
Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance



Abstract
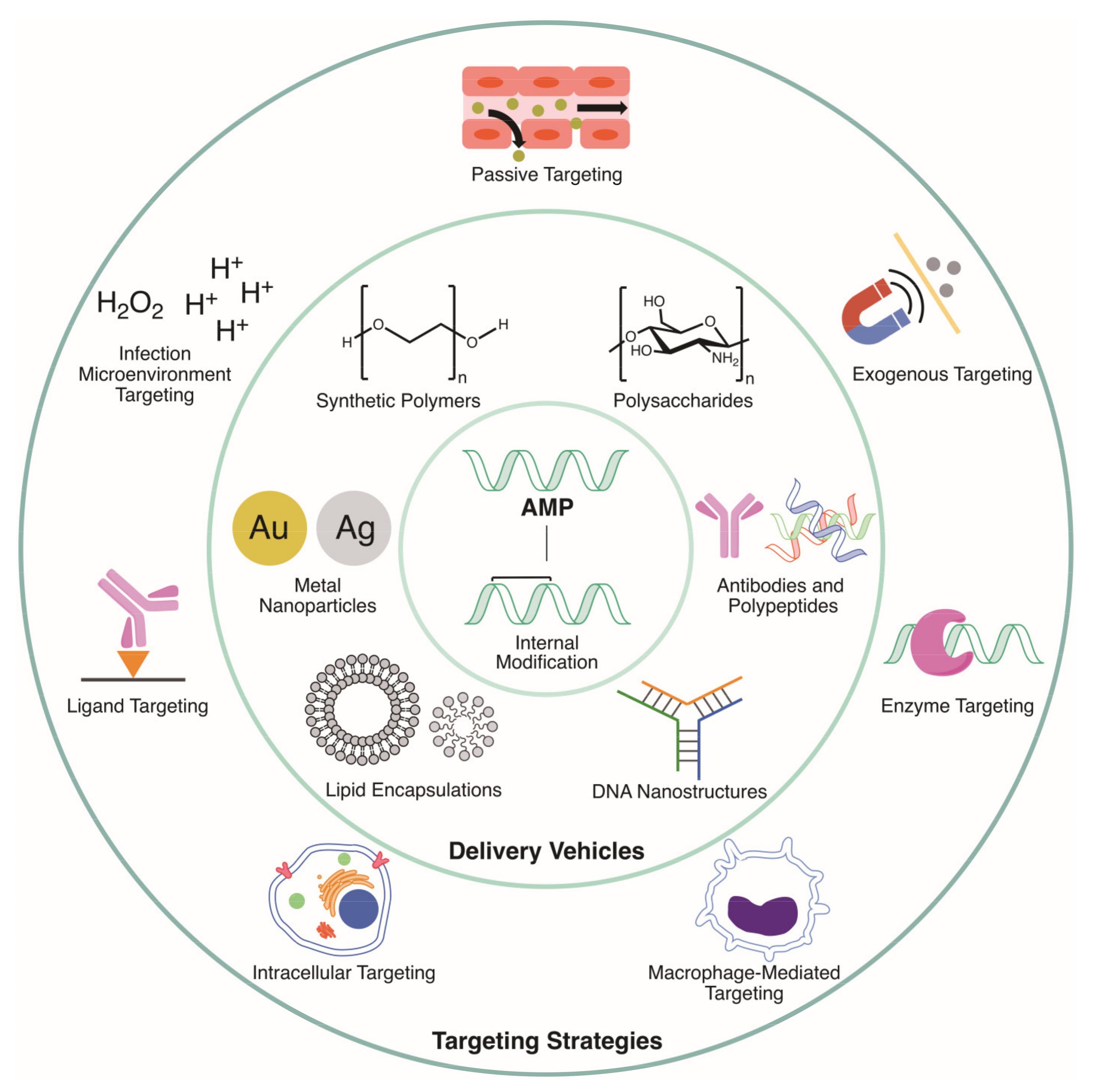
:1. Introduction
2. Targeting of Delivery Vehicles
2.1. Passive Targeting
2.2. Endogenous Targeting
Targeting Intracellular Bacteria
2.3. Exogenous Targeting
3. Delivery Vehicles
3.1. Lipid Encapsulations
3.1.1. Liposomes
3.1.2. Micelles
3.1.3. Liquid Crystalline Nanoparticles
3.2. Metal Nanoparticles
3.2.1. Gold Nanoparticles
3.2.2. Silver Nanoparticles
3.3. Synthetic Polymers
3.4. Natural Biomolecules
3.4.1. Polysaccharides
3.4.2. Polypeptides
3.4.3. Antibodies
3.4.4. DNA Nanostructures
4. Alternatives: Internal Modification of AMPs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides With Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Falla, T.J. Antimicrobial peptides: Broad-spectrum antibiotics from nature. Clin. Microbiol. Infect. 1996, 1, 226–229. [Google Scholar] [CrossRef] [Green Version]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The interaction of antimicrobial peptides with membranes. Adv. Colloid Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta -Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Chernysh, S.; Kim, S.I.; Bekker, G.; Pleskach, V.A.; Filatova, N.A.; Anikin, V.B.; Platonov, V.G.; Bulet, P. Antiviral and antitumor peptides from insects. Proc. Natl. Acad. Sci. USA 2002, 99, 12628–12632. [Google Scholar] [CrossRef] [Green Version]
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef]
- Bowdish, D.M.E.; Davidson, D.J.; Scott, M.G.; Hancock, R.E.W. Immunomodulatory Activities of Small Host Defense Peptides. Antimicrob. Agents Chemother. 2005, 49, 1727–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowdish, D.M.E.; Davidson, D.J.; Hancock, R.E.W. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 2005, 6, 35–51. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente-Núñez, C.; Cardoso, M.H.; De Souza Cândido, E.; Franco, O.L.; Hancock, R.E.W. Synthetic antibiofilm peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of antimicrobial peptides against bacterial biofilms. Materials (Basel) 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Raheem, N.; Kumar, P.; Lee, E.; Cheng, J.T.J.; Hancock, R.E.W.; Straus, S.K. Insights into the mechanism of action of two analogues of aurein 2.2. Biochim. Biophys. Acta Biomembr. 2020, 1862. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updat. 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. New approaches in peptide antibiotics. Curr. Opin. Pharmacol. 2009, 9, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, Á.; Gómez, R.; Ortega, P.; Mata, F.J.D. La Nanosystems as vehicles for the delivery of antimicrobial peptides (Amps). Pharmaceutics 2019, 11, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Makowski, M.; Silva, Í.C.; Do Amaral, C.P.; Gonçalves, S.; Santos, N.C. Advances in lipid and metal nanoparticles for antimicrobial peptide delivery. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111. [Google Scholar] [CrossRef]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Abbina, S.; Abbasi, U.; Gill, A.; Wong, K.; Kalathottukaren, M.T.; Kizhakkedathu, J.N. Design of Safe Nanotherapeutics for the Excretion of Excess Systemic Toxic Iron. ACS Cent. Sci. 2019, 5, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Thamphiwatana, S.; Angsantikul, P.; Zhang, L. Nanoparticle approaches against bacterial infections. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 532–547. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.L.; Liles, W.C. Endothelial activation, dysfunction and permeability during severe infections. Curr. Opin. Hematol. 2011, 18, 191–196. [Google Scholar] [CrossRef]
- Boerman, O.C.; Storm, G.; Oyen, W.J.G.; Van Bloois, L.; Van der Meer, J.W.M.; Claessens, R.A.M.J.; Crommelin, D.J.A.; Corstens, F.H.M. Sterically stabilized liposomes labeled with Indium-111 to image focal infection. J. Nucl. Med. 1995, 36, 1639–1644. [Google Scholar] [PubMed]
- Boerman, O.C.; Oyen, W.J.G.; Van Bloois, L.; Koenders, E.B.; Van Der Meer, J.W.M.; Corstens, F.H.M.; Storm, G. Optimization of technetium-99m-labeled PEG liposomes to image focal infection: Effects of particle size and circulation time. J. Nucl. Med. 1997, 38, 489–493. [Google Scholar] [PubMed]
- Laverman, P.; Dams, E.T.M.; Storm, G.; Hafmans, T.G.; Croes, H.J.; Oyen, W.J.G.; Corstens, F.H.M.; Boerman, O.C. Microscopic localization of PEG-liposomes in a rat model of focal infection. J. Control. Release 2001, 75, 347–355. [Google Scholar] [CrossRef]
- Kell, A.J.; Stewart, G.; Ryan, S.; Peytavi, R.; Boissinot, M.; Huletsky, A.; Bergeron, M.G.; Simard, B. Vancomycin-modified nanoparticles for efficient targeting and preconcentration of gram-positive and gram-negative bacteria. ACS Nano 2008, 2, 1777–1788. [Google Scholar] [CrossRef]
- Chen, M.; Xie, S.; Wei, J.; Song, X.; Ding, Z.; Li, X. Antibacterial Micelles with Vancomycin-Mediated Targeting and pH/Lipase-Triggered Release of Antibiotics. ACS Appl. Mater. Interfaces 2018, 10, 36814–36823. [Google Scholar] [CrossRef]
- Meeker, D.G.; Jenkins, S.V.; Miller, E.K.; Beenken, K.E.; Loughran, A.J.; Powless, A.; Muldoon, T.J.; Galanzha, E.I.; Zharov, V.P.; Smeltzer, M.S.; et al. Synergistic Photothermal and Antibiotic Killing of Biofilm-Associated Staphylococcus aureus Using Targeted Antibiotic-Loaded Gold Nanoconstructs. ACS Infect. Dis. 2016, 2, 241–250. [Google Scholar] [CrossRef]
- Yeom, J.H.; Lee, B.; Kim, D.; Lee, J.K.; Kim, S.; Bae, J.; Park, Y.; Lee, K. Gold nanoparticle-DNA aptamer conjugate-assisted delivery of antimicrobial peptide effectively eliminates intracellular Salmonella enterica serovar Typhimurium. Biomaterials 2016, 104, 43–51. [Google Scholar] [CrossRef]
- Lee, B.; Park, J.; Ryu, M.; Kim, S.; Joo, M.; Yeom, J.H.; Kim, S.; Park, Y.; Lee, K.; Bae, J. Antimicrobial peptide-loaded gold nanoparticle-DNA aptamer conjugates as highly effective antibacterial therapeutics against Vibrio vulnificus. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Liu, Q.; Shao, X.; Feng, C.; Shen, Y.; Zhang, Q.; Jiang, X. Solanum tuberosum lectin-conjugated PLGA nanoparticles for nose-to-brain delivery: In vivo and in vitro evaluations. J. Drug Target. 2012, 20, 174–184. [Google Scholar] [CrossRef]
- Umamaheshwari, R.B.; Jain, N.K. Receptor mediated targeting of lectin conjugated gliadin nanoparticles in the treatment of Helicobacter pylori. J. Drug Target. 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Radovic-Moreno, A.F.; Lu, T.K.; Puscasu, V.A.; Yoon, C.J.; Langer, R.; Farokhzad, O.C. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano 2012, 6, 4279–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, F.; Hu, X.; Lu, J.; Sun, X.; Gao, J.; Ling, D. Responsive Assembly of Silver Nanoclusters with a Biofilm Locally Amplified Bactericidal Effect to Enhance Treatments against Multi-Drug-Resistant Bacterial Infections. ACS Cent. Sci. 2019, 5, 1366–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Das, R.; Yesilbag Tonga, G.; Mizuhara, T.; Rotello, V.M. Charge-Switchable Nanozymes for Bioorthogonal Imaging of Biofilm-Associated Infections. ACS Nano 2018, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Gao, H.; Cheng, T.; Zhang, Y.; Liu, J.; Huang, F.; Yang, C.; Shi, L.; Liu, J. A charge-adaptive nanosystem for prolonged and enhanced: In vivo antibiotic delivery. Chem. Commun. 2016, 52, 6265–6268. [Google Scholar] [CrossRef] [PubMed]
- Coessens, V.; Schacht, E.; Domurado, D. Synthesis of polyglutamine and dextran conjugates of streptomycin with an acid-sensitive drug-carrier linkage. J. Control. Release 1996, 38, 141–150. [Google Scholar] [CrossRef]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, M.J.; Murthy, N. Polyketal nanoparticles: A new pH-sensitive biodegradable drug delivery vehicle. Bioconjug. Chem. 2005. [Google Scholar] [CrossRef]
- Shenoi, R.A.; Narayanannair, J.K.; Hamilton, J.L.; Lai, B.F.L.; Horte, S.; Kainthan, R.K.; Varghese, J.P.; Rajeev, K.G.; Manoharan, M.; Kizhakkedathu, J.N. Branched multifunctional polyether polyketals: Variation of ketal group structure enables unprecedented control over polymer degradation in solution and within cells. J. Am. Chem. Soc. 2012. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Li, L.L.; Ma, H.L.; Qi, G.B.; Zhang, D.; Yu, F.; Hu, Z.; Wang, H. Pathological-Condition-Driven Construction of Supramolecular Nanoassemblies for Bacterial Infection Detection. Adv. Mater. 2016, 28, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.-B.; Zhang, D.; Liu, F.-H.; Qiao, Z.-Y.; Wang, H. An “On-Site Transformation” Strategy for Treatment of Bacterial Infection. Adv. Mater. 2017, 1703461, 1703461. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.H.; Bao, Y.; Yang, X.Z.; Wang, Y.C.; Sun, B.; Wang, J. Lipase-sensitive polymeric triple-layered nanogel for “on-demand” drug delivery. J. Am. Chem. Soc. 2012, 134, 4355–4362. [Google Scholar] [CrossRef] [PubMed]
- Vanlaere, I.; Libert, C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clin. Microbiol. Rev. 2009, 22, 224–239. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Peyron, P.; Maridonneau-Parini, I.; Stegmann, T. Fusion of human neutrophil phagosomes with lysosomes in vitro: Involvement of tyrosine kinases of the Src family and inhibition by mycobacteria. J. Biol. Chem. 2001, 276, 35512–35517. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Zhang, X.; Zhao, W.; Zeng, C.; Deng, B.; McComb, D.W.; Du, S.; Zhang, C.; Li, W.; Dong, Y. Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat. Nanotechnol. 2020, 15, 41–46. [Google Scholar] [CrossRef]
- Kamaruzzaman, N.F.; Kendall, S.; Good, L. Targeting the hard to reach: Challenges and novel strategies in the treatment of intracellular bacterial infections. Br. J. Pharmacol. 2017, 174, 2225–2236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Tan, M.W. Antibody–Antibiotic Conjugates: A Novel Therapeutic Platform against Bacterial Infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef]
- Noore, J.; Noore, A.; Li, B. Cationic antimicrobial peptide ll-37 is effective against both extraand intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 1283–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonawane, A.; Santos, J.C.; Mishra, B.B.; Jena, P.; Progida, C.; Sorensen, O.E.; Gallo, R.; Appelberg, R.; Griffiths, G. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell. Microbiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Gutsmann, T. Interaction between antimicrobial peptides and mycobacteria. Biochim. Biophys. Acta Biomembr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.H.; Li, Y.J.; Bao, Y.; Yang, X.Z.; Hu, B.; Wang, J. Bacteria-responsive multifunctional nanogel for targeted antibiotic delivery. Adv. Mater. 2012, 24, 6175–6180. [Google Scholar] [CrossRef] [PubMed]
- Canaparo, R.; Foglietta, F.; Giuntini, F.; Pepa, C.D.; Dosio, F.; Serpe, L. Recent developments in antibacterial therapy: Focus on stimuli-responsive drug-delivery systems and therapeutic nanoparticles. Molecules 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Zhou, Y.; He, W.; Hua, D. A strategy for enhanced antibacterial activity against Staphylococcus aureus by the assembly of alamethicin with a thermo-sensitive polymeric carrier. Chem. Commun. 2016, 52, 896–899. [Google Scholar] [CrossRef]
- Mohapatra, A.; Harris, M.A.; LeVine, D.; Ghimire, M.; Jennings, J.A.; Morshed, B.I.; Haggard, W.O.; Bumgardner, J.D.; Mishra, S.R.; Fujiwara, T. Magnetic stimulus responsive vancomycin drug delivery system based on chitosan microbeads embedded with magnetic nanoparticles. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2018, 106, 2169–2176. [Google Scholar] [CrossRef]
- Maleki, H.; Rai, A.; Pinto, S.; Evangelista, M.; Cardoso, R.M.S.; Paulo, C.; Carvalheiro, T.; Paiva, A.; Imani, M.; Simchi, A.; et al. High Antimicrobial Activity and Low Human Cell Cytotoxicity of Core-Shell Magnetic Nanoparticles Functionalized with an Antimicrobial Peptide. ACS Appl. Mater. Interfaces 2016, 8, 11366–11378. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Irimie, A.I.; Sonea, L.; Jurj, A.; Mehterov, N.; Zimta, A.A.; Budisan, L.; Braicu, C.; Berindan-Neagoe, I. Future trends and emerging issues for nanodelivery systems in oral and oropharyngeal cancer. Int. J. Nanomedicine 2017, 12, 4593–4606. [Google Scholar] [CrossRef] [Green Version]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid lipid nanoparticles and nanostructured lipid carriers: Structure preparation and application. Adv. Pharm. Bull. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, G.S.; Kataoka, K. Block copolymer micelles as long-circulating drug vehicles. Adv. Drug Deliv. Rev. 2012, 64, 237–245. [Google Scholar] [CrossRef]
- Jhaveri, A.M.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5 APR, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Rajchakit, U.; Sarojini, V. Recent Developments in Antimicrobial-Peptide-Conjugated Gold Nanoparticles. Bioconjug. Chem. 2017, 28, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Abbina, S.; Vappala, S.; Kumar, P.; Siren, E.M.J.; La, C.C.; Abbasi, U.; Brooks, D.E.; Kizhakkedathu, J.N. Hyperbranched polyglycerols: Recent advances in synthesis, biocompatibility and biomedical applications. J. Mater. Chem. B 2017, 5, 9249–9277. [Google Scholar] [CrossRef]
- Petrin, T.H.C.; Fadel, V.; Martins, D.B.; Dias, S.A.; Cruz, A.; Sergio, L.M.; Arcisio-Miranda, M.; Castanho, M.A.R.B.; Dos Santos Cabrera, M.P. Synthesis and Characterization of Peptide-Chitosan Conjugates (PepChis) with Lipid Bilayer Affinity and Antibacterial Activity. Biomacromolecules 2019, 20, 2743–2753. [Google Scholar] [CrossRef]
- Shelke, N.B.; James, R.; Laurencin, C.T.; Kumbar, S.G. Polysaccharide biomaterials for drug delivery and regenerative engineering. Polym. Adv. Technol. 2014, 25, 448–460. [Google Scholar] [CrossRef]
- Miao, T.; Wang, J.; Zeng, Y.; Liu, G.; Chen, X. Polysaccharide-Based Controlled Release Systems for Therapeutics Delivery and Tissue Engineering: From Bench to Bedside. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, T.N.; Capecchi, A.; Gan, B.H.; Jin, X.; He, R.; Wei, D.; Ma, L.; Köhler, T.; van Delden, C.; Javor, S.; et al. Optimizing Antimicrobial Peptide Dendrimers in Chemical Space. Angew. Chemie Int. Ed. 2018, 57, 8483–8487. [Google Scholar] [CrossRef] [PubMed]
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial dendrimeric peptides: Structure, activity and new therapeutic applications. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurawski, D.V.; McLendon, M.K. Monoclonal antibodies as an antibacterial approach against bacterial pathogens. Antibiotics 2020, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Madhanagopal, B.R.; Zhang, S.; Demirel, E.; Wady, H.; Chandrasekaran, A.R. DNA Nanocarriers: Programmed to Deliver. Trends Biochem. Sci. 2018, 43, 997–1013. [Google Scholar] [CrossRef]
- Rao, L.; Tian, R.; Chen, X. Cell-Membrane-Mimicking Nanodecoys against Infectious Diseases. ACS Nano 2020, 14, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Silveira, S.A.; Shorr, A.F. Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections — A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug. Antibiotics 2020, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Liu, J.; Gao, J.; Chen, S.; Huang, G. Chitosan coated vancomycin hydrochloride liposomes: Characterizations and evaluation. Int. J. Pharm. 2015, 495, 508–515. [Google Scholar] [CrossRef]
- Cantor, S.; Vargas, L.; Rojas, O.E.A.; Yarce, C.J.; Salamanca, C.H.; Oñate-Garzón, J. Evaluation of the antimicrobial activity of cationic peptides loaded in surface-modified nanoliposomes against foodborne bacteria. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Lopes, N.A.; Pinilla, C.M.B.; Brandelli, A. Pectin and polygalacturonic acid-coated liposomes as novel delivery system for nisin: Preparation, characterization and release behavior. Food Hydrocoll. 2017, 70, 1–7. [Google Scholar] [CrossRef]
- Pu, C.; Tang, W. A chitosan-coated liposome encapsulating antibacterial peptide, Apep10: Characterisation, triggered-release effects and antilisterial activity in thaw water of frozen chicken. Food Funct. 2016, 7, 4310–4322. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of Proteins and Liposomes: A Powerful and Flexible Strategy to Improve the Drug Delivery. Curr. Drug Metab. 2011, 13, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, M.-R.; Chiu, G.N.C. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J. Pharm. Sci. 2013, 8, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Gdowski, A.; Johnson, K.; Shah, S.; Gryczynski, I.; Vishwanatha, J.; Ranjan, A. Optimization and scale up of microfluidic nanolipomer production method for preclinical and potential clinical trials. J. Nanobiotechnol. 2018, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.M.; Nguyen, D.X.; Patel, P.; Cote, B.; Al-Fatease, A.; Pham, Y.; Huynh, M.G.; Woo, Y.; Alani, A.W. Liposomes produced by microfluidics and extrusion: A comparison for scale-up purposes. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 146–156. [Google Scholar] [CrossRef]
- Webb, C.; Forbes, N.; Roces, C.B.; Anderluzzi, G.; Lou, G.; Abraham, S.; Ingalls, L.; Marshall, K.; Leaver, T.J.; Watts, J.A.; et al. Using microfluidics for scalable manufacturing of nanomedicines from bench to GMP: A case study using protein-loaded liposomes. Int. J. Pharm. 2020, 582, 119266. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.E.W.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infect. Dis. 2019, 5, 443–453. [Google Scholar] [CrossRef]
- Lee, W.; Park, E.J.; Min, G.; Choi, J.; Na, D.H.; Bae, J.S. Dual functioned PEGylated phospholipid micelles containing cationic antimicrobial decapeptide for treating sepsis. Theranostics 2017, 7, 3759–3767. [Google Scholar] [CrossRef]
- Zhai, J.; Fong, C.; Tran, N.; Drummond, C.J. Non-Lamellar Lyotropic Liquid Crystalline Lipid Nanoparticles for the Next Generation of Nanomedicine. ACS Nano 2019, 13, 6178–6206. [Google Scholar] [CrossRef] [PubMed]
- Boge, L.; Umerska, A.; Matougui, N.; Bysell, H.; Ringstad, L.; Davoudi, M.; Eriksson, J.; Edwards, K.; Andersson, M. Cubosomes post-loaded with antimicrobial peptides: Characterization, bactericidal effect and proteolytic stability. Int. J. Pharm. 2017, 526, 400–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boge, L.; Hallstensson, K.; Ringstad, L.; Johansson, J.; Andersson, T.; Davoudi, M.; Larsson, P.T.; Mahlapuu, M.; Håkansson, J.; Andersson, M. Cubosomes for topical delivery of the antimicrobial peptide LL-37. Eur. J. Pharm. Biopharm. 2019, 134, 60–67. [Google Scholar] [CrossRef]
- Garcia-Orue, I.; Gainza, G.; Girbau, C.; Alonso, R.; Aguirre, J.J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M. LL37 loaded nanostructured lipid carriers (NLC): A new strategy for the topical treatment of chronic wounds. Eur. J. Pharm. Biopharm. 2016, 108, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Fumakia, M.; Ho, E.A. Nanoparticles encapsulated with LL37 and serpin A1 promotes wound healing and synergistically enhances antibacterial activity. Mol. Pharm. 2016, 13, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Sans-Serramitjana, E.; Fusté, E.; Martínez-Garriga, B.; Merlos, A.; Pastor, M.; Pedraz, J.L.; Esquisabel, A.; Bachiller, D.; Vinuesa, T.; Viñas, M. Killing effect of nanoencapsulated colistin sulfate on Pseudomonas aeruginosa from cystic fibrosis patients. J. Cyst. Fibros. 2016, 15, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Casciaro, B.; Moros, M.; Rivera-Fernández, S.; Bellelli, A.; de la Fuente, J.M.; Mangoni, M.L. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomater. 2017, 47, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Wadhwani, P.; Heidenreich, N.; Podeyn, B.; Bürck, J.; Ulrich, A.S. Antibiotic gold: Tethering of antimicrobial peptides to gold nanoparticles maintains conformational flexibility of peptides and improves trypsin susceptibility. Biomater. Sci. 2017, 5, 817–827. [Google Scholar] [CrossRef]
- Chowdhury, R.; Ilyas, H.; Ghosh, A.; Ali, H.; Ghorai, A.; Midya, A.; Jana, N.R.; Das, S.; Bhunia, A. Multivalent gold nanoparticle-peptide conjugates for targeting intracellular bacterial infections. Nanoscale 2017, 9, 14074–14093. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, W.; Chen, Y.; Li, C.; Jiang, H.; Wang, X. Conjugating gold nanoclusters and antimicrobial peptides: From aggregation-induced emission to antibacterial synergy. J. Colloid Interface Sci. 2019, 546, 1–10. [Google Scholar] [CrossRef]
- Palmieri, G.; Tatè, R.; Gogliettino, M.; Balestrieri, M.; Rea, I.; Terracciano, M.; Proroga, Y.T.; Capuano, F.; Anastasio, A.; De Stefano, L. Small Synthetic Peptides Bioconjugated to Hybrid Gold Nanoparticles Destroy Potentially Deadly Bacteria at Submicromolar Concentrations. Bioconjug. Chem. 2018, 29, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Ryou, S.M.; Yeom, J.H.; Kang, H.J.; Won, M.; Kim, J.S.; Lee, B.; Seong, M.J.; Ha, N.C.; Bae, J.; Lee, K. Gold nanoparticle-DNA aptamer composites as a universal carrier for in vivo delivery of biologically functional proteins. J. Control. Release 2014, 196, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Le Ouay, B.; Stellacci, F. Antibacterial activity of silver nanoparticles: A surface science insight. Nano Today 2015, 10, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Poblete, H.; Agarwal, A.; Thomas, S.S.; Bohne, C.; Ravichandran, R.; Phospase, J.; Comer, J.; Alarcon, E.I. New Insights into Peptide-Silver Nanoparticle Interaction: Deciphering the Role of Cysteine and Lysine in the Peptide Sequence. Langmuir 2016, 32, 265–273. [Google Scholar] [CrossRef]
- Pal, I.; Brahmkhatri, V.P.; Bera, S.; Bhattacharyya, D.; Quirishi, Y.; Bhunia, A.; Atreya, H.S. Enhanced stability and activity of an antimicrobial peptide in conjugation with silver nanoparticle. J. Colloid Interface Sci. 2016, 483, 385–393. [Google Scholar] [CrossRef]
- Pandit, R.; Rai, M.; Santos, C.A. Enhanced antimicrobial activity of the food-protecting nisin peptide by bioconjugation with silver nanoparticles. Environ. Chem. Lett. 2017, 15, 443–452. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Xie, J.; Luo, Z.; Jiang, J.; Yang, Y.Y.; Liu, S. The potent antimicrobial properties of cell penetrating peptide-conjugated silver nanoparticles with excellent selectivity for Gram-positive bacteria over erythrocytes. Nanoscale 2013, 5, 3834–3840. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Na, H.; Zhong, R.; Yuan, M.; Guo, J.; Zhao, L.; Wang, Y.; Wang, L.; Zhang, F. One step synthesis of antimicrobial peptide protected silver nanoparticles: The core-shell mutual enhancement of antibacterial activity. Coll. Surfaces B Biointerfaces 2020, 186, 110704. [Google Scholar] [CrossRef]
- Pal, I.; Bhattacharyya, D.; Kar, R.K.; Zarena, D.; Bhunia, A.; Atreya, H.S. A Peptide-Nanoparticle System with Improved Efficacy against Multidrug Resistant Bacteria. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Imura, Y.; Nishida, M.; Matsuzaki, K. Action mechanism of PEGylated magainin 2 analogue peptide. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2578–2585. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Papareddy, P.; Mörgelin, M.; Schmidtchen, A.; Malmsten, M. Effects of PEGylation on membrane and lipopolysaccharide interactions of host defense peptides. Biomacromolecules 2014, 15, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J.; Beck, K.; Fox, M.A.; Ulaeto, D.; Clark, G.C.; Gumbleton, M. Pegylation of antimicrobial peptides maintains the active peptide conformation, model membrane interactions, and antimicrobial activity while improving lung tissue biocompatibility following airway delivery. Antimicrob. Agents Chemother. 2012, 56, 3298–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chereddy, K.K.; Her, C.H.; Comune, M.; Moia, C.; Lopes, A.; Porporato, P.E.; Vanacker, J.; Lam, M.C.; Steinstraesser, L.; Sonveaux, P.; et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J. Control. Release 2014, 194, 138–147. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Water, J.J.; Smart, S.; Franzyk, H.; Foged, C.; Nielsen, H.M. Nanoparticle-mediated delivery of the antimicrobial peptide plectasin against Staphylococcus aureus in infected epithelial cells. Eur. J. Pharm. Biopharm. 2015, 92, 65–73. [Google Scholar] [CrossRef]
- Ko, J.C.; Chuang, S.C.; Yang, C. Da Amidated pleurocidin peptide encapsulated in biodegradable poly(lactide-co-glycolide) microparticles sustains its antibacterial activity against Photobacterium damselae subsp. piscicida in cobia. Aquaculture 2019, 512, 734290. [Google Scholar] [CrossRef]
- Vijayan, A.; James, P.P.; Nanditha, C.K.; Vinod Kumar, G.S. Multiple cargo deliveries of growth factors and antimicrobial peptide using biodegradable nanopolymer as a potential wound healing system. Int. J. Nanomedicine 2019, 14, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Casciaro, B.; D’Angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.P.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide- co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: In Vitro and in Vivo Studies. Biomacromolecules 2019, 20, 1876–1888. [Google Scholar] [CrossRef]
- Kumar, P.; Shenoi, R.A.; Lai, B.F.L.; Nguyen, M.; Kizhakkedathu, J.N.; Straus, S.K. Conjugation of Aurein 2.2 to HPG Yields an Antimicrobial with Better Properties. Biomacromolecules 2015, 16. [Google Scholar] [CrossRef]
- Kumar, P.; Takayesu, A.; Abbasi, U.; Kalathottukaren, M.T.; Abbina, S.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptide-Polymer Conjugates with High Activity: Influence of Polymer Molecular Weight and Peptide Sequence on Antimicrobial Activity, Proteolysis, and Biocompatibility. ACS Appl. Mater. Interfaces 2017, 9. [Google Scholar] [CrossRef]
- Haney, E.F.; Wuerth, K.C.; Rahanjam, N.; Nikouei, N.S.; Ghassemi, A.; Noghani, M.A.; Boey, A.; Hancock, R.E.W. Identification of an IDR peptide formulation candidate that prevents peptide aggregation and retains immunomodulatory activity. Pept. Sci. 2019. [Google Scholar] [CrossRef]
- Piras, A.M.; Maisetta, G.; Sandreschi, S.; Gazzarri, M.; Bartoli, C.; Grassi, L.; Esin, S.; Chiellini, F.; Batoni, G. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front. Microbiol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaaytah, A.; Mohammed, G.K.; Abualhaijaa, A.; Al-Balas, Q. Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug Des. Devel. Ther. 2017, 11, 3159–3170. [Google Scholar] [CrossRef] [Green Version]
- Sahariah, P.; Sørensen, K.K.; Hjálmarsdóttir, M.A.; Sigurjónsson, Ó.E.; Jensen, K.J.; Másson, M.; Thygesen, M.B. Antimicrobial peptide shows enhanced activity and reduced toxicity upon grafting to chitosan polymers. Chem. Commun. 2015, 51, 11611–11614. [Google Scholar] [CrossRef]
- Hou, Z.; Shankar, Y.V.; Liu, Y.; Ding, F.; Subramanion, J.L.; Ravikumar, V.; Zamudio-Vázquez, R.; Keogh, D.; Lim, H.; Tay, M.Y.F.; et al. Nanoparticles of Short Cationic Peptidopolysaccharide Self-Assembled by Hydrogen Bonding with Antibacterial Effect against Multidrug-Resistant Bacteria. ACS Appl. Mater. Interfaces 2017, 9, 38288–38303. [Google Scholar] [CrossRef]
- Li, J.F.; Zhang, J.X.; Wang, Z.G.; Yao, Y.J.; Han, X.; Zhao, Y.L.; Liu, J.P.; Zhang, S.Q. Identification of a cyclodextrin inclusion complex of antimicrobial peptide CM4 and its antimicrobial activity. Food Chem. 2017, 221, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.K.; Lyu, Y.; Zhu, X.; Wang, J.P.; Jin, Z.Y.; Narsimhan, G. Enhanced solubility and antimicrobial activity of alamethicin in aqueous solution by complexation with γ-cyclodextrin. J. Funct. Foods 2018, 40, 700–706. [Google Scholar] [CrossRef]
- Jiao, J.B.; Wang, G.Z.; Hu, X.L.; Zang, Y.; Maisonneuve, S.; Sedgwick, A.C.; Sessler, J.L.; Xie, J.; Li, J.; He, X.P.; et al. Cyclodextrin-Based Peptide Self-Assemblies (Spds) That Enhance Peptide-Based Fluorescence Imaging and Antimicrobial Efficacy. J. Am. Chem. Soc. 2020, 142, 1925–1932. [Google Scholar] [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.L.; Ong, H.K.; Teo, J.; Ow, D.S.W.; Chao, S.H. HEXIM1 peptide exhibits antimicrobial activity against antibiotic resistant bacteria through guidance of cell penetrating peptide. Front. Microbiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umstätter, F.; Domhan, C.; Hertlein, T.; Ohlsen, K.; Mühlberg, E.; Kleist, C.; Zimmermann, S.; Beijer, B.; Klika, K.D.; Haberkorn, U.; et al. Vancomycin Resistance is Overcome by Conjugation of Polycationic Peptides. Angew. Chemie Int. Ed. 2020, 59, 8823–8827. [Google Scholar] [CrossRef] [Green Version]
- Pini, A.; Falciani, C.; Mantengoli, E.; Bindi, S.; Brunetti, J.; Iozzi, S.; Rossolini, G.M.; Bracci, L. A novel tetrabranched antimicrobial peptide that neutralizes bacterial lipopolysaccharide and prevents septic shock in vivo. FASEB J. 2010, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, J.; Falciani, C.; Roscia, G.; Pollini, S.; Bindi, S.; Scali, S.; Arrieta, U.C.; Gomez-Vallejo, V.; Quercini, L.; Ibba, E.; et al. In vitro and in vivo efficacy, toxicity, bio-distribution and resistance selection of a novel antibacterial drug candidate. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Falciani, C.; Lozzi, L.; Pollini, S.; Luca, V.; Carnicelli, V.; Brunetti, J.; Lelli, B.; Bindi, S.; Scali, S.; Di Giulio, A.; et al. Isomerization of an Antimicrobial Peptide Broadens Antimicrobial Spectrum to Gram-Positive Bacterial Pathogens. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Franzman, M.R.; Burnell, K.K.; Dehkordi-Vakil, F.H.; Guthmiller, J.M.; Dawson, D.V.; Brogden, K.A. Targeted antimicrobial activity of a specific IgG-SMAP28 conjugate against Porphyromonas gingivalis in a mixed culture. Int. J. Antimicrob. Agents 2009, 33, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Peschen, D.; Li, H.P.; Fischer, R.; Kreuzaler, F.; Liao, Y.C. Fusion proteins comprising a Fusarium-specific antibody linked to antifungal peptides protect plants against a fungal pathogen. Nat. Biotechnol. 2004, 22, 732–738. [Google Scholar] [CrossRef]
- Touti, F.; Lautrette, G.; Johnson, K.D.; Delaney, J.C.; Wollacott, A.; Tissire, H.; Viswanathan, K.; Shriver, Z.; Mong, S.K.; Mijalis, A.J.; et al. Antibody–Bactericidal Macrocyclic Peptide Conjugates To Target Gram-Negative Bacteria. ChemBioChem 2018, 19, 2039–2044. [Google Scholar] [CrossRef]
- Gautam, S.; Kim, T.; Lester, E.; Deep, D.; Spiegel, D.A. Wall teichoic acids prevent antibody binding to epitopes within the cell wall of Staphylococcus aureus. ACS Chem. Biol. 2016, 11, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Obuobi, S.; Tay, H.K.L.; Tram, N.D.T.; Selvarajan, V.; Khara, J.S.; Wang, Y.; Ee, P.L.R. Facile and efficient encapsulation of antimicrobial peptides via crosslinked DNA nanostructures and their application in wound therapy. J. Control. Release 2019, 313, 120–130. [Google Scholar] [CrossRef]
- Mela, I.; Endo, M.; Sugiyama, H.; Henderson, R.M.; Kaminski, C.F. DNA Origami as a Tool in the Targeted Destruction of Bacteria. Biophys. J. 2019, 116, 324a. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. 2017, 49, 916–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, M.; Qiu, S.; Wang, J.; Peng, J.; Zhao, P.; Zhu, R.; Wang, H.; Li, Y.; Wang, K.; et al. Antimicrobial activity and stability of the d-amino acid substituted derivatives of antimicrobial peptide polybia-MPI. AMB Express 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; De Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, T.N.; Stach, M.; He, R.; Gan, B.H.; Javor, S.; Heitz, M.; Ma, L.; Cai, X.; Chen, P.; Wei, D.; et al. Lipidated Peptide Dendrimers Killing Multidrug-Resistant Bacteria. J. Am. Chem. Soc. 2018, 140, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Chu-Kung, A.F.; Nguyen, R.; Bozzelli, K.N.; Tirrell, M. Chain length dependence of antimicrobial peptide-fatty acid conjugate activity. J. Colloid Interface Sci. 2010, 345, 160–167. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Gera, L.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides to Target Gram-negative Pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of Charge, “Specificity Determinants,” Total Hydrophobicity, Hydrophobe Type and Location as Design Para. Chem. Biol. Drug Des. 2011, 77, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.D.T.; Pedron, C.N.; Higashikuni, Y.; Kramer, R.M.; Cardoso, M.H.; Oshiro, K.G.N.; Franco, O.L.; Silva Junior, P.I.; Silva, F.D.; Oliveira Junior, V.X.; et al. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Biol. 2018, 1, 1–16. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.J.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migoń, D.; Neubauer, D.; Kamysz, W. Hydrocarbon Stapled Antimicrobial Peptides. Protein J. 2018, 37, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.S.; Pentelute, B.L. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautrette, G.; Touti, F.; Lee, H.G.; Dai, P.; Pentelute, B.L. Nitrogen Arylation for Macrocyclization of Unprotected Peptides. J. Am. Chem. Soc. 2016, 138, 8340–8343. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef]
- Méndez-Samperio, P. Peptidomimetics as a new generation of antimicrobial agents: Current progress. Infect. Drug Resist. 2014, 7, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Kuppusamy, R.; Willcox, M.; Black, D.S.C.; Kumar, N. Short cationic peptidomimetic antimicrobials. Antibiotics 2019, 8, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, R.P.; Romanowski, E.G.; Yates, K.A.; Mah, F.S. An independent evaluation of a novel peptide mimetic, Brilacidin (PMX30063), for ocular anti-infective. J. Ocul. Pharmacol. Ther. 2016, 32, 23–27. [Google Scholar] [CrossRef]
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Piddock, L.J.V. Non-traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Rex, J.H.; Fernandez Lynch, H.; Cohen, I.G.; Darrow, J.J.; Outterson, K. Designing development programs for non-traditional antibacterial agents. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
| Delivery Vehicle | Advantages | Disadvantages | References |
|---|---|---|---|
| Liposomes |
|
| [22,23,70,71] |
| Polymeric Micelles |
|
| [72,73,74,75] |
| Liquid Crystalline Nanoparticles |
|
| [22,23] |
| Nano-structured Lipid Carriers and Solid Lipid Nanoparticles |
|
| [23,71,76] |
| Metal Nanoparticles |
|
| [23,77] |
| Synthetic polymers |
|
| [22,70,78] |
| Poly-saccharides |
|
| [79,80,81] |
| Polypeptide Dendrimers |
|
| [82,83] |
| Antibodies |
|
| [84] |
| DNA Nano-structures |
|
| [85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drayton, M.; Kizhakkedathu, J.N.; Straus, S.K. Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance. Molecules 2020, 25, 3048. https://doi.org/10.3390/molecules25133048
Drayton M, Kizhakkedathu JN, Straus SK. Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance. Molecules. 2020; 25(13):3048. https://doi.org/10.3390/molecules25133048
Chicago/Turabian StyleDrayton, Matthew, Jayachandran N. Kizhakkedathu, and Suzana K. Straus. 2020. "Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance" Molecules 25, no. 13: 3048. https://doi.org/10.3390/molecules25133048