1. Introduction
Gas sensing is an essential and crucial facet of cell signaling, regulating a vast array of cellular and physiological functions including respiration, cytoprotection, gas storage, neurotransmission, inflammation, gene transcription and cardiovascular homeostasis [
1,
2,
3,
4]. The binding of small diatomic gas molecules such as nitric oxide (NO), carbon monoxide (CO) and oxygen (O
2) to intracellular sensing proteins triggers a conformational change that leads to versatile changes in the cellular metabolism [
2,
5].
NO is a key signaling molecule of human physiology which activates its receptor, soluble guanylate cyclase (sGC) [
6]; impairment of the sGC-based signaling cascade has been linked to cardiovascular, neurodegenerative and inflammatory diseases. Nitroglycerol, a compound capable of releasing NO, has been used for more than a century for the treatment of angina pectoris, although its mode of action was not understood for long. This example highlights the therapeutic potential of small signaling molecules, but their clinical use has remained limited due to their gaseous nature, extensive reactivity, short half-life and systemic toxicity [
7,
8]. While small-molecule NO precursor compounds offer improvements over the direct delivery of NO, the practical application of these NO donor compounds remain hindered by their inability to target NO to a specific site [
7]. Consequently, therapeutic use of gases like NO/CO/O
2 requires specific delivery systems.
Selective binders can be used for trafficking gaseous ligands: H-NOX proteins (named Heme-Nitric oxide and OXygen binding domain) are one of the six major groups of heme sensor proteins whose bindings with the gaseous messengers cause changes in the downstream effector proteins, evoking various responses [
9]. H-NOXs were already shown to be able to deliver O
2 to hypoxic regions of solid tumors [
10] and accumulate as a multimeric pattern in multiple intracranial glioblastoma models, thus improving the efficacy of radiation therapy [
10,
11].
However, in order to fully exploit the capacity of H-NOX proteins for delivering ligands, their selectivity and affinity need to be maximized. This requires a clear understanding of how ligand differentiation and affinity are governed by the protein matrix surrounding the heme. H-NOXs have a remarkably stable fold with moderate sequence identities (~30–35%), allowing for optimizing cavity structure and polarity for fine-tuning ligand preference—if the principles of such were clarified.
Coordination of gaseous ligands to the Fe (II)-protoporphyrin IX unit is governed by backbonding between the metal d and the ligand antibonding π* orbitals [
12], resulting in an affinity ordering of NO > CO > O
2 with K
D values of 1:~10
3:~10
6 commonly referred to as the sliding scale rule [
13]. This affinity ordering, however, can eventually be modulated by the surrounding protein matrix. Further complication arises from the fact that these ligands bind to the heme group in a spin-forbidden process: in the course of the chemical binding step, the high-spin (quintet, with four unpaired electrons) ground state of the five-coordinate iron(II) system is converted to a low spin, six-coordinate system: doublet (one unpaired electron) in the case of NO and singlet (all electrons are paired) in the case of O
2 and CO. The inversion of spins is formally forbidden and has a profound effect on the kinetics of binding [
14,
15].
Distal pocket size, shape and polarity are—unquestioningly—also distinct determining factors in ligand binding (in the case of heme proteins, the pocket containing the amino acid that directly anchors the heme to the protein matrix is called the proximal pocket, while the usually more spacious ligand-binding cavity at the opposing side of the heme is called the distal pocket). However, the polarity of the diffusing ligands is just the opposite of that of the bound ligands. Free NO has the largest dipole moment of the three, while O
2 is completely apolar; but in contrast, iron-bound NO carries minimal charge on the oxygen, while O
2 ligated to the heme is decidedly polarized. Thus, a distal pocket that is welcoming for the incoming gas molecule will necessarily be less apt to stabilize the ligand once it is coordinated to the heme iron. For example, the apolar pocket of facultative anaerobes like H-NOX from
Nostoc sp. (
Ns) or sGC is ideal for stabilizing O
2 the gas molecule, but does not support the formation of stable Fe-O
2 adducts. In contrast, the formation of a stable Fe-O
2 bond can be observed in systems having a close reaching proton donor that is capable of stabilizing the partial negative charge appearing on the terminal oxygen once the Fe-O
2 bond is formed. Examples include histidine in globins, or tyrosine in the case of obligate anaerobe H-NOXs from
Caldanaerobacter subterraneus (Cs) (formally known as
Thermoanaerobacter tengcongensis (
Tt)) or
Clostridium botulinum (Cb) [
16]. However, again, the distal pocket in itself cannot be the sole determinant of the ligand-binding process, since, for example,
Cb and
Cs H-NOXs, having the same residues reaching closest to the heme bound ligand, show characteristically differing affinities toward O
2 with a K
D(O
2) of 5.3 × 10
−5 M, K
D(O
2) = 4.4 × 10
−8 M, respectively [
17].
The proximal pocket, and especially the conformation and conformational freedom of the proximal residue as well as its basicity also have a direct effect on the reactivity of heme iron. In the case of sGC, for example, the proximal strain was shown to reduce affinity for O
2 [
18], which in itself might be a formidable contribution to the mechanism of O
2 exclusion, and the NO-bound system is predicted to behave very differently from other related systems [
19].
The tunnel system interlacing the entire protein matrix is the primary route of ligand entry to the binding site of H-NOXs and as such is of special importance in the design of ligand transport systems. However, controversial results have been obtained concerning the significance of the various diffusion routes. On the experimental side, xenon (Xe) saturation experiments in conjunction with X-Ray diffraction studies found bifurcated Y-shaped tunnels in
Ns H-NOX, but no continuous pathway connecting the heme site and the solvent in
Cs H-NOX showing that the fold of the protein is not the only determining factor of the ligand migration mechanism [
20]. In contrast, based on the shape of the molecular surfaces, Wu et al. proposed that the major and most significant route for ligand entry to the heme group in all H-NOXs is the wide gateway in the plane of the heme group on the side of its propionates [
9]; however, no such route was identified in the case of
Cs H-NOX by molecular dynamics simulations [
21]. These studies demonstrated that ligand access does not necessarily require additional channel systems to connect the heme site and the solvent, and that the different protein matrices of H-NOXs, in spite of the identical fold, possess different cavity structures, in which differing sidechain compositions lead to the formation of pockets of distinct positions, sizes and polarities, which could be important for their divergent functions.
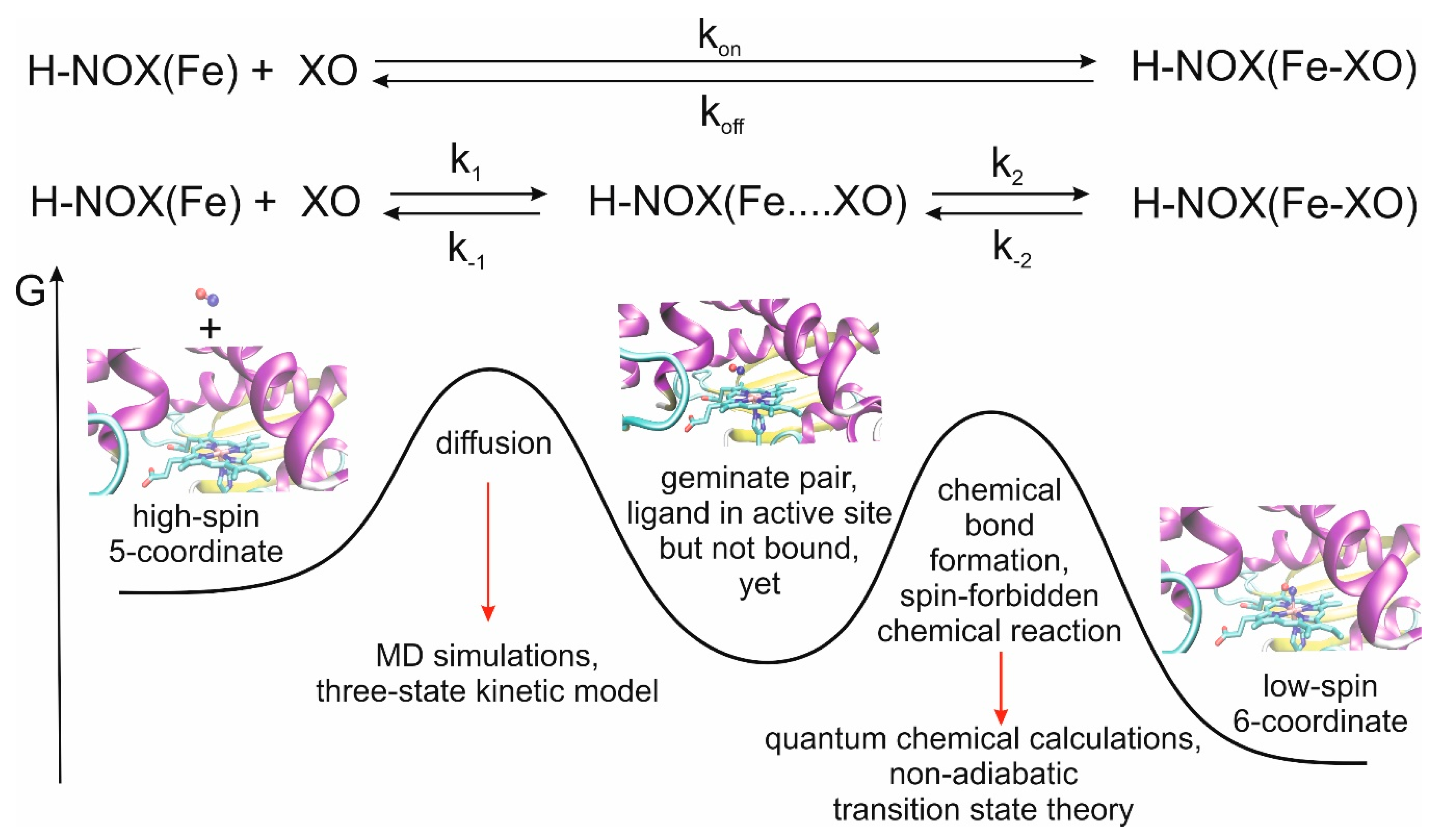
We recently developed a multiscale model for the theoretical description of the CO and NO ligand-binding events of myoglobin, another histidine-ligated Fe(II)-heme protein and obtained very good agreement between calculated and measured rate constants [
22]. A meaningful characterization of the overall process can be obtained by using a classical approach for the study of ligand diffusion and, in a separate step, quantum mechanical approach for bond formation between the ligand and the iron(II) center, which is a significant factor for the stabilization of the ligand in the distal pocket (
Figure 1).
Here, we consider the first step of NO, CO and O2 binding of different bacterial H-NOXs and analyze in detail the diffusion kinetics, access and exit routes, and the cavities that may serve as traps or reservoirs for various ligands—to provide guidelines for the design of selective and effective transport machinery for diatomic gases, but do not consider the formation of the covalent bond between the ligand and the Fe ion. More specifically, we aim to answer the following questions: (1) How similar are the kinetics of the diffusion of the various gases in the studied proteins? (2) Is it sufficient to consider the direct route (opening at the heme propionates) for ligand entrance/exit OR should the tunnel system be also taken into account? (3) Are the channels specific to the fold (practically identical in all H-NOXs) OR matrix specific? (4) Are the ligand-binding pockets selective toward specific ligands, and if so, is this selectivity created by the local sidechain pattern or the global fold? (5) What kind of guidelines can be given for the design of NO/CO/O2-specific H-NOX transport systems in terms of pockets/cavities?
We chose three bacterial H-NOXs (
Ns,
Ka and
Cs) for our study that differ in their ligand specificity and flexibility.
Ns is an example of a facultative anaerobe, thus a selective NO binder.
Cs is an obligate anaerobe that binds O
2 with large affinity and qualifies as an O
2-sensor [
17], although it can also bind CO and NO.
Ka is another example of facultative anaerobe microbes, however, it contains a unique glycine-rich hinge region, that plays a major role in flexibility and protein-protein interactions [
23].
In order to answer the questions, we carried out 300–500 ns long molecular dynamics (MD) simulations on the three bacterial H-NOXs in solutions containing O2 or NO or CO or an NO-O2 mixture. We determined the rate constant for diffusion, analyzed the sensitivity of protein structure to ligand diffusion and identified diffusion routes and ligand storage pockets, and compared the various systems to get an overall picture of the major factors influencing the ligand transport properties.
2. Results and Discussion
First, we are going to describe our results obtained on the kinetics of the diffusion followed by the investigation of the tunnel systems and of the cavity structure of the studied proteins.
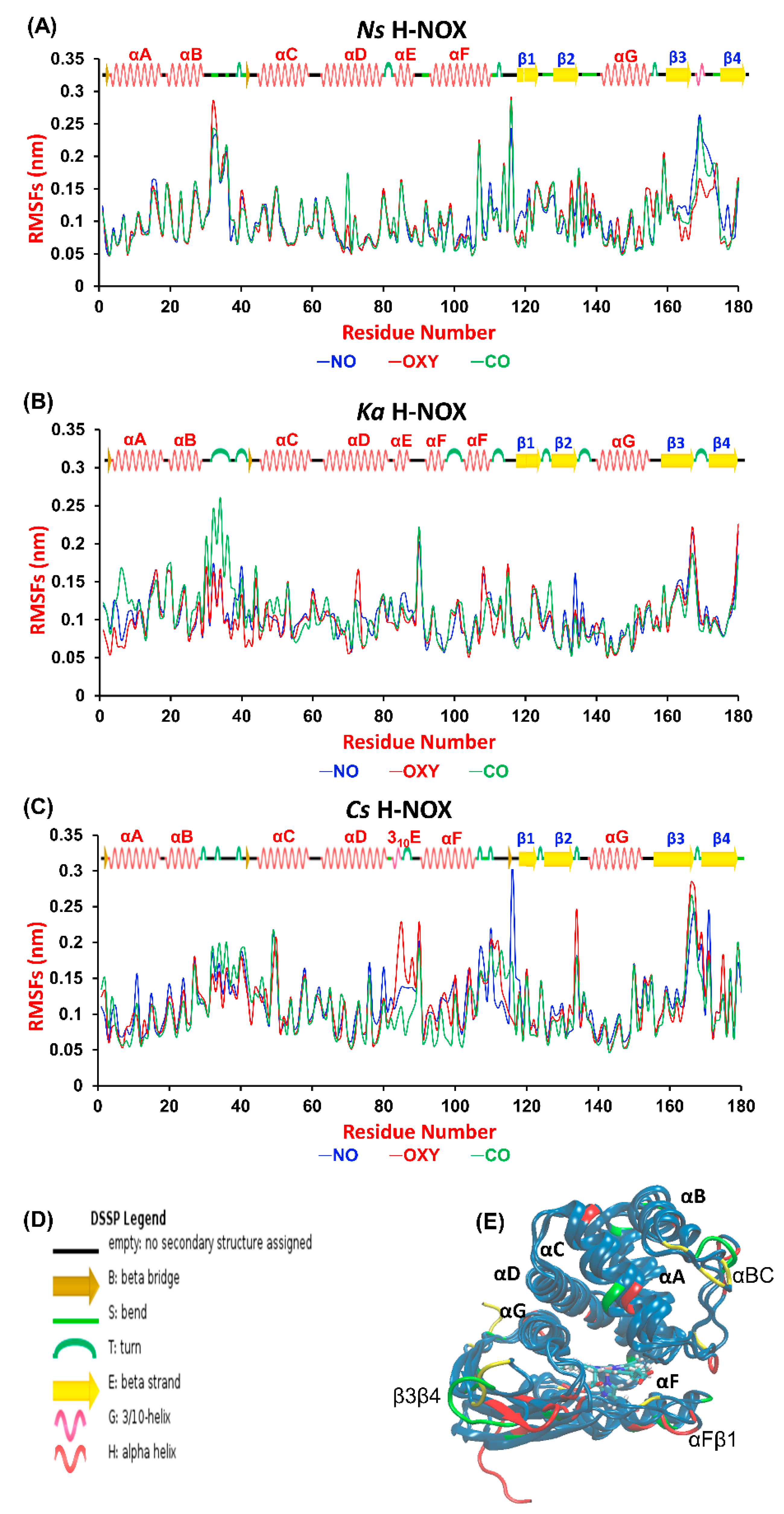
2.1. Structural Response of the Protein Structure to Gas Ligand Migration
A wide variety of experimental techniques (e.g., site-directed mutagenesis, EPR and Xe-binding X-ray diffraction) have been exploited to describe gas migration in the protein matrix [
20,
24,
25,
26]. Theoretical approaches, such as molecular dynamics (MD) simulations can provide complementary information on the dynamics of gas permeation through the protein matrix to obtain a full molecular-scale understanding of ligand transport [
21,
22,
26,
27]. First, we aimed at identifying those regions of the proteins that are most sensitive to gas ligand permeation. Therefore, we calculated the root mean square fluctuation (RMSF) per-residue averaged over time along the trajectories and plotted it against the residue number (
Figure 2A, overall RMSD values are given in
Table S1 in the Supplementary Materials). The most fluctuating regions (>0.15 nm) are identical for the three proteins: the αBC loop, the αFβ1 loop and the β3β4 loop, which connect the corresponding helices and β-sheets (see
Figure 2C). These elements have been suggested to be catalytically significant, possibly regulating the entrance gate for gas ligands [
21,
28]. Our results do not show any systematic difference between the various systems, although we could identify sensitive regions in each protein to gas migration. In
Ns ARG107 in αF seems to respond to the presence of all ligands, as its RMSF value exceeded 0.2 nm in all cases. Furthermore, the turn between β3 and β4 is very sensitive to the presence of CO and NO in
Ns, but not to that of O
2. The flexibility of the tiny 3
10E helix, which is only found in
Cs—the only oxygen sensor protein studied herein—increases considerably in the presence of oxygen. Out of the three proteins
Ka H-NOX seems to exhibit the smallest flexibility, but its loop connecting αB and αC responds strongly to the presence of CO.
2.2. Diffusion Rate Constants for Gas Ligand Migration
After studying the response of the overall protein structure to the presence of gas ligands, we wanted to see whether any systematic difference exists between the rate constants for diffusion of the various gases in the three proteins. Due to the extended timescales needed for the modeling of the diffusion process, its realistic study only became possible since the appearance of GPU-accelerated MD simulations techniques. In recent years, reasonable rate constants for ligand diffusion to myoglobin [
29], and to nitrogenase/hydrogenase enzymes [
30,
31,
32] have been calculated.
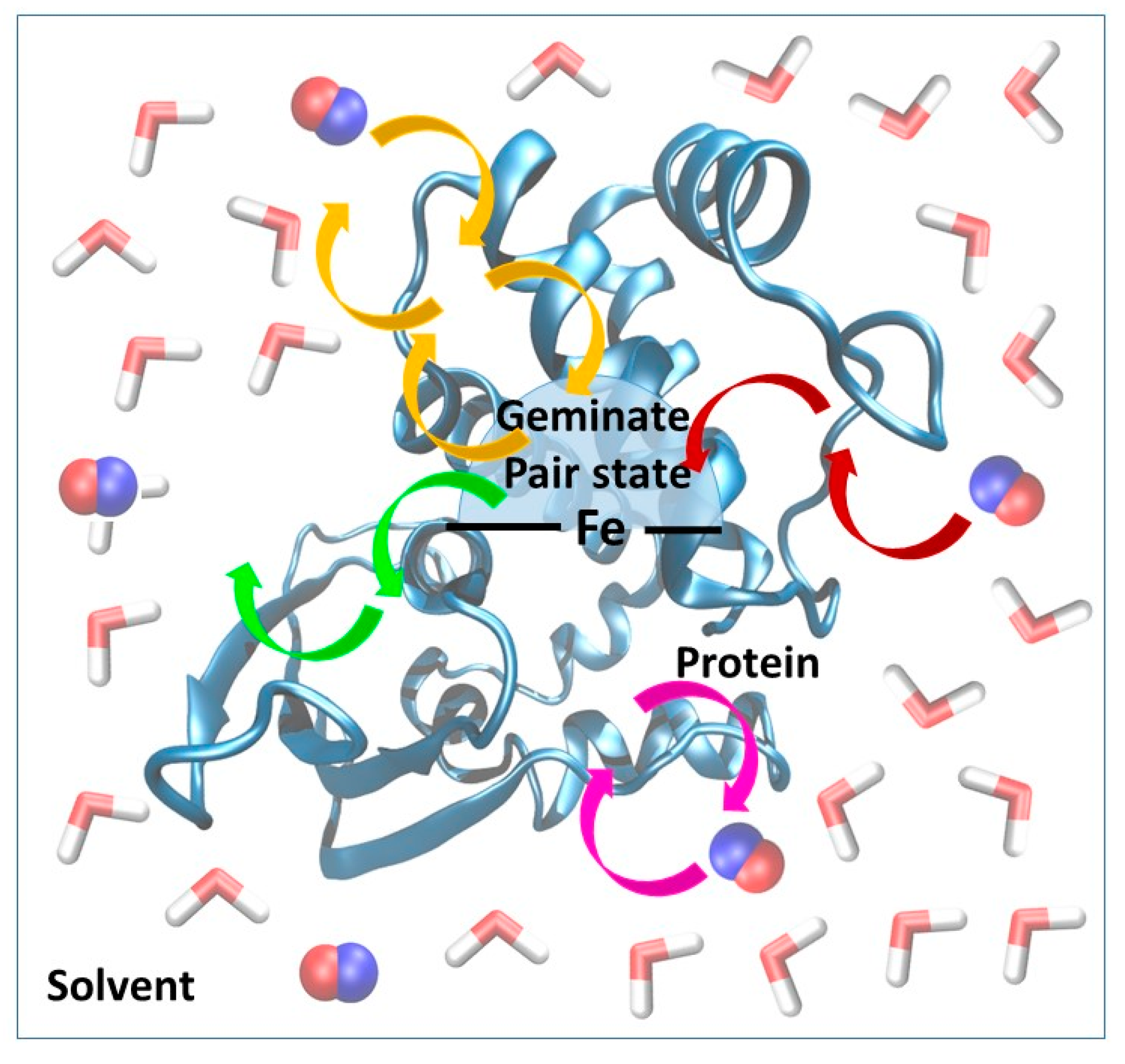
We recently developed and validated against experimental results a three-state kinetic model for the study of ligand diffusion [
22] that is much simpler than the complex Markov model used by Blumberger et al. [
30,
31,
32]. The model groups the instantaneous position of the ligands into three different categories: in the solvent phase, protein matrix or distal pocket (
Figure 3). During diffusion, ligand molecules enter the protein matrix from the solvent and gradually reach the distal pocket and could also return from the distal pocket to the solvent via the protein matrix. We call this a global in-and-out event. In equilibrium, the number of molecules reaching the active site from the solvent and returning to the solvent is equal per unit time. Thus, from the number of global in-and-out events and the length of the trajectory, the rate constants for diffusion into (k
1) and out (k
−1) of the protein can be derived. The equations used for the calculation and the definition of the various states (protein, distal pocket and solvent) are described in more detail in the method section. Naturally, the higher number of events observed, the more reliable the derived rate constants are. To this end, in the simulations, we used a relatively high gas concentration to ensure better sampling, as it was shown not to affect the rate constant values up to a concentration of about 0.5 M [
26]. Furthermore, the reliability of the observed events can be checked by comparing trajectory sections: if the equilibrium has been reached one should observe twice as many events in 200 ns than in 100 ns, and the rate constants should be identical. This is what we did, and observed less than 15% variability in the rate of constant values.
Table 1 summarizes the calculated rate constants for diffusion as well as the experimental data for ligand binding. It is worth noting at this point that the experimental numbers can only be compared to the calculated results after careful consideration of the reaction mechanisms. In experiments, k
on and k
off are usually determined, which include both ligand migration and chemical bond formation. In contrast, here, we only address the ligand migration step (k
1 and k
−1, see also
Figure 1). Depending on the relative values of k
1, k
−1, k
2 and k
−2, the bottleneck of the reactions can be determined. For example, in the case of myoglobin, we showed that diffusion is rate-limiting in the case of NO binding, while for CO binding both processes are partially rate-limiting and influence the overall value of k
on [
22]. In contrast to binding, bond scission is the rate-limiting step of ligand release in all cases, as a consequence, no agreement between k
−1 and k
off can be expected.
First, let us consider the reliability of our obtained rate constants. The case of NO binding provides an excellent opportunity for validation: it is diffusion-limited, as was suggested both by experiments and our own calculations on myoglobin. As a consequence, the overall rate constant is determined by the diffusion process, thus kon = k1. Indeed, we find good agreement between calculated k1 and experimental kon values: 6.5 × 108 M−1 s−1 vs. 6.5 × 108 M−1 s−1 for Ns and 3.8 × 107 M−1 s−1 vs. 1.5 × 108 M1 s−1 for Cs, respectively. In the case of CO, the second step is surely partially rate-limiting which makes a direct comparison impossible, while O2 only binds to Cs.
After having seen that the model is capable of yielding results in agreement with the experiment, we can compare the behavior of the various proteins and systems. A clear trend can be observed: the rate of ligand diffusion into the protein (k1) decreases in the O2 > NO > CO in all proteins, which most likely originates from the increasing size of the ligands and possibly increasing polarity. Contrarily, the k-1 values show no such trend arising from the fact that k−1 is influenced by the overall time that the ligand spends in the distal pocket, and this value varies a lot by protein and gas molecule. For example, the nonpolar distal pocket of Ns seems to stabilize O2 efficiently showed by the large occupancy ratio (in 91 ns out of the 300 ns long trajectory the pocket was occupied by O2), and as the polarity of the gas increases the time spent in the pocket decreases, in accordance with our expectations. The active site of Ka H-NOX prefers NO and that of Cs has an overwhelming preference for O2, again; this could be related to the O2-sensing character of Cs and the presence of a hydrogen-bond donor (TYR140) in the distal pocket.
If we analyze the behavior of a certain gas towards the various proteins, we see that the rates decrease in the
Ns >
Ka > myoglobin >
Cs order. We note that the sampling of the in-and-out event is quite low for
Cs which strongly suggests that migration into this protein is the slowest but also warns that the numbers should be treated with caution. The effect might be linked to the number of salt bridges found in the various systems.
Cs is a heat resistant microbe and its H-NOX domain contains 20 salt bridges, significantly more than the five salt bridges found in H-NOXs of
Ns and
Ka. In addition, the smaller volume of the distal pocket of
Cs compared to
Ns [
21] and
Ka, and the presence of hydrogen bond triad found in
Cs (H-bond of Trp9 and Asn74 to Tyr140 [
16,
21,
33]) may contribute to the computationally predicted slower diffusion into
Cs, which is in agreement with the slightly lower k
on value of NO in
Cs than in
Ns.
Until now, we have focused on the diffusion of a certain gas into a certain protein, but in reality, different sensor molecules might face a mixture of gases at the same time. We wondered if the presence of competitor molecules affects the diffusion properties of gases. This is an especially important question in the case of the human H-NOX protein, sGC, as it selectively binds NO even in the presence of multiple magnitudes of excess O
2. Therefore, we carried out MD simulations in the presence of solutions containing both NO and O
2. According to the best of our knowledge, this has not been done before. In this mixture, the concentration of the individual gases was half of that in the single-gas simulations, as we wanted to keep the overall gas concentration constant. Therefore, we expected half as many events per unit time compared to the one-gas simulations. We observed (see
Table 1) that the value of k
1 is not particularly sensitive to the presence of the other gas; in contrast, k
−1 and the equilibrium constant K are strongly affected. This arises from the fact that the gases compete for the distal pocket and strongly influence each other’s residential time. Here, we can clearly observe that in the case of
Ns H-NOX, if both O
2 and NO are present, the preference for O
2 decreases, compared to the single-gas simulations. Unfortunately, for
Cs, we did not manage to observe any reasonable number of global in-and-out events that made the derivation of the rate constant values impossible. This probably arose as a consequence of several factors: (1) diffusion is slowest to
Cs out of the three proteins; (2) we used a lower individual gas concentration than in the one-gas simulations; (3) as it will be shown later, in the case of
Cs, the widest tunnel that could most easily channel various ligands at the same time is blocked, and therefore, gases, forced to choose from fewer possible inlets, especially hinder each other’s diffusion in this system.
2.3. Gas Migration Routes in H-NOX Proteins
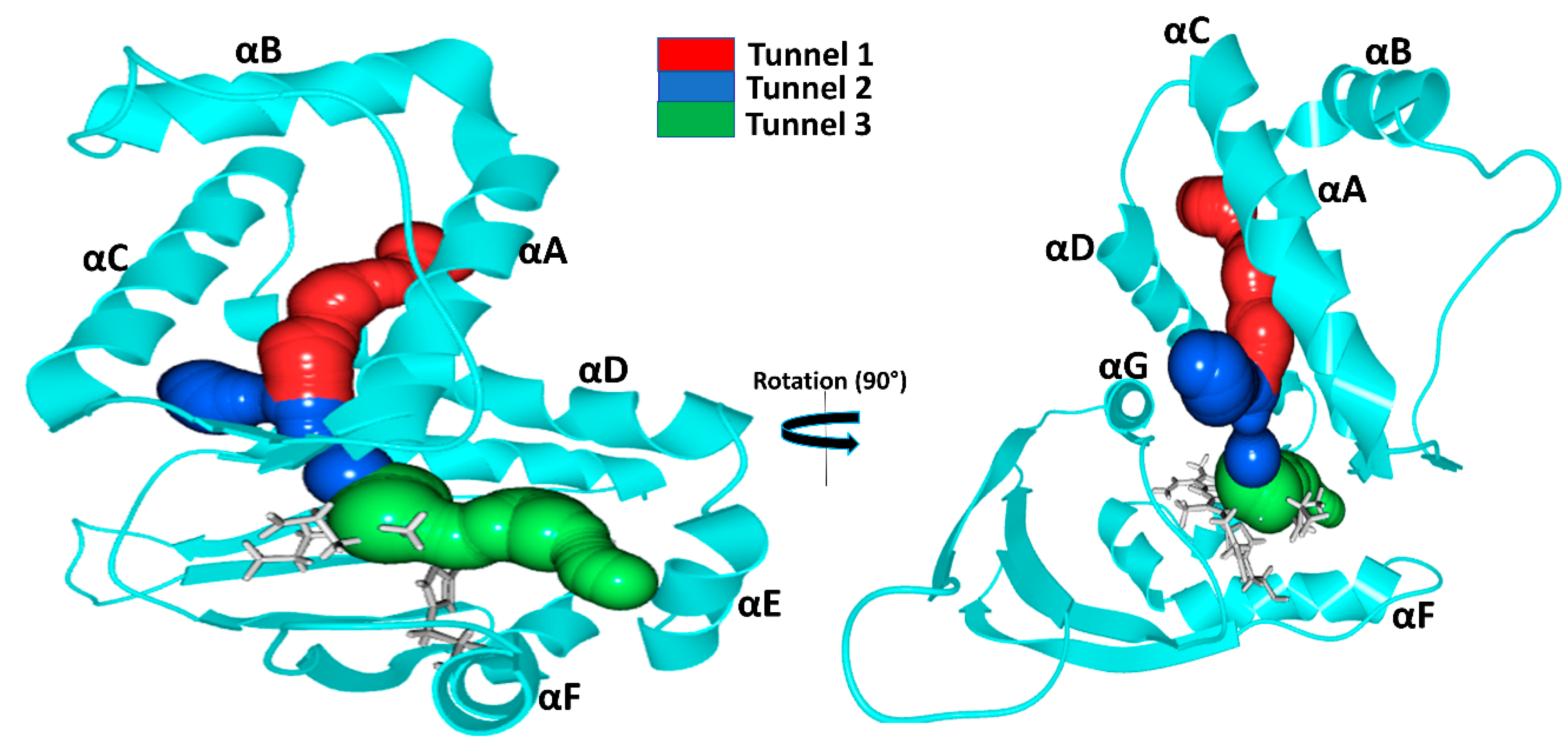
The tunnel system and protein dynamics together profoundly determine the routes taken by gas molecules from the solvent to the distal pocket. To get a clear picture, we visually inspected the inward/outward transport events that had previously been identified in our MD simulations and summarized in the previous chapter. We found three main diffusion routes that seem to be determined by the overall fold of the H-NOX proteins. Interestingly, each protein seems to use only two of the three possible routes and very rarely the third (
Figure 4).
Tunnel 1 extends between helices αA and αD from the solvent to the distal heme pocket. It is primarily composed of the sidechains of hydrophobic amino acids (residue numbers for
Ns: VAL5, ILE9, MET12, ILE13, THR48, VAL52, LEU66, LEU67, PHE70 and LEU141) and has been previously suggested by Xe-binding experiments [
20].
Tunnel 2 is the shortest route to the heme lined by helices αC, αD and αG and tunnels the gas ligand directly from the external solvent environment towards the heme propionic moiety and then to heme-FE. It is primarily bordered by polar residues (residues numbers for
Ns: MET1, THR48, TYR49, LEU67, ARG138, CYS139, GLY140 and LEU141). These routes intersect at LEU67 position forming a bifurcated Y-shape channel leading to the heme-binding cavity [
21].
Tunnel 3 is bordered by three helices: αA, αD and αF (
Ka: ILE4. VAL77, ILE78, SER81, TYR82, VAL107 and TYR111).
In the case of
Ns H-NOX, we observed, in accordance with previous results, ligand migration primarily through
Tunnels 1 and
2 (
Figure 4).
Tunnel 3 is blocked by the bulky residue TRP74 bordering the distal pocket: the indole ring of TRP74 is perpendicular to the heme plane, creating enough steric hindrance to hamper gas access. Additional hindrance arises from the hydrogen bond between TRP74 and heme group (see
Figure S1 in the Supplementary Materials). In silico mutation of TRP74 to ASN was shown to open this tunnel for ligand migration as well [
21].
Ka H-NOX is the least studied among crystallized H-NOXs. Our results show that ligands primarily use
Tunnels 1 and
3 for migration. Similarly to
Ns, it has an apolar elongated access tunnel (
Tunnel 1: ILE5, PHE9, LEU12, VAL13, PHE17, MET48, LEU52, LEU66, LEU67, THR69, TYR70, MET139) and similarly to
Cs a slightly polar short route lined by αA, αD and αF (ILE4. VAL77, ILE78, SER81, TYR82, VAL107 and TYR111 see
Figure 4) leading to one side of the heme group. For this system, the direct
Tunnel 2, previously suggested to be universal for all H-NOXs, was not observed [
9]. Closer inspection showed that this tunnel is blocked at both ends in
Ka. At the entrance from the solvent phase, PHE45 establishes a cation–π with the vertically aligned ARG137, and the exit of the tunnel at the distal pocket is blocked by two interacting methionine residues (MET 1and139) (see
Figure S2 in the Supplementary Materials).
In
Cs H-NOX ligands primarily move through
Tunnels 2 and
3 (
Figure 4), but not the longer, apolar tunnel (
Tunnel 1). Both of these routes are short and provide easy access to both the distal and the proximal sides of the heme. These tunnels may play a major role in the reactivity of sGC [
36],
Shewanella oneidensis (
So) H-NOX [
9] and
Vibrio cholera (
Vc) H-NOX for which ligand-binding to the proximal side has also been suggested.
Tunnel 1, the long apolar tunnel has been visited by ligands at its entrance towards the solvent phase; however, the exit to the distal pocket remained fully blocked by a hydrogen bonding triad (TYR140 with sidechains of TRP9 and ASN74). Earlier in silico efforts to open this tunnel by double mutation (TYR140PHE and ASN74LEU) of the residues failed. When we further examined this tunnel, we found that there are two bulky residues along this tunnel: TRP9 and TRP67 forming extensive hydrophobic interactions, and we speculate that their presence also significantly contributes to the blocking of the tunnel beside the hydrogen-bonded triad (see
Figure S3 in Supplementary Materials).
Comparison of Ka with Cs and Ns H-NOXs systems clearly shows that the overall fold primarily determines the tunnels, but the specific protein matrix has a significant effect on their accessibility, through hydrogen bonding networks, steric hindrance, cation–π interactions or a combination of these. Potentially salt bridges may block tunnels as well, but we could not find any direct proof for this in our systems.
Our results also contradict the suggestion that the major place of ligand entry is the entrance at the propionic groups of the heme group in all H-NOXs. We clearly see that it is not the length of the tunnel that primarily determines the rate constant for diffusion, much more so the spaciousness of the tunnel and the presence of spacious pockets along the route which provide opportunities for ligands to pass next to each other similarly to railroads with one track.
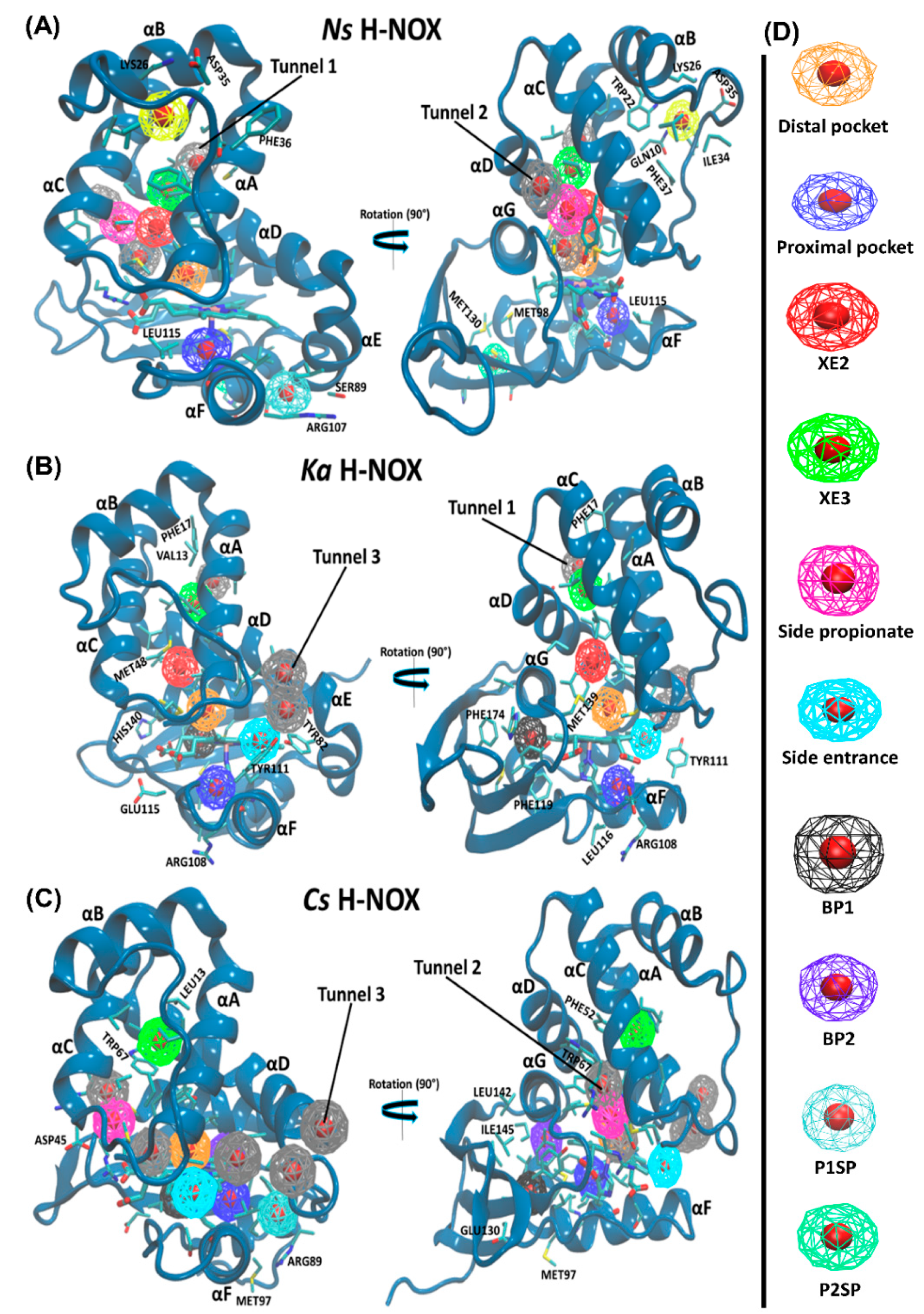
2.4. Pockets and Cavities Identified in H-NOX Proteins
There are two major types of protein concavities: pockets (in principle found on the surface, but the distal pocket is buried) and cavities (usually buried). However, considerable controversy is found in the literature regarding the naming; therefore, we will call all concavities “pockets”. Pockets play a significant role in biological functions such as ligand-binding and trapping. The active site (or in the case of heme proteins the distal pocket) is usually in the center of interest; however, other cavities/pockets may play a role in gas storage. Therefore, we first obtained global descriptors of the interactions between gas molecules and the protein matrix (see
Table 2), then the properties of individual pockets have been analyzed (selected properties are given in
Table 3, while an extensive comparison of the pockets for all systems are given in the
Supplementary Materials in Tables S2–S4).
The average number of gas molecules found in the protein matrix shows distinct differences between proteins: the capacity to store gas molecules decreases in the Ns > Ka > Cs order, and this is most pronounced for NO. It is in accordance with the native function of the gas sensors: Ks and Ns are NO-sensors that do not coordinate oxygen. For these two proteins the amount of gas stored in the protein decreases in the NO > O2 > CO order, but interestingly, and possibly related to its O2-sensor function, Cs H-NOX has a preference for O2. Similar conclusions can be deduced from the average number of protein contacts summed over all gas molecules. A higher number of protein contact/gas molecules implies that gas molecules are trapped in pockets more buried within the matrix, which is highest in the case of Cs. Moreover, we found that the ratio between the number of protein contacts to a single-gas molecule was higher for Cs H-NOX than that of Ka and Ns H-NOXs which suggest the presence of less spacious pockets and possibly higher packing density.
We quantitatively analyzed the accessible-binding spots positioned through the three gas migration routes (
Table 3). Altogether eight pockets have been identified for
Ns H-NOX, six for
Ka and seven for
Cs H-NOX. For the trajectories of NO-diffusion, we have added the maximum and the average number of gas molecules in the pockets as well. It is obvious that
Ns and
Ka H-NOX have several spacious pockets that can accommodate several ligands at the same time, while
Cs has only smaller pockets. This is in line with our previous proposition that wide spots facilitating ligand exchange in the two directions are needed for faster diffusion. It is also worth estimating the average number of gas molecules found in the pockets (thus summing the numbers in
Table 3) and comparing them with the average number of gas molecules in the various proteins in
Table 2. The comparison of NO molecules shows a larger difference for
Ns: ~10 vs. 17.6 and for
Ka: ~9 vs. 16.4, but a reasonable agreement for
Cs: 7.2 vs. 9.4. The question arises where the molecules could be found that are not in the identified pockets. We believe that molecules temporarily can access other parts of the protein but do not reside there long enough to call those parts pockets (e.g., the route towards the buried pocket in
Ka) or they can be temporarily associated with the surface of the proteins, but quickly changing position.
Let us turn our attention towards the identified pockets. Along
Tunnel 1 (lined by αA and αD) in
Ns and
Ka, two hydrophobic binding spots have been identified that are not only shared by all three gas molecules of this study, although with differing occupancies, but were confirmed as binding sites by Xe trapping experiments (Xe2 and Xe3) in the case of
Ns H-NOX (PBD id: 3TFA and 3TF9). A polar entrance spot is associated to
Tunnel 2 (lined by αC, αD and αG) in
Ns and
Cs, which directly connects to both the proximal and distal pockets, and was also confirmed experimentally (in the case of
Cs H-NOX (PBD id: 3TF1, “Xe2”)).
Tunnel 3, similarly to
Tunnel 2 is very short and it is lined by αA, αD and αF. It has been frequently taken ligands both in
Ka and
Cs H-NOX as shown by the data in
Table 3.
Ka and
Cs possess a buried spacious hydrophobic pocket situated between the distal and proximal lobes near the proximal pocket (formed by αF, αG and the proximal β-sheet). This spacious pocket (BP1) in
Cs is most frequently occupied by O
2 (69.5% of snapshots there is at least one oxygen molecule inside); neither NO, nor CO binds to this pocket. While in the
Ka system, BP1 was most selective toward the NO ligand (94.8% of the snapshots). Two small hydrophobic pockets (BP2 and P1SP) in
Cs H-NOX were demonstrated to be CO-specific with 99.3% and 84.4% of snapshots. BP2 is near the distal, while P1SP is near the proximal pocket. Another buried hydrophobic pocket has been recognized in
Cs H-NOX, which is mostly sampled by the NO ligand. Interestingly, this pocket contained one of the two Xe molecules trapped in
Cs H-NOX under 6 atm of pressure (PBD id: 3TF1, “Xe1”) [
20,
37].
Apart from the accessible-binding sites that are located along the diffusion routes, only the Ns H-NOX system displayed three surface pockets occupied by various gas ligands. These pockets are more polar than buried ones thus water molecules can also access them. Two (P1SP and P2SP) are in the proximal side, and another one is positioned at the distal side (DSP) lined by αBC loop, αB and αC. P2SP appeared to be NO-specific, trapping an NO molecule in 99.3% of trajectory frames.
In the
Supplementary Materials, in Tables S2–S4, we give detailed information on several parameters of ligand diffusion and binding. It is obvious that there is an inverse relationship between the time spent in the pocket and the number of in-and-out events. In most cases, ligands spend little time in pockets which can be easily accessed, and spend more time in pockets with hindered access. On average, the residential time is in the range of 20–200 ps, but in the case of the proximal pocket of
Ka, much longer time has been found: in the range of 2–8 ns for CO and NO. One might expect that buried pockets are better places for storing ligands in terms of residential time, but interestingly, this is not fully proven. In the case of the distal surface pocket of
Ns H-NOX, the residential time is 2–4 ns for CO and NO. This pocket is bordered by amino acid residues of various characters. Hydrophobic and/or bulky groups such as PHE36 and PHE37 are included as well as amino acids with positively or negatively charged sidechains such as LYS26 and ASP35. This suggests that composite pockets are better for storing ligands, which is also supported by the fact that the residential time of ligands in the non-polar cavities along
Tunnel 1 is significantly shorter both in
Ns and in
Ka H-NOXs (on average shorter than 150 ps).
2.5. Guidelines for Designing Gas-Transporting Proteins
We asked in the introduction whether guidelines could be given for the design of protein systems in order to transport gas molecules and whether such systems could be designed. The results discussed until now show that all of the studied H-NOX proteins are capable of storing a reasonable number of gas molecules at the same time. It is also apparent that the residential time of ligands in these spots varies greatly: from ~10 ps to 8 ns and that composite pockets including both charged residues and bulky groups are most suited for storing ligands for longer periods of time, thus this property is designable. However, would this be enough for the efficient transport of gases?
In order to answer such a question, let us consider the gas release by hemoglobin, which is the most prominent gas-transporting protein in the human body. The rate constant for the release of oxygen (k
off) of hemoglobin 1 is 61.1 s
−1 [
38]. Let us make a thought experiment and suppose that the rate constant is due to ligand release only from the pocket and no bond breaking takes place. How long residential time in a pocket would result in a rate constant of 61.1 s
−1? In order to answer this question, we could use Equation (5) of the method section and use the data given for
Ns H-NOX in the
Supplementary Materials. By considering the number of events to be one, and substituting Avogadro’s number, protein concentration (0.044 M), cell volume (3.78 × 10
−22 dm
3), we get 1.64 ms for the residential time, which is orders of magnitude bigger than the longest time we observed (around 8 ns).
Therefore, the major guideline for designing transport proteins is to increase the residence time of ligands in the pockets into the region of at least ms. This might be achieved by the selective design of the pockets, but the covalent linking of the ligand to the heme group could also be considered and creating e.g., fused proteins with several heme groups could increase the transport capacity, as was exploited in a recent patent [
39].
4. Conclusions
Gas sensing is a complex process that involves the migration of the gaseous ligand to the active site of the protein from the environment, the chemical bond formation and finally also ligand release. As gas sensing H-NOX proteins were suggested as suitable candidates for transporting gaseous molecules, we set out to investigate their dynamic response to the presence of gas molecules, focusing on the migration events of gaseous ligands into and out of the H-NOX protein matrices.
We compared three systems with decidedly different ligand affinities (and flexibility) in a comprehensive analysis and we concluded that the basic fold is quite similar and the flexible regions are also in the same segments in the three systems despite the low homology of their sequences. We could identify three conserved tunnels in all three proteins, the accessibility of which, however, differ in the three. Neither protein relies on a single short access route connecting the heme and the solvent, as was suggested earlier [
21], but utilizes at least two of the three tunnels.
We also showed—for the first time—that the rate of ligand migration follows the same ordering (O2 > NO > CO) in the three systems. Diffusion into the protein, characterized by k1 is much less sensitive to the specific protein matrix than ligand egress (k−1), the differences created in both the size and polarity of the cavities by the different sidechain composition of the three proteins significantly influence the kinetics of the diffusion out of the protein. This is nicely demonstrated by the oxygen ligand spending the longest time within the protein matrix of the oxygen sensing Cs protein while Ns and Ka, which sense mainly NO, are more effective in retaining both CO and NO than oxygen.
By modeling the gas-binding event using a gas mixture we were the first to show that they influence each other’s trapping in the distal pocket.
We have also shown that the overall rate of gas diffusion is influenced by the total number of intramolecular H-bond/salt bridges of an H-NOX protein: there are many more salt bridges in Cs H-NOX than in the other two systems and this has a remarkable effect on ligand migration. Actually, this is a very simple parameter, but has a profound effect and should be considered.
By searching for reasons for the selectivity differences between the oxygen sensing
Cs H-NOX and NO-sensing
Ka and
Ns H-NOXs we found that in the case of
Cs H-NOX the whole protein matrix seems to accommodate oxygen much better than the other ligands (as shown in
Table 2).
Cs H-NOX is the only protein matrix that slightly prefers oxygen over NO (or CO), while the other proteins show a very strong preference for NO. We found several factors that probably contribute to creating this difference.
Cs H-NOX is the most tightly woven protein matrix of the three, rigidified by a great number of intramolecular interactions, which creates narrower, less pliable channels and thus a preference for the smallest ligand: oxygen. Additionally, in
Cs H-NOX, the spacious, apolar
Tunnel 1 which would provide an ideal escape route for the apolar oxygen ligand is blocked, so O
2 is forced to enter and exit through the decidedly more polar side tunnels. Oxygen is fast to reach the distal binding spot in all three proteins, but in the case of
Ns H-NOX and
Ka H-NOX, it also leaves the distal pocket rapidly. In the case of
Cs H-NOX, its detainment provides enough time for oxygen—the weakest binder of the three studied gases—to chemically bind to the heme group.
Our results suggest that several properties of H-NOX proteins could be tuned when the design of gas transport proteins is considered. The highly conserved global fold creates rather similar tunnel systems in H-NOX proteins, but the accessibility of these is determined by the individual sequences—thus it is a designable property. We identified several ligand-selective pockets in the various protein matrices, demonstrating that the local environment can be tailored to enhance selectivity. We also showed that an intricate sidechain-based H-bond system could greatly hinder the movement of ligand molecules within the protein matrix. Our results indicate, that since the fold of H-NOX proteins is remarkably stable, it should be possible, through selecting or blocking channels and redesigning the nature of surface-close polar and buried apolar pockets, to create selective and effective gas transport vehicles based on the proteins of the H-NOX family, and the design of systems including various heme groups is advised for longer storage times.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}