Anticancer Attributes of Cantharidin: Involved Molecular Mechanisms and Pathways


Abstract
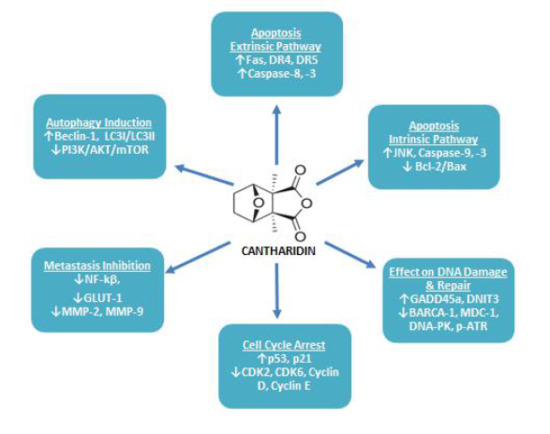
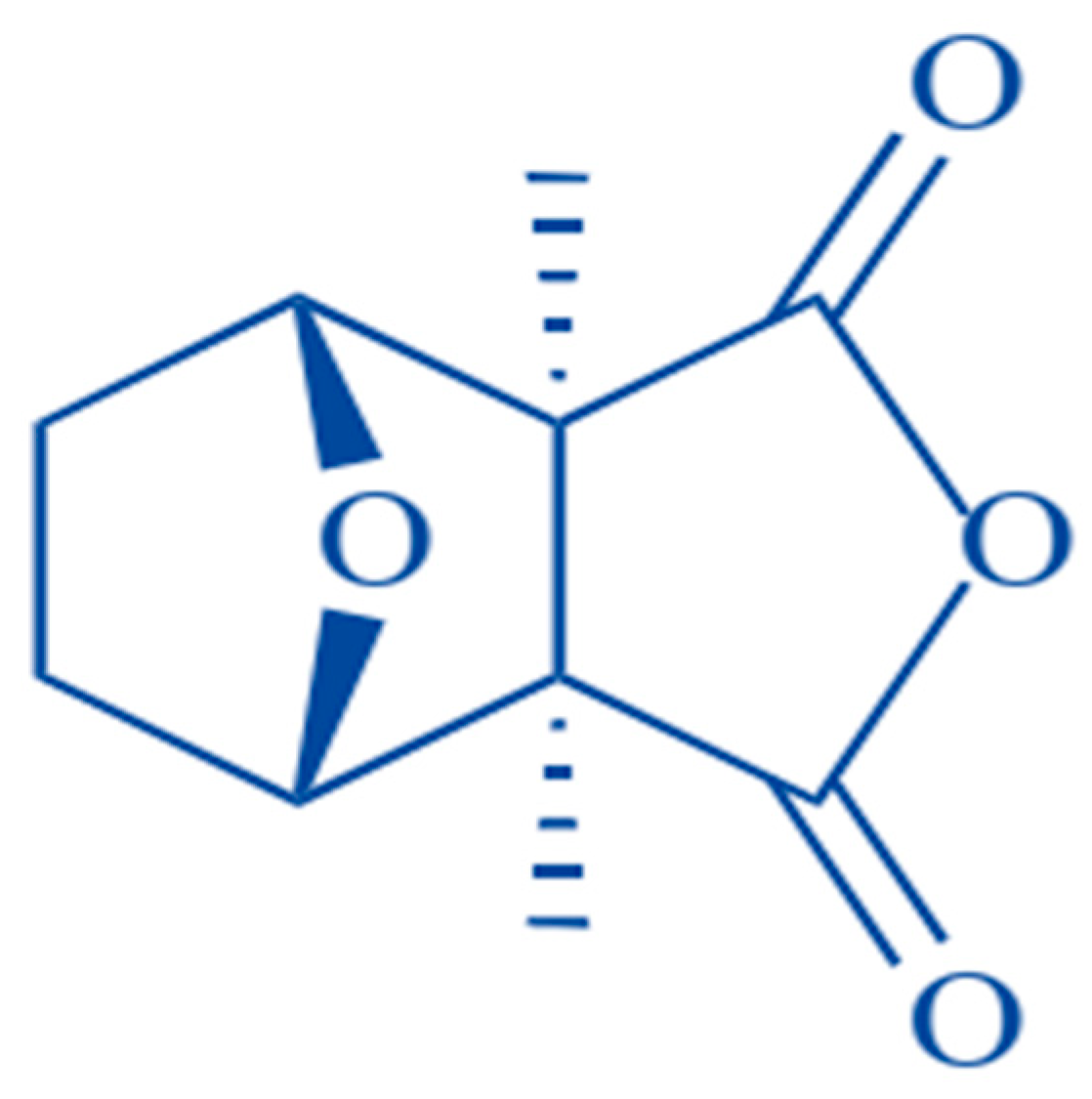
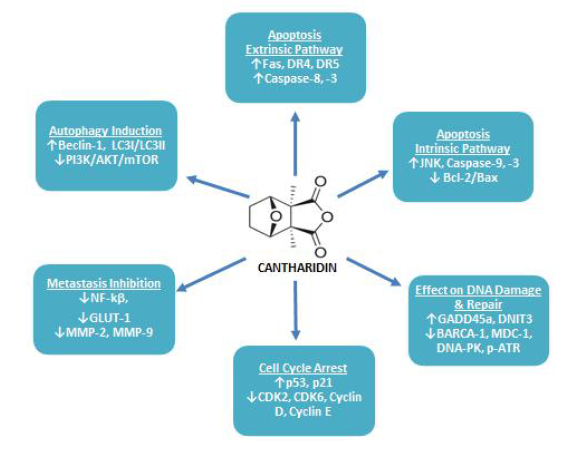
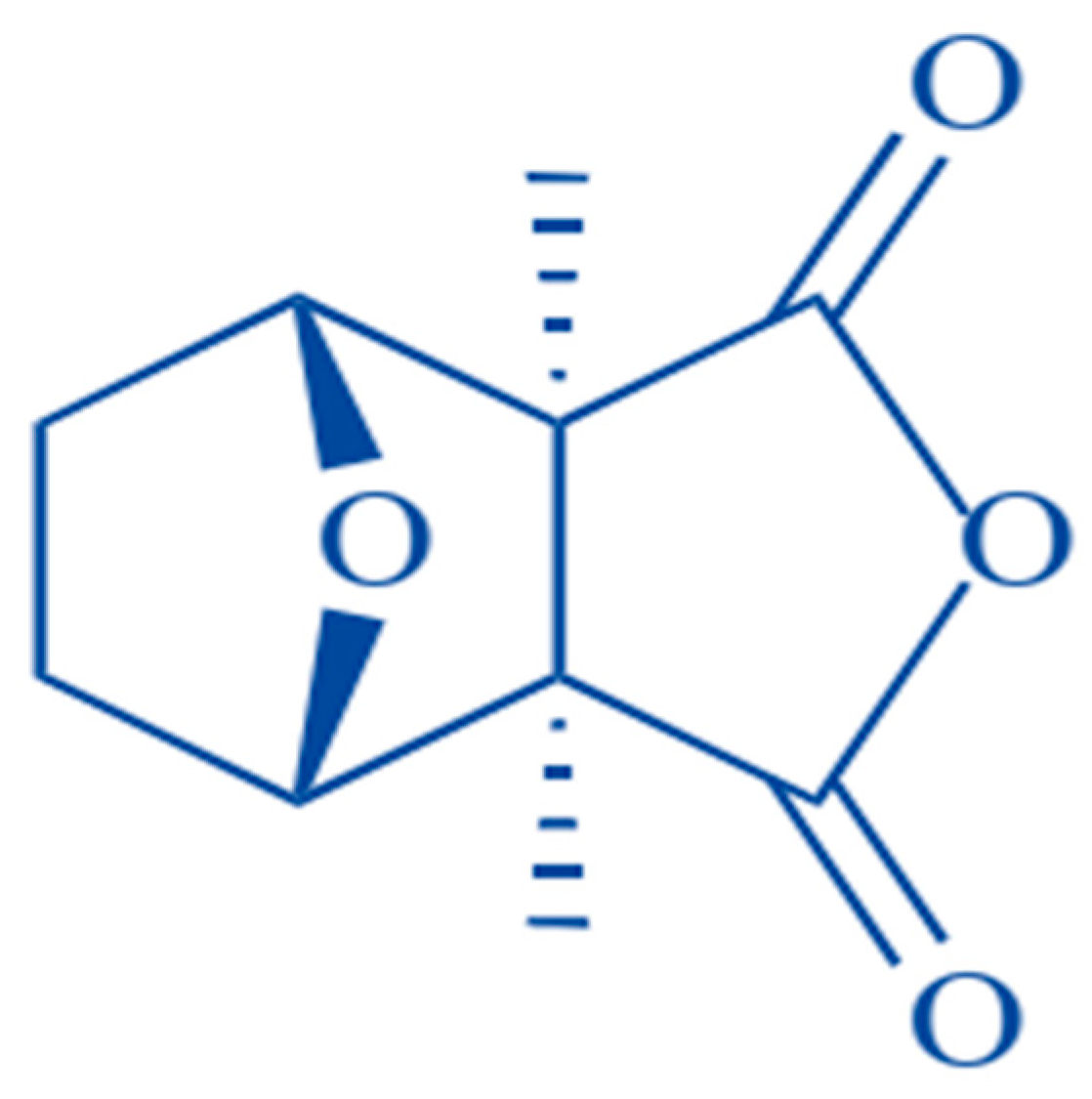
:
1. Introduction
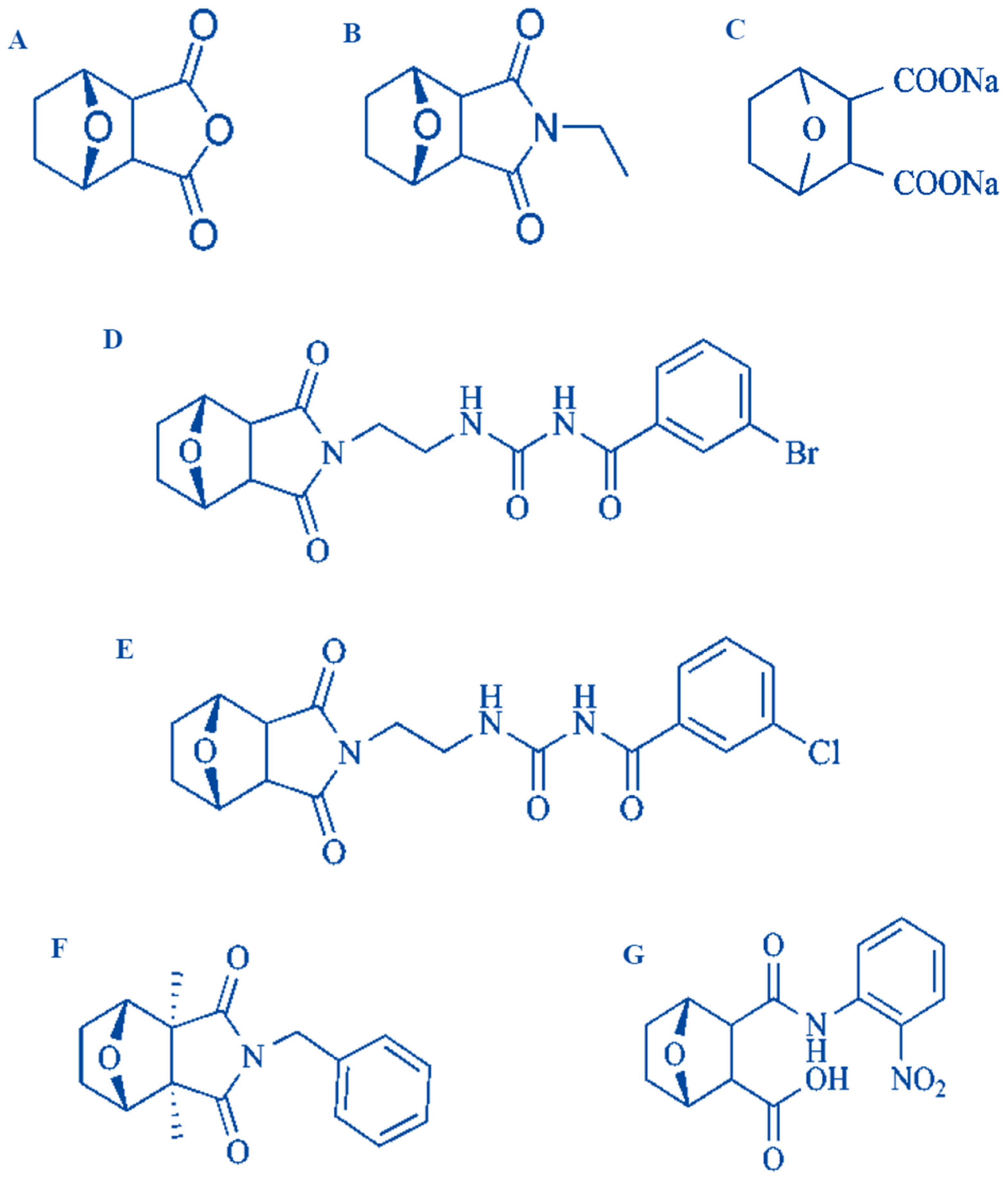
2. Natural Sources of CTD and Its Synthetic Derivatives
3. Biologic Features of CTD
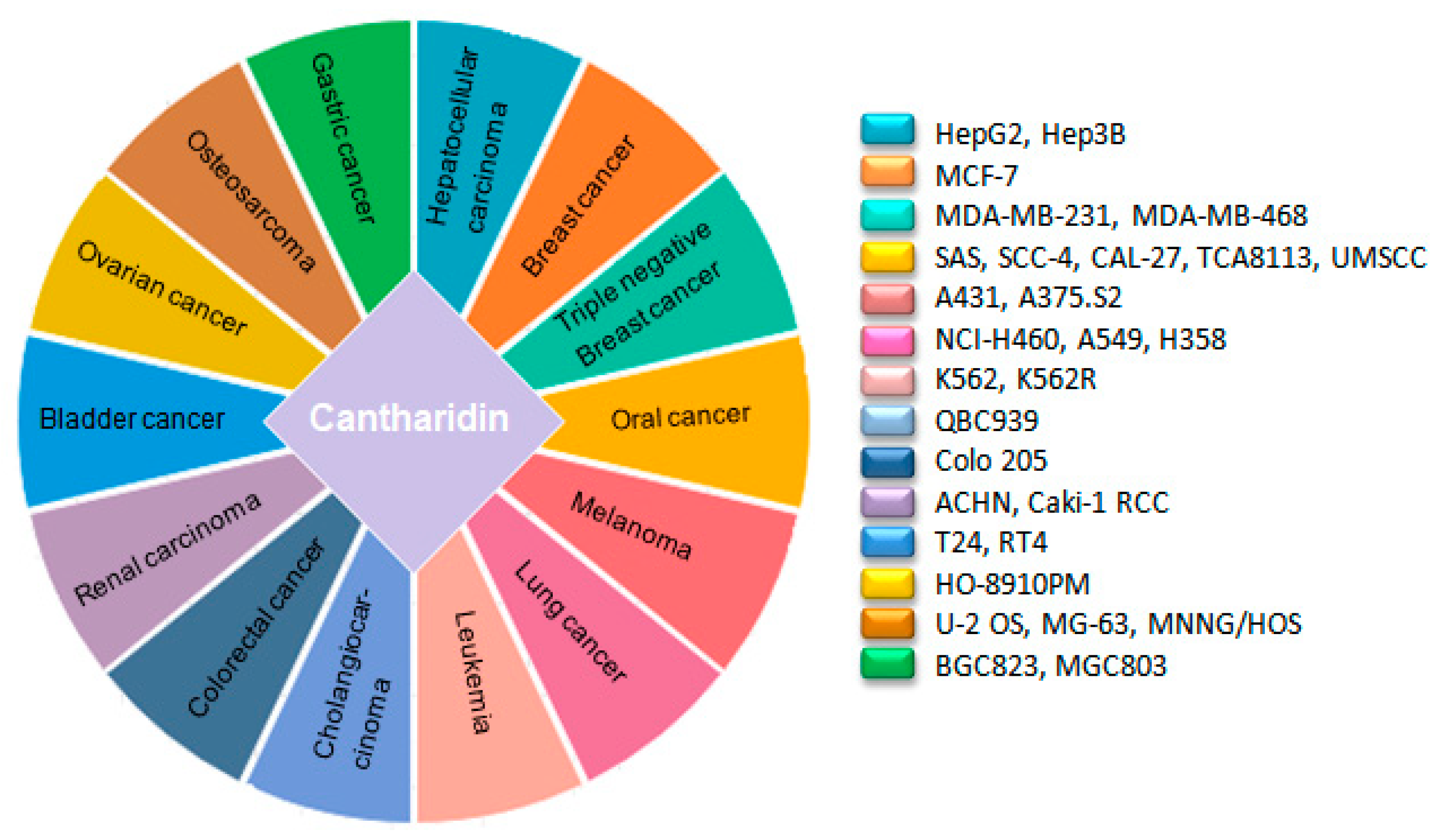
4. Anticancer Attributes of CTD
4.1. Repression of Cancerous Cell Growth & Proliferation
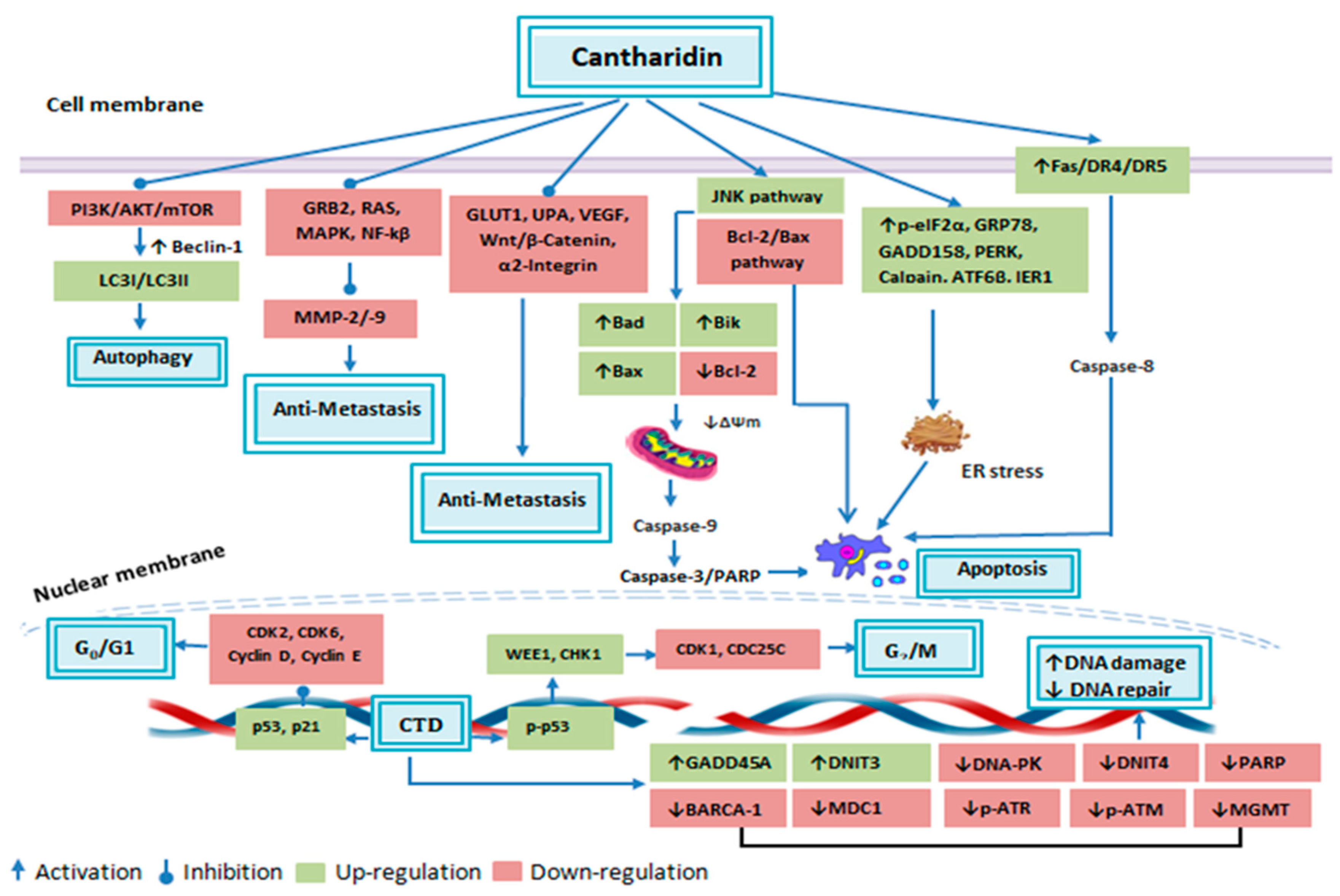
4.2. Induction of Cancerous Cell Apoptosis
4.2.1. Apoptosis: Extrinsic Pathway
4.2.2. Apoptosis: Intrinsic Pathway
4.3. Effect of CTD on Cancerous Cell DNA Damaging and Repair Associated Proteins
4.4. Induction of Cancerous Cell Cycle Arrest
4.5. Inhibition of Cancer Cell Metastasis
4.6. Induction of Cancerous Cell’s Autophagy
4.7. Cytotoxic Effects of CTD in Xenograft Mice Model
5. CTD in Combined Therapy
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abotaleb, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Therapeutic Potential of Plant Phenolic Acids in the Treatment of Cancer. Biomolecules 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancet, T. Cancer Now Leading Cause of Death in High-Income Countries—While Heart Disease Burden Persists in Low-Income and Middle-Income Countries. Available online: www.sciencedaily.com/releases/2019/09/190903084037.htm (accessed on 20 February 2019).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, M. why Is Cancer So Difficult to Cure? Available online: https://www.jax.org/news-and-insights/2015/december/why-no-cure-for-cancer (accessed on 21 February 2020).
- Liang, X.J.; Chen, C.; Zhao, Y.; Wang, P.C. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol. Biol. 2010, 596, 467–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Duan, H.; Luo, Q.; Liu, W.; Liang, L.; Wan, H.; Chang, S.; Hu, J.; Shi, H. Inhibition Mechanism of Indoleamine 2, 3-Dioxygenase 1 (IDO1) by Amidoxime Derivatives and Its Revelation in Drug Design: Comparative Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.; Sibaud, V. Toxic Side Effects of Targeted Therapies and Immunotherapies Affecting the Skin, Oral Mucosa, Hair, and Nails. Am. J. Clin. Derm. 2018, 19, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.; Jafari, B.; Asgharian, P.; Martorell, M.; Sharifi-Rad, J. Medicinal plants used in the treatment of Malaria: A key emphasis to Artemisia, Cinchona, Cryptolepis, and Tabebuia genera. Phytother. Res. 2020. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Scaccabarozzi, D.; Signorini, L.; Perego, F.; Ilboudo, D.P.; Ferrante, P.; Delbue, S. The Use of Antimalarial Drugs against Viral Infection. Microorganisms 2020, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.; Danesi, F.; Del Rio, D.; Silva, P. Resveratrol and inflammatory bowel disease: The evidence so far. Nutr. Res. Rev. 2018, 31, 85–97. [Google Scholar] [CrossRef]
- Dutta, P.; Sahu, R.K.; Dey, T.; Lahkar, M.D.; Manna, P.; Kalita, J. Beneficial role of insect-derived bioactive components against inflammation and its associated complications (colitis and arthritis) and cancer. Chem. Biol. Interact. 2019, 313, 108824. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.P.; Dong, J.; Cai, H.; Wang, W. Cantharidin as an antitumor agent: A retrospective review. Curr. Med. Chem. 2013, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Torbeck, R.; Pan, M.; DeMoll, E.; Levitt, J. Cantharidin: A comprehensive review of the clinical literature. Derm. Online J. 2014, 20. Available online: https://escholarship.org/uc/item/45r512w0 (accessed on 14 July 2020).
- Chen, R.T.; Hua, Z.; Yang, J.L.; Han, J.X.; Zhang, S.Y.; Lü, F.L.; Xü, B. Studies on antitumor actions of cantharidin. Chin. Med. J. 1980, 93, 183–187. [Google Scholar] [PubMed]
- Pan, Y.; Zheng, Q.; Ni, W.; Wei, Z.; Yu, S.; Jia, Q.; Wang, M.; Wang, A.; Chen, W.; Lu, Y. Breaking Glucose Transporter 1/Pyruvate Kinase M2 Glycolytic Loop Is Required for Cantharidin Inhibition of Metastasis in Highly Metastatic Breast Cancer. Front. Pharm. 2019, 10, 590. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Kim, Y.; Kwon, B.M.; Han, D.C. The natural compound cantharidin induces cancer cell death through inhibition of heat shock protein 70 (HSP70) and Bcl-2-associated athanogene domain 3 (BAG3) expression by blocking heat shock factor 1 (HSF1) binding to promoters. J. Biol. Chem. 2013, 288, 28713–28726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, D.; Maura, M.; Pan, Z.; Bologna, M. Phylogenetic systematics of Mylabris blister beetles (Coleoptera, Meloidae): A molecular assessment using species trees and total evidence. Cladistics 2019. [Google Scholar] [CrossRef]
- Liu, D.; Chen, Z. The Effects of Cantharidin and Cantharidin Derivates on Tumour Cells. Anticancer. Agents Med. Chem. 2009, 9, 392–396. [Google Scholar] [CrossRef]
- Jiang, M.; Lü, S.; Zhang, Y. The Potential Organ Involved in Cantharidin Biosynthesis in Epicauta chinensis Laporte (Coleoptera: Meloidae). J. Insect Sci. 2017, 17, 52. [Google Scholar] [CrossRef]
- Nikbakhtzadeh, M.R.; Hemp, C.; Ebrahimi, B. Further evidence for the role of Cantharidin in the mating behaviour of blister beetles (Coleoptera: Meloidae). Integr. Biosci. 2007, 11, 141–146. [Google Scholar] [CrossRef]
- Wilson, C.R. Methods for Analysis of Gastrointestinal Toxicants. Available online: https://scinapse.io/papers/2265534266 (accessed on 17 July 2020).
- Ghoneim, K.; Ghoneim, K.S. Embryonic and postembryonic development of blister beetles (Coleoptera: Meloidae) in the world: A synopsis. International Journal of Biology and Biological Sciences. Int. J. Biol. Biol. Sci. 2013, 2, 6–18. [Google Scholar]
- Bologna, M.A.; Oliverio, M.; Pitzalis, M.; Mariottini, P. Phylogeny and evolutionary history of the blister beetles (Coleoptera, Meloidae). Mol. Phylogenet. Evol. 2008, 48, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Bologna, M.A.; Pinto, J.D. The Old World genera of Meloidae (Coleoptera): A key and synopsis. J. Nat. Hist. 2002, 36, 2013–2102. [Google Scholar] [CrossRef]
- Wang, G.; Dong, J.; Deng, L. Overview of Cantharidin and its Analogues. Curr. Med. Chem. 2018, 25, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Tomar, R.; Sahu, P. Cantharidin Alters GPI-Anchored Protein Sorting by Targeting Cdc1 Mediated Remodeling in Endoplasmic Reticulum. Available online: https://www.biorxiv.org/content/10.1101/460253v1 (accessed on 17 July 2020).
- Bajsa, J.; McCluskey, A.; Gordon, C.P.; Stewart, S.G.; Hill, T.A.; Sahu, R.; Duke, S.O.; Tekwani, B.L. The antiplasmodial activity of norcantharidin analogs. Bioorg. Med. Chem. Lett. 2010, 20, 6688–6695. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S. Medical uses of mylabris in ancient China and recent studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef]
- Moed, L.; Shwayder, T.A.; Chang, M.W. Cantharidin Revisited: A Blistering Defense of an Ancient Medicine. Arch. Derm. 2001, 137, 1357–1360. [Google Scholar] [CrossRef]
- Recanati, M.A.; Kramer, K.J.; Maggio, J.J.; Chao, C.R. Cantharidin is Superior to Trichloroacetic Acid for the Treatment of Non-mucosal Genital Warts: A Pilot Randomized Controlled Trial. Clin. Exp. Obs. Gynecol. 2018, 45, 383–386. [Google Scholar] [CrossRef]
- Maroufi, Y.; Ghaffarifar, F.; Abdolhosein, D.; Sharifi, Z.; Zuhair, H. Effect of Cantharidin on Apoptosis of the Leishmania major and on Parasite Load in BALB/c Mice. Res. J. Parasitol. 2013, 8, 14–25. [Google Scholar] [CrossRef]
- Whitman, D.W.; Andrés, M.F.; Martínez-Díaz, R.A.; Ibáñez-Escribano, A.; Olmeda, A.S.; González-Coloma, A. Antiparasitic Properties of Cantharidin and the Blister Beetle Berberomeloe majalis (Coleoptera: Meloidae). Toxins (Basel) 2019, 11, 234. [Google Scholar] [CrossRef] [Green Version]
- Li, C.C.; Yu, F.S.; Fan, M.J.; Chen, Y.Y.; Lien, J.C.; Chou, Y.C.; Lu, H.F.; Tang, N.Y.; Peng, S.F.; Huang, W.W.; et al. Anticancer effects of cantharidin in A431 human skin cancer (Epidermoid carcinoma) cells in vitro and in vivo. Environ. Toxicol. 2017, 32, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.C.; Hsiao, Y.P.; Tsai, C.H.; Chang, S.J.; Hsu, S.C.; Liu, H.C.; Huang, Y.P.; Lien, J.C.; Chung, J.G. Cantharidin impairs cell migration and invasion of A375.S2 human melanoma cells by suppressing MMP-2 and -9 through PI3K/NF-κB signaling pathways. Anticancer Res. 2015, 35, 729–738. [Google Scholar]
- Su, C.C.; Liu, S.H.; Lee, K.I.; Huang, K.T.; Lu, T.H.; Fang, K.M.; Wu, C.C.; Yen, C.C.; Lai, C.H.; Su, Y.C.; et al. Cantharidin Induces Apoptosis Through the Calcium/PKC-Regulated Endoplasmic Reticulum Stress Pathway in Human Bladder Cancer Cells. Am. J. Chin. Med. 2015, 43, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Hsia, T.C.; Yu, C.C.; Hsu, S.C.; Tang, N.Y.; Lu, H.F.; Yu, C.S.; Wu, S.H.; Lin, J.G.; Chung, J.G. cDNA microarray analysis of the effect of cantharidin on DNA damage, cell cycle and apoptosis-associated gene expression in NCI-H460 human lung cancer cells in vitro. Mol. Med. Rep. 2015, 12, 1030–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, T.C.; Yu, C.C.; Hsu, S.C.; Tang, N.Y.; Lu, H.F.; Huang, Y.P.; Wu, S.H.; Lin, J.G.; Chung, J.G. Cantharidin induces apoptosis of H460 human lung cancer cells through mitochondria-dependent pathways. Int. J. Oncol. 2014, 45, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Li, L.; Xu, L.; Dai, E.N.; Chen, W.D. Cantharidin suppresses cell growth and migration, and activates autophagy in human non-small cell lung cancer cells. Oncol. Lett. 2018, 15, 6527–6532. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, F.S.; Hung, M.H.; Wang, C.Y.; Chen, Y.L.; Hsiao, Y.J.; Tsai, M.H.; Li, J.R.; Chen, L.J.; Shih, C.T.; Chao, T.I.; et al. Inhibition of protein phosphatase 5 suppresses non-small cell lung cancer through AMP-activated kinase activation. Lung Cancer 2017, 112, 81–89. [Google Scholar] [CrossRef]
- Huang, W.W.; Ko, S.W.; Tsai, H.Y.; Chung, J.G.; Chiang, J.H.; Chen, K.T.; Chen, Y.C.; Chen, H.Y.; Chen, Y.F.; Yang, J.S. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int. J. Oncol. 2011, 38, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Wang, X.; Luo, Y.; Liu, Z.; Tan, W.; Ye, P.; Fu, Z.; Lu, F.; Xiang, W.; Tang, L.; et al. Cantharidin suppresses gastric cancer cell migration/invasion by inhibiting the PI3K/Akt signaling pathway via CCAT1. Chem. Biol. Interact. 2020, 317, 108939. [Google Scholar] [CrossRef]
- Ma, Q.; Feng, Y.; Deng, K.; Shao, H.; Sui, T.; Zhang, X.; Sun, X.; Jin, L.; Ma, Z.; Luo, G. Unique Responses of Hepatocellular Carcinoma and Cholangiocarcinoma Cell Lines toward Cantharidin and Norcantharidin. J. Cancer 2018, 9, 2183–2190. [Google Scholar] [CrossRef] [Green Version]
- Shou, L.M.; Zhang, Q.Y.; Li, W.; Xie, X.; Chen, K.; Lian, L.; Li, Z.Y.; Gong, F.R.; Dai, K.S.; Mao, Y.X.; et al. Cantharidin and norcantharidin inhibit the ability of MCF-7 cells to adhere to platelets via protein kinase C pathway-dependent downregulation of α2 integrin. Oncol. Rep. 2013, 30, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.-D.; Liu, L.; Wu, M.-Y.; Jiang, M.; Shou, L.-M.; Wang, W.-J.; Wu, J.; Zhang, Y.; Gong, F.-R.; Chen, K.; et al. The combination of cantharidin and antiangiogenic therapeutics presents additive antitumor effects against pancreatic cancer. Oncogenesis 2018, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wu, M.Y.; Shen, M.; Zhi, Q.; Liu, Z.Y.; Gong, F.R.; Tao, M.; Li, W. Cantharidin and norcantharidin impair stemness of pancreatic cancer cells by repressing the β-catenin pathway and strengthen the cytotoxicity of gemcitabine and erlotinib. Int. J. Oncol. 2015, 47, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Lee, K.I.; Chen, M.K.; Kuo, C.Y.; Tang, C.H.; Liu, S.H. Cantharidin Induced Oral Squamous Cell Carcinoma Cell Apoptosis via the JNK-Regulated Mitochondria and Endoplasmic Reticulum Stress-Related Signaling Pathways. PLoS ONE 2016, 11, e0168095. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zeng, G.; Li, X.; Wu, Z.; Wang, L. Cantharidin inhibits cell proliferation and promotes apoptosis in tongue squamous cell carcinoma through suppression of miR-214 and regulation of p53 and Bcl-2/Bax. Oncol. Rep. 2015, 33, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Garshott, D.M.; Brownell, A.L.; Yoo, G.H.; Lin, H.S.; Freeburg, T.L.; Yoo, N.G.; Kaufman, R.J.; Callaghan, M.U.; Fribley, A.M. Cantharidins induce ER stress and a terminal unfolded protein response in OSCC. J. Dent. Res. 2015, 94, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taiping, H.; Mo, L.-E.; Liang, N.-C. [Inhibitory effect of cantharidin on invasion and metastasis of highly metastatic ovarian carcinoma cell line HO-8910PM]. Ai Zheng Aizheng Chin. J. Cancer 2005, 24, 443–447. [Google Scholar]
- Sun, X.; Cai, X.; Yang, J.; Chen, J.; Guo, C.; Cao, P. Cantharidin Overcomes Imatinib Resistance by Depleting BCR-ABL in Chronic Myeloid Leukemia. Mol. Cells 2016, 39, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.D.; Xu, L.L.; Zhao, H.; Gu, J.Z.; Xie, X.H. Cantharidin suppressed breast cancer MDA-MB-231 cell growth and migration by inhibiting MAPK signaling pathway. Braz. J. Med Biol. Res. Rev. Bras. De Pesqui. Med. E Biol. 2017, 50, e5920. [Google Scholar] [CrossRef] [Green Version]
- Li, H.C.; Xia, Z.H.; Chen, Y.F.; Yang, F.; Feng, W.; Cai, H.; Mei, Y.; Jiang, Y.M.; Xu, K.; Feng, D.X. Cantharidin Inhibits the Growth of Triple-Negative Breast Cancer Cells by Suppressing Autophagy and Inducing Apoptosis in Vitro and in Vivo. Cell. Physiol. Biochem. 2017, 43, 1829–1840. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany N. Y.) 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Wang, C.; Zheng, J.; Paudyal, N.; Zhu, Y.; Sun, H. The potential role of tubeimosides in cancer prevention and treatment. Eur. J. Med. Chem. 2019, 162, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Park, M.K.; Ko, H.; Lee, K.; Kim, Y.S. Bioassay-guided isolation of cantharidin from blister beetles and its anticancer activity through inhibition of epidermal growth factor receptor-mediated STAT3 and Akt pathways. J. Nat. Med. 2018, 72, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wu, M.-Y.; Chen, L.-P.; Zhi, Q.; Gong, F.-R.; Chen, K.; Li, D.-M.; Wu, Y.; Tao, M.; Li, W. Cantharidin represses invasion of pancreatic cancer cells through accelerated degradation of MMP2 MRNA. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-Y.; Xie, X.; Xu, Z.-K.; Xie, L.; Chen, Z.; Shou, L.-M.; Gong, F.-R.; Xie, Y.-F.; Li, W.; Tao, M. PP2A inhibitors suppress migration and growth of PANC-1 pancreatic cancer cells through inhibition on the Wnt/β-catenin pathway by phosphorylation and degradation of β-catenin. Oncol. Rep. 2014, 32, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xie, L.; Chen, Z.; Zhu, Y.; Sun, Y.; Miao, Y.; Xu, Z.; Han, X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010, 101, 1226–1233. [Google Scholar] [CrossRef]
- Li, W.; Chen, Z.; Gong, F.R.; Zong, Y.; Chen, K.; Li, D.M.; Yin, H.; Duan, W.M.; Miao, Y.; Tao, M.; et al. Growth of the pancreatic cancer cell line PANC-1 is inhibited by protein phosphatase 2A inhibitors through overactivation of the c-Jun N-terminal kinase pathway. Eur. J. Cancer 2011, 47, 2654–2664. [Google Scholar] [CrossRef]
- Li, W.; Chen, Z.; Zong, Y.; Gong, F.; Zhu, Y.; Zhu, Y.; Lv, J.; Zhang, J.; Xie, L.; Sun, Y.; et al. PP2A inhibitors induce apoptosis in pancreatic cancer cell line PANC-1 through persistent phosphorylation of IKKα and sustained activation of the NF-κB pathway. Cancer Lett. 2011, 304, 117–127. [Google Scholar] [CrossRef]
- Kuo, J.H.; Chu, Y.L.; Yang, J.S.; Lin, J.P.; Lai, K.C.; Kuo, H.M.; Hsia, T.C.; Chung, J.G. Cantharidin induces apoptosis in human bladder cancer TSGH 8301 cells through mitochondria-dependent signal pathways. Int. J. Oncol. 2010, 37, 1243–1250. [Google Scholar] [CrossRef]
- Huang, Y.P.; Ni, C.H.; Lu, C.C.; Chiang, J.H.; Yang, J.S.; Ko, Y.C.; Lin, J.P.; Kuo, J.H.; Chang, S.J.; Chung, J.G. Suppressions of Migration and Invasion by Cantharidin in TSGH-8301 Human Bladder Carcinoma Cells through the Inhibitions of Matrix Metalloproteinase-2/-9 Signaling. Evid. Based Complement. Altern. Med. 2013, 2013, 190281. [Google Scholar] [CrossRef]
- Hsia, T.C.; Yu, C.C.; Hsiao, Y.T.; Wu, S.H.; Bau, D.T.; Lu, H.F.; Huang, Y.P.; Lin, J.G.; Chang, S.J.; Chung, J.G. Cantharidin Impairs Cell Migration and Invasion of Human Lung Cancer NCI-H460 Cells via UPA and MAPK Signaling Pathways. Anticancer Res. 2016, 36, 5989–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Ku, M.J.; Son, Y.J.; Yun, J.M.; Kim, S.H.; Lee, S.Y. Anti-metastatic effect of cantharidin in A549 human lung cancer cells. Arch. Pharm. Res. 2013, 36, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, S.W.; Xie, Z.H.; Xu, X.M.; Chen, L.L.; Lou, Z.G.; Weng, G.B.; Yao, X.P. Cantharidin induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Mol. Med. Rep. 2016, 14, 5614–5618. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chueh, F.S.; Peng, S.F.; Huang, W.W.; Tsai, C.H.; Tsai, F.J.; Huang, C.Y.; Tang, C.H.; Yang, J.S.; Hsu, Y.M.; et al. Cantharidin decreased viable cell number in human osteosarcoma U-2 OS cells through G(2)/M phase arrest and induction of cell apoptosis. Biosci. Biotechnol. Biochem. 2019, 83, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhu, J.; Xia, K.; Yu, W.; Wang, Y.; Wang, J.; Li, F.; Yang, Z.; Yang, X.; Liu, B.; et al. Cantharidin Inhibits Anti-Apoptotic Bcl-2 Family Proteins and Induces Apoptosis in Human Osteosarcoma Cell Lines MG-63 and MNNG/HOS via Mitochondria-Dependent Pathway. Med. Sci. Monit. 2018, 24, 6742–6749. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, J.; Wang, S.; Peng, J. Role of cantharidin in the activation of IKKα/IκBα/NF-κB pathway by inhibiting PP2A activity in cholangiocarcinoma cell lines. Mol. Med. Rep. 2018, 17, 7672–7682. [Google Scholar] [CrossRef]
- Le, A.P.; Zhang, L.L.; Liu, W.; Shi, Y.F. Cantharidin inhibits cell proliferation and induces apoptosis through G2/M phase cell cycle arrest in hepatocellular carcinoma stem cells. Oncol. Rep. 2016, 35, 2970–2976. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.-R.; Wu, M.-Y.; Shen, M.; Zhi, Q.; Xu, Z.-K.; Wang, R.; Wang, W.-J.; Zong, Y.; Li, Z.-L.; Wu, Y.; et al. PP2A inhibitors arrest G2/M transition through JNK/Sp1- dependent down-regulation of CDK1 and autophagy-dependent up-regulation of p21. Oncotarget 2015, 6, 18469–18483. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Loane, D.J.; Stoica, B.A.; Faden, A.I. Chapter 22-Neuroprotection for traumatic brain injury. In Handbook of Clinical Neurology; Grafman, J., Salazar, A.M., Eds.; Elsevier: Cambridge, MA, USA, 2015; Volume 127, pp. 343–366. [Google Scholar]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [Green Version]
- Hsia, T.C.; Lin, J.H.; Hsu, S.C.; Tang, N.Y.; Lu, H.F.; Wu, S.H.; Lin, J.G.; Chung, J.G. Cantharidin induces DNA damage and inhibits DNA repair-associated protein levels in NCI-H460 human lung cancer cells. Environ. Toxicol. 2015, 30, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Future Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.-D.; Liu, S.-L.; Zheng, B.-B.; Wu, J.; Wu, M.-Y.; Zhang, Y.; Gong, F.-R.; Tao, M.; Zhang, J.; Li, W. The radiotherapy-sensitization effect of cantharidin: Mechanisms involving cell cycle regulation, enhanced DNA damage, and inhibited DNA damage repair. Pancreatology 2018, 18, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Ying, K.; Xiao, Z.Y.; Zhou, B.; Huang, Q.S.; Wu, H.M.; Yin, M.; Xie, Y.; Mao, Y.M.; Rui, Y.C. Analysis of gene expression profiles in human HL-60 cell exposed to cantharidin using cDNA microarray. Int. J. Cancer 2004, 108, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.H.; Shih, T.Y.; Lin, J.P.; Lai, K.C.; Lin, M.L.; Yang, M.D.; Chung, J.G. Cantharidin induces DNA damage and inhibits DNA repair-associated protein expressions in TSGH8301 human bladder cancer cell. Anticancer Res. 2015, 35, 795–804. [Google Scholar] [PubMed]
- Kuo, J.H.; Huang, A.C.; Lin, J.-J.; Lai, K.-C.; Wu, R.S.-C.; Yang, J.-L.; Ji, B.-C.; Yang, M.-D.C.; Yang, J.-L.; Ji, B.-C.; et al. Cantharidin alters the expression of genes associated with the NKG2D-associated immune response in TSGH-8301 human bladder carcinoma cells. Oncol. Lett. 2017, 14, 234–240. [Google Scholar]
- Li, Y.; Fan, J.; Ju, D. 15-Neurotoxicity concern about the brain targeting delivery systems. In Brain Targeted Drug Delivery System; Gao, H., Gao, X., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 377–408. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Chapter 27-The Molecular Response to Renal Injury: How Does Chronic Renal Damage Suppress Normal Repair Processes? In Kidney Development, Disease, Repair and Regeneration; Little, M.H., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 367–379. [Google Scholar] [CrossRef]
- Lanza, F.; Bi, S. Role of p53 in leukemogenesis of chronic myeloid leukemia. Stem Cells 1995, 13, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Tahtamouni, L.; Ahram, M.; Koblinski, J.; Rolfo, C. Molecular Regulation of Cancer Cell Migration, Invasion, and Metastasis. Anal. Cell. Pathol. (Amst.) 2019, 2019, 1356508. [Google Scholar] [CrossRef] [Green Version]
- Das, C.K.; Banerjee, I.; Mandal, M. Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Chifenti, B.; Locci, M.T.; Lazzeri, G.; Guagnozzi, M.; Dinucci, D.; Chiellini, F.; Filice, M.E.; Salerno, M.G.; Battini, L. Autophagy-related protein LC3 and Beclin-1 in the first trimester of pregnancy. Clin. Exp. Reprod. Med. 2013, 40, 33–37. [Google Scholar] [CrossRef]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggiò, M.A.; Racanelli, V.; Vacca, A.; Frassanito, M.A. Autophagy: A New Mechanism of Prosurvival and Drug Resistance in Multiple Myeloma. Transl. Oncol. 2018, 11, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Prasad, S.B. Antitumor effect of blister beetles: An ethno-medicinal practice in Karbi community and its experimental evaluation against a murine malignant tumor model. J. Ethnopharmacol. 2013, 148, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Wu, M.-Y.; Shou, L.-M.; Chen, L.-P.; Gong, F.-R.; Chen, K.; Li, D.-M.; Duan, W.-M.; Xie, Y.-F.; Mao, Y.-X.; et al. Tamoxifen enhances the anticancer effect of cantharidin and norcantharidin in pancreatic cancer cell lines through inhibition of the protein kinase C signaling pathway. Oncol. Lett. 2015, 9, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Wang, C.; Chen, L.; Tang, X.; Li, L.; Li, N.; Li, J.; Gong, Q.; Tang, F.; Feng, J.; et al. Has aidi injection the attenuation and synergistic efficacy to gemcitabine and cisplatin in non-small cell lung cancer? A meta-analysis of 36 randomized controlled trials. Oncotarget 2017, 8, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.Q.; Huang, X.E.; Wu, X.Y.; Liu, J.; Wang, L.; Tang, J.H. Safety of Brucea javanica and cantharidin combined with chemotherapy for treatment of NSCLC patients. Asian Pac. J. Cancer Prev. 2014, 15, 8603–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Huang, X.E.; Cao, J. Clinical study on safety of cantharidin sodium and shenmai injection combined with chemotherapy in treating patients with breast cancer postoperatively. Asian Pac. J. Cancer Prev. 2014, 15, 5597–5600. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.P.; Huang, X.E.; Cao, J.; Lu, Y.Y.; Wu, X.Y.; Liu, J.; Xu, X.; Xu, L.; Xiang, J.; Ye, L.H. Clinical study on safety and efficacy of Qinin® (cantharidin sodium) injection combined with chemotherapy in treating patients with gastric cancer. Asian Pac. J. Cancer Prev. 2012, 13, 4773–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, S.L.; Zhang, H.R.; Gao, L.; Gao, X.; Liu, P.J.; Yi, Z.Y.; Li, N.; Xu, Z.Q. Combination radiotherapy and cantharidin inhibits lung cancer growth through altering tumor infiltrating lymphocytes. Future Oncol. 2017, 13, 1173–1180. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | Cell Line | Consequence | Molecular Mechanism | Observation Model | Ref |
|---|---|---|---|---|---|
| CML | K562, K562R | Growth inhibition, cell cycle arrest, DNA damage | Downregulation of BCR-ABL protein expression | In vitro | [52] |
| Melanoma | A431 | Apoptosis, cell cycle arrest, DNA damage | Caspase-8,-9 & -3 activation, decreased ΔΨm to release Cyc C, Endo G & AIF, increased expression level of DR4, DR5 & TRAIL, /G1 phase arrest via elevation of p21 while reduction of cyclin D, cyclin E and CDK6 expression level | In vitro, In vivo | [35] |
| Bladder | T24, RT4 | Apoptosis | Induction of apoptosis by calcium/PKC regulated ER stress pathway that involves upregulation of Grp78 & phospho-eIF2a | In vitro, In vivo | [37] |
| Lung | H460 | Apoptosis, cell cycle arrest, DNA damage | Upregulation of DNA damaging genes DNIT3 & GADD45A while downregulation of DdiT4, alteration of cell cycle progression genes (RASA4, CCND2, CDKL3 upregulation, CDC42EP3 downregulation) upregulation of apoptosis-associated genes including CARD6 | In vitro | [38] |
| Lung | H460 | Apoptosis, cell cycle arrest, DNA damage | Increased Ca2+ & ROS production, initiation of caspase-3, -8, decreased ΔΨm, increased expression of Cyc C, AIF & Bax & induction of ER stress via upregulation of IRE1σ, IRE1β, GRP78, ATF6α, caspase-4, calpain 2 & XBP1 | In vitro | [39] |
| Melanoma | A375.S2 | Growth inhibition, invasion & migration inhibition | Inhibition of migration & invasion via MAPK signaling pathway through NF-ĸB and AKT downregulation resulting in reduction of MMP-2/-9 enzymatic activity and expression level | In vitro | [36] |
| TNBC | MDA-MB-231, MDA-MB-468 | Apoptosis, inhibition of pro-survival autophagy | Inhibition of LC3-I to LC3-II conversion and autophagosome formation through suppression of beclin-1 | In vitro, In vivo | [54] |
| Colorectal | Colo 205 | Apoptosis, cell cycle arrest, DNA damage | Elevated activities of caspase-8,-9 & -3, decreased ΔΨm, increased ROS production, stimulation of Cyc C, Fas/CD95 and Bax expression whereas inhibition of Bcl-2 expression, Induction of phase via CDK1, cyclin A, cyclin B decreased expression and p21 and CHK1 increased expression, induction of apoptosis through increased ROS production & decreased ΔΨm | In vitro | [42] |
| Breast | MCF-7 | Apoptosis, Adhesion inhibition | Adhesion inhibition by α2 integrin downregulation through PKC dependent-pathway | [45] | |
| TNBC | MDA-MB-231 | Inhibition of growth, cell cycle arrest, Inhibition of migration & invasion | Suppression of growth & migration via inhibition of MAPK signaling pathway | In vitro, In vivo | [53] |
| TNBC | MDA-MB-231 | Apoptosis | Inhibition of PI3k/Akt & STAT3 signaling pathways by EGF receptor phosphorylation, downregulation of COX-2, Bcl-2 & cyclin | In vitro | [57] |
| Pancreatic | PANC-1, CFPAC-1 | Inhibition of invasion | Post-transcriptional degradation of MMP2 via NF-κB, PKC, JNK, ERK & β-catenin pathways | In vitro | [58] |
| Pancreatic | PANC-1 | Growth & migration inhibition | Suppression of Wnt/β-catenin pathway through β-catenin phosphorylation & degradation | In vitro | [59] |
| Pancreatic | PANC-1, CFPAC-1, BxPC-3, Capan-1, Human Pancreatic duct cells, Rat Pancreatic duct cells | Apoptosis, cell cycle arrest | JNK pathway-dependent growth inhibition, Activation of caspase-8 & -9, elevation of TRAILR1, TRAILR2, TNF-α, Bak, Bad & Bik while repression of Bcl-2, /M phase arrest via p21 upregulation & CDK1 downregulation | In vitro | [60] |
| Pancreatic | PANC-1 | Growth inhibition | Over-activation of JNK pathway | In vitro | [61] |
| Pancreatic | PANC-1 | Apoptosis | NF-κB pathway activation leading to overexpression of TNF-α, TRAIL-1 & TRAIL-2 | In vitro | [62] |
| Tongue squamous cell carcinoma | TCA8113 | Apoptosis | Weakened expression of miR-214 leading to p53 upregulation and Bcl-2/Bax pathway downregulation | In vitro | [49] |
| Oral Squamous Cell Carcinoma | SAS, SSC-4, CAL-27 | Apoptosis | JNK-mediated mitochondria & ER stress pathways involving increased expression of caspase-9, -7, & -3, decreased ΔΨm, induction of Cyc C & AIF release, elevated level of Bax, Bak & Bid, reduced expression of Bcl-2, increased expression of p-eIF2 & CHOP, & reduction of pro-caspase-12 expression level | [48] | |
| Bladder | TSGH 8301 | Apoptosis, cell cycle arrest, DNA damage | caspase-8, -9, & -3 activation, increased ROS and Cageneration, decreased ΔΨm, increased AIF & Endo G release, upregulation of Bax & PARP, downregulation of Bcl-2, /G1 phase arrest in association with decreased cyclin E & Cdc25c, but elevation of p21 & p-p53 | In vitro | [63] |
| Bladder | TSGH 8301 | Inhibition of migration, invasion & adhesion | Reduction of MMP-2 & MMP-9 through p38 & JNK1/2 MAPK pathway | In vitro | [64] |
| Oral squamous cell carcinoma | UMSCC | Apoptosis, DNA damage | Induction of ER stress and activation of UPP | In vitro | [50] |
| NSCLC | A549 | Inhibition of growth, migration & invasion, induction of autophagy | Growth & migration inhibition through induction of autophagy and apoptosis which is consorted with PI3 K/Akt/mTOR pathway repression | In vitro | [40] |
| NSCLC | NCI-H460 | Inhibition of migration, invasion & adhesion | Attenuation of MAPK pathway by reducing NF-ĸB & AKT, leading to down of MMP-2/-9 & UPA | In vitro | [65] |
| NSCLC | A549 | Inhibition of metastasis | Alteration of PIk3/Akt pathway activation resulting in the inhibition of MMP-2 activity | In vitro | [66] |
| Renal cell carcinoma | ACHN, Caki-1 RCC | Apoptosis, cell cycle arrest | Upregulation of Notch-1 & Jagged1 | In vitro | [67] |
| Osteosarcoma | U-2 OS | Apoptosis, cell cycle arrest, DNA damage | Apoptosis induction through both extrinsic & intrinsic pathways, /M phase arrest via upregulation of CHK-1, WEE-1, CDK-1, p-p53, CDC25C & p21 | In vitro | [68] |
| Osteosarcoma | MG-63 MNNG/HOS | Apoptosis | Increased Bax, PARP whereas reduced Bcl-2 p-Cdc2 & p-Akt expression level | In vitro | [69] |
| Cholangiocarcinoma | QBC939 | Inhibition of migration & invasion | Inhibition of migration and invasion through activation of IKKα/IĸBα/NF-ĸB pathway resulting in suppression of MMP-2 & MMP-9 expression level | In vitro | [70] |
| Gastric cancer | BGC823, MGC803 | Apoptosis, inhibition of metastasis | Suppression of growth & migration by suppressing PI3k/Akt signaling pathway which was mediated by CCAT1 downregulation | In vitro | [43] |
| Hepatocellular carcinoma | HepG2 CD133+ | Apoptosis, cell cycle arrest, inhibition of self-renew ability | Halted self-renewable ability by upregulation of β-catenin & cyclin D1, arrested /M phase by upregulation Myt1, p53, histone H2AX, cyclin A2, Cyclin B1 | In vitro | [71] |
| TNBC | MDA-MB-231 | Apoptosis, Inhibition of migration & invasion, induction of angiogenesis | Transformation of aerobic glycolysis to oxidation by breaking GLUT1/PKM glycolytic loop | In vitro, In vivo | [17] |
| Pancreatic, Breast, NSCLC | PANC-1, T47D, MCF-7, NCI-H292, NCI-H1650 | cell cycle arrest | /M phase arrest via autophagy-dependent upregulation of p21 & JNK/Sp1-dependent downregulation of CDK1 | In vitro | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naz, F.; Wu, Y.; Zhang, N.; Yang, Z.; Yu, C. Anticancer Attributes of Cantharidin: Involved Molecular Mechanisms and Pathways. Molecules 2020, 25, 3279. https://doi.org/10.3390/molecules25143279
Naz F, Wu Y, Zhang N, Yang Z, Yu C. Anticancer Attributes of Cantharidin: Involved Molecular Mechanisms and Pathways. Molecules. 2020; 25(14):3279. https://doi.org/10.3390/molecules25143279
Chicago/Turabian StyleNaz, Faiza, Yixin Wu, Nan Zhang, Zhao Yang, and Changyuan Yu. 2020. "Anticancer Attributes of Cantharidin: Involved Molecular Mechanisms and Pathways" Molecules 25, no. 14: 3279. https://doi.org/10.3390/molecules25143279
APA StyleNaz, F., Wu, Y., Zhang, N., Yang, Z., & Yu, C. (2020). Anticancer Attributes of Cantharidin: Involved Molecular Mechanisms and Pathways. Molecules, 25(14), 3279. https://doi.org/10.3390/molecules25143279