
Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design


Abstract
:1. Introduction
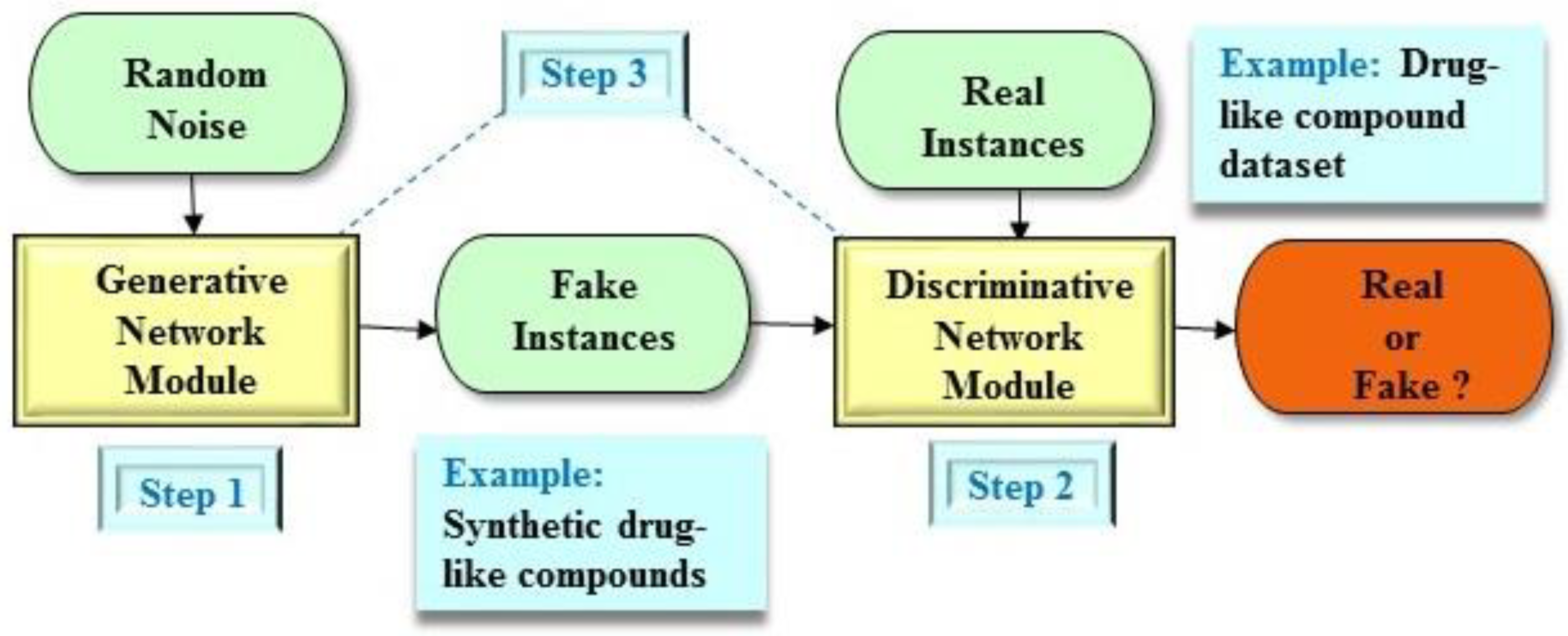
2. Generative Adversarial Network (GAN) Architecture
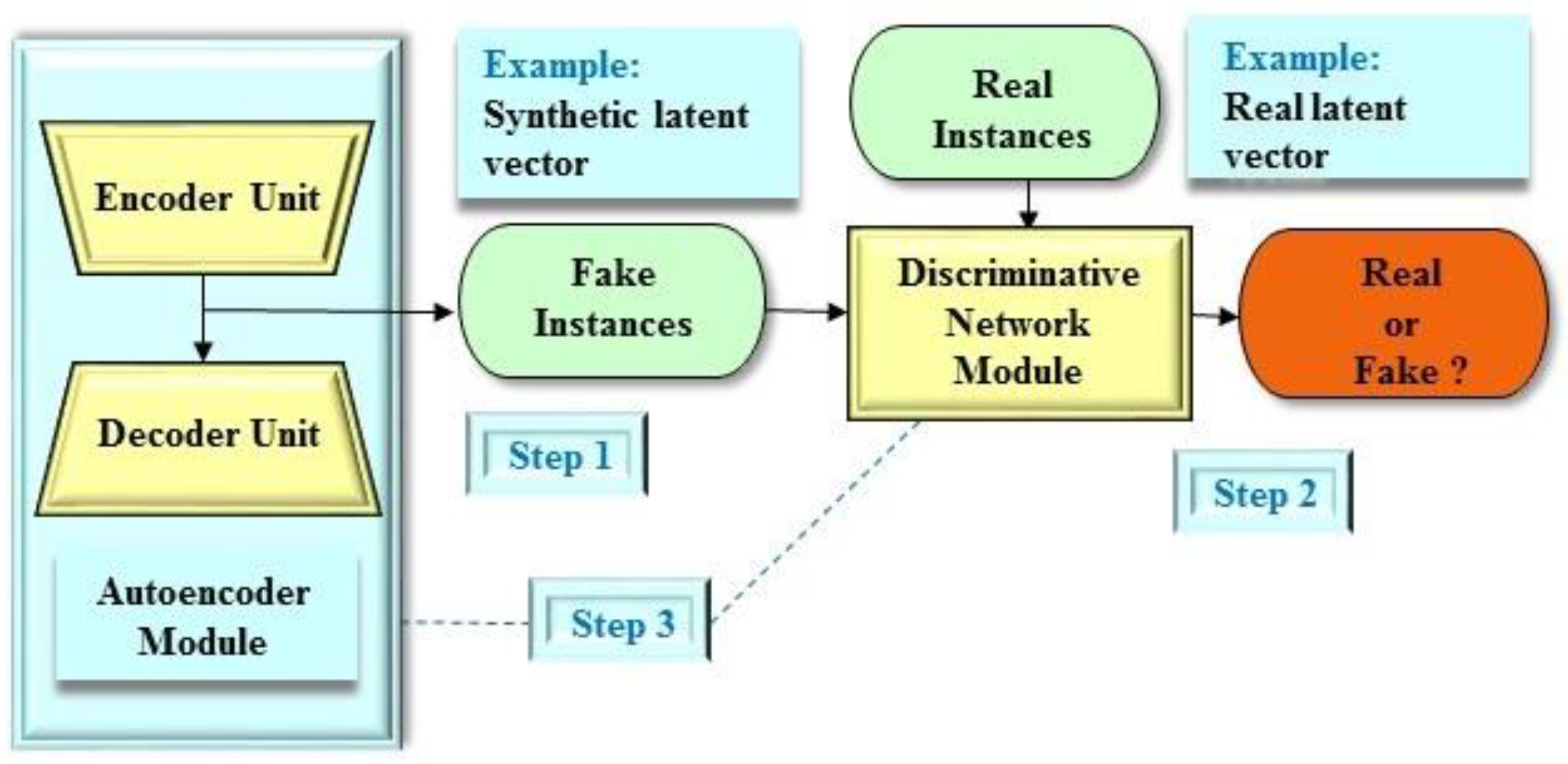
2.1. Brief Description of the GAN Architecture
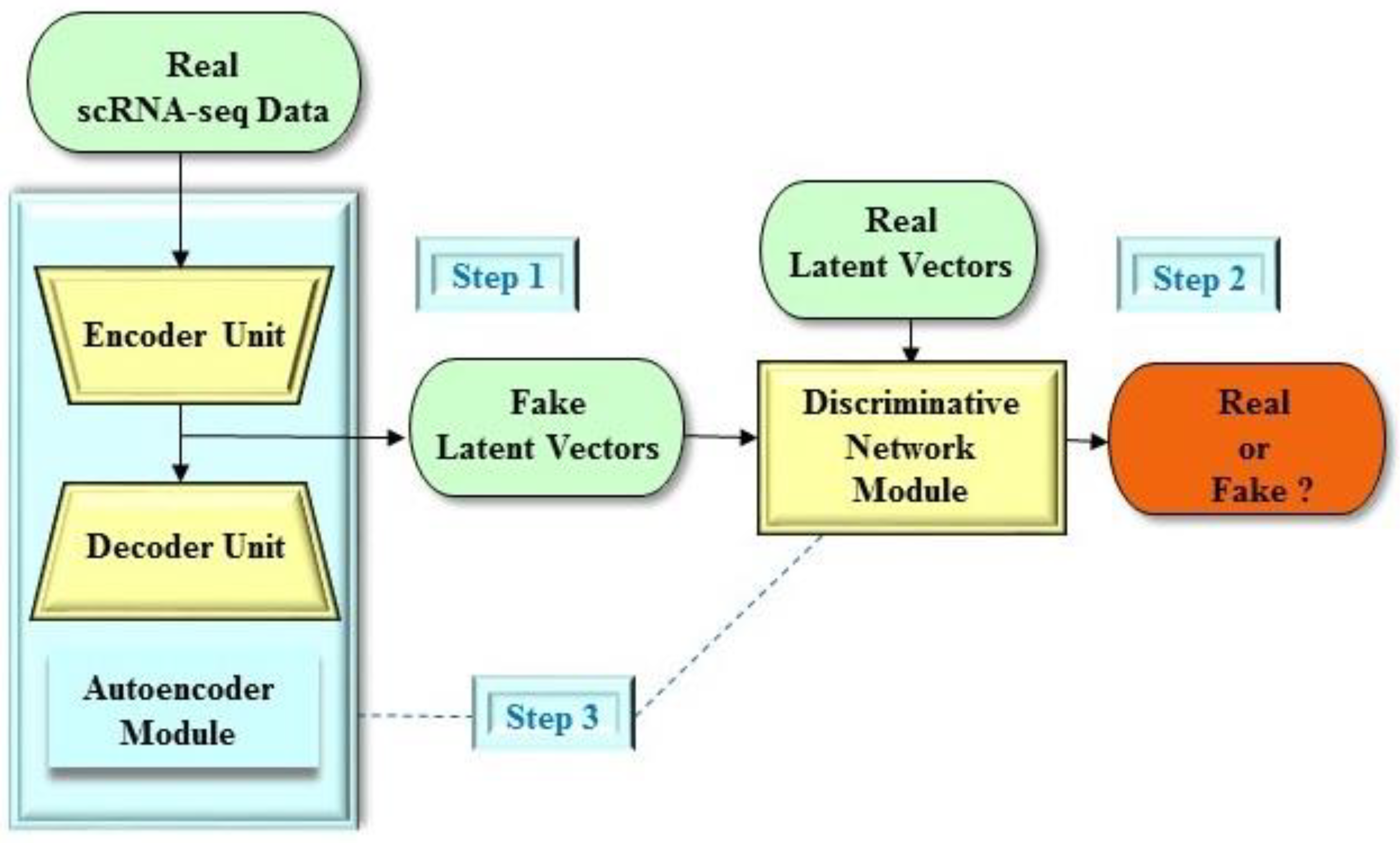
2.2. Applications of the GAN Architecture
2.3. Variants of the GAN Architecture
2.3.1. Wasserstein GAN
2.3.2. Conditional GAN
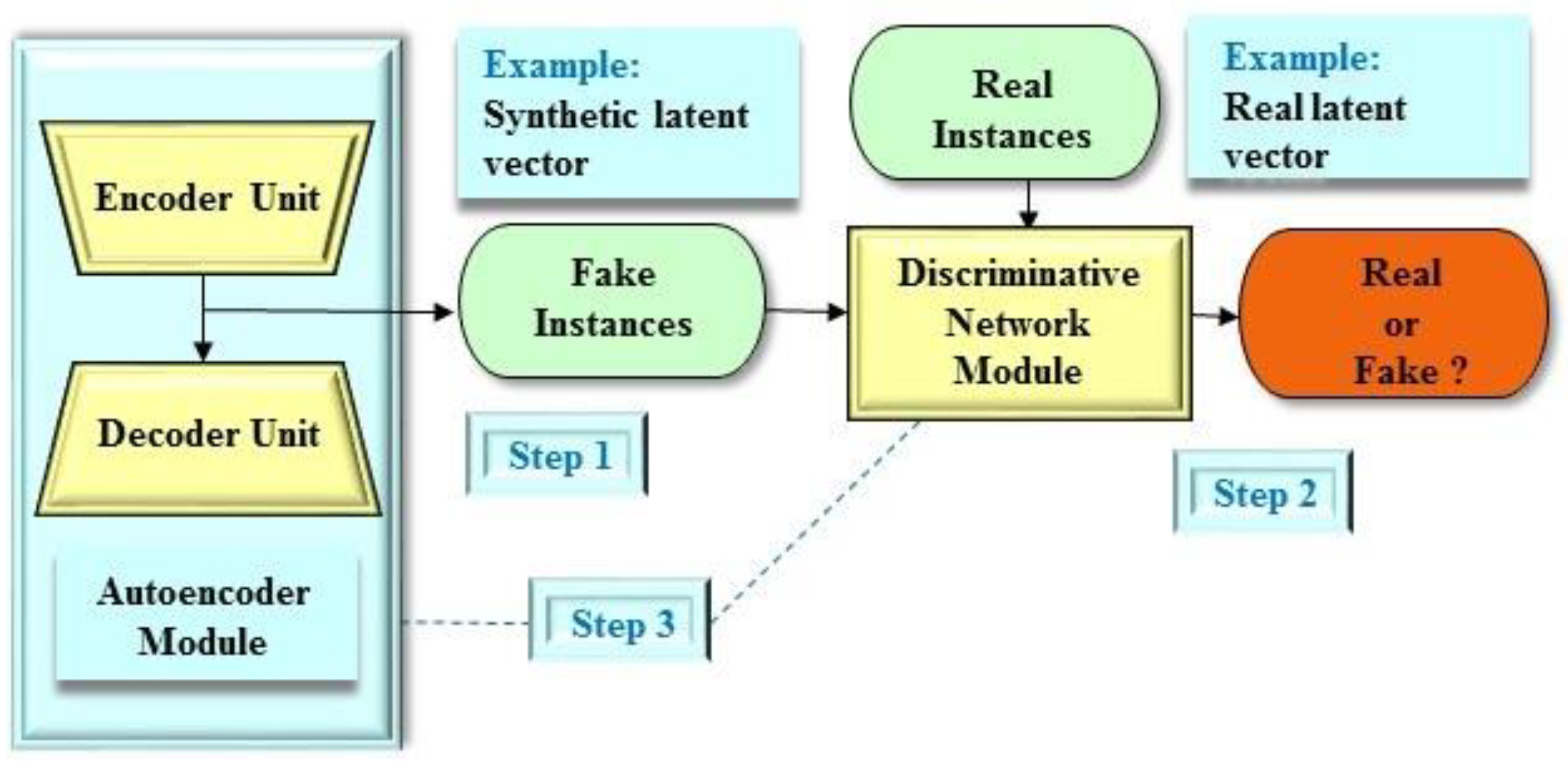
2.3.3. Adversarial Autoencoder
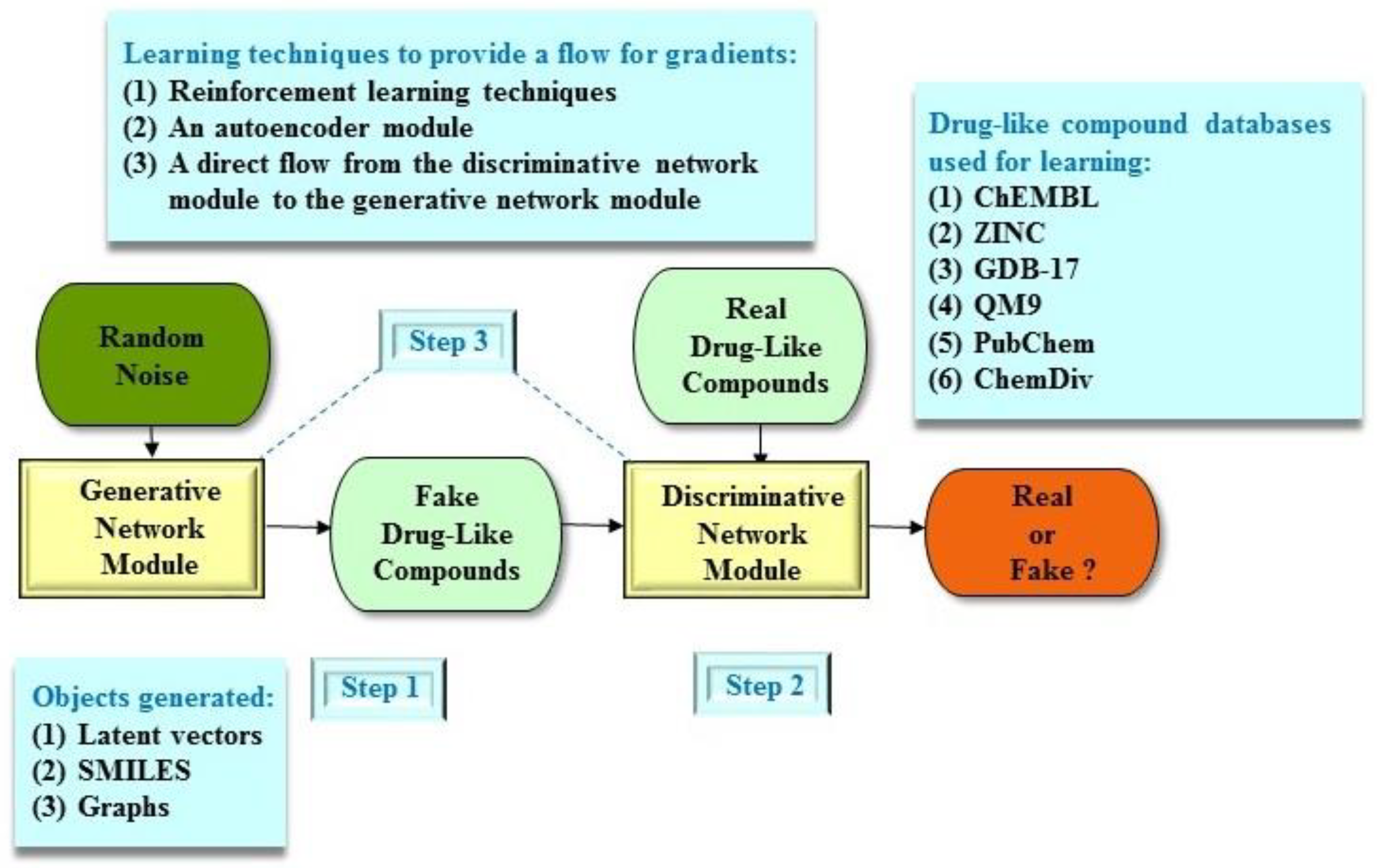
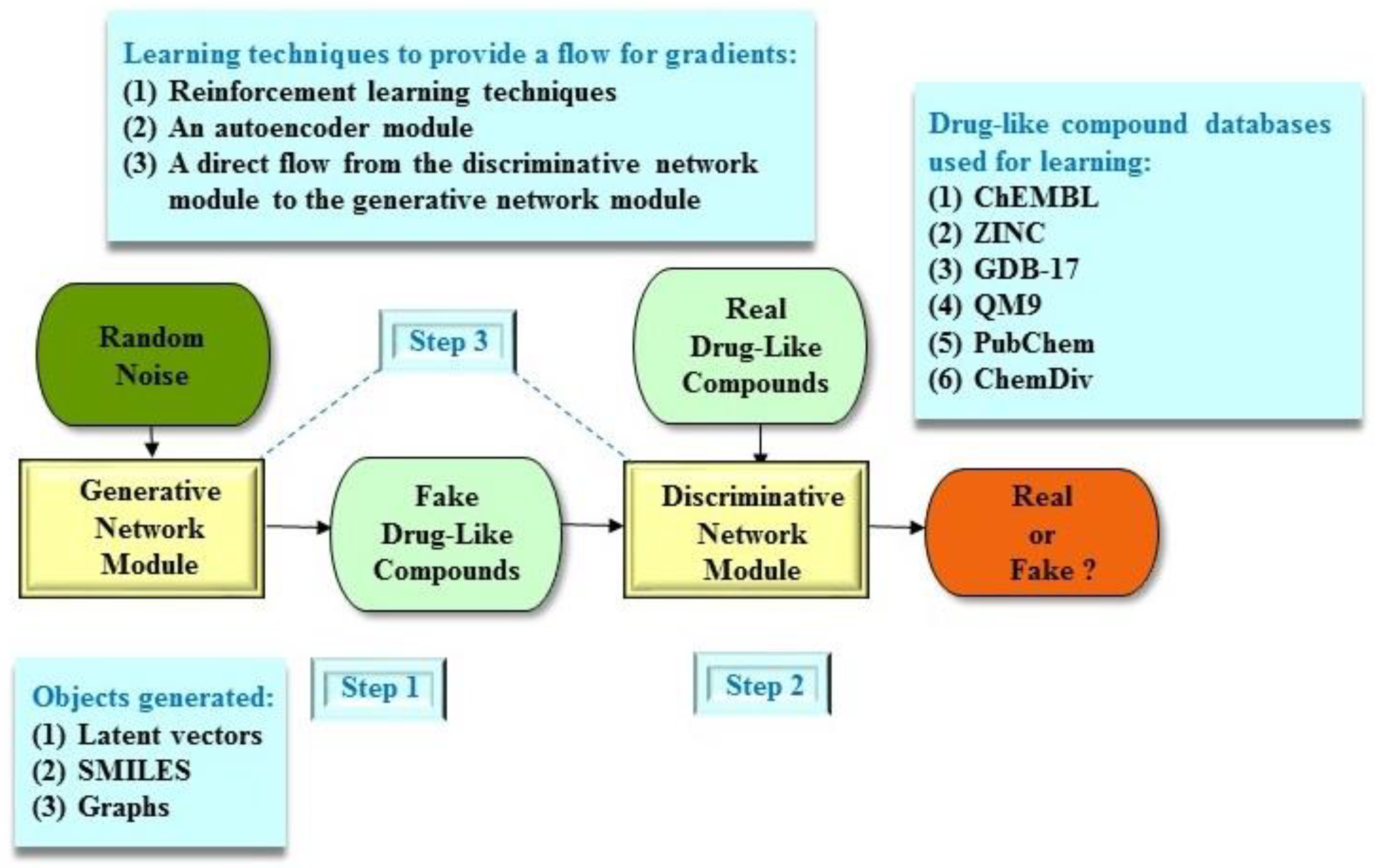
3. Molecular De Novo Design
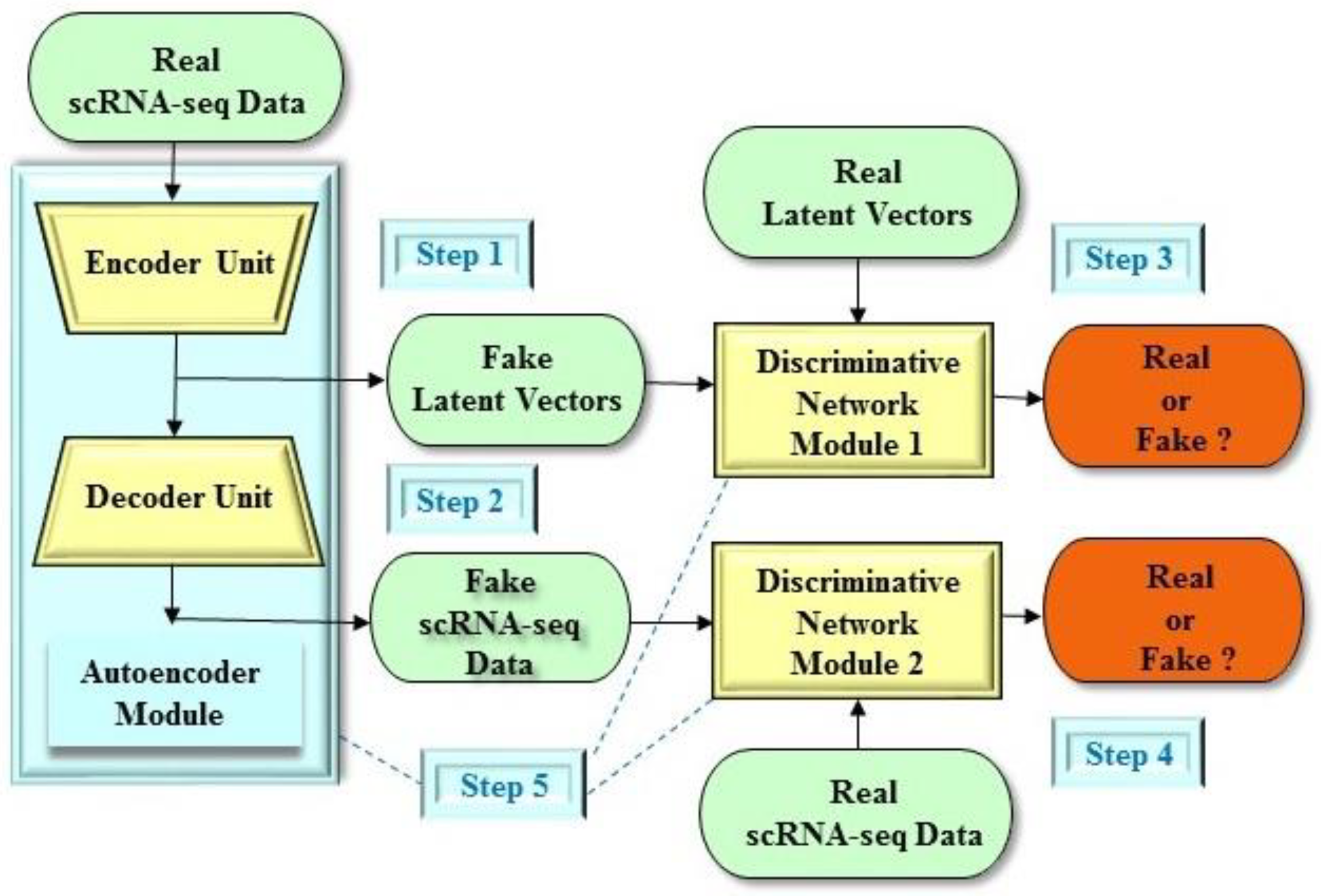
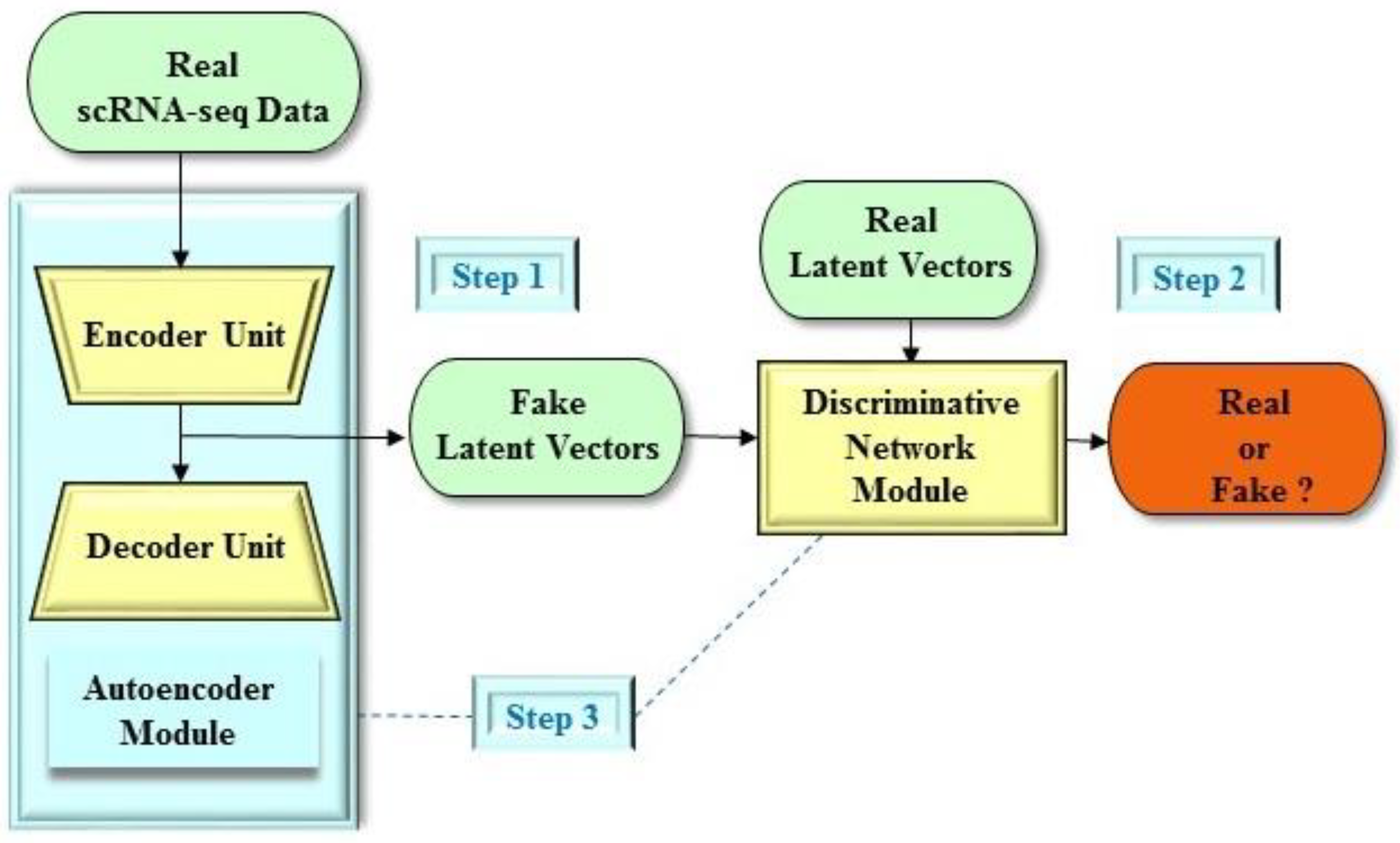
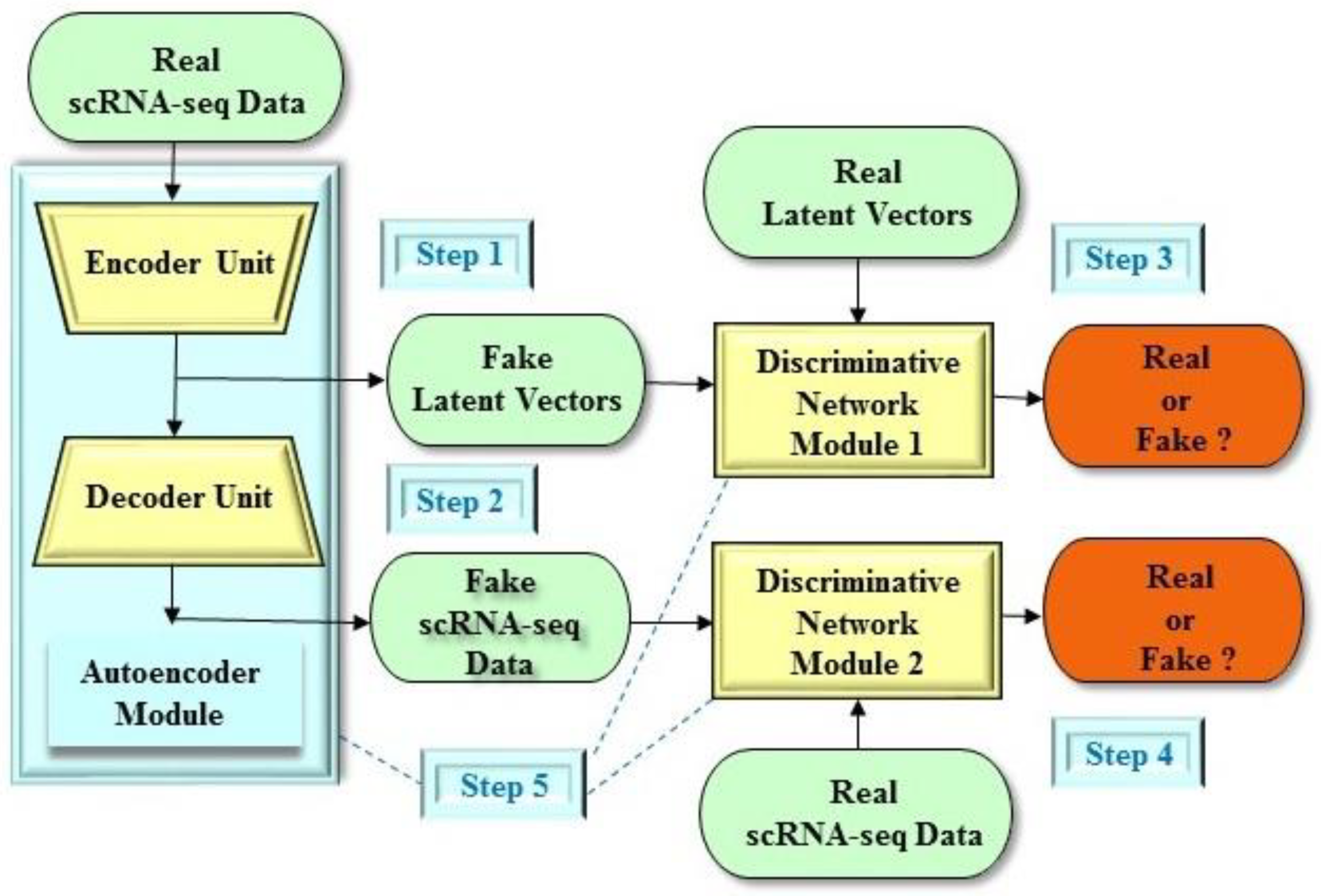
4. Dimension Reduction of Single-Cell Data in Preclinical Development
5. De Novo Peptide and Protein Design
6. Limitations
7. Other Relevant Applications in Drug Design and Discovery
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.-H. Artificial intelligence in drug design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Segler, M.H.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskin, I.I.; Winkler, D.; Tetko, I.V. A renaissance of neural networks in drug discovery. Expert Opin. Drug Discov. 2016, 11, 785–795. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Hinton, G. Deep learning—a technology with the potential to transform health care. Jama 2018, 320, 1101–1102. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q.S. Deep learning for drug design: An artificial intelligence paradigm for drug discovery in the big data era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinf. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of deep learning in biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S. The next era: Deep learning in pharmaceutical research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018, 9, 1–13. [Google Scholar]
- Wang, D.; Gu, J. VASC: Dimension reduction and visualization of single-cell RNA-seq data by deep variational autoencoder. Genom. Proteom. Bioinf. 2018, 16, 320–331. [Google Scholar] [CrossRef]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inf. 2018, 37, 1700123. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, F.; Mehridehnavi, A.; Perez-Garrido, A.; Perez-Sanchez, H. Neural network and deep-learning algorithms used in QSAR studies: Merits and drawbacks. Drug Discov. Today 2018, 23, 1784–1790. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef] [Green Version]
- Dana, D.; Gadhiya, S.V.; St Surin, L.G.; Li, D.; Naaz, F.; Ali, Q.; Paka, L.; Yamin, M.A.; Narayan, M.; Goldberg, I.D.; et al. Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules 2018, 23, 2384. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Kuo, P.H.; Liu, Y.L.; Yu, Y.W.; Yang, A.C.; Tsai, S.J. A Deep Learning Approach for Predicting Antidepressant Response in Major Depression Using Clinical and Genetic Biomarkers. Front. Psychiatr. 2018, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.; Tsai, S.-J. Machine Learning in Neural Networks. In Frontiers in Psychiatry; Springer: Singapore, 2019; pp. 127–137. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Cananda, 8–13 December 2014; pp. 2672–2680. [Google Scholar]
- Zhao, H.; Li, H.; Maurer-Stroh, S.; Cheng, L. Synthesizing retinal and neuronal images with generative adversarial nets. Med. Image Anal. 2018, 49, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Tang, Y.; Chang, E.I.; Fan, Y.; Lai, M.; Xu, Y. Unsupervised Learning For Cell-level Visual Representation with Generative Adversarial Networks. IEEE J. Biomed. Health Inform. 2018, 23, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardani, M.; Gong, E.; Cheng, J.Y.; Vasanawala, S.S.; Zaharchuk, G.; Xing, L.; Pauly, J.M. Deep Generative Adversarial Neural Networks for Compressive Sensing (GANCS) MRI. IEEE Trans. Med. Imaging 2018, 38, 167–179. [Google Scholar] [CrossRef]
- Lin, E.; Mukherjee, S.; Kannan, S. A deep adversarial variational autoencoder model for dimensionality reduction in single-cell RNA sequencing analysis. BMC Bioinf. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883–10890. [Google Scholar] [CrossRef] [Green Version]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Alqahtani, H.; Kavakli-Thorne, M.; Kumar, G. Applications of generative adversarial networks (gans): An updated review. Arch. Comput. Methods Eng. 2019, 1–28. [Google Scholar] [CrossRef]
- Lan, L.; You, L.; Zhang, Z.; Fan, Z.; Zhao, W.; Zeng, N.; Chen, Y.; Zhou, X. Generative Adversarial Networks and Its Applications in Biomedical Informatics. Front. Public Health 2020, 8, 164. [Google Scholar] [CrossRef]
- Arjovsky, M.; Chintala, S.; Bottou, L. Wasserstein gan. arXiv 2017, arXiv:1701.07875. [Google Scholar]
- Mirza, M.; Osindero, S. Conditional generative adversarial nets. arXiv 2014, arXiv:1411.1784. [Google Scholar]
- Makhzani, A.; Shlens, J.; Jaitly, N.; Goodfellow, I.; Frey, B. Adversarial autoencoders. arXiv 2015, arXiv:1511.05644. [Google Scholar]
- Rezende, D.J.; Mohamed, S.; Wierstra, D. Stochastic backpropagation and approximate inference in deep generative models. In Proceedings of the 31st International Conference on Machine Learning, Beijing, China, 21 June 2014; pp. 1278–1286. [Google Scholar]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC). ChemRxiv 2017, 530968. [Google Scholar] [CrossRef] [Green Version]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef]
- Putin, E.; Asadulaev, A.; Vanhaelen, Q.; Ivanenkov, Y.; Aladinskaya, A.V.; Aliper, A.; Zhavoronkov, A. Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 2018, 15, 4386–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polykovskiy, D.; Zhebrak, A.; Vetrov, D.; Ivanenkov, Y.; Aladinskiy, V.; Mamoshina, P.; Bozdaganyan, M.; Aliper, A.; Zhavoronkov, A.; Kadurin, A. Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 2018, 15, 4398–4405. [Google Scholar] [CrossRef]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Guarino, M.; Shah, A.; Rivas, P. DiPol-GAN: Generating Molecular Graphs Adversarially with Relational Differentiable Pooling. under review.
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminf. 2019, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminf. 2020, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.-A.; Rouquié, D.; Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020, 11, 1–10. [Google Scholar]
- Lipton, Z.C.; Berkowitz, J.; Elkan, C. A critical review of recurrent neural networks for sequence learning. arXiv 2015, arXiv:1506.00019. [Google Scholar]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Bjerrum, E.J.; Sattarov, B. Improving chemical autoencoder latent space and molecular de novo generation diversity with heteroencoders. Biomolecules 2018, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv 2017, arXiv:1706.06689. [Google Scholar]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph Convolutional Policy Network for Goal-Directed Molecular Graph Generation. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 13 December 2018; pp. 6410–6421. [Google Scholar]
- Simonovsky, M.; Komodakis, N. Graphvae: Towards generation of small graphs using variational autoencoders. In International Conference on Artificial Neural Networks; Springer: Cham, Switzerland, 2018; pp. 412–422. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; Von Lilienfeld, O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Mnih, V.; Kavukcuoglu, K.; Silver, D.; Rusu, A.A.; Veness, J.; Bellemare, M.G.; Graves, A.; Riedmiller, M.; Fidjeland, A.K.; Ostrovski, G. Human-level control through deep reinforcement learning. Nature 2015, 518, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Henderson, P.; Islam, R.; Bachman, P.; Pineau, J.; Precup, D.; Meger, D. Deep reinforcement learning that matters. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Yu, L.; Zhang, W.; Wang, J.; Yu, Y. Seqgan: Sequence generative adversarial nets with policy gradient. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, Marina del Rey, CA, USA, 1–3 June 2005. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, Nevada, 3–6 December 2012; pp. 1097–1105. [Google Scholar]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.; Wayne, G.; Reynolds, M.; Harley, T.; Danihelka, I.; Grabska-Barwińska, A.; Colmenarejo, S.G.; Grefenstette, E.; Ramalho, T.; Agapiou, J. Hybrid computing using a neural network with dynamic external memory. Nature 2016, 538, 471–476. [Google Scholar] [CrossRef]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar variational autoencoder. In Proceedings of the 34th International Conference on Machine Learning-Volume 70, Sydney, Australia, 6–11 August 2017; pp. 1945–1954. [Google Scholar]
- Schlichtkrull, M.; Kipf, T.N.; Bloem, P.; Van Den Berg, R.; Titov, I.; Welling, M. Modeling relational data with graph convolutional networks. In European Semantic Web Conference; Springer: Cham, Switzerland, 2018; pp. 593–607. [Google Scholar]
- Zhu, J.-Y.; Park, T.; Isola, P.; Efros, A.A. Unpaired image-to-image translation using cycle-consistent adversarial networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 2223–2232. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. arXiv 2018, arXiv:1802.04364. [Google Scholar]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Gulrajani, I.; Ahmed, F.; Arjovsky, M.; Dumoulin, V.; Courville, A.C. Improved training of wasserstein gans. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 5767–5777. [Google Scholar]
- Wang, S.; Jiang, J. A compare-aggregate model for matching text sequences. arXiv 2016, arXiv:1611.01747. [Google Scholar]
- Willett, P. The calculation of molecular structural similarity: Principles and practice. Mol. Inf. 2014, 33, 403–413. [Google Scholar] [CrossRef]
- Zeisel, A.; Munoz-Manchado, A.B.; Codeluppi, S.; Lonnerberg, P.; La Manno, G.; Jureus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zhang, Y.; Fan, J.; Seelig, G.; Kannan, S. Scalable preprocessing for sparse scRNA-seq data exploiting prior knowledge. Bioinformatics 2018, 34, i124–i132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Asnani, H.; Lin, E.; Kannan, S. Clustergan: Latent space clustering in generative adversarial networks. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HA, USA, 27 January–1 February 2019; pp. 4610–4617. [Google Scholar]
- Zheng, J.; Wang, K. Emerging deep learning methods for single-cell RNA-seq data analysis. Quant. Biol. 2019, 7, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Jolliffe, I. Principal component analysis. In International Encyclopedia of Statistical Science; Springer: New York, NY, USA, 2011; pp. 1094–1096. [Google Scholar]
- Grün, D.; Kester, L.; Van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 2014, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053. [Google Scholar] [CrossRef]
- Pierson, E.; Yau, C. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015, 16, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, M.; Van Dijk, D.; Srinivasan, K.; Chen, W.S.; Mohsen, H.; Moon, K.R.; Campbell, A.; Zhao, Y.; Wang, X.; Venkataswamy, M. Exploring single-cell data with deep multitasking neural networks. BioRxiv 2019, 237065. [Google Scholar] [CrossRef] [PubMed]
- Maaten, L.v.d.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Risso, D.; Perraudeau, F.; Gribkova, S.; Dudoit, S.; Vert, J.-P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 2018, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Ghahramani, A.; Watt, F.M.; Luscombe, N.M. Generative adversarial networks uncover epidermal regulators and predict single cell perturbations. bioRxiv 2018, 262501. [Google Scholar] [CrossRef] [Green Version]
- Radford, A.; Metz, L.; Chintala, S. Unsupervised representation learning with deep convolutional generative adversarial networks. arXiv 2015, arXiv:1511.06434. [Google Scholar]
- Huang, P.-S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Sabban, S.; Markovsky, M. RamaNet: Computational de novo helical protein backbone design using a long short-term memory generative adversarial neural network. F1000Research 2020, 9, 298. [Google Scholar] [CrossRef]
- Karimi, M.; Zhu, S.; Cao, Y.; Shen, Y. De novo Protein Design for Novel Folds using Guided Conditional Wasserstein Generative Adversarial Networks (gcWGAN). bioRxiv 2019, 769919. [Google Scholar] [CrossRef]
- Anand, N.; Huang, P. Generative modeling for protein structures. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 3–8 December 2018; pp. 7494–7505. [Google Scholar]
- Bian, Y.; Wang, J.; Jun, J.J.; Xie, X.-Q. Deep convolutional generative adversarial network (dcGAN) models for screening and design of small molecules targeting cannabinoid receptors. Mol. Pharm. 2019, 16, 4451–4460. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, A.M.; Zhou, W. GANDALF: A Prototype of a GAN-based Peptide Design Method. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 61–66. [Google Scholar]
- Gupta, A.; Zou, J. Feedback GAN (FBGAN) for DNA: A novel feedback-loop architecture for optimizing protein functions. arXiv 2018, arXiv:1804.01694. [Google Scholar]
- Palangi, H.; Deng, L.; Shen, Y.; Gao, J.; He, X.; Chen, J.; Song, X.; Ward, R. Deep sentence embedding using long short-term memory networks: Analysis and application to information retrieval. IEEE/ACM Trans. Audio Speech Lang. Proc. 2016, 24, 694–707. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Jackel, L.; Bottou, L.; Brunot, A.; Cortes, C.; Denker, J.; Drucker, H.; Guyon, I.; Muller, U.; Sackinger, E. Comparison of learning algorithms for handwritten digit recognition. In Proceedings of the International conference on artificial neural networks, Perth, Australia, 26–28 June 1995; pp. 53–60. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gönen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Hintsanen, P.; Khan, S.A.; Mpindi, J.-P. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking models for de novo molecular design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M. Molecular sets (MOSES): A benchmarking platform for molecular generation models. arXiv 2018, arXiv:1811.12823. [Google Scholar]
- Preuer, K.; Renz, P.; Unterthiner, T.; Hochreiter, S.; Klambauer, G. Fréchet ChemNet distance: A metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 2018, 58, 1736–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimans, T.; Goodfellow, I.; Zaremba, W.; Cheung, V.; Radford, A.; Chen, X. Improved techniques for training gans. In Proceedings of the Advances in neural information processing systems, Barcelona, Spain, 5–10 December 2016; pp. 2234–2242. [Google Scholar]
- Srivastava, A.; Valkov, L.; Russell, C.; Gutmann, M.U.; Sutton, C. Veegan: Reducing mode collapse in gans using implicit variational learning. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 3308–3318. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V. ExCAPE-DB: An integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminf. 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep learning in drug discovery. Mol. Inf. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.-H.; Hung, C.-C.; Lane, H.-Y. An ensemble approach to predict schizophrenia using protein data in the N-methyl-D-aspartate receptor (NMDAR) and tryptophan catabolic pathways. Front. Bioeng. Biotechnol. 2020, 8, 569. [Google Scholar] [CrossRef]
- Jiao, Y.; Du, P. Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 2016, 4, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.-C.; Hsu, S.-Y.; Lin, E. A comparison of classification methods for predicting Chronic Fatigue Syndrome based on genetic data. J. Transl. Med. 2009, 7, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.-T. Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation. Pattern Recog. 2015, 48, 2839–2846. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, J. Chemoinformatics: Achievements and challenges, a personal view. Molecules 2016, 21, 151. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—a review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.; Gong, Y.; Yang, Z.; Chuai, G.; Qu, S.; Shen, A.; Yu, J.; Liu, Q. Advances and challenges in deep generative models for de novo molecule generation. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019, 9, e1395. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Vanhaelen, Q.; Oprea, T.I. Will Artificial Intelligence for Drug Discovery Impact Clinical Pharmacology? Clin. Pharmacol. Ther. 2020, 107, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Hwang, Y.; Wang, S.-C.; Gu, Z.J.; Chen, E.Y. An artificial neural network approach to the drug efficacy of interferon treatments. Future Med. 2006. [Google Scholar] [CrossRef]
- Janowczyk, A.; Madabhushi, A. Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases. J. Pathol. Inf. 2016, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Wei, G.-W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLoS Comput. Boil. 2017, 13, e1005690. [Google Scholar] [CrossRef]
- Lin, E.; Lane, H.-Y. Machine learning and systems genomics approaches for multi-omics data. Biomarker Res. 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Lin, C.-H.; Lai, Y.-L.; Huang, C.-H.; Huang, Y.-J.; Lane, H.-Y. Combination of G72 genetic variation and G72 protein level to detect schizophrenia: Machine learning approaches. Front. Psychiatr. 2018, 9, 566. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.-H.; Lane, H.-Y. Precision psychiatry applications with pharmacogenomics: Artificial intelligence and machine learning approaches. Int. J. Mol. Sci. 2020, 21, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Boil. 2016, 12, 878. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.-J. Multi-omics and machine learning applications in precision medicine. Curr. Pharm. Pers. Med. (Former. Curr. Pharm.) 2017, 15, 97–104. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.-J. Diagnostic Prediction Based on Gene Expression Profiles and Artificial Neural Networks. In Soft Computing for Biological Systems; Springer: Singapore, 2018; pp. 13–22. [Google Scholar]
- Litjens, G.; Sánchez, C.I.; Timofeeva, N.; Hermsen, M.; Nagtegaal, I.; Kovacs, I.; Hulsbergen-Van De Kaa, C.; Bult, P.; Van Ginneken, B.; Van Der Laak, J. Deep learning as a tool for increased accuracy and efficiency of histopathological diagnosis. Sci. Rep. 2016, 6, 26286. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Structure | Architecture | Object Generated | Learning Technique | Databases | Results |
|---|---|---|---|---|---|---|
| Kadurin et al. [28,29] | druGAN | AAE | latent vector | autoencoder | PubChem | druGAN generated novel molecular compounds which can be considered as potential anticancer agents. |
| Guimaraes et al. [36] | ORGAN | GAN | SMILES | RL | ZINC, GDB-17 | ORGAN performed better than recurrent neural networks or GAN alone. |
| Sanchez-Lengeling et al. [37] | ORGANIC | GAN | SMILES | RL | ZINC, GDB-17 | ORGANIC showed good performance in terms of the quantitative estimate of drug-likeness, but not the Lipinski’s Rule-of-Five. |
| Putin et al. [38] | RANC | GAN | SMILES | RL | ZINC, ChemDiv | RANC was superior to ORGANIC in terms of several drug discovery metrics. |
| Putin et al. [39] | ATNC | GAN | SMILES | RL | ChemDiv | ATNC performed better than ORGANIC in terms of various functions. |
| Polykovskiy et al. [40] | ECAAE | AAE | latent vector | autoencoder | ZINC | ECAAE generated novel molecular compounds which can be considered as target drugs in rheumatoid arthritis, psoriasis, and vitiligo. |
| Cao and Kipf [41] | MolGAN | GAN | graph | RL | QM9 | MolGAN outperformed ORGAN and variational autoencoder-based structures. |
| Guarino et al. [42] | DiPol-GAN | GAN | graph | RL | QM9 | DiPol-GAN had 1.3 times higher drug-likeliness scores than MolGAN. |
| Prykhodko et al. [43] | LatentGAN | GAN | SMILES | autoencoder | ChEMBL | LatentGAN created novel drug-like compounds and was compatible to recurrent neural networks. |
| Maziarka et al. [44] | Mol-CycleGAN | GAN | latent vector | direct flow | ZINC, ChEMBL | Mol-CycleGAN outperformed the junction tree variational autoencoder and the graph convolutional policy network structures. |
| Méndez-Lucio et al. [45] | Conditioned GAN | GAN | latent vector | direct flow | L1000 | Conditioned GAN produced molecular compounds with desired gene expression signatures. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, E.; Lin, C.-H.; Lane, H.-Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules 2020, 25, 3250. https://doi.org/10.3390/molecules25143250
Lin E, Lin C-H, Lane H-Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules. 2020; 25(14):3250. https://doi.org/10.3390/molecules25143250
Chicago/Turabian StyleLin, Eugene, Chieh-Hsin Lin, and Hsien-Yuan Lane. 2020. "Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design" Molecules 25, no. 14: 3250. https://doi.org/10.3390/molecules25143250
APA StyleLin, E., Lin, C.-H., & Lane, H.-Y. (2020). Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules, 25(14), 3250. https://doi.org/10.3390/molecules25143250