Charge Accumulation of Amplified Spontaneous Emission in a Conjugated Polymer Chain and Its Dynamical Phonon Spectra

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
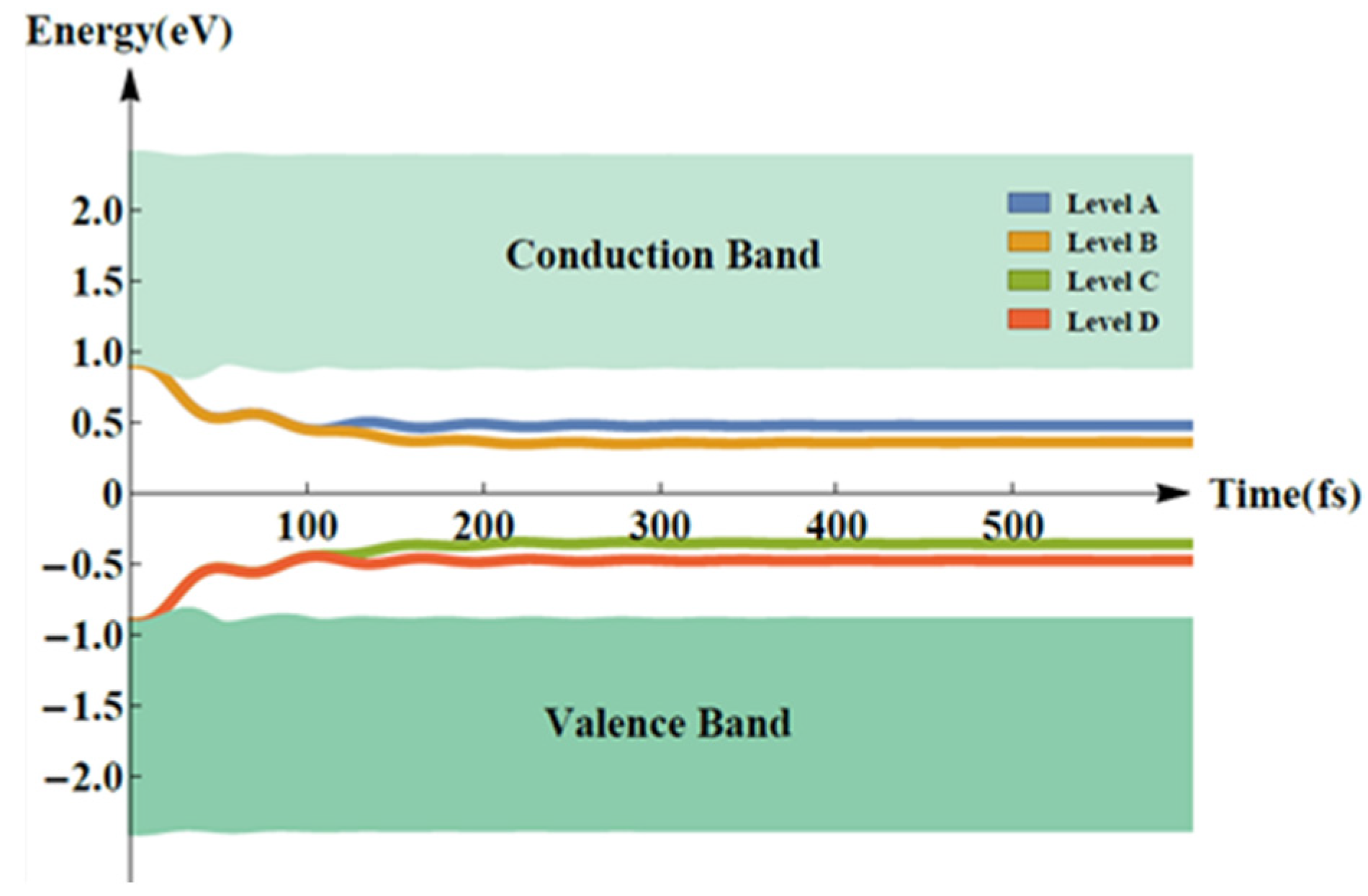
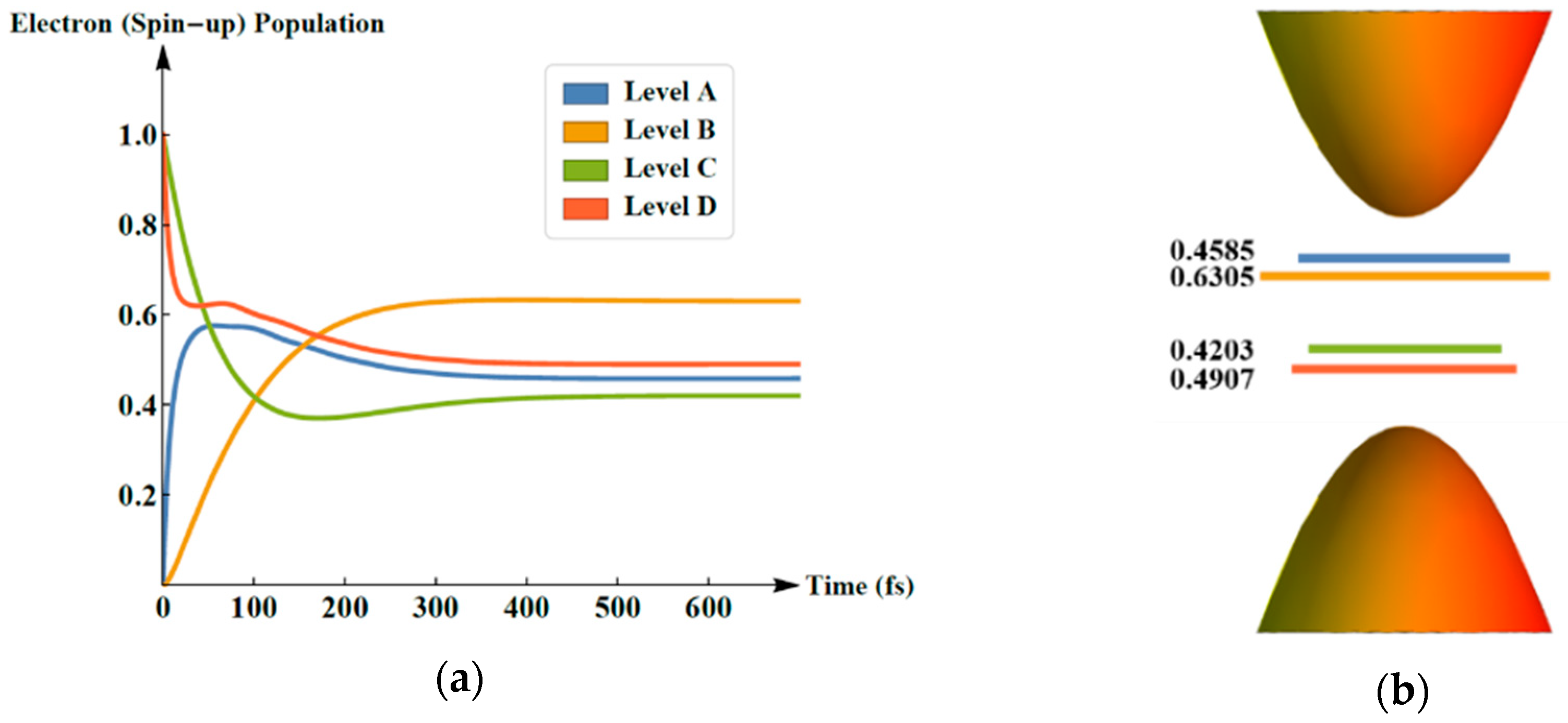
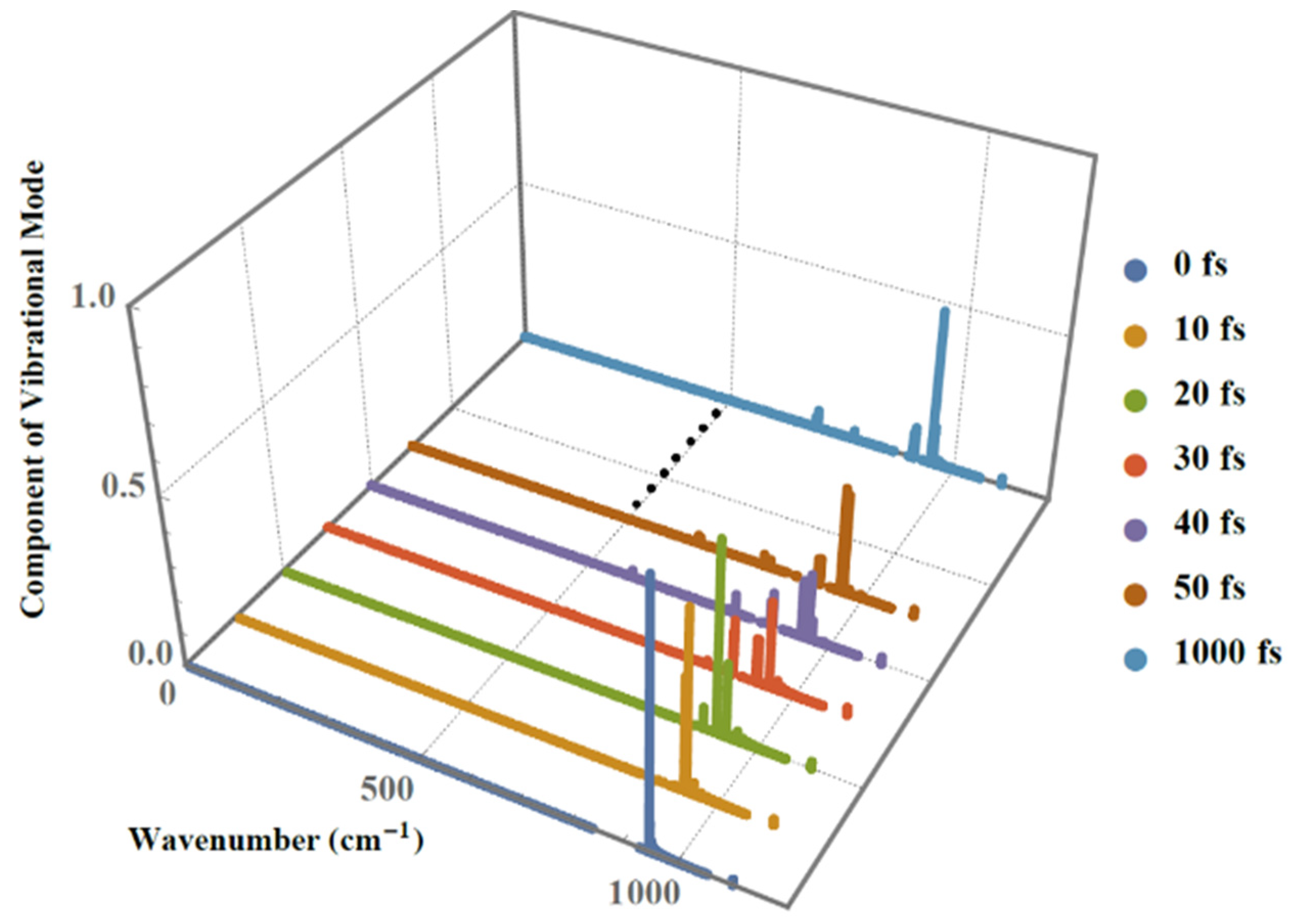
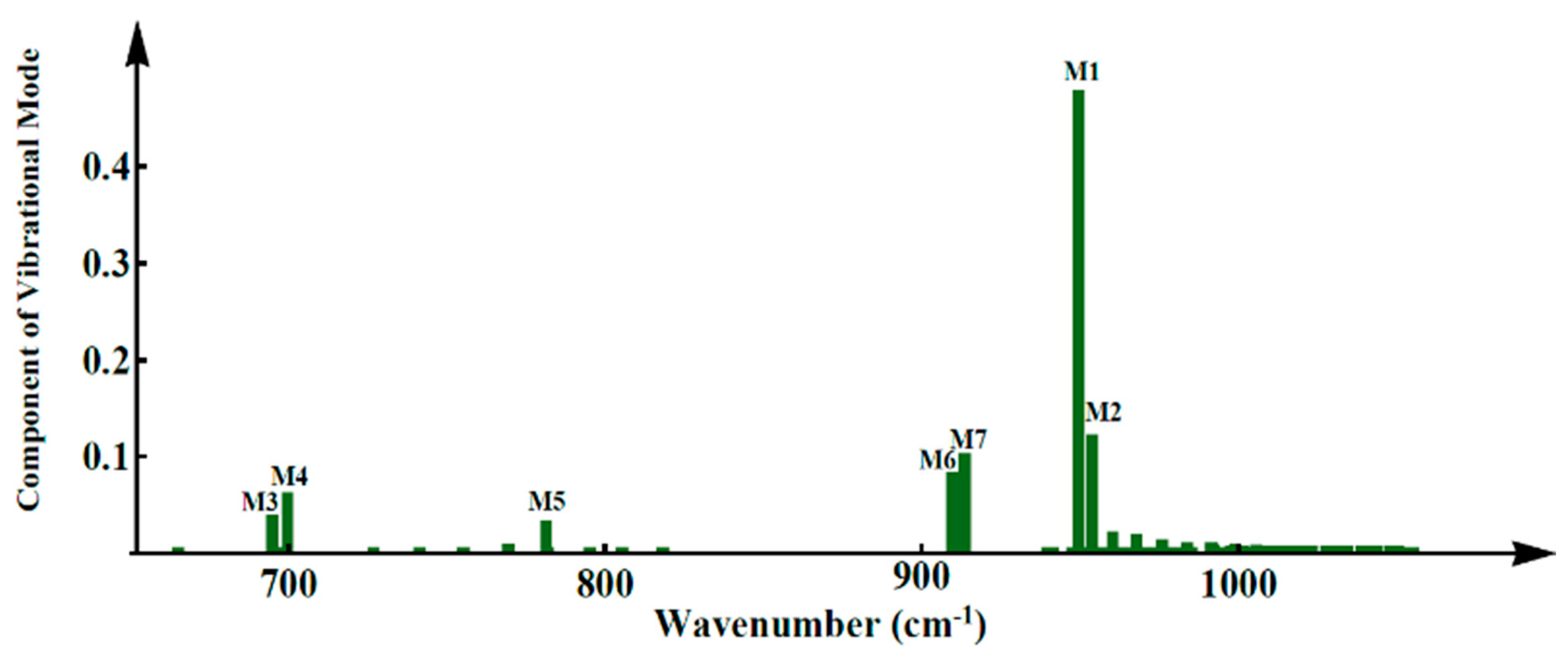
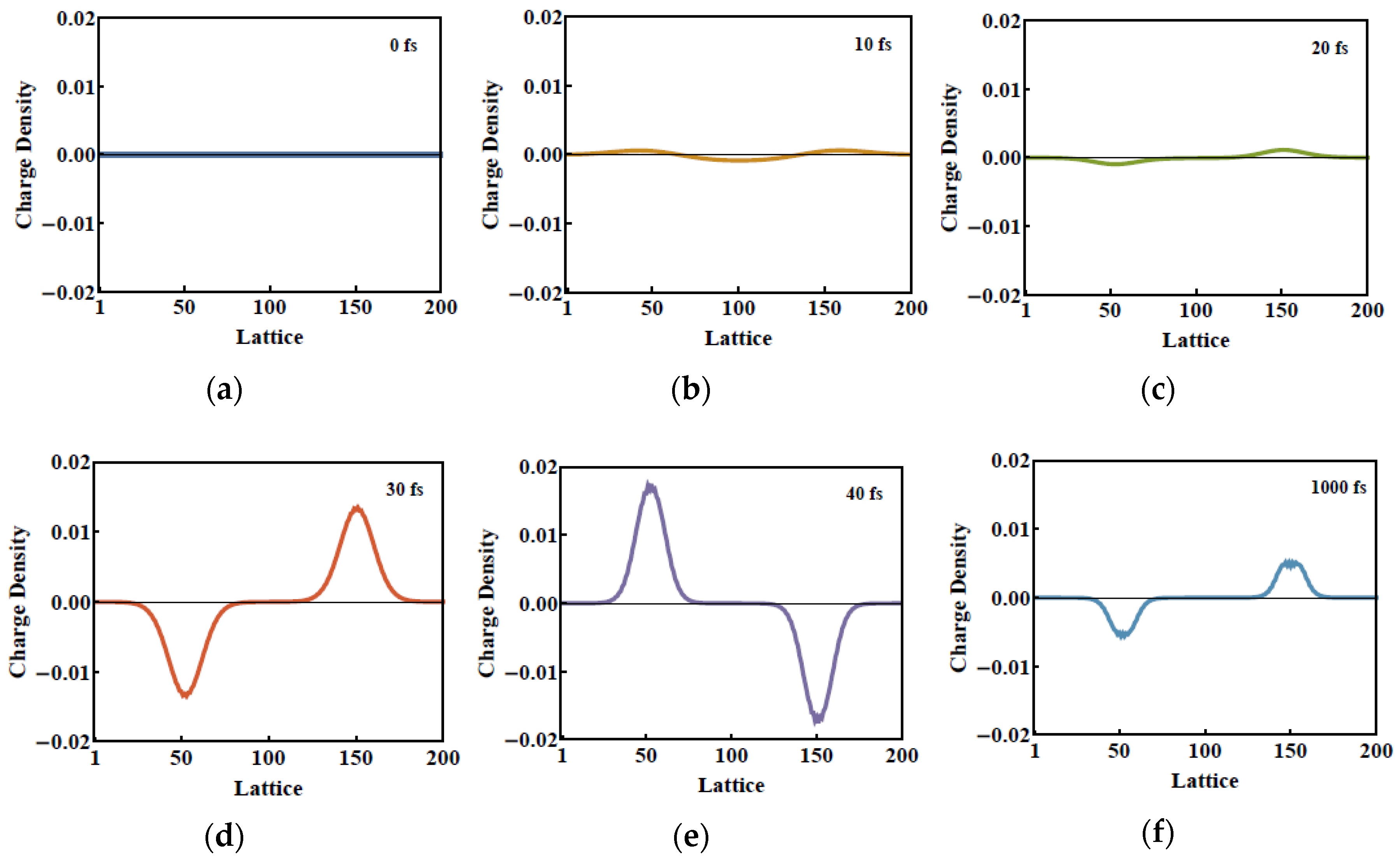
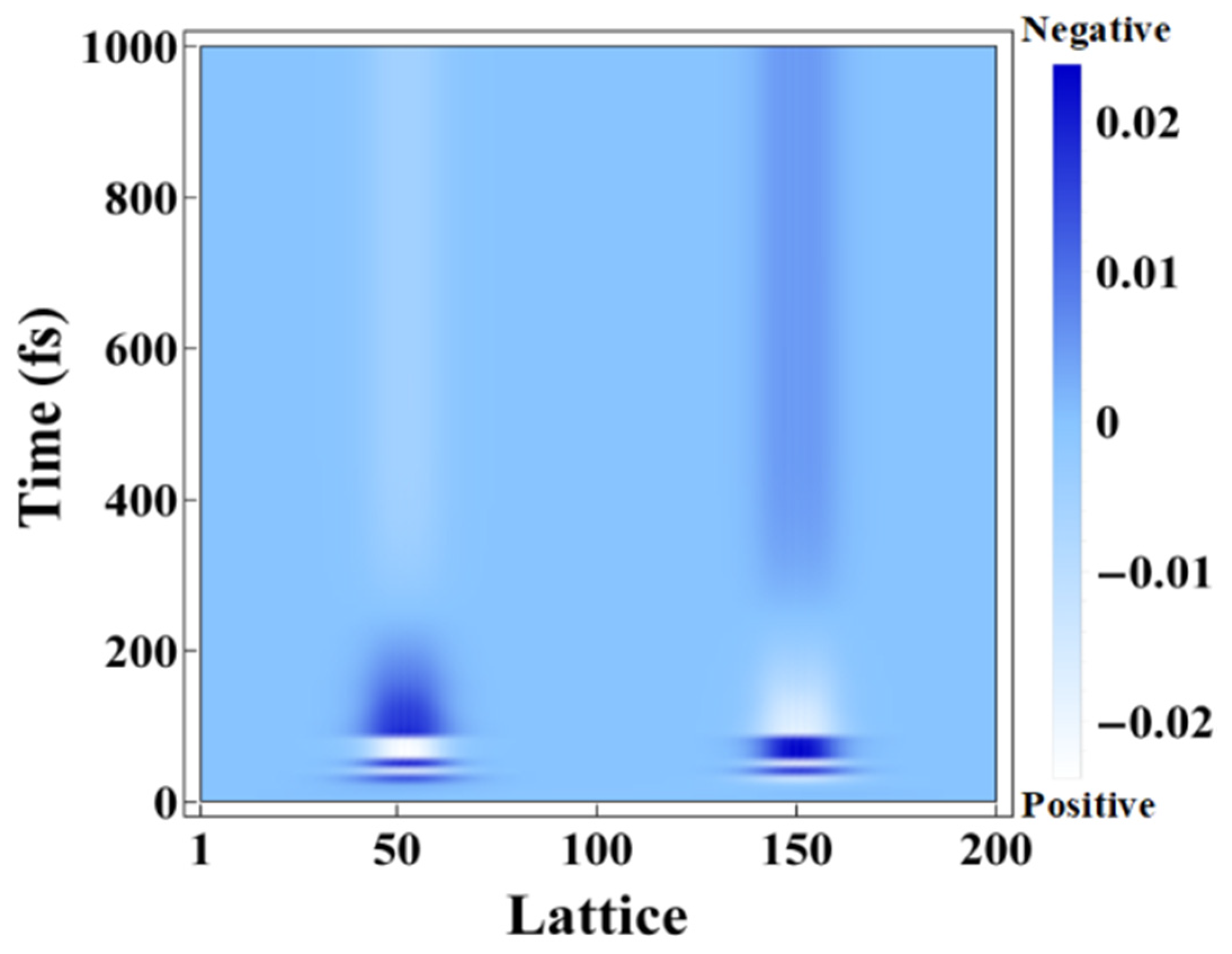
2. Results and Discussion
3. Methods
3.1. Hamiltonian
3.2. Non-Adiabatic Dynamics
3.3. Lattice Vibrational Modes and Phonon Spectrum
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moses, D. High quantum efficiency luminescence from a conducting polymer in solution: A novel polymer laser dye. Appl. Phys. Lett. 1992, 60, 3215–3216. [Google Scholar] [CrossRef]
- Burroughes, J.H.; Bradley, D.D.; Brown, A.; Marks, R.; Mackay, K.; Friend, R.H.; Burns, P.; Holmes, A. Light-emitting diodes based on conjugated polymers. Nature 1990, 347, 539. [Google Scholar] [CrossRef]
- Friend, R.H.; Gymer, R.W.; Holmes, A.B.; Burroughes, J.H.; Marks, R.N.; Taliani, C.; Bradley, D.D.C.; Santos, D.A.D.; Brédas, J.L.; Lögdlund, M.; et al. Electroluminescence in conjugated polymers. Nature 1999, 397, 121–128. [Google Scholar] [CrossRef]
- Tessler, N.; Denton, G.J.; Friend, R.H. Lasing from conjugated-polymer microcavities. Nature 1996, 382, 695–697. [Google Scholar] [CrossRef]
- Hide, F.; Diaz-Garcia, M.A.; Schwartz, B.J.; Andersson, M.R.; Pei, Q.; Heeger, A.J. Semiconducting polymers: A new class of solid-state laser materials. Science 1996, 273, 1833–1836. [Google Scholar] [CrossRef]
- Holzer, W.; Penzkofer, A.; Gong, S.-H.; Bleyer, A.; Bradley, D.D.C. Laser action in poly (m-phenylenevinylene-co-2,5-dioctoxy-p-phenylenevinylene). Adv. Mater. 1996, 8, 974–978. [Google Scholar] [CrossRef]
- Frolov, S.V.; Ozaki, M.; Gellermann, W.; Vardeny, Z.V.; Yoshino, K. Mirrorless lasing in conducting polymer poly(2,5-dioctyloxy- p-phenylenevinylene) films. Jpn. J. Appl. Phys. 1996, 35, L1371–L1373. [Google Scholar] [CrossRef]
- Akselrod, G.M.; Young, E.R.; Bradley, M.S.; Bulović, V. Lasing through a strongly-coupled mode by intra-cavity pumping. Opt. Express 2013, 21, 12122–12128. [Google Scholar] [CrossRef]
- Berggren, M.; Dodabalapur, A.; Slusher, R. Stimulated emission and lasing in dye-doped organic thin films with Forster transfer. Appl. Phys. Lett. 1997, 71, 2230–2232. [Google Scholar] [CrossRef]
- Kéna-Cohen, S.; Forrest, S. Room-temperature polariton lasing in an organic single-crystal microcavity. Nat. Photonics 2010, 4, 371. [Google Scholar] [CrossRef]
- Bulović, V.; Kozlov, V.; Khalfin, V.; Forrest, S. Transform-limited, narrow-linewidth lasing action in organic semiconductor microcavities. Science 1998, 279, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Lee, W.; Caruge, J.-M.; Bawendi, M.; Thomas, E.L.; Kooi, S.; Prasad, P.N. Defect-mode mirrorless lasing in dye-doped organic/inorganic hybrid one-dimensional photonic crystal. Appl. Phys. Lett. 2006, 88, 091102. [Google Scholar] [CrossRef] [Green Version]
- Gourdon, F.; Chakaroun, M.; Fabre, N.; Solard, J.; Cambril, E.; Yacomotti, A.-M.; Bouchoule, S.; Fischer, A.; Boudrioua, A. Optically pumped lasing from organic two-dimensional planar photonic crystal microcavity. Appl. Phys. Lett. 2012, 100, 117. [Google Scholar] [CrossRef]
- Meier, M.; Mekis, A.; Dodabalapur, A.; Timko, A.; Slusher, R.; Joannopoulos, J.; Nalamasu, O. Laser action from two-dimensional distributed feedback in photonic crystals. Appl. Phys. Lett. 1999, 74, 7–9. [Google Scholar] [CrossRef]
- Mekis, A.; Meier, M.; Dodabalapur, A.; Slusher, R.; Joannopoulos, J. Lasing mechanism in two-dimensional photonic crystal lasers. Appl. Phys. A 1999, 69, 111–114. [Google Scholar] [CrossRef]
- Riechel, S.; Kallinger, C.; Lemmer, U.; Feldmann, J.; Gombert, A.; Wittwer, V.; Scherf, U. A nearly diffraction limited surface emitting conjugated polymer laser utilizing a two-dimensional photonic band structure. Appl. Phys. Lett. 2000, 77, 2310–2312. [Google Scholar] [CrossRef]
- Shapira, O.; Kuriki, K.; Orf, N.D.; Abouraddy, A.F.; Benoit, G.; Viens, J.F.; Rodriguez, A.; Ibanescu, M.; Joannopoulos, J.D.; Fink, Y.; et al. Surface-emitting fiber lasers. Opt. Express 2006, 14, 3929–3935. [Google Scholar] [CrossRef]
- Vietze, U.; Krauss, O.; Laeri, F.; Ihlein, G.; Schüth, F.; Limburg, B.; Abraham, M. Zeolite-dye microlasers. Phys. Rev. Lett. 1998, 81, 4628. [Google Scholar] [CrossRef] [Green Version]
- Kushida, S.; Braam, D.; Pan, C.; Dao, T.D.; Tabata, K.; Sugiyasu, K.; Takeuchi, M.; Ishii, S.; Nagao, T.; Lorke, A.; et al. Whispering gallery resonance from self-assembled microspheres of highly fluorescent isolated conjugated polymers. Macromolecules 2015, 48, 3928–3933. [Google Scholar] [CrossRef]
- Wang, X.; Liao, Q.; Li, H.; Bai, S.; Wu, Y.; Lu, X.; Hu, H.; Shi, Q.; Fu, H. Near-infrared lasing from small-molecule organic hemispheres. J. Am. Chem. Soc. 2015, 137, 9289–9295. [Google Scholar] [CrossRef]
- Kowalsky, W.; Rabe, T.; Schneider, D.; Johannes, H.-H.; Karnutsch, C.; Gerken, M.; Lemmer, U.; Wang, J.; Weimann, T.; Hinze, P.; et al. Organic semiconductor distributed feedback lasers. In Proceedings of the Nanosensing: Materials and devices II; SPIE: Boston, MA, USA, 2005; Volume 6008, p. 60080Z. [Google Scholar]
- Schneider, D.; Rabe, T.; Riedl, T.; Dobbertin, T.; Kröger, M.; Becker, E.; Johannes, H.-H.; Kowalsky, W.; Weimann, T.; Wang, J.; et al. Laser threshold reduction in an all-spiro guest–host system. Appl. Phys. Lett. 2004, 85, 1659–1661. [Google Scholar] [CrossRef]
- Rose, A.; Zhu, Z.; Madigan, C.F.; Swager, T.M.; Bulović, V. Sensitivity gains in chemosensing by lasing action in organic polymers. Nature 2005, 434, 876. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ta, V.D.; Sun, H. Bending-induced bidirectional tuning of whispering gallery mode lasing from flexible polymer fibers. ACS Photonics 2014, 1, 11–16. [Google Scholar] [CrossRef]
- Oh-E, M.; Yokoyama, H.; Yorozuya, S.; Akagi, K.; Belkin, M.A.; Shen, Y.R. Sum-frequency vibrational spectroscopy of a helically structured conjugated polymer. Phys. Rev. Lett. 2004, 93, 267402.1–267402.4. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Lupton, J.M.; Becker, K.; Feldmann, J.; Gaefke, G.; Höger, S. Simultaneous Raman and fluorescence spectroscopy of single conjugated polymer chains. Phys. Rev. Lett. 2007, 98, 137401. [Google Scholar] [CrossRef] [PubMed]
- Giebink, N.C.; Forrest, S. Temporal response of optically pumped organic semiconductor lasers and its implication for reaching threshold under electrical excitation. Phys. Rev. B 2009, 79, 073302. [Google Scholar] [CrossRef]
- Ma, L.; Wu, Z.; Lei, T.; Yu, Y.; Yuan, F.; Ning, S.; Jiao, B.; Hou, X. Theoretical insight into the deep-blue amplified spontaneous emission of new organic semiconductor molecules. Org. Electron. 2014, 15, 3144–3153. [Google Scholar] [CrossRef]
- Jensen, S.A.; Mics, Z.; Ivanov, I.; Varol, H.S.; Turchinovich, D.; Koppens, F.; Bonn, M.; Tielrooij, K.-J. Competing ultrafast energy relaxation pathways in photoexcited graphene. Nano Lett. 2014, 14, 5839–5845. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Xie, S.; Li, Y.; Xia, C. Realization of the population inversion in a conjugated polymer by a single or double stimulating pulse. Org. Electron. 2014, 15, 1965–1971. [Google Scholar] [CrossRef]
- Galindo, J.F.; Atas, E.; Altan, A.; Kuroda, D.G.; Fernandez-Alberti, S.; Tretiak, S.; Roitberg, A.E.; Kleiman, V.D. Dynamics of Energy Transfer in a Conjugated Dendrimer Driven by Ultrafast Localization of Excitations. J. Am. Chem. Soc. 2015, 137, 11637–11644. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, W.; Zhang, Y.; Li, S.; George, T.F. Photoinduced localized phonons and instantaneous structure contributing to amplified spontaneous emission of conjugated polymers. J. Phys. Chem. C 2017, 121, 1055–1061. [Google Scholar] [CrossRef]
- Gao, K.; Liu, X.; Liu, D.; Xie, S. Charge carrier generation through reexcitations of an exciton in poly(p-phenylene vinylene) molecules. Phys Rev B 2007, 75, 205412–205417. [Google Scholar] [CrossRef]
- Choi, H.Y.; Rice, M. Excited polarons in poly(phenylene vinylene) and poly(diacetylene). Phys. Rev. B 1991, 44, 10521–10524. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.; Schrieffer, J.R.; Heeger, A.J. Solitons in polyacetylene. Phys. Rev. Lett. 1979, 42, 1698–1701. [Google Scholar] [CrossRef]
- Бpaзoвcкий, C.; Kиpoвa, H. Экcитoны, пoляpoны и бипoляpoны в пpoвoдящиx пoлимepax. Пиcьмa B Жypн Экcпepим И Teopeт Φизики 1981, 33, 6. [Google Scholar]
- Kivelson, S.; Su, W.-P.; Schrieffer, J.R.; Heeger, A.J. Missing bond-charge repulsion in the extended Hubbard model: Effects in polyacetylene. Phys. Rev. Lett. 1987, 58, 1899–1902. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Z.; Chen, J.; Zhang, Y.; Shen, J.; Li, S.; George, T.F. Charge Accumulation of Amplified Spontaneous Emission in a Conjugated Polymer Chain and Its Dynamical Phonon Spectra. Molecules 2020, 25, 3003. https://doi.org/10.3390/molecules25133003
Lin Z, Chen J, Zhang Y, Shen J, Li S, George TF. Charge Accumulation of Amplified Spontaneous Emission in a Conjugated Polymer Chain and Its Dynamical Phonon Spectra. Molecules. 2020; 25(13):3003. https://doi.org/10.3390/molecules25133003
Chicago/Turabian StyleLin, Zhe, Jiahao Chen, Yusong Zhang, Jianguo Shen, Sheng Li, and Thomas F. George. 2020. "Charge Accumulation of Amplified Spontaneous Emission in a Conjugated Polymer Chain and Its Dynamical Phonon Spectra" Molecules 25, no. 13: 3003. https://doi.org/10.3390/molecules25133003