
4,5-Diazafluorene and 9,9’-Dimethyl-4,5-Diazafluorene as Ligands Supporting Redox-Active Mn and Ru Complexes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
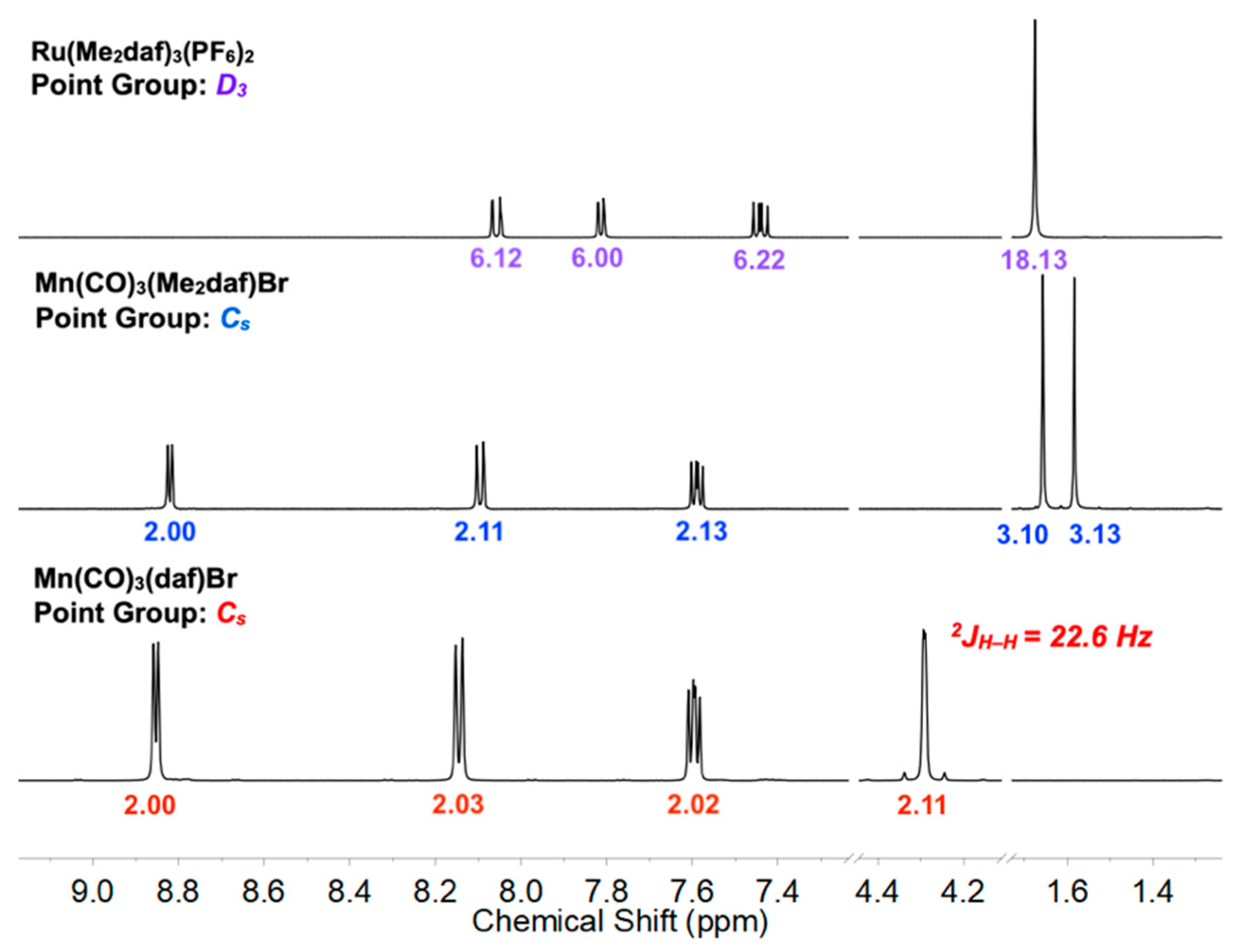
2.1. Synthesis and NMR Characterization of Complexes 2, 3, and 5
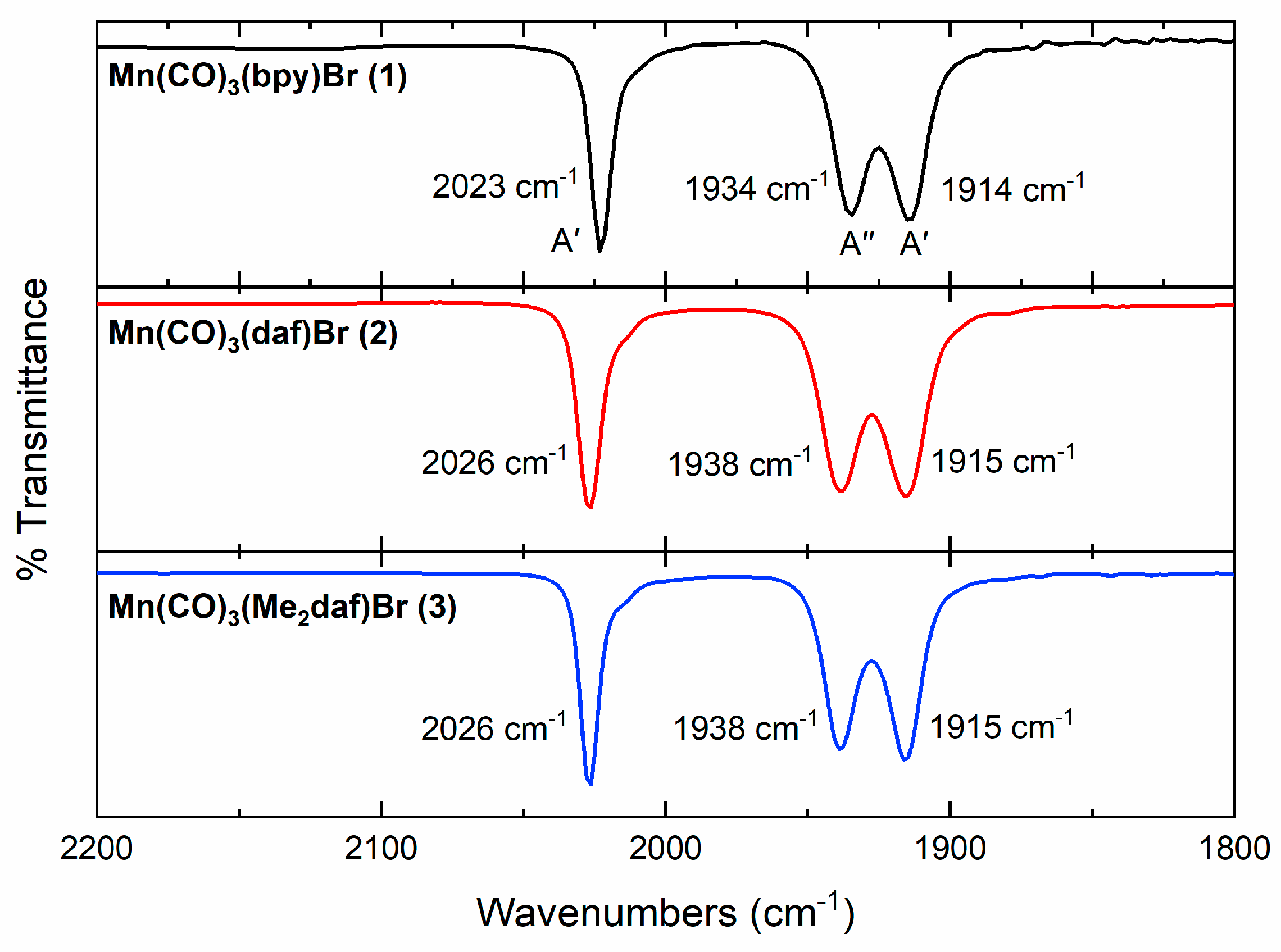
2.2. Electronic Absorption, IR, and X-ray Diffraction Studies
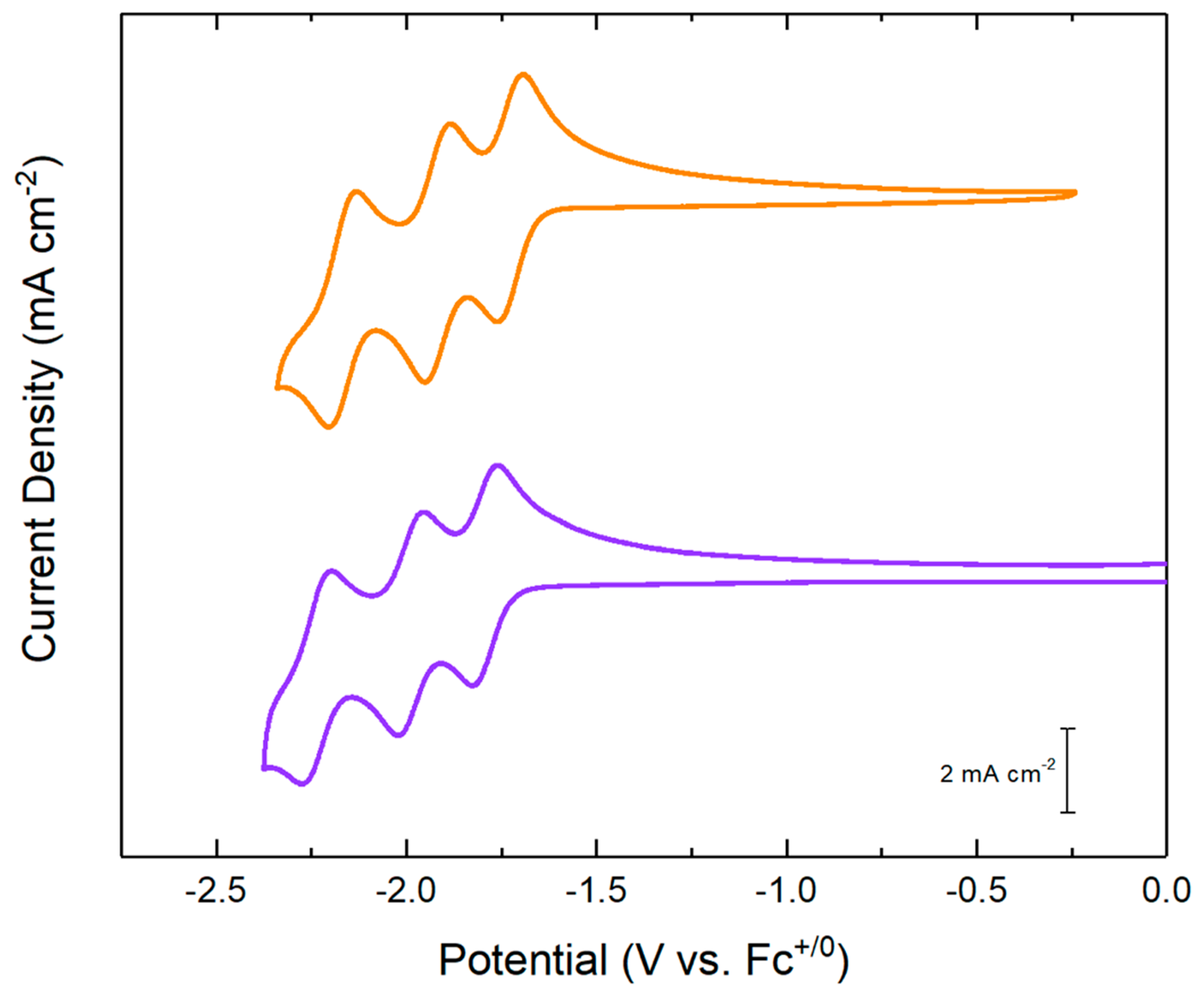
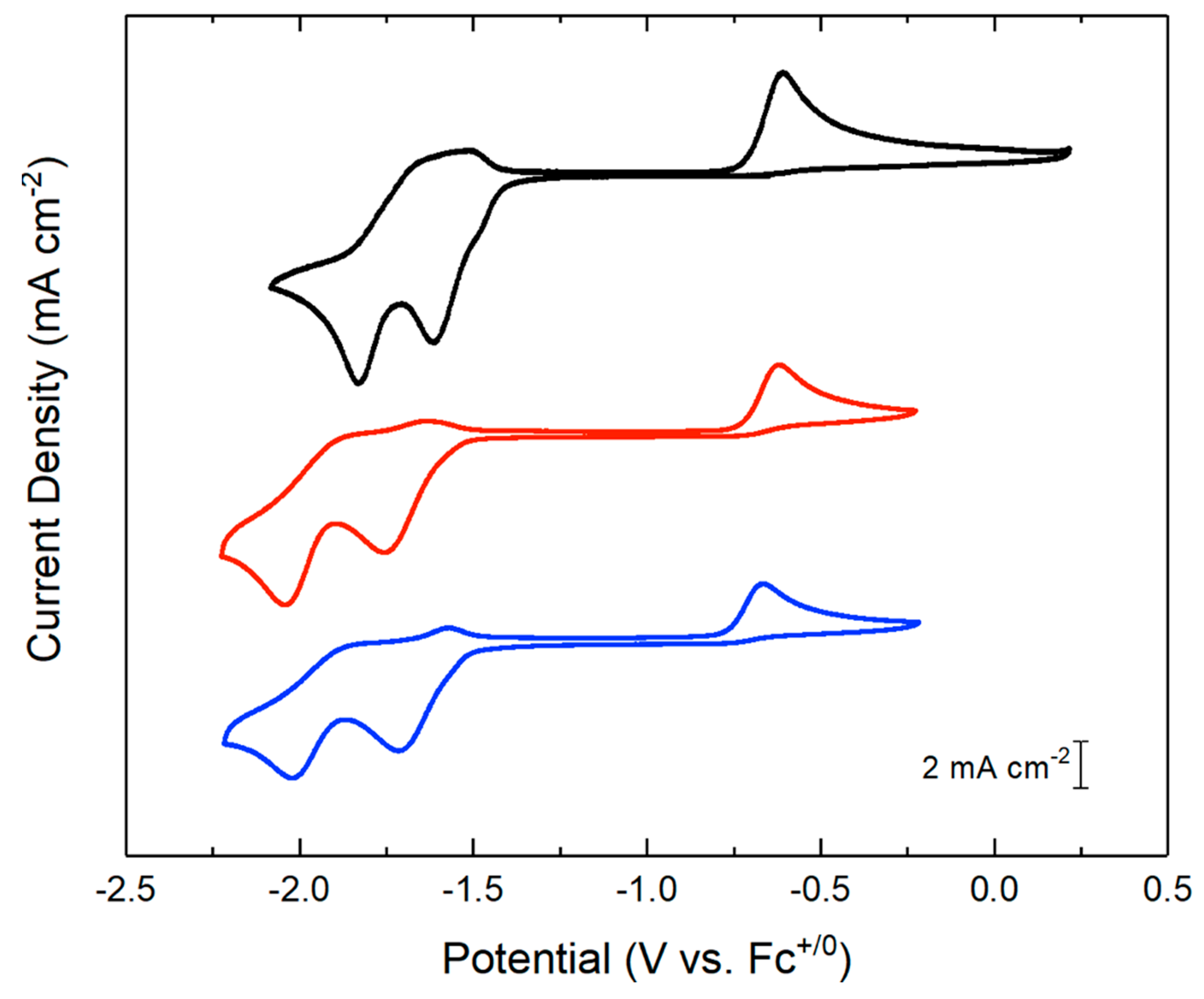
2.3. Electrochemical Studies
3. Conclusions
4. Materials and Methods
4.1. General Considerations
4.2. X-Ray Crystallography
4.3. Electrochemistry
4.4. Gas Chromatography
4.5. Preparation of Mn(CO)3(4,5-diazafluorene)Br (2)
4.6. Preparation of Mn(CO)3(9,9’-dimethyl-4,5-diazafluorene)Br (3)
4.7. Preparation of [Tris(9,9’-dimethyl-4,5-diazafluorene)Ruthenium](PF6)2 (5)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- English, A.M.; Delaive, P.J.; Gray, H.B.; Lum, V.R. Metalloprotein electron-transfer mechanisms. Quenching of electronically excited tris(2,2’-bipyridine)ruthenium(II) by reduced blue copper proteins. J. Am. Chem. Soc. 1982, 104, 870–871. [Google Scholar] [CrossRef]
- Brunschwig, B.S.; Delaive, P.J.; English, A.M.; Goldberg, M.; Gray, H.B.; Mayo, S.L.; Sutin, N. Kinetics and mechanisms of electron transfer between blue copper proteins and electronically excited chromium and ruthenium polypyridine complexes. Inorg. Chem. 1985, 24, 3743–3749. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Bolinger, C.M.; Conrad, D.; Vining, W.J.; Meyer, T.J. One- and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2’-bipyridine). J. Chem. Soc. Chem. Commun. 1985, 1414. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J. Electron Transfer Reactions Induced by Excited State Quenching. Isr. J. Chem. 1976, 15, 200–205. [Google Scholar] [CrossRef]
- Moyer, B.A.; Meyer, T.J. Properties of the oxo/aqua system (bpy)2(py)RuO2+/(bpy)2(py)Ru(OH2)2+. Inorg. Chem. 1981, 20, 436–444. [Google Scholar] [CrossRef]
- Imayoshi, R.; Tanaka, H.; Matsuo, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Cobalt-Catalyzed Transformation of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. A Eur. J. 2015, 21, 8905–8909. [Google Scholar] [CrossRef]
- Henke, W.C.; Otolski, C.J.; Moore, W.N.G.; Elles, C.G.; Blakemore, J.D. Ultrafast Spectroscopy of [Mn(CO)3] Complexes: Tuning the Kinetics of Light-Driven CO Release and Solvent Binding. Inorg. Chem. 2020, 59, 2178–2187. [Google Scholar] [CrossRef]
- Henke, W.; Lionetti, D.; Moore, W.N.G.; Hopkins, J.A.; Day, V.; Blakemore, J.D. Ligand Substituents Govern the Efficiency and Mechanistic Path of Hydrogen Production with [Cp*Rh] Catalysts. ChemSusChem 2017, 10, 4589–4598. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.L.; Cheung, P.L.; Lessio, M.; Carter, E.A.; Kubiak, C.P. Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. [Google Scholar] [CrossRef]
- Tignor, S.E.; Kuo, H.-Y.; Lee, T.S.; Scholes, G.; Bocarsly, A.B. Manganese-Based Catalysts with Varying Ligand Substituents for the Electrochemical Reduction of CO2 to CO. Organometallics 2018, 38, 1292–1299. [Google Scholar] [CrossRef]
- Henderson, L.J.; Fronczek, F.R.; Cherry, W.R. Selective perturbation of ligand field excited states in polypyridine ruthenium(II) complexes. J. Am. Chem. Soc. 1984, 106, 5876–5879. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Wang, K.-Z.; Wang, C.-R.; Yang, Y.; Cheung, K.-K. Synthesis, Characterization, and Second-Harmonic Generation Studies of Surfactant Rhenium(I) Diimine Complexes in Langmuir−Blodgett Films. X-ray Crystal Structure offac-ClRe(CO)3L (L = 9-Heptylamino-4,5-diazafluorene). Organometallics 1998, 17, 2440–2446. [Google Scholar] [CrossRef]
- Sykora, M.; Kincaid, J.R. Synthetic Manipulation of Excited State Decay Pathways in a Series of Ruthenium(II) Complexes Containing Bipyrazine and Substituted Bipyridine Ligands. Inorg. Chem. 1995, 34, 5852–5856. [Google Scholar] [CrossRef]
- Annibale, V.T.; Song, D. Coordination chemistry and applications of versatile 4,5-diazafluorene derivatives. Dalton Trans. 2016, 45, 32–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Song, D. Syntheses, Characterizations, and Reactivities of 4,5-Diazafluorenide Complexes of Palladium(II) and Rhodium(I). Organometallics 2008, 27, 3587–3592. [Google Scholar] [CrossRef]
- Jiang, H.; Stepowska, E.; Song, D. Syntheses, Structures and Reactivities of Rhodium 4,5-Diazafluorene Derivatives. Eur. J. Inorg. Chem. 2009, 2009, 2083–2089. [Google Scholar] [CrossRef]
- Stepowska, E.; Jiang, H.; Song, D. Reversible H2 splitting between Ru(II) and a remote carbanion in a zwitterionic compound. Chem. Commun. 2010, 46, 556–558. [Google Scholar] [CrossRef]
- Annibale, V.T.; Batcup, R.; Bai, T.; Hughes, S.J.; Song, D. RuCp* Complexes of Ambidentate 4,5-Diazafluorene Derivatives: From Linkage Isomers to Coordination-Driven Self-Assembly. Organometallics 2013, 32, 6511–6521. [Google Scholar] [CrossRef]
- Batcup, R.; Chiu, F.S.N.; Annibale, V.T.; Huh, J.-E.U.; Tan, R.; Song, D. Selective one-pot syntheses of PtII–CuI heterobimetallic complexes of 4,5-diazafluorenide derivatives. Dalton Trans. 2013, 42, 16343. [Google Scholar] [CrossRef]
- Campbell, A.N.; White, P.B.; Guzei, I.A.; Stahl, S.S. Allylic C−H Acetoxylation with a 4,5-Diazafluorenone-Ligated Palladium Catalyst: A Ligand-Based Strategy To Achieve Aerobic Catalytic Turnover. J. Am. Chem. Soc. 2010, 132, 15116–15119. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.N.; Meyer, E.B.; Stahl, S.S. Regiocontrolled aerobic oxidative coupling of indoles and benzene using Pd catalysts with 4,5-diazafluorene ligands. Chem. Commun. 2011, 47, 10257–10259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.B.; Jaworski, J.N.; Fry, C.G.; Dolinar, B.S.; Guzei, I.A.; Stahl, S.S. Structurally Diverse Diazafluorene-Ligated Palladium(II) Complexes and Their Implications for Aerobic Oxidation Reactions. J. Am. Chem. Soc. 2016, 138, 4869–4880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsevier, C.J. Catalytic and stoichiometric C–C bond formation employing palladium compounds with nitrogen ligands. Co-ord. Chem. Rev. 1999, 185, 809–822. [Google Scholar] [CrossRef]
- Baran, M.F.; Durap, F.; Aydemir, M.; Baysal, A. Transfer hydrogenation of aryl ketones with homogeneous ruthenium catalysts containing diazafluorene ligands. Appl. Organomet. Chem. 2016, 30, 1030–1035. [Google Scholar] [CrossRef]
- Klein, R.A.; Witte, P.; Van Belzen, R.; Fraanje, J.; Goubitz, K.; Numan, M.; Schenk, H.; Ernsting, J.M.; Elsevier, C.J. Monodentate and Bridging Coordination of 3,3′-Annelated 2,2′-Bipyridines in Zerovalent Palladium- and Platinum-p-quinone Complexes. Eur. J. Inorg. Chem. 1998, 319–330. [Google Scholar] [CrossRef]
- Druey, J.; Schmidt, P. Phenanthrolinchinone und Diazafluorene. Helvetica Chim. Acta 1950, 33, 1080–1087. [Google Scholar] [CrossRef]
- Ohrui, H.; Senoo, A.; Tetsuya, K. Diazafluorene Compound. U.S. Patent US20080161574A1, 2008. [Google Scholar]
- Luong, J.C.; Faltynek, R.A.; Wrighton, M.S. Ground- and excited-state oxidation-reduction chemistry of (triphenyltin)- and (triphenylgermanium)tricarbonyl(1,10-phenanthroline)rhenium and related compounds. J. Am. Chem. Soc. 1980, 102, 7892–7900. [Google Scholar] [CrossRef]
- Caspar, J.V.; Meyer, T.J. Application of the energy gap law to nonradiative, excited-state decay. J. Phys. Chem. 1983, 87, 952–957. [Google Scholar] [CrossRef]
- Miguel, D.; Riera, V. Synthesis of manganese(I) carbonyls with σ-bonded alkynyl ligands. J. Organomet. Chem. 1985, 293, 379–390. [Google Scholar] [CrossRef]
- Günther, H. NMR Spectroscopy; John Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Türschmann, P.; Colell, J.; Theis, T.; Blumich, B.; Appelt, S. Analysis of parahydrogen polarized spin system in low magnetic fields. Phys. Chem. Chem. Phys. 2014, 16, 15411–15421. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J. Photochemistry of metal coordination complexes:metal to ligand charge transfer excited states. Pure Appl. Chem. 1986, 58, 1193–1206. [Google Scholar]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Co-ord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Witte, P.T.; Klein, R.; Kooijman, H.; Spek, A.L.; Polášek, M.; Varga, V.; Mach, K. Electron transfer in the reactions of titanocene-bis(trimethylsilyl) acetylene complexes with 2,2’-bipyridine and 4,5-diazafluorene. The crystal structure of (4,5-diazafluorenyl) bis(pentamethylcyclopentadienyl) titanium(III). J. Organomet. Chem. 1996, 519, 195–204. [Google Scholar] [CrossRef]
- Machan, C.; Sampson, M.; Chabolla, S.A.; Dang, T.; Kubiak, C.P. Developing a Mechanistic Understanding of Molecular Electrocatalysts for CO2 Reduction using Infrared Spectroelectrochemistry. Organometallics 2014, 33, 4550–4559. [Google Scholar] [CrossRef]
- Rillema, D.P.; Jones, D.S. Structure of tris(2,2′-bipyridyl)ruthenium(II) hexafluorophosphate, [Ru(bipy)3][PF6]2. J. Chem. Soc. Chem. Commun. 1979, 849. [Google Scholar] [CrossRef]
- Rillema, D.P.; Jones, D.S.; Woods, C.; Levy, H.A. Comparison of the crystal structures of tris heterocyclic ligand complexes of ruthenium(II). Inorg. Chem. 1992, 31, 2935–2938. [Google Scholar] [CrossRef]
- Breu, J.; Domel, H.; Stoll, A. Racemic Compound Formation versus Conglomerate Formation with [M(bpy)3](PF6)2 (M = Ni, Zn, Ru); Molecular and Crystal Structures. Eur. J. Inorg. Chem. 2000, 2401–2408. [Google Scholar] [CrossRef]
- Fronczek, F.R. CCDC 287572: Experimental Crystal Structure Determination. CCDC 2006. [Google Scholar]
- Constable, E.C. ChemInform Abstract: Homoleptic Complexes of 2,2′-Bipyridine. Chemin 1990, 34, 1–63. [Google Scholar] [CrossRef]
- Kaim, W. The transition metal coordination chemistry of anion radicals. Co-ord. Chem. Rev. 1987, 76, 187–235. [Google Scholar] [CrossRef]
- England, J.; Scarborough, C.C.; Weyhermüller, T.; Sproules, S.; Wieghardt, K. Electronic Structures of the Electron Transfer Series [M(bpy)3]n, [M(tpy)2]n, and [Fe(tbpy)3]n(M = Fe, Ru;n= 3+, 2+, 1+, 0, 1-): A Mössbauer Spectroscopic and DFT Study. Eur. J. Inorg. Chem. 2012, 4605–4621. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; De Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Corcos, A.R.; Villanueva, O.; Walroth, R.C.; Sharma, S.K.; Bacsa, J.; Lancaster, K.M.; Macbeth, C.E.; Berry, J.F. Oxygen Activation by Co(II) and a Redox Non-Innocent Ligand: Spectroscopic Characterization of a Radical–Co(II)–Superoxide Complex with Divergent Catalytic Reactivity. J. Am. Chem. Soc. 2016, 138, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Sampson, M.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Concepts Magn. Reson. 2002, 14, 326–346. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further Conventions for NMR Shielding and Chemical Shifts. IUPAC Standards Online 2016, 80, 59–84. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds 2, 3, and 5 are available from the authors upon request. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henke, W.C.; Hopkins, J.A.; Anderson, M.L.; Stiel, J.P.; Day, V.W.; Blakemore, J.D. 4,5-Diazafluorene and 9,9’-Dimethyl-4,5-Diazafluorene as Ligands Supporting Redox-Active Mn and Ru Complexes. Molecules 2020, 25, 3189. https://doi.org/10.3390/molecules25143189
Henke WC, Hopkins JA, Anderson ML, Stiel JP, Day VW, Blakemore JD. 4,5-Diazafluorene and 9,9’-Dimethyl-4,5-Diazafluorene as Ligands Supporting Redox-Active Mn and Ru Complexes. Molecules. 2020; 25(14):3189. https://doi.org/10.3390/molecules25143189
Chicago/Turabian StyleHenke, Wade C., Julie A. Hopkins, Micah L. Anderson, Jonah P. Stiel, Victor W. Day, and James D. Blakemore. 2020. "4,5-Diazafluorene and 9,9’-Dimethyl-4,5-Diazafluorene as Ligands Supporting Redox-Active Mn and Ru Complexes" Molecules 25, no. 14: 3189. https://doi.org/10.3390/molecules25143189