroban | ChEMBL1166 | Approved 2002 | Eagle Pharmaceuticals | Bleeding from the bladder, blurred vision, chest pain, dizziness, fever [53] |
| Inogatran | ChEMBL114715 | Approved 2016 | AstraZeneca | Well tolerated | |
| Melagatran | AstraZeneca | ||||
| Dabigatran | ChEMBL539697 | Approved 2010 | Boehringer-Ingelheim | Gastrointestinal [54] | |
| Apixaban | ChEMBL231779 | Approved 2014 | Bristol-Myers Squibb and Pfizer | Bleeding, bausea [55] | |
| Betrixaban | ChEMBL512351 | Approved 2017 | Millennium Pharmaceuticals, Merk | Bleeding [56] | |
| Edoxaban | CHEBI:85973 | Approved 2015 | Daiichi Sankyo | Stomach ache, abnormal results of blood tests that measure liver function, anemia [57] | |
| Otamixaban | - | Ended at phase III | Sanofi | - | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eleftheriou, P.; Amanatidou, D.; Petrou, A.; Geronikaki, A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules 2020, 25, 2529. https://doi.org/10.3390/molecules25112529
Eleftheriou P, Amanatidou D, Petrou A, Geronikaki A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules. 2020; 25(11):2529. https://doi.org/10.3390/molecules25112529
Chicago/Turabian StyleEleftheriou, Phaedra, Dionysia Amanatidou, Anthi Petrou, and Athina Geronikaki. 2020. "In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus" Molecules 25, no. 11: 2529. https://doi.org/10.3390/molecules25112529
APA StyleEleftheriou, P., Amanatidou, D., Petrou, A., & Geronikaki, A. (2020). In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules, 25(11), 2529. https://doi.org/10.3390/molecules25112529