
Recent Advances in the Chemistry of Metal Carbamates



Abstract
| Index: | |
| 1. Introduction | 2 |
| 2. The Reactivity of Carbon Dioxide with Amines and other N-Donors | 2 |
| 2.1. CO2/Amine Equilibria in Aqueous Solution | 3 |
| 2.2. Amine/CO2 Interaction: Isolation and Characterization of Carbamato Salts | 5 |
| 2.3. Stabilization of Carbamates by Superbases | 6 |
| 3. Synthesis, Structure and Reactivity of Metal Carbamates | 10 |
| 3.1. Homoleptic Carbamato Complexes | 11 |
| 3.2. Heteroleptic Carbamato Complexes | 15 |
| 3.3. Dynamics and Reactivity of Metal Carbamato Complexes | 22 |
| 3.4. Crystallographic and Spectroscopic Features of Carbamato Ligands | 26 |
| 4. Catalysis with Metal Carbamates | 29 |
| 4.1. CO2 Activation Routes | 29 |
| 4.2. Other Catalytic Processes | 32 |
| 5. Other Applications | 33 |
| 6. Conclusions | 38 |
| References | 39 |
1. Introduction
2. The Reactivity of Carbon Dioxide with Amines and other N-Donors
2.1. CO2/Amine Equilibria in Aqueous Solution
2.2. Amine/CO2 Interaction: Isolation and Characterization of Carbamato Salts
2.3. Stabilization of Carbamates by Superbases
3. Synthesis, Structure and Reactivity of Metal Carbamates
3.1. Homoleptic Carbamato Complexes
3.2. Heteroleptic Carbamato Complexes
3.3. Dynamics and Reactivity of Metal Carbamato Complexes
3.4. Crystallographic and Spectroscopic Features of Carbamato Ligands
4. Catalysis with Metal Carbamates
4.1. CO2 Activation Routes
4.2. Other Catalytic Processes
5. Other Applications
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Smith, P.; Davis, S.J.; Creutzig, F.; Fuss, S.; Minx, J.; Gabrielle, B.; Kato, E.; Jackson, R.B.; Cowie, A.; Kriegler, E.; et al. Biophysical and economic limits to negative CO2 emissions. Nat. Clim. Chang. 2016, 6, 42–50. [Google Scholar] [CrossRef]
- Aresta, M. Carbon Dioxide Recovery and Utilization; Springer: Dordrecht, The Netherlands, 2003; ISBN 978-90-481-6335-9. [Google Scholar]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef] [PubMed]
- Marques Mota, F.; Kim, D.H. From CO2 methanation to ambitious long-chain hydrocarbons: Alternative fuels paving the path to sustainability. Chem. Soc. Rev. 2019, 48, 205–259. [Google Scholar] [CrossRef] [PubMed]
- Cherubini-Celli, A.; Mateos, J.; Bonchio, M.; Dell’Amico, L.; Companyó, X. Transition Metal-Free CO2 Fixation into New Carbon-Carbon Bonds. Chem. Sus. Chem. 2018, 11, 3056–3070. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, J.W. Toward ideal carbon dioxide functionalization. Chem. Sci. 2019, 10, 3905–3926. [Google Scholar] [CrossRef]
- Lu, X.-B. CO2-Mediated Formation of Chiral Fine Chemicals. In Carbon Dioxide and Organometallics; Springer International Publishing: New York, NY, USA, 2015; pp. 171–197. ISBN 9783319220789. [Google Scholar]
- Yu, B.; He, L.-N. Upgrading Carbon Dioxide by Incorporation into Heterocycles. Chem. Sus. Chem. 2015, 8, 52–62. [Google Scholar] [CrossRef]
- Aresta, M.; Forti, G. Carbon Dioxide as a Source of Carbon; Springer: Dordrecht, The Netherlands, 1987; ISBN 978-94-010-8240-2. [Google Scholar]
- Behr, A. Activation of Carbon Dioxide via Coordination to Transition Metal Complexes. In Catalysis in C1 Chemistry; Keim, W., Ed.; Catalysis by Metal Complexes; Springer: Dordrecht, The Netherlands, 1983; Volume 3, p. 169. ISBN 978-94-009-7042-7. [Google Scholar]
- Braunstein, P.; Matt, D.; Nobel, D. Reactions of carbon dioxide with carbon-carbon bond formation catalyzed by transition-metal complexes. Chem. Rev. 1988, 88, 747–764. [Google Scholar] [CrossRef]
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef]
- Li, Y.-N.; He, L.-N.; Diao, Z.-F.; Yang, Z.-Z. Carbon Capture with Simultaneous Activation and Its Subsequent Transformation. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2014; pp. 289–345. [Google Scholar]
- Tappe, N.A.; Reich, R.M.; D’Elia, V.; Kühn, F.E. Current advances in the catalytic conversion of carbon dioxide by molecular catalysts: An update. Dalton Trans. 2018, 47, 13281–13313. [Google Scholar] [CrossRef]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Aresta, M.; Nobile, C.F. (Carbon dioxide)bis(trialkylphosphine)nickel complexes. J. Chem. Soc. Dalt. Trans. 1977, 7, 708. [Google Scholar] [CrossRef]
- Viasus, C.J.; Gabidullin, B.; Gambarotta, S. Linear End—On Coordination Modes of CO2. Angew. Chem. Int. Ed. 2019, 58, 14887–14890. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-E.; Kim, J.; Lee, Y. Formation of a nickel carbon dioxide adduct and its transformation mediated by a Lewis acid. Chem. Commun. 2014, 50, 11458–11461. [Google Scholar] [CrossRef] [PubMed]
- Devillard, M.; Declercq, R.; Nicolas, E.; Ehlers, A.W.; Backs, J.; Saffon-Merceron, N.; Bouhadir, G.; Slootweg, J.C.; Uhl, W.; Bourissou, D. A Significant but Constrained Geometry Pt→Al Interaction: Fixation of CO2 and CS2, Activation of H2 and PhCONH2. J. Am. Chem. Soc. 2016, 138, 4917–4926. [Google Scholar] [CrossRef]
- Aresta, M. Carbon Dioxide Reduction and Uses as a Chemical Feedstock. In Activation of Small Molecules; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–41. ISBN 3527313125. [Google Scholar]
- Alvarez, R.; Carmona, E.; Poveda, M.L.; Sanchez-Delgado, R. Carbon dioxide chemistry. The synthesis and properties of trans-bis(carbon dioxide)tetrakis(trimethylphosphine)molybdenum (trans-[Mo(CO2)2(PMe3)4]): The first stable bis(carbon dioxide) adduct of a transition me. J. Am. Chem. Soc. 1984, 106, 2731–2732. [Google Scholar] [CrossRef]
- Janes, T.; Yang, Y.; Song, D. Chemical reduction of CO2 facilitated by C-nucleophiles. Chem. Commun. 2017, 53, 11390–11398. [Google Scholar] [CrossRef]
- Baisch, U.; Schnick, W. Synthese und Kristallstruktur vonbis-1, 3-Dimethoxyethan-trichloro-samarium(III) undtris-N,N-Diisopropylcarbamato-samarium(III). Z. Anorg. Allg. Chem. 2003, 629, 2073–2078. [Google Scholar] [CrossRef]
- Murphy, L.J.; Robertson, K.N.; Kemp, R.A.; Tuononen, H.M.; Clyburne, J.A.C. Structurally simple complexes of CO2. Chem. Commun. 2015, 51, 3942–3956. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, X.; Fei, H. N-heterocyclic carbene-functionalized metal–organic frameworks for the chemical fixation of CO2. Dalton Trans. 2020, 49, 6548–6552. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Xue, X.-S.; Ji, P. Acidity Scale of N-Heterocyclic Carbene Precursors: Can We Predict the Stability of NHC–CO2 Adducts? Org. Lett. 2018, 20, 6041–6045. [Google Scholar] [CrossRef]
- Hazari, N.; Heimann, J.E. Carbon Dioxide Insertion into Group 9 and 10 Metal–Element σ Bonds. Inorg. Chem. 2017, 56, 13655–13678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hanna, B.S.; Dineen, A.; Williard, P.G.; Bernskoetter, W.H. Functionalization of Carbon Dioxide with Ethylene at Molybdenum Hydride Complexes. Organometallics 2013, 32, 3969–3979. [Google Scholar] [CrossRef]
- Chen, J.-H.; Deng, C.-H.; Fang, S.; Ma, J.-G.; Cheng, P. Binuclear molybdenum alkoxide as the versatile catalyst for the conversion of carbon dioxide. Green Chem. 2018, 20, 989–996. [Google Scholar] [CrossRef]
- Field, L.D.; Jurd, P.M.; Magill, A.M.; Bhadbhade, M.M. Reactions of CO2 and CS2 with [RuH(η2-CH2PMe2)(PMe3)3]. Organometallics 2013, 32, 636–642. [Google Scholar] [CrossRef]
- Braunstein, P.; Matt, D.; Dusausoy, Y.; Fischer, J.; Mitschler, A.; Ricard, L. Complexes of functional phosphines. 4. Coordination properties of (diphenylphosphino)acetonitrile, ethyl (diphenylphosphino)acetate and corresponding carbanions. Characterization of a new facile reversible carbon dioxide insertion into palladium(II) compl. J. Am. Chem. Soc. 1981, 103, 5115–5125. [Google Scholar] [CrossRef]
- Braunstein, P.; Matt, D.; Nobel, D. Carbon dioxide activation and catalytic lactone synthesis by telomerization of butadiene and carbon dioxide. J. Am. Chem. Soc. 1988, 110, 3207–3212. [Google Scholar] [CrossRef]
- Varghese, A.M.; Karanikolos, G.N. CO2 capture adsorbents functionalized by amine–bearing polymers: A review. Int. J. Greenh. Gas Control 2020, 96, 103005. [Google Scholar] [CrossRef]
- Mazari, S.A.; Ghalib, L.; Sattar, A.; Bozdar, M.M.; Qayoom, A.; Ahmed, I.; Muhammad, A.; Abro, R.; Abdulkareem, A.; Nizamuddin, S.; et al. Review of modelling and simulation strategies for evaluating corrosive behavior of aqueous amine systems for CO2 capture. Int. J. Greenh. Gas Control 2020, 96, 103010. [Google Scholar] [CrossRef]
- Gelles, T.; Lawson, S.; Rownaghi, A.A.; Rezaei, F. Recent Advances in Development of Amine Functionalized Adsorbents for CO2 Capture; Springer: New York, NY, USA, 2020; Volume 26, ISBN 0123456789. [Google Scholar]
- Zhao, T.-X.; Zhai, G.-W.; Liang, J.; Li, P.; Hu, X.-B.; Wu, Y.-T. Catalyst-free N-formylation of amines using BH3 NH3 and CO2 under mild conditions. Chem. Commun. 2017, 53, 8046–8049. [Google Scholar] [CrossRef]
- Hulla, M.; Nussbaum, S.; Bonnin, A.R.; Dyson, P.J. The dilemma between acid and base catalysis in the synthesis of benzimidazole from o-phenylenediamine and carbon dioxide. Chem. Commun. 2019, 55, 13089–13092. [Google Scholar] [CrossRef]
- Hao, L.; Zhang, H.; Luo, X.; Wu, C.; Zhao, Y.; Liu, X.; Gao, X.; Chen, Y.; Liu, Z. Reductive formylation of amines with CO2 using sodium borohydride: A catalyst-free route. J. CO2 Util. 2017, 22, 208–211. [Google Scholar] [CrossRef]
- Hulla, M.; Dyson, P.J. Pivotal Role of the Basic Character of Organic and Salt Catalysts in C−N Bond Forming Reactions of Amines with CO2. Angew. Chem. Int. Ed. 2020, 59, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Stec, B. Structural mechanism of RuBisCO activation by carbamylation of the active site lysine. Proc. Natl. Acad. Sci. USA 2012, 109, 18785–18790. [Google Scholar] [CrossRef] [PubMed]
- Cleland, W.W.; Andrews, T.J.; Gutteridge, S.; Hartman, F.C.; Lorimer, G.H. Mechanism of Rubisco: The Carbamate as General Base. Chem. Rev. 1998, 98, 549–562. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F.; Pampaloni, G. Converting Carbon Dioxide into Carbamato Derivatives. Chem. Rev. 2003, 103, 3857–3898. [Google Scholar] [CrossRef]
- Hampe, E.M.; Rudkevich, D.M. Reversible covalent chemistry of CO2. Chem. Commun. 2002, 14, 1450–1451. [Google Scholar] [CrossRef]
- Terlouw, J.K.; Schwarz, H. The Generation and Characterization of Molecules by Neutralization-Reionization Mass Spectrometry (NRMS). New Analytical Methods (33). Angew. Chem. Int. Ed. 1987, 26, 805–815. [Google Scholar] [CrossRef]
- Khanna, R.K.; Moore, M.H. Carbamic acid: Molecular structure and IR spectra. Spectrochim. Acta. Part. A Mol. Biomol. Spectrosc. 1999, 55, 961–967. [Google Scholar] [CrossRef]
- Radeglia, R.; Andersch, J.; Schroth, W. Zum dynamischen Strukturverhalten des Dimethylamin—Kohlendioxid-Komplexes (Dimcarb)/On the Dynamic Structure Behaviour of the Dimethylamine—Carbondioxide Complex (Dimcarb). Zeitschrift für Naturforsch. B 1989, 44, 181–186. [Google Scholar] [CrossRef]
- Aresta, M.; Ballivet-Tkatchenko, D.; Belli Dell’Amico, D.; Bonnet, M.C.; Boschi, D.; Calderazzo, F.; Faure, R.; Labella, L.; Marchetti, F. Isolation and structural determination of two derivatives of the elusive carbamic acid. Chem. Commun. 2000, 8, 1099–1100. [Google Scholar] [CrossRef]
- McGhee, W.; Riley, D.; Christ, K.; Pan, Y.; Parnas, B. Carbon Dioxide as a Phosgene Replacement: Synthesis and Mechanistic Studies of Urethanes from Amines, CO2, and Alkyl Chlorides. J. Org. Chem. 1995, 60, 2820–2830. [Google Scholar] [CrossRef]
- Ishikawa, T. Superbases for Organic Synthesis; Ishikawa, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009; ISBN 9780470740859. [Google Scholar]
- Hu, X.; Yu, Q.; Barzagli, F.; LI, C.; Fan, M.; Gasem, K.A.M.; Zhang, X.; Shiko, E.; Tian, M.; Luo, X.; et al. NMR techniques and prediction models for the analysis of the species formed in CO2 capture processes with amine-based sorbents: A critical review. ACS Sustain. Chem. Eng. 2020, 8, 6173–6193. [Google Scholar] [CrossRef]
- Bavoh, C.B.; Lal, B.; Osei, H.; Sabil, K.M.; Mukhtar, H. A review on the role of amino acids in gas hydrate inhibition, CO2 capture and sequestration, and natural gas storage. J. Nat. Gas Sci. Eng. 2019, 64, 52–71. [Google Scholar] [CrossRef]
- Conway, W.; Wang, X.; Fernandes, D.; Burns, R.; Lawrance, G.; Puxty, G.; Maeder, M. Toward the understanding of chemical absorption processes for post-combustion capture of carbon dioxide: Electronic and steric considerations from the kinetics of reactions of CO2 (aq) with sterically hindered amines. Environ. Sci. Technol. 2013, 47, 1163–1169. [Google Scholar] [CrossRef]
- Ermatchkov, V.; Pérez-Salado Kamps, Á.; Maurer, G. Chemical equilibrium constants for the formation of carbamates in (carbon dioxide + piperazine + water) from 1H-NMR-spectroscopy. J. Chem. Thermodyn. 2003, 35, 1277–1289. [Google Scholar] [CrossRef]
- Fernandes, D.; Conway, W.; Burns, R.; Lawrance, G.; Maeder, M.; Puxty, G. Investigations of primary and secondary amine carbamate stability by 1H NMR spectroscopy for post combustion capture of carbon dioxide. J. Chem. Thermodyn. 2012, 54, 183–191. [Google Scholar] [CrossRef]
- Conway, W.; Wang, X.; Fernandes, D.; Burns, R.; Lawrance, G.; Puxty, G.; Maeder, M. Toward rational design of amine solutions for PCC applications: The kinetics of the reaction of CO2 (aq) with cyclic and secondary amines in aqueous solution. Environ. Sci. Technol. 2012, 46, 7422–7429. [Google Scholar] [CrossRef]
- Li, L.; Clifford, S.; Puxty, G.; Maeder, M.; Burns, R.; Yu, H.; Conway, W. Kinetic and Equilibrium Reactions of a New Heterocyclic Aqueous 4-Aminomethyltetrahydropyran (4-AMTHP) Absorbent for Post Combustion Carbon Dioxide (CO2) Capture Processes. ACS Sustain. Chem. Eng. 2017, 5, 9200–9206. [Google Scholar] [CrossRef]
- Kumar, P.S.; Hogendoorn, J.A.; Timmer, S.J.; Feron, P.H.M.; Versteeg, G.F. Equilibrium Solubility of CO2 in Aqueous Potassium Taurate Solutions: Part 2. Experimental VLE Data and Model. Ind. Eng. Chem. Res. 2003, 42, 2841–2852. [Google Scholar] [CrossRef]
- Coulier, Y.; Lowe, A.R.; Coxam, J.Y.; Ballerat-Busserolles, K. Thermodynamic Modeling and Experimental Study of CO2 Dissolution in New Absorbents for Post-Combustion CO2 Capture Processes. ACS Sustain. Chem. Eng. 2018, 6, 918–926. [Google Scholar] [CrossRef]
- Jensen, A.; Jensen, J.B.; Faurholt, C.; Finsnes, E.; Sörensen, J.S.; Sörensen, N.A. Studies on Carbamates. VI. The Carbamate of Glycine. Acta Chem. Scand. 1952, 6, 395–397. [Google Scholar] [CrossRef]
- Xiang, Q.; Fang, M.; Yu, H.; Maeder, M. Kinetics of the reversible reaction of CO2 (aq) and HCO3- with sarcosine salt in aqueous solution. J. Phys. Chem. A 2012, 116, 10276–10284. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Faurholt, C.; Faurholt, C.; Finsnes, E.; Sörensen, J.S.; Sörensen, N.A. Studies on Carbamates. V. The Carbamates of alpha-Alanine and beta-Alanine. Acta Chem. Scand. 1952, 6, 385–394. [Google Scholar] [CrossRef]
- Majchrowicz, M.E.; Brilman, D.W.F. Solubility of CO2 in aqueous potassium L-prolinate solutions-absorber conditions. Chem. Eng. Sci. 2012, 72, 35–44. [Google Scholar] [CrossRef]
- Shen, S.; Zhao, Y.; Bian, Y.; Wang, Y.; Guo, H.; Li, H. CO2 absorption using aqueous potassium lysinate solutions: Vapor–liquid equilibrium data and modelling. J. Chem. Thermodyn. 2017, 115, 209–220. [Google Scholar] [CrossRef]
- Jensen, A.; Jensen, M.B.; Faurholt, C. Studies on Carbamates. VIII. The Carbamates of Benzylamine, Piperidine and Aniline. Acta Chem. Scand. 1952, 6, 1073–1085. [Google Scholar] [CrossRef]
- Jensen, A.; Jensen, M.B.; Faurholt, C.; Sörensen, N.A. Studies on Carbamates. X. The Carbamates of Di-n-Propylamine and Di-iso-Propylamine. Acta Chem. Scand. 1954, 8, 1129–1136. [Google Scholar] [CrossRef][Green Version]
- Jensen, M.B. Studies on Carbamates. XII. The Carbamates of the Butylamines. Acta Chem. Scand. 1957, 11, 499–505. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Martens, J.A.; Bogaerts, A.; De Kimpe, N.; Jacobs, P.A.; Marin, G.B.; Rabaey, K.; Saeys, M.; Verhelst, S. The Chemical Route to a Carbon Dioxide Neutral World. Chem. Sus. Chem. 2017, 10, 1039–1055. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Smith, K.H.; Wu, Y.; Mumford, K.A.; Kentish, S.E.; Stevens, G.W. Carbon dioxide capture by solvent absorption using amino acids: A review. Chinese J. Chem. Eng. 2018, 26, 2229–2237. [Google Scholar] [CrossRef]
- Fernandes, D.; Conway, W.; Wang, X.; Burns, R.; Lawrance, G.; Maeder, M.; Puxty, G. Protonation constants and thermodynamic properties of amines for post combustion capture of CO2. J. Chem. Thermodyn. 2012, 51, 97–102. [Google Scholar] [CrossRef]
- Christensen, J.J.; Izatt, R.M.; Wrathall, D.P.; Hansen, L.D. Thermodynamics of proton ionization in dilute aqueous solution. Part XI. pK, ΔH°, and ΔS° values for proton ionization from protonated amines at 25°. J. Chem. Soc. A 1969, 1212–1223. [Google Scholar] [CrossRef]
- Dey, A.; Desiraju, G.R.; Mondal, R.; Howard, J.A.K. A 1:1 molecular complex of 4-aminocyclohexanol and (4-hydroxycyclohexyl)carbamic acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, o857–o859. [Google Scholar] [CrossRef]
- Mondal, R.; Bhunia, M.K. Crystal Chemistry of 1:1 Molecular Complexes of Carbamate Salts Formed by Slow Aerial Carbonation of Amines. J. Chem. Crystallogr. 2008, 38, 787–792. [Google Scholar] [CrossRef]
- Zhang, W.H.; Li, K.; Li, Q.; Mak, T.C.W. Two inclusion compounds of guanylthiourea and 1,3,5-thiadiazole-5-amido-2-carbamate. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 353–359. [Google Scholar] [CrossRef]
- Jiang, H.; Novak, I. Piperidine–CO2–H2O molecular complex. J. Mol. Struct. 2003, 645, 177–183. [Google Scholar] [CrossRef]
- Madarász, J.; Székely, E.; Halász, J.; Bánsághi, G.; Varga, D.; Simándi, B.; Pokol, G. Ammonium carbamate type self-derivative salts of (R-)- and racemic α-methylbenzylamine: Composition and thermal stability by evolved gas analyses (TG-FTIR and TG/DTA-MS). J. Therm. Anal. Calorim. 2013, 111, 567–574. [Google Scholar] [CrossRef]
- Shi, P.-F.; Xu, T.-T.; Xu, X.-Y.; Niu, S.-R. (3-Ammonio-2-hydroxypropyl)carbamate monohydrate. Acta. Crystallogr. Sect. E Struct. Rep. Online 2006, 62, o5191–o5193. [Google Scholar] [CrossRef]
- Neda, I.; Kaukorat, T.; Fischer, A.K. Unusual Stabilization of 1,2-Diamino Derivatives of Quincorine and Quincoridine by Carbon Dioxide: Persistent Crystallineprim-Ammonium-Carbamate Salts and Their Reactivity towards Isatoic Acid Anhydride. Eur. J. Org. Chem. 2003, 2003, 3784–3790. [Google Scholar] [CrossRef]
- Meijide, F.; Antelo, A.; Alvarez, M.; Soto, V.H.; Trillo, J.V.; Jover, A.; Vázquez Tato, J. Spontaneous Formation in the Solid State of Carbamate Derivatives of Bile Acids. Cryst. Growth Des. 2011, 11, 356–361. [Google Scholar] [CrossRef]
- Jo, E.; Jhon, Y.H.; Choi, S.B.; Shim, J.G.; Kim, J.H.; Lee, J.H.; Lee, I.Y.; Jang, K.R.; Kim, J. Crystal structure and electronic properties of 2-amino-2-methyl-1-propanol (AMP) carbamate. Chem. Commun. 2010, 46, 9158–9160. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Schröder, M.; Sklorz, C.A.; Tabanella, S.; Vogl, N.; Lex, J.; Neudörfl, J.M. Enantioselective Synthesis of DIANANE, a Novel C2 -Symmetric Chiral Diamine for Asymmetric Catalysis. J. Org. Chem. 2004, 69, 3050–3056. [Google Scholar] [CrossRef]
- Berkessel, A.; Roland, K.; Schröder, M.; Neudörfl, J.M.; Lex, J. Enantiomerically Pure Isophorone Diamine [3-(Aminomethyl)-3,5,5-trimethylcyclohexylamine]: A Chiral 1,4-Diamine Building Block Made Available on Large Scale. J. Org. Chem. 2006, 71, 9312–9318. [Google Scholar] [CrossRef]
- Fonari, M.S.; Antal, S.; Castañeda, R.; Ordonez, C.; Timofeeva, T.V. Crystalline products of CO2 capture by piperazine aqueous solutions. Cryst. Eng. Comm. 2016, 18, 6282–6289. [Google Scholar] [CrossRef]
- Garbauskas, M.F.; Goehner, R.P.; Davis, A.M. The structure of two polymorphs of N-(2-ammonioethyl)carbamate, C3H8N2O2. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1983, 39, 1684–1686. [Google Scholar] [CrossRef]
- Katchalski, E.; Berliner-Klibanski, C.; Berger, A. The Chemical Structure of Some Diamine Carbamates. J. Am. Chem. Soc. 1951, 73, 1829–1831. [Google Scholar] [CrossRef]
- Baidya, M.; Mayr, H. Nucleophilicities and carbon basicities of DBU and DBN. Chem. Commun. 2008, 15, 1792–1794. [Google Scholar] [CrossRef]
- Pereira, F.S.; Lincon Da Silva Agostini, D.; Do Espírito Santo, R.D.; Deazevedo, E.R.; Bonagamba, T.J.; Job, A.E.; González, E.R.P. A comparative solid state 13C NMR and thermal study of CO2 capture by amidines PMDBD and DBN. Green Chem. 2011, 13, 2146–2153. [Google Scholar] [CrossRef]
- Pereira, F.S.; DeAzevedo, E.R.; da Silva, E.F.; Bonagamba, T.J.; da Silva Agostíni, D.L.; Magalhães, A.; Job, A.E.; Pérez González, E.R. Study of the carbon dioxide chemical fixation—activation by guanidines. Tetrahedron 2008, 64, 10097–10106. [Google Scholar] [CrossRef]
- Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. An Isolated CO2 Adduct of a Nitrogen Base: Crystal and Electronic Structures. Angew. Chem. Int. Ed. 2010, 49, 3465–3468. [Google Scholar] [CrossRef] [PubMed]
- Wilm, L.F.B.; Eder, T.; Mück-Lichtenfeld, C.; Mehlmann, P.; Wünsche, M.; Buß, F.; Dielmann, F. Reversible CO2 fixation by N-heterocyclic imines forming water-stable zwitterionic nitrogen-base-CO2 adducts. Green Chem. 2019, 21, 640–648. [Google Scholar] [CrossRef]
- Pérez, E.R.; Santos, R.H.A.; Gambardella, M.T.P.; De Macedo, L.G.M.; Rodrigues-Filho, U.P.; Launay, J.C.; Franco, D.W. Activation of carbon dioxide by bicyclic amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef]
- Ramkumar, V.; Gardas, R.L. Thermophysical Properties and Carbon Dioxide Absorption Studies of Guanidinium-Based Carboxylate Ionic Liquids. J. Chem. Eng. Data 2019, 64, 4844–4855. [Google Scholar] [CrossRef]
- Nicholls, R.; Kaufhold, S.; Nguyen, B.N. Observation of guanidine-carbon dioxide complexation in solution and its role in the reaction of carbon dioxide and propargylamines. Catal. Sci. Technol. 2014, 4, 3458–3462. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The reaction of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) with carbon dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef]
- Valera Lauridsen, J.M.; Cho, S.Y.; Bae, H.Y.; Lee, J.-W. CO2 (De)Activation in Carboxylation Reactions: A Case Study Using Grignard Reagents and Nucleophilic Bases. Organometallics 2020, 39, 1652–1657. [Google Scholar] [CrossRef]
- Lv, M.; Wang, P.; Yuan, D.; Yao, Y. Conversion of Carbon Dioxide into Oxazolidinones Mediated by Quaternary Ammonium Salts and DBU. Chem. Cat. Chem. 2017, 9, 4451–4455. [Google Scholar] [CrossRef]
- Yoshida, M.; Mizuguchi, T.; Shishido, K. Synthesis of oxazolidinones by efficient fixation of atmospheric CO2 with propargylic amines by using a silver/1,8-diazabicyclo[5.4.0] undec-7-ene (DBU) dual-catalyst system. Chem. Eur. J. 2012, 18, 15578–15581. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Komatsuzaki, Y.; Ihara, M. Synthesis of 5-Vinylideneoxazolidin-2-ones by DBU-Mediated CO2-Fixation Reaction of 4-(Benzylamino)-2-butynyl Carbonates and Benzoates. Org. Lett. 2008, 10, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Kaupmees, K.; Trummal, A.; Leito, I. Basicities of strong bases in water: A computational study. Croat. Chem. Acta 2014, 87, 385–395. [Google Scholar] [CrossRef]
- McGhee, W.D.; Riley, D.P. Palladium-Mediated Synthesis of Urethanes from Amines, Carbon Dioxide, and Cyclic Diolefins. Organometallics 1992, 11, 900–907. [Google Scholar] [CrossRef]
- McGhee, W.D.; Pan, Y.; Riley, D.P. Highly selective generation of urethanes from amines, carbon dioxide and alkyl chlorides. J. Chem. Soc. Chem. Commun. 1994, 25, 699–700. [Google Scholar] [CrossRef]
- Jessop, P.G.; Heldebrant, D.J.; Li, X.; Eckert, C.A.; Liotta, C.L. Reversible nonpolar-to-polar solvent. Nature 2005, 436, 1102. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Lukac, P.J.; George, M.; Weiss, R.G. Reversible, room-temperature ionic liquids. Amidinium carbamates derived from amidines and aliphatic primary amines with carbon dioxide. Chem. Mater. 2007, 19, 967–969. [Google Scholar] [CrossRef]
- Yu, T.; Yamada, T.; Gaviola, G.C.; Weiss, R.G. Carbon dioxide and molecular nitrogen as switches between ionic and uncharged room-temperature liquids comprised of amidines and chiral amino alcohols. Chem. Mater. 2008, 20, 5337–5344. [Google Scholar] [CrossRef]
- Yamada, T.; Lukac, P.J.; Yu, T.; Weiss, R.G. Reversible, room-temperature, chiral ionic liquids. Amidinium carbamates derived from amidines and amino-acid esters with carbon dioxide. Chem. Mater. 2007, 19, 4761–4768. [Google Scholar] [CrossRef]
- Biancalana, L.; Bresciani, G.; Chiappe, C.; Marchetti, F.; Pampaloni, G. Synthesis and study of the stability of amidinium/guanidinium carbamates of amines and α-amino acids. New J. Chem. 2017, 41, 1798–1805. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Bresciani, G.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Synthesis and structural characterization of mixed halide–N,N-diethylcarbamates of group 4 metals, including a case of unusual tetrahydrofuran activation. New J. Chem. 2017, 41, 1781–1789. [Google Scholar] [CrossRef]
- Carrera, G.V.S.M.; da Ponte, M.N.; Branco, L.C. Synthesis and properties of reversible ionic liquids using CO2, mono- to multiple functionalization. Tetrahedron 2012, 68, 7408–7413. [Google Scholar] [CrossRef]
- Carrera, G.V.S.M.; Jordão, N.; Santos, M.M.; Da Ponte, M.N.; Branco, L.C. Reversible systems based on CO2, amino-acids and organic superbases. RSC Adv. 2015, 5, 35564–35571. [Google Scholar] [CrossRef]
- Nicholls, R.L.; McManus, J.A.; Rayner, C.M.; Morales-Serna, J.A.; White, A.J.P.; Nguyen, B.N. Guanidine-Catalyzed reductive amination of carbon dioxide with silanes: Switching between pathways and suppressing catalyst deactivation. ACS Catal. 2018, 8, 3678–3687. [Google Scholar] [CrossRef]
- Liu, X.; Wang, M.-Y.; Wang, S.-Y.; Wang, Q.; He, L.-N. In Situ Generated Zinc(II) Catalyst for Incorporation of CO2 into 2-Oxazolidinones with Propargylic Amines at Atmospheric Pressure. Chem. Sus. Chem. 2017, 10, 1210–1216. [Google Scholar] [CrossRef]
- Das Neves Gomes, C.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. A Diagonal Approach to Chemical Recycling of Carbon Dioxide: Organocatalytic Transformation for the Reductive Functionalization of CO2. Angew. Chem. 2012, 124, 191–194. [Google Scholar] [CrossRef]
- Cerveri, A.; Pace, S.; Monari, M.; Lombardo, M.; Bandini, M. Redox-Neutral Metal-Free Three-Component Carbonylative Dearomatization of Pyridine Derivatives with CO2. Chem. -Eur. J. 2019, 25, 15272–15276. [Google Scholar] [CrossRef]
- Matulková, I.; Charvátová, H.; Císařová, I.; Štěpnička, P. The crystal structure of the inner salt of 2-[(aminoiminomethyl)amino]ethylcarbamic acid [systematic name: (2-((diaminomethylene)ammonio)ethyl)carbamate], C4H10N4O2. Z. Kristallogr. NCS 2017, 232, 685–687. [Google Scholar] [CrossRef]
- Wang, C.; Luo, H.; Jiang, D.E.; Li, H.; Dai, S. Carbon dioxide capture by superbase-derived protic ionic liquids. Angew. Chem. Int. Ed. 2010, 49, 5978–5981. [Google Scholar] [CrossRef]
- Wang, C.; Luo, X.; Luo, H.; Jiang, D.E.; Li, H.; Dai, S. Tuning the basicity of ionic liquids for equimolar CO2 capture. Angew. Chem. Int. Ed. 2011, 50, 4918–4922. [Google Scholar] [CrossRef]
- Gao, F.; Wang, Z.; Ji, P.; Cheng, J.P. CO2 absorption by DBU-based protic ionic liquids: Basicity of anion dictates the absorption capacity and mechanism. Front. Chem. 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Zhu, X.; Song, M.; Xu, Y. DBU-Based Protic Ionic Liquids for CO2 Capture. ACS Sustain. Chem. Eng. 2017, 5, 8192–8198. [Google Scholar] [CrossRef]
- Zhu, X.; Song, M.; Ling, B.; Wang, S.; Luo, X. The Highly Efficient Absorption of CO2 by a Novel DBU Based Ionic Liquid. J. Solution Chem. 2020, 49, 257–271. [Google Scholar] [CrossRef]
- Chandra, G.; Lappert, M.F. Reactions of amino-derivatives of transition-metals. Inorg. Nucl. Chem. Lett. 1965, 1, 83–84. [Google Scholar] [CrossRef]
- Horley, G.A.; Mahon, M.F.; Molloy, K.C.; Haycock, P.W.; Myers, C.P. Synthesis and Characterization of Novel Homoleptic N,N -Dialkylcarbamato Complexes of Antimony: Precursors for the Deposition of Antimony Oxides. Inorg. Chem. 2002, 41, 5052–5058. [Google Scholar] [CrossRef]
- Chandra, G.; Jenkins, A.D.; Lappert, M.F.; Srivastava, R.C. Amido-derivatives of metals and metalloids. Part X. Reactions of titanium(IV), zirconium(IV), and hafnium(IV) amides with unsaturated substrates, and some related experiments with amides of boron, silicon, germanium, and tin(IV). J. Chem. Soc. A Inorg. Phys. Theor. 1970, 2550. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Extine, M. Tris(dimethylaminato)tris(N,N-dimethylcarbamato)tungsten(VI). Product of the remarkable reaction between hexakis(dimethylaminato)tungsten and carbon dioxide. J. Am. Chem. Soc. 1974, 96, 6214–6216. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Extine, M.W. Reactions of transition metal-nitrogen. sigma. bonds. 4. Mechanistic studies of carbon dioxide insertion and carbon dioxide exchange reactions involving early transition metal dimethylamido and N,N-dimethylcarbamato compounds. J. Am. Chem. Soc. 1977, 99, 792–802. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Extine, M.W. Reactions of transition metal-nitrogen. sigma. bonds. 3. Early transition metal N,N-dimethylcarbamates. Preparation, properties, and carbon dioxide exchange reactions. J. Am. Chem. Soc. 1977, 99, 782–792. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Cotton, F.A.; Extine, M.W.; Rideout, D.C. Reactions of transition-metal-nitrogen.sigma.-bonds. 5. Carbonation of tetrakis(diethylamido)chromium(IV) to yield binuclear chromium(III) and -(II) carbamato complexes. Inorg. Chem. 1978, 17, 3536–3540. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Haitko, D.A.; Murillo, C.A. New. sigma.-ethyl compounds of dimolybdenum (M.tplbond.M) and evidence for dinuclear reductive elimination with a concomitant metal-metal triple to quadruple bond transformation: Et-M. tplbond. M-Et. fwdarw. M::M + C2H4 + C2. J. Am. Chem. Soc. 1978, 100, 6262–6263. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Extine, M. Reactions of transition metal-nitrogen. sigma.-bonds. II. Pentakis(N,N-dimethylcarbamato)niobium(V) and its facile exchange reaction with carbon dioxide. J. Am. Chem. Soc. 1975, 97, 1623–1625. [Google Scholar] [CrossRef]
- Forte, C.; Pampaloni, G.; Pinzino, C.; Renili, F. N,N-Dialkylcarbamato derivatives of niobium and tantalum as precursors to metal-functionalized silica surfaces. Inorg. Chim. Acta 2011, 365, 251–255. [Google Scholar] [CrossRef]
- Bagnall, K.W.; Yanir, E. Thorium(IV) and uranium(IV) carbamates. J. Inorg. Nucl. Chem. 1974, 36, 777–779. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Mulvey, R.E.; Oliver, D.E.; Robertson, S.D. Lithium and aluminium carbamato derivatives of the utility amide 2,2,6,6-tetramethylpiperidide. Dalton Trans. 2010, 39, 6190–6197. [Google Scholar] [CrossRef]
- Bayer, U.; Werner, D.; Maichle-Mössmer, C.; Anwander, R. Effective and Reversible Carbon Dioxide Insertion into Cerium Pyrazolates. Angew. Chem. Int. Ed. 2020, 59, 5830–5836. [Google Scholar] [CrossRef]
- Cosham, S.D.; Hill, M.S.; Horley, G.A.; Johnson, A.L.; Jordan, L.; Molloy, K.C.; Stanton, D.C. Synthesis and materials chemistry of bismuth Tris-(di-i-propylcarbamate): Deposition of photoactive Bi2O3 thin films. Inorg. Chem. 2014, 53, 503–511. [Google Scholar] [CrossRef]
- Stewart, C.A.; Dickie, D.A.; Tang, Y.; Kemp, R.A. Insertion reactions of CO2, OCS, and CS2 into the Sn-N bonds of (Me2N)2Sn: NMR and X-ray structural characterization of the products. Inorg. Chim. Acta. 2011, 376, 73–79. [Google Scholar] [CrossRef]
- Calderazzo, F.; Dell’Amico, G.; Netti, R.; Pasquali, M. Dialkylcarbamato Complexes of Transition Elements. 1. A New Method for the Synthesis of N,N-Dialkylcarbamato and N,N-Dialkyldithiocarbamato Complexes of Uranium(IV). Inorg. Chem. 1978, 17, 471–473. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Calderazzo, F.; Dell’Innocenti, M.; Guldenpfennig, B.; Ianelli, S.; Pelizzi, G.; Robino, P. N,N-Dialkylcarbamates of Silicon and Aluminium. Gazz. Chim. Ital. 1993, 123, 283–288. [Google Scholar]
- Hayatifar, M.; Forte, C.; Pampaloni, G.; Kissin, Y.V.; Maria Raspolli Galletti, A.; Zacchini, S. Titanium complexes bearing carbamato ligands as catalytic precursors for propylene polymerization reactions. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4095–4102. [Google Scholar] [CrossRef]
- Albinati, A.; Carraro, M.L.; Gross, S.; Rancan, M.; Rizzato, S.; Tondello, E.; Venzo, A. Synthesis and Characterisation of a New Cu(O2CNAllyl2)2 Carbamato Complex and an Unusual Polymeric CuI Complex [CuI2Cl4(NHAllyl2)4]n: New Insigh. Eur. J. Inorg. Chem. 2009, 35, 5346–5351. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F. N,N-Di-iso-propylcarbamato Complexes of Boron. Inorg. Chem. 2008, 47, 5372–5376. [Google Scholar] [CrossRef] [PubMed]
- Levashov, A.S.; Buryi, D.S.; Latypova, A.R. Indium tris(N,N-diethylcarbamate), Method of Its Production and Obtaining of Films of Indium Oxide on Its Basis. 2018. RU 2649147 C1 20180330. Available online: https://www.elibrary.ru/item.asp?id=38150566 (accessed on 3 July 2020).
- Belli Dell’Amico, D.; Labella, L.; Marchetti, F.; Mastrorilli, P.; Samaritani, S.; Todisco, S. Oxidation by dioxygen of manganese(II) and iron(II) complexes. Polyhedron 2013, 65, 275–281. [Google Scholar] [CrossRef]
- Baisch, U.; Dell’ Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F.; Vitali, D. Reaction of a tetranuclear N,N-di-iso-propylcarbamato complex of cerium(III) with dioxygen: Synthesis and X-ray characterization of both the oxidation product and its precursor. J. Mol. Catal. A Chem. 2003, 204–205, 259–265. [Google Scholar] [CrossRef]
- Baisch, U.; Dell’Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F.; Merigo, A. N,N-dialkylcarbamato lanthanide complexes, a series of isotypical coordination compounds. Eur. J. Inorg. Chem. 2004, 6, 1219–1224. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Biagini, P.; Chiaberge, S.; Falchi, L.; Labella, L.; Lezzerini, M.; Marchetti, F.; Samaritani, S. Partial and exhaustive hydrolysis of lanthanide N,N-dialkylcarbamato complexes. A viable access to lanthanide mixed oxides. Polyhedron 2015, 102, 452–461. [Google Scholar] [CrossRef]
- Pineda, E.M.; Lan, Y.; Fuhr, O.; Wernsdorfer, W.; Ruben, M. Exchange-bias quantum tunnelling in a CO2-based Dy4-single molecule magnet. Chem. Sci. 2017, 8, 1178–1185. [Google Scholar] [CrossRef]
- Bradley, D.C.; Thomas, I.M. Metallo-Organic compounds containing metal-nitrogen bonds: Part III. Dialkylamino compounds of tantalum. Can. J. Chem. 1962, 40, 1355–1360. [Google Scholar] [CrossRef]
- Bradley, D.C.; Thomas, I.M. Metallo-Organic compounds containing metal-nitrogen bonds: Part II. Some dialkylamino derivatives of Nb(V) and Nb(IV). Can. J. Chem. 1962, 40, 449–454. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Farnocchi, S.; Labella, L.; Marchetti, F. The NHR2/CO2 system as a metal ion extraction reagent from aqueous solution into hydrocarbons: Copper(II) and zinc(II). Inorg. Chem. Commun. 2002, 5, 848–852. [Google Scholar] [CrossRef]
- Armelao, L.; Belli Dellamico, D.; Biagini, P.; Bottaro, G.; Chiaberge, S.; Falvo, P.; Labella, L.; Marchetti, F.; Samaritani, S. Preparation of N,N -dialkylcarbamato lanthanide complexes by extraction of lanthanide ions from aqueous solution into hydrocarbons. Inorg. Chem. 2014, 53, 4861–4871. [Google Scholar] [CrossRef] [PubMed]
- Belli Dell’Amico, D.; De Sanctis, M.; Ishak, R.; Dolci, S.; Labella, L.; Lezzerini, M.; Marchetti, F. Cerium(III) N,N-dibutylcarbamate as precursor to nanocrystalline cerium dioxide. Polyhedron 2015, 99, 170–176. [Google Scholar] [CrossRef]
- Armelao, L.; Belli Dellamico, D.; Bellucci, L.; Bottaro, G.; Labella, L.; Marchetti, F.; Samaritani, S. Smart Grafting of Lanthanides onto Silica via N,N-Dialkylcarbamato Complexes. Inorg. Chem. 2016, 55, 939–947. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Biagini, P.; Bongiovanni, G.; Chiaberge, S.; Di Giacomo, A.; Labella, L.; Marchetti, F.; Marra, G.; Mura, A.; Quochi, F.; et al. A convenient preparation of nano-powders of Y2O3, Y3Al5O12 and Nd:Y3Al5O12 and study of the photoluminescent emission properties of the neodymium doped oxide. Inorg. Chim. Acta. 2018, 470, 149–157. [Google Scholar] [CrossRef]
- Belforte, A.; Calderazzo, F. Formation of alkylurethanes from carbon dioxide by regioselective O-alkylation of alkali-metal N,N-diethylcarbamates in the presence of complexing agents. J. Chem. Soc. Dalt. Trans. 1989, 5, 1007–1009. [Google Scholar] [CrossRef]
- Belforte, A.; Dell’Amico, D.B.; Calderazzo, F. Incorporation and deoxygenation of carbon dioxide: A metal-assisted facile conversion of carbon dioxide and primary amines to isocyanates. Chem. Ber. 1988, 121, 1891–1897. [Google Scholar] [CrossRef]
- Klunker, J.; Biedermann, M.; Schäfer, W.; Hartung, H. N,N-Dimethylcarbamato-Komplexe von Kupfer und Zink. Z. Anorg. Allg. Chem. 1998, 624, 1503–1508. [Google Scholar]
- Kim, K.A.; Cha, J.R.; Yun, S.W.; Gong, M.S. Preparation of zinc oxide nanoparticles at low temperature using new organometallic zinc carbamate precursor. Bull. Korean Chem. Soc. 2015, 36, 1426–1432. [Google Scholar] [CrossRef]
- Alessio, R.; Dell’Amico, D.B.; Calderazzo, F.; Englert, U.; Guarini, A.; Labella, L.; Strasser, P. N,N-Dialkylcarbamato Complexes of the d10 cations of copper, silver, and gold. Helv. Chim. Acta. 1998, 81, 219–230. [Google Scholar] [CrossRef]
- Tyler Caudle, M. Carboxylate-shift-like dynamic equilibrium in bromomagnesium diethylcarbamate. Inorg. Chem. 1999. [Google Scholar] [CrossRef]
- Srivastava, R.S.; Singh, G.; Nakano, M.; Osakada, K.; Ozawa, F.; Yamamoto, A. Synthesis, characterization, and carbonylation reactions of methylpalladium amide, carbamate, and alkyl carbonate complexes. J. Organomet. Chem. 1993, 451, 221–229. [Google Scholar] [CrossRef]
- Ozawa, F.; Ito, T.; Yamamoto, A. Reactions of Carbon Dioxide with Palladium Ccomplexes. Synthesis and Characterization of Carbamato Complexes of Palladium(II). Chem. Lett. 1979, 8, 735–738. [Google Scholar] [CrossRef]
- Belforte, A.; Calderazzo, F.; Zanazzi, P.F. Synthesis and reactivity of N,N-dialkylcarbamato complexes of manganese(II). Crystal and molecular structure of [Mn6(O2CNEt2)12], a hexamer with four five-co-ordinated manganese(II) atoms. J. Chem. Soc. Dalt. Trans. 1988, 2921–2926. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F.; Mazzoncini, I. N,N-dimethylcarbamato complexes of zinc. Inorg. Chim. Acta 2006, 359, 3371–3374. [Google Scholar] [CrossRef]
- Gauld, R.M.; Kennedy, A.R.; McLellan, R.; Barker, J.; Reid, J.; Mulvey, R.E. Diverse outcomes of CO2 fixation using alkali metal amides including formation of a heterobimetallic lithium-sodium carbamato-anhydride via lithium-sodium bis-hexamethyldisilazide. Chem. Commun. 2019, 55, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Bortoluzzi, M.; Zacchini, S.; Gabbani, A.; Pineider, F.; Marchetti, F.; Pampaloni, G. Synthesis and Structural Characterization of Non-Homoleptic Carbamato Complexes of V V and W VI and Their Facile Implantation onto Silica Surfaces. Eur. J. Inorg. Chem. 2018, 10, 1176–1184. [Google Scholar] [CrossRef]
- Calderazzo, F.; Pampaloni, G.; Sperrlea, M.; Englert, U. Electron-and Ligand-Transfer Reactions Involving N,N-Dialkylcarbamates. Synthesis and Molecular Structure of V2[η5-(C5H5)]2[O2CN(C2H5)2]4. Zeitschrift für Naturforsch. B 1992, 47, 389–394. [Google Scholar] [CrossRef]
- Arimondo, P.B.; Calderazzo, F.; Englert, U.; Maichle-Moessmer, C.; Pampaloni, G.; Straehle, J. Preparation and characterization of dialkylcarbamato derivatives of niobium and tantalum. J. Chem. Soc. Dalt. Trans. 1996, 3, 311–319. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Calderazzo, F.; Gingl, F.; Labella, L.; Straehle, J. N,N-dialkylcarbamato complexes of chromium(III). Crystal and molecular structure of two non-homoleptic products of chromium(III), Cr3(µ3-O)(O2CNEt2)6Cl(NHEt2) and Cr2(O2CNiPr2)5Cl. Gazz. Chim. Ital. 1994, 124, 375–380. [Google Scholar]
- Arimondo, P.B.; Calderazzo, F.; Hiemeyer, R.; Maichle-Mössmer, C.; Marchetti, F.; Pampaloni, G.; Strähle, J. Synthesis and Crystal Structure of a Self-Assembled, Octanuclear Oxo-Tantalum(V) Derivative Containing the First Example of a Transition Metal M8(μ-O)12 Cage. Inorg. Chem. 1998, 37, 5507–5511. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, M.; Ghini, F.; Hayatifar, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Oxido- and sulfidoniobium(V) N,N-diethylcarbamates: Synthesis, characterization and DFT study. Eur. J. Inorg. Chem. 2013, 3112–3118. [Google Scholar] [CrossRef]
- Forte, C.; Hayatifar, M.; Pampaloni, G.; Galletti, A.M.R.; Renili, F.; Zacchini, S. Ethylene polymerization using novel titanium catalytic precursors bearing N,N-dialkylcarbamato ligands. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 3338–3345. [Google Scholar] [CrossRef]
- Straessler, N.A.; Caudle, M.T.; Groy, T.L. Tetrakis (N,N-diethylcarbamato)titanium(IV). Acta. Crystallogr. Sect. E Struct. Rep. Online 2008, 64, m48. [Google Scholar] [CrossRef] [PubMed]
- Dell’Amico, D.B.; Calderazzo, F.; Ianelli, S.; Labella, L.; Marchetti, F.; Pelizzi, G. Stepwise substitution of N,N-di-isopropylcarbamato groups of Ti(O2CNiPr2)4 by triphenylsilanol leading from eight- to six- and four-co-ordinated titanium(IV). J. Chem. Soc. Dalt. Trans. 2000, 4339–4342. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Cotton, F.A.; Extine, M.W.; Stults, B.R. The tungsten-tungsten triple bond. 6. Hexakis(N,N-dimethylcarbamato)ditungsten and dimethyltetrakis(N,N-diethylcarbamato)ditungsten. Structures and dynamical solution behavior. Inorg. Chem. 1977, 16, 603–611. [Google Scholar] [CrossRef]
- Belforte, A.; Belli Dell’Amico, D.; Calderazzo, F.; Devillers, M.; Englert, U. Chromium(II) and molybdenum(II) N,N-diethylcarbamato complexes from metal halides/carbon dioxide/diethylamine: Crystal and molecular structure of the quadruply metal-metal-bonded Mo2(O2CNEt2)4. Inorg. Chem. 1993, 32, 2282–2286. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. Reaction of alkali-metal tetraphenylborates with amines in the presence of CO2: A new easy way to aliphatic and aromatic alkali-metal carbamates. J. Chem. Soc. Dalt. Trans. 1995, 20, 3359. [Google Scholar] [CrossRef]
- Esparza-Ruiz, A.; Herrmann, C.; Chen, J.; Patrick, B.O.; Polishchuk, E.; Orvig, C. Synthesis and in vitro anticancer activity of ferrocenyl-aminoquinoline-carboxamide conjugates. Inorg. Chim. Acta. 2012, 393, 276–283. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Reichert, W.W. Bis(dimethylamido)tetrakis(N,N-dimethylcarbamato)dimolybdenum. Inorg. Chem. 1978, 17, 767–769. [Google Scholar] [CrossRef]
- Mendiratta, A.; Cummins, C.C.; Cotton, F.A.; Ibragimov, S.A.; Murillo, C.A.; Villagrán, D. A Diamagnetic Dititanium(III) Paddlewheel Complex with No Direct Metal−Metal Bond. Inorg. Chem. 2006, 45, 4328–4330. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Bortoluzzi, M.; Zacchini, S.; Marchetti, F.; Pampaloni, G. Structural Characterization of a Fluorido-Amide of Niobium, and Facile CO2 Incorporation Affording a Fluorido-Carbamate. Eur. J. Inorg. Chem. 2018, 8, 999–1006. [Google Scholar] [CrossRef]
- Habereder, T.; Nöth, H.; Paine, R.T. Synthesis and Reactivity of New Bis(tetramethylpiperidino)(phosphanyl)alumanes. Eur. J. Inorg. Chem. 2007, 27, 4298–4305. [Google Scholar] [CrossRef]
- Normand, A.T.; Daniliuc, C.G.; Wibbeling, B.; Kehr, G.; Le Gendre, P.; Erker, G. Phosphido- and Amidozirconocene Cation-Based Frustrated Lewis Pair Chemistry. J. Am. Chem. Soc. 2015, 137, 10796–10808. [Google Scholar] [CrossRef] [PubMed]
- McNevin, M.J.; Hagadorn, J.R. Dititanium Complexes of Preorganized Binucleating Bis(amidinates). Inorg. Chem. 2004, 43, 8547–8554. [Google Scholar] [CrossRef]
- Chakraborty, S.; Blacque, O.; Berke, H. Ligand assisted carbon dioxide activation and hydrogenation using molybdenum and tungsten amides. Dalton Trans. 2015, 44, 6560–6570. [Google Scholar] [CrossRef]
- Dickie, D.A.; Parkes, M.V.; Kemp, R.A. Insertion of Carbon Dioxide into Main-Group Complexes: Formation of the [N(CO2)3]3− Ligand. Angew. Chem. Int. Ed. 2008, 47, 9955–9957. [Google Scholar] [CrossRef]
- Hartwig, J.F.; Bergman, R.G.; Andersen, R.A. Insertion reactions of carbon monoxide and carbon dioxide with ruthenium benzyl, arylamido, and aryloxide complexes: A comparison of the reactivity of ruthenium-carbon, ruthenium-nitrogen, and ruthenium-oxygen bonds. J. Am. Chem. Soc. 1991, 113, 6499–6508. [Google Scholar] [CrossRef]
- Park, S.; Rheingold, A.L.; Roundhill, D.M. Synthesis and reaction chemistry of monomeric and dimeric amide complexes of platinum(II). Organometallics 1991, 10, 615–623. [Google Scholar] [CrossRef]
- Rahim, M.; White, C.; Rheingold, A.L.; Ahmed, K.J. Mononuclear arylamido complexes of iridium(I). Organometallics 1993, 12, 2401–2403. [Google Scholar] [CrossRef]
- Dobereiner, G.E.; Wu, J.; Manas, M.G.; Schley, N.D.; Takase, M.K.; Crabtree, R.H.; Hazari, N.; Maseras, F.; Nova, A. Mild, Reversible Reaction of Iridium(III) Amido Complexes with Carbon Dioxide. Inorg. Chem. 2012, 51, 9683–9693. [Google Scholar] [CrossRef] [PubMed]
- Truscott, B.J.; Nelson, D.J.; Slawin, A.M.Z.; Nolan, S.P. CO2 fixation employing an iridium(I)-hydroxide complex. Chem. Commun. 2014, 50, 286–288. [Google Scholar] [CrossRef]
- Schmeier, T.J.; Nova, A.; Hazari, N.; Maseras, F. Synthesis of PCP-Supported Nickel Complexes and their Reactivity with Carbon Dioxide. Chem. Eur. J. 2012, 18, 6915–6927. [Google Scholar] [CrossRef]
- Mousa, A.H.; Bendix, J.; Wendt, O.F. Synthesis, Characterization, and Reactivity of PCN Pincer Nickel Complexes. Organometallics 2018, 37, 2581–2593. [Google Scholar] [CrossRef]
- Cámpora, J.; Matas, I.; Palma, P.; Álvarez, E.; Graiff, C.; Tiripicchio, A. Monomeric Alkoxo and Amido Methylnickel(II) Complexes. Synthesis and Heterocumulene Insertion Chemistry. Organometallics 2007, 26, 3840–3849. [Google Scholar] [CrossRef]
- Hao, J.; Vabre, B.; Mougang-Soumé, B.; Zargarian, D. Small Molecule Activation by POCsp3OP-Nickel Complexes. Chem. -Eur. J. 2014, 20, 12544–12552. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, K.; Lee, Y. Synthesis and characterization of a four-coordinate nickel carbamato species (MeSiPiPr2)Ni(OC(O)NHMes) generated from the reaction of (MeSiPiPr2)Ni(NHMes) with CO2. Inorg. Chim. Acta 2017, 460, 55–62. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.-E.; Park, K.; Lee, Y. A Silyl-Nickel Moiety as a Metal–Ligand Cooperative Site. Inorg. Chem. 2019, 58, 11534–11545. [Google Scholar] [CrossRef]
- Seul, J.-M.; Park, S. Palladium(II) p-Tolylamide and Reaction with CO2 to Generate a Carbamato Derivative. Bull. Korean Chem. Soc. 2010, 31, 3745–3748. [Google Scholar] [CrossRef]
- Comanescu, C.C.; Iluc, V.M. E-H (E = N, O) bond activation by a nucleophilic palladium carbene. Polyhedron 2018, 143, 176–183. [Google Scholar] [CrossRef]
- Johnson, M.W.; Shevick, S.L.; Toste, F.D.; Bergman, R.G. Preparation and reactivity of terminal gold(I) amides and phosphides. Chem. Sci. 2013, 4, 1023–1027. [Google Scholar] [CrossRef]
- Tang, Y.; Kassel, W.S.; Zakharov, L.N.; Rheingold, A.L.; Kemp, R.A. Insertion reactions of carbon dioxide into Zn-N bonds: Syntheses and structures of tetrameric and dimeric alkylzinc carbamato complexes. Inorg. Chem. 2005, 44, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.A.; O’Brien, P.; Motevalli, M.; Abrahams, I. The adoption of the beryllium acetate structural motif in zinc oxycarbamates, oxythiocarbamates and oxythiophosphinates. Polyhedron 2006, 25, 241–250. [Google Scholar] [CrossRef]
- Domide, D.; Kaifer, E.; Mautz, J.; Walter, O.; Behrens, S.; Himmel, H. Synthesis and Characterisation of Some New Zinc Carbamate Complexes Formed by CO2 Fixation and Their Use as Precursors for ZnO Particles under Mild Conditions. Eur. J. Inorg. Chem. 2008, 20, 3177–3185. [Google Scholar] [CrossRef]
- Kahnes, M.; Görls, H.; Westerhausen, M. Synthesis of Dimeric Methylzinc N,N-Bis(2-pyridylmethyl)carbamate via Addition of CO2 to a Methylzinc Amide. Z. Anorg. Allg. Chem. 2011, 637, 397–400. [Google Scholar] [CrossRef]
- Webster, C.L.; Langeslay, R.R.; Ziller, J.W.; Evans, W.J. Synthetic Utility of Tetrabutylammonium Salts in Uranium Metallocene Chemistry. Organometallics 2016, 35, 520–527. [Google Scholar] [CrossRef]
- Higgins Frey, J.A.; Cloke, F.G.N.; Roe, S.M. Synthesis and Reactivity of a Mixed-Sandwich Uranium(IV) Primary Amido Complex. Organometallics 2015, 34, 2102–2105. [Google Scholar] [CrossRef]
- Schmidt, A.-C.; Heinemann, F.W.; Maron, L.; Meyer, K. A Series of Uranium (IV, V, VI) Tritylimido Complexes, Their Molecular and Electronic Structures and Reactivity with CO2. Inorg. Chem. 2014, 53, 13142–13153. [Google Scholar] [CrossRef]
- Matson, E.M.; Fanwick, P.E.; Bart, S.C. Formation of Trivalent U–C, U–N, and U–S Bonds and Their Reactivity toward Carbon Dioxide and Acetone. Organometallics 2011, 30, 5753–5762. [Google Scholar] [CrossRef]
- Bart, S.C.; Anthon, C.; Heinemann, F.W.; Bill, E.; Edelstein, N.M.; Meyer, K. Carbon Dioxide Activation with Sterically Pressured Mid- and High-Valent Uranium Complexes. J. Am. Chem. Soc. 2008, 130, 12536–12546. [Google Scholar] [CrossRef]
- Harris, L.A.M.; Coles, M.P.; Fulton, J.R. Synthesis and reactivity of tin amide complexes. Inorg. Chim. Acta 2011, 369, 97–102. [Google Scholar] [CrossRef]
- Feier-Iova, O.; Linti, G. Synthesis and Structure of a Carbamato-bridgded Digallyl-ferrocenophane—Fixation of Carbon Dioxide with Aminogallanes. Z. Anorg. Allg. Chem. 2008, 634, 559–564. [Google Scholar] [CrossRef]
- Uhl, W.; Willeke, M.; Hepp, A.; Pleschka, D.; Layh, M. A Dimeric Gallium Hydrazide as an Active Lewis Pair—Complexation and Activation of Me2GaH and Various Heterocumulenes. Z. Anorg. Allg. Chem. 2017, 643, 387–397. [Google Scholar] [CrossRef]
- Hill, M.R.; Jensen, P.; Russell, J.J.; Lamb, R.N. Synthesis and properties of Zn-Mg heterobimetallic carbamates. Crystal structures of the first reported single source precursors for ZnxMg1-xO thin films. J. Chem. Soc. Dalt. Trans. 2008, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Vummaleti, S.V.C.; Talarico, G.; Nolan, S.P.; Cavallo, L.; Poater, A. Mechanism of CO2 Fixation by Ir I-X Bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 2015, 4653–4657. [Google Scholar] [CrossRef]
- Truscott, B.J.; Kruger, H.; Webb, P.B.; Bühl, M.; Nolan, S.P. The Mechanism of CO2 Insertion into Iridium(I) Hydroxide and Alkoxide Bonds: A Kinetics and Computational Study. Chem. -Eur. J. 2015, 21, 6930–6935. [Google Scholar] [CrossRef]
- Vadivelu, P.; Suresh, C.H. Metal- and Ligand-Assisted CO2 Insertion into Ru–C, Ru–N, and Ru–O Bonds of Ruthenium(II) Phosphine Complexes: A Density Functional Theory Study. Inorg. Chem. 2015, 54, 502–512. [Google Scholar] [CrossRef]
- Chu, J.; Lu, E.; Liu, Z.; Chen, Y.; Leng, X.; Song, H. Reactivity of a Scandium Terminal Imido Complex Towards Unsaturated Substrates. Angew. Chem. Int. Ed. 2011, 50, 7677–7680. [Google Scholar] [CrossRef]
- Boyd, C.L.; Clot, E.; Guiducci, A.E.; Mountford, P. Pendant Arm Functionalized Benzamidinate Titanium Imido Compounds: Experimental and Computational Studies of Their Reactions with CO2. Organometallics 2005, 24, 2347–2367. [Google Scholar] [CrossRef]
- Dubberley, S.R.; Friedrich, A.; Willman, D.A.; Mountford, P.; Radius, U. Synthesis and Reactivity of Calix[4]arene-Supported Group 4 Imido Complexes. Chem. -Eur. J. 2003, 9, 3634–3654. [Google Scholar] [CrossRef]
- Guiducci, A.E.; Boyd, C.L.; Clot, E.; Mountford, P. Reactions of cyclopentadienyl-amidinate titanium imido compounds with CO2: Cycloaddition-extrusion vs. cycloaddition-insertion. Dalton Trans. 2009, 30, 5960. [Google Scholar] [CrossRef] [PubMed]
- Mindiola, D.J.; Waterman, R.; Iluc, V.M.; Cundari, T.R.; Hillhouse, G.L. Carbon–Hydrogen Bond Activation, C–N Bond Coupling, and Cycloaddition Reactivity of a Three-Coordinate Nickel Complex Featuring a Terminal Imido Ligand. Inorg. Chem. 2014, 53, 13227–13238. [Google Scholar] [CrossRef] [PubMed]
- Goodner, S.J.; Grünwald, A.; Heinemann, F.W.; Munz, D. Carbon Dioxide Activation by a Palladium Terminal Imido Complex. Aust. J. Chem. 2019, 72, 900. [Google Scholar] [CrossRef]
- Glueck, D.S.; Wu, J.; Hollander, F.J.; Bergman, R.G. Monomeric (pentamethylcyclopentadienyl)iridium imido compounds: Synthesis, structure, and reactivity. J. Am. Chem. Soc. 1991, 113, 2041–2054. [Google Scholar] [CrossRef]
- Kinauer, M.; Diefenbach, M.; Bamberger, H.; Demeshko, S.; Reijerse, E.J.; Volkmann, C.; Würtele, C.; Van Slageren, J.; De Bruin, B.; Holthausen, M.C.; et al. An iridium(III/IV/V) redox series featuring a terminal imido complex with triplet ground state. Chem. Sci. 2018, 9, 4325–4332. [Google Scholar] [CrossRef]
- Falcone, M.; Chatelain, L.; Mazzanti, M. Nucleophilic Reactivity of a Nitride-Bridged Diuranium(IV) Complex: CO2 and CS2 Functionalization. Angew. Chem. Int. Ed. 2016, 55, 4074–4078. [Google Scholar] [CrossRef]
- Falcone, M.; Poon, L.N.; Fadaei Tirani, F.; Mazzanti, M. Reversible Dihydrogen Activation and Hydride Transfer by a Uranium Nitride Complex. Angew. Chem. Int. Ed. 2018, 57, 3697–3700. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Lobkovsky, E.; Chirik, P.J. Nitrogen–Carbon Bond Formation from N2 and CO2 Promoted by a Hafnocene Dinitrogen Complex Yields a Substituted Hydrazine. Angew. Chem. Int. Ed. 2007, 46, 2858–2861. [Google Scholar] [CrossRef]
- Knobloch, D.J.; Toomey, H.E.; Chirik, P.J. Carboxylation of an ansa -Zirconocene Dinitrogen Complex: Regiospecific Hydrazine Synthesis from N2 and CO2. J. Am. Chem. Soc. 2008, 130, 4248–4249. [Google Scholar] [CrossRef]
- Ma, X.; Tang, Y.; Lei, M. Mechanistic Studies on the Carboxylation of Hafnocene and ansa -Zirconocene Dinitrogen Complexes with CO2. Organometallics 2013, 32, 7077–7082. [Google Scholar] [CrossRef]
- McCowan, C.S.; Buss, C.E.; Young, V.G.; McDonnell, R.L.; Caudle, M.T. Chloro(diethylamino)tris(μ-diethylcarbamato)dizinc(II): An example of the generality of the threefold paddlewheel structure in carbamatozinc chemistry. Acta. Crystallogr. Sect. E Struct. Rep. Online 2004, 60, 285–287. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Englert, U.; Labella, L.; Marchetti, F.; Specos, M. New N,N-diisopropylcarbamato complexes of ruthenium(II) as catalytic precursors for olefin hydrogenation. Eur. J. Inorg. Chem. 2004, 19, 3938–3945. [Google Scholar] [CrossRef]
- Mochizuki, K.; Kondou, H.; Ando, K.; Kawasumi, T.; Takahashi, J. Degradation of urea mediated by dinickel(II) complexes with the binucleating ligand N,N′-bis[2-(N,N-dimethyl)aminoethyl]-N,N′-dimethyl-1,3-diamino-2-hydroxypropane (HL). Inorg. Chim. Acta 2016, 441, 50–56. [Google Scholar] [CrossRef]
- Lozan, V.; Holldorf, J.; Kersting, B. Preparation and characterization of macrocyclic dinickel complexes coligated by monoalkyl- and dialkylcarbamates. Inorg. Chim. Acta 2009, 362, 793–798. [Google Scholar] [CrossRef]
- Calucci, L.; Forte, C.; Pampaloni, G.; Pinzino, C.; Renili, F. Chemical implantation of Group 4 cations on silica via cyclopentadienyl- and N,N-dialkylcarbamato derivatives. Inorg. Chim. Acta 2010, 363, 33–40. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, F.-S.; Wang, Y.-Y. Two {Dy2} single-molecule magnets formed via an in situ reaction by capturing CO2 from atmosphere under ambient conditions. Dalton Trans. 2017, 46, 1753–1756. [Google Scholar] [CrossRef]
- Neis, C.; Weyhermüller, T.; Bill, E.; Stucky, S.; Hegetschweiler, K. Carbamates of Polyamines–Versatile Building Blocks for the Construction of Polynuclear Metal Complexes. Eur. J. Inorg. Chem. 2008, 7, 1019–1021. [Google Scholar] [CrossRef]
- Bramsen, F.; Bond, A.D.; McKenzie, C.J.; Hazell, R.G.; Moubaraki, B.; Murray, K.S. Self-Assembly of the Octanuclear Cluster [Cu8(OH)10(NH2(CH2)2CH3)12]6+ and the One-Dimensional N-Propylcarbamate-Linked Coordination Polymer {[Cu(O2CNH(CH2)2CH3(NH2(CH2)2CH3)3](ClO4)}n. Chem. Eur. J. 2005, 11, 825–831. [Google Scholar] [CrossRef]
- García-España, E.; Gaviña, P.; Latorre, J.; Soriano, C.; Verdejo, B. CO2 Fixation by Copper(II) Complexes of a Terpyridinophane Aza Receptor. J. Am. Chem. Soc. 2004, 126, 5082–5083. [Google Scholar] [CrossRef]
- Vo, T.T.; Parrish, D.A.; Shreeve, J.M. 1,1-Diamino-2,2-dintroethene (FOX-7) in Copper and Nickel Diamine Complexes and Copper FOX-7. Inorg. Chem. 2012, 51, 1963–1968. [Google Scholar] [CrossRef]
- Kalia, S.B.; Kumar, R.; Bharti, M.; Christopher, J. Experimental investigations of thermal stability of some morpholinecarbamic acid complexes of copper(II) and zinc(II). J. Therm. Anal. Calorim. 2017, 127, 1291–1306. [Google Scholar] [CrossRef]
- Bedeković, N.; Stilinović, V. Morpholine-N-carboxylate as a ligand in coordination chemistry–Syntheses and structures of three heteroleptic copper(ii) and zinc complexes. J. Mol. Struct. 2020, 1205, 127627. [Google Scholar] [CrossRef]
- Notni, J.; Schenk, S.; Görls, H.; Breitzke, H.; Anders, E. Formation of a Unique Zinc Carbamate by CO2 Fixation: Implications for the Reactivity of Tetra-Azamacrocycle Ligated Zn(II) Complexes. Inorg. Chem. 2008, 47, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.A.; Sava, D.F.; Nenoff, T.M. catena -Poly[zinc-tris(μ-dimethylcarbamato-κ2O:O′)-zinc-μ-(2-phenylbenzimidazolido-κ2 N:N′]. Acta. Crystallogr. Sect. E Struct. Rep. Online 2012, 68, m59–m60. [Google Scholar] [CrossRef]
- Masci, B.; Thuéry, P. A Tetrahomodioxacalix[6]arene as a Ditopic Ligand for Uranyl Ions with Carbonate or Carbamate Bridges. Supramol. Chem. 2003, 15, 101–108. [Google Scholar] [CrossRef]
- Anillo, A.; Dell’Amico, D.B.; Calderazzo, F.; Nardelli, M.; Pelizzi, G.; Rocchi, L. Dialkylcarbamato complexes of palladium(II). Crystal structure of trans-[Pd(O2CNEt2)2(NHEt2)2]. J. Chem. Soc. Dalt. Trans. 1991, 11, 2845. [Google Scholar] [CrossRef]
- El Rez, B.; Costes, J.-P.P.; Duhayon, C.; Vendier, L.; Sutter, J.-P.P. Structural determinations of carbamato-bridging ligands derived from atmospheric CO2 in 3d-4f complexes. Polyhedron 2015, 89, 213–218. [Google Scholar] [CrossRef]
- Yin, C.-L.; Hu, Z.-B.; Long, Q.-Q.; Wang, H.-S.; Li, J.; Song, Y.; Zhang, Z.-C.; Zhang, Y.-Q.; Pan, Z.-Q. Single molecule magnet behaviors of Zn4Ln2 (Ln = Dy III, Tb III) complexes with multidentate organic ligands formed by absorption of CO2 in air through in situ reactions. Dalton Trans. 2019, 48, 512–522. [Google Scholar] [CrossRef]
- Caudle, M.T.; Brennessel, W.W.; Young, V.G. Structural variability and dynamics in carboxylato- and carbamatomagnesium bromides. Relationship to the carboxylate shift. Inorg. Chem. 2005, 44, 3233–3240. [Google Scholar] [CrossRef]
- Dell’ Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F.; Martini, M.; Mazzoncini, I. N,N-Dimethylcarbamato derivatives of magnesium starting from the metal oxide. C. R. Chim. 2004, 7, 877–884. [Google Scholar] [CrossRef]
- Yin, S.-F.; Maruyama, J.; Yamashita, T.; Shimada, S. Efficient Fixation of Carbon Dioxide by Hypervalent Organobismuth Oxide, Hydroxide, and Alkoxide. Angew. Chem. Int. Ed. 2008, 47, 6590–6593. [Google Scholar] [CrossRef] [PubMed]
- Bloodworth, A.J.; Serlin, J. Organometallic oxides, alkoxides, and peroxides. Part IV. Reaction of phenylmercury(II) alkoxides, oxide, and hydroxide with organic isocyanates. J. Chem. Soc. Perkin Trans. 1 1973, 7, 261–267. [Google Scholar] [CrossRef]
- Edwards, A.J.; Elipe, S.; Esteruelas, M.A.; Lahoz, F.J.; Oro, L.A.; Valero, C. Synthesis and Reactivity of the Unusual Five-Coordinate Hydrido−Hydroxo Complex OsH(OH)(CO)(PiPr3)2. Organometallics 1997, 16, 3828–3836. [Google Scholar] [CrossRef]
- Giandomenico, C.M.; Abrams, M.J.; Murrer, B.A.; Vollano, J.F.; Rheinheimer, M.I.; Wyer, S.B.; Bossard, G.E.; Higgins, J.D. Carboxylation of Kinetically Inert Platinum(IV) Hydroxy Complexes. An Entr.acte.ee into Orally Active Platinum(IV) Antitumor Agents. Inorg. Chem. 1995, 34, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, R.; Zhang, J.; Chen, Z.; Zhou, X. Reactivity of lanthanocene hydroxides toward ketene, isocyanate, lanthanocene alkyl, and triscyclopentadienyllanthanide complexes. Inorg. Chem. 2006, 45, 5867–5877. [Google Scholar] [CrossRef]
- Xu, X.P.; Qi, R.P.; Xu, B.; Yao, Y.M.; Nie, K.; Zhang, Y.; Shen, Q. Synthesis, reactivity and structural characterization of lanthanide hydroxides stabilized by a carbon-bridged bis(phenolate) ligand. Polyhedron 2009, 28, 574–578. [Google Scholar] [CrossRef]
- Cuesta, L.; Gerbino, D.C.; Hevia, E.; Morales, D.; Navarro Clemente, M.E.; Pérez, J.; Riera, L.; Riera, V.; Miguel, D.; Del Río, I.; et al. Reactivity of Molybdenum and Rhenium Hydroxo-Carbonyl Complexes toward Organic Electrophiles. Chem. Eur. J. 2004, 10, 1765–1777. [Google Scholar] [CrossRef]
- Jackson, W.G.; McKeon, J.A.; Balahura, R.J. N -Methylmonothiocarbamatopentamminecobalt(III): Restricted C–N Bond Rotation and the Acid-Catalyzed O- to S-Bonded Rearrangement. Inorg. Chem. 2004, 43, 4889–4896. [Google Scholar] [CrossRef]
- Yao, C.; Chakraborty, P.; Aresu, E.; Li, H.; Guan, C.; Zhou, C.; Liang, L.-C.; Huang, K.-W. Monomeric nickel hydroxide stabilized by a sterically demanding phosphorus–nitrogen PN 3 P-pincer ligand: Synthesis, reactivity and catalysis. Dalton Trans. 2018, 47, 16057–16065. [Google Scholar] [CrossRef]
- Martínez-Prieto, L.M.; Palma, P.; Cámpora, J. Monomeric alkoxide and alkylcarbonate complexes of nickel and palladium stabilized with the iPr PCP pincer ligand: A model for the catalytic carboxylation of alcohols to alkyl carbonates. Dalton Trans. 2019, 48, 1351–1366. [Google Scholar] [CrossRef]
- Wilson, J.J.; Lippard, S.J. Synthesis, Characterization, and Cytotoxicity of Platinum(IV) Carbamate Complexes. Inorg. Chem. 2011, 50, 3103–3115. [Google Scholar] [CrossRef] [PubMed]
- Pichler, V.; Mayr, J.; Heffeter, P.; Dömötör, O.; Enyedy, É.A.; Hermann, G.; Groza, D.; Köllensperger, G.; Galanksi, M.; Berger, W.; et al. Maleimide-functionalised platinum(IV) complexes as a synthetic platform for targeted drug delivery. Chem. Commun. 2013, 49, 2249. [Google Scholar] [CrossRef] [PubMed]
- Pichler, V.; Göschl, S.; Meier, S.M.; Roller, A.; Jakupec, M.A.; Galanski, M.; Keppler, B.K. Bulky N,N-((Di)alkylethane-1,2-diamineplatinum(II) Compounds as Precursors for Generating Unsymmetrically Substituted Platinum(IV) Complexes. Inorg. Chem. 2013, 52, 8151–8162. [Google Scholar] [CrossRef]
- Zheng, Y.-R.R.; Suntharalingam, K.; Johnstone, T.C.; Yoo, H.; Lin, W.; Brooks, J.G.; Lippard, S.J. Pt(IV) prodrugs designed to bind non-covalently to human serum albumin for drug delivery. J. Am. Chem. Soc. 2014, 136, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Lu, Z.; Lang, W.H.; Guo, L.; Ma, C.G.; Sun, G.H. Binding of a potential anti-hepatoma drug cis,cis,trans-[Pt(NH3)2Cl2(O2CCH2CH2COOH)-(OCONHC16H33)] with serum albumin—Thermodynamic and conformational investigations. New J. Chem. 2015, 39, 9234–9241. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Yiu, S.-M.; Zhu, G. Mono- and di-bromo platinum(IV) prodrugs via oxidative bromination: Synthesis, characterization, and cytotoxicity. Dalton Trans. 2015, 44, 19918–19926. [Google Scholar] [CrossRef]
- Li, W.; Jiang, M.; Cao, Y.; Yan, L.; Qi, R.; Li, Y.; Jing, X. Turning Ineffective Transplatin into a Highly Potent Anticancer Drug via a Prodrug Strategy for Drug Delivery and Inhibiting Cisplatin Drug Resistance. Bioconjug. Chem. 2016, 27, 1802–1806. [Google Scholar] [CrossRef]
- Feng, B.; Zhou, F.; Xu, Z.; Wang, T.; Wang, D.; Liu, J.; Fu, Y.; Yin, Q.; Zhang, Z.; Yu, H.; et al. Versatile Prodrug Nanoparticles for Acid-Triggered Precise Imaging and Organelle-Specific Combination Cancer Therapy. Adv. Funct. Mater. 2016, 26, 7431–7442. [Google Scholar] [CrossRef]
- Mayr, J.; Heffeter, P.; Groza, D.; Galvez, L.; Koellensperger, G.; Roller, A.; Alte, B.; Haider, M.; Berger, W.; Kowol, C.R.; et al. An albumin-based tumor-targeted oxaliplatin prodrug with distinctly improved anticancer activity in vivo. Chem. Sci. 2017, 8, 2241–2250. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Takahashi, T.; Wada, A.; Funahashi, Y.; Ozawa, T.; Jitsukawa, K.; Masuda, H. Fixation of CO2 by Hydroxozinc(II) Complex with Pyridylamino Type Ligand. Chem. Lett. 2007, 36, 842–843. [Google Scholar] [CrossRef]
- Norris, M.R.; Flowers, S.E.; Mathews, A.M.; Cossairt, B.M. H2 Production Mediated by CO2 via Initial Reduction to Formate. Organometallics 2016, 35, 2778–2781. [Google Scholar] [CrossRef]
- Jayarathne, U.; Hazari, N.; Bernskoetter, W.H. Selective Iron-Catalyzed N -Formylation of Amines using Dihydrogen and Carbon Dioxide. ACS Catal. 2018, 8, 1338–1345. [Google Scholar] [CrossRef]
- Babu, T.; Sarkar, A.; Karmakar, S.; Schmidt, C.; Gibson, D. Multiaction Pt(IV) Carbamate Complexes Can Codeliver Pt(II) Drugs and Amine Containing Bioactive Molecules. Inorg. Chem. 2020, 59, 5182–5193. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Neumann, R. Coordination of Carbon Dioxide to the Lewis Acid Site of a Zinc-Substituted Polyoxometalate and Formation of an Adduct Using a Polyoxometalate-2,4,6-Trimethylpyridine Frustrated Lewis Pair. Eur. J. Inorg. Chem. 2018, 6, 791–794. [Google Scholar] [CrossRef]
- Cristóbal, C.; Hernández, Y.A.; López-Serrano, J.; Paneque, M.; Petronilho, A.; Poveda, M.L.; Salazar, V.; Vattier, F.; Álvarez, E.; Maya, C.; et al. Reactivity Studies of Iridium Pyridylidenes [TpMe2Ir(C6H5)2(C(CH)2C(R)NH] (R=H, Me, Ph). Chem. Eur. J. 2013, 19, 4003–4020. [Google Scholar] [CrossRef]
- Zhu, Y.; Smith, D.A.; Herbert, D.E.; Gatard, S.; Ozerov, O.V. C–H and C–O oxidative addition in reactions of aryl carboxylates with a PNP pincer-ligated Rh(I) fragment. Chem. Commun. 2012, 48, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.L.; Geary, J.; Ardon, Y.; Reese, M.S.; Philliber, M.A.; VanderLinden, R.T.; Saouma, C.T. Thermodynamic Analysis of Metal–Ligand Cooperativity of PNP Ru Complexes: Implications for CO2 Hydrogenation to Methanol and Catalyst Inhibition. J. Am. Chem. Soc. 2019, 141, 14317–14328. [Google Scholar] [CrossRef]
- Ueno, A.; Kayaki, Y.; Ikariya, T. Heterolysis of NH-indoles by bifunctional amido complexes and applications to carboxylation with carbon dioxide. Organometallics 2014, 33, 4479–4485. [Google Scholar] [CrossRef]
- Roth, C.E.; Dibenedetto, A.; Aresta, M. Synthesis and Characterization of Chloro- and Alkyliron Complexes with N-Donor Ligands and Their Reactivity towards CO2. Eur. J. Inorg. Chem. 2015, 30, 5066–5073. [Google Scholar] [CrossRef]
- Neumüller, B.; Esser, M.; Petz, W. Formation of Unusual Complexes from the Reaction of [C(NMe2)3][(CO)4Fe{C(O)NMe2}] with InMe3; Crystal Structures of [C(NMe2)3]3[Fe2(CO)6(μ-CO){μ- InFe(CO)4(μ-O2CNMe2)InFe(CO)4}] and [C(NMe2)3]2[{(CO)4Fe}2In(O2CNMe2)]·THF. Z. Anorg. Allg. Chem. 2007, 633, 314–319. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Patil, Y.; Galletti, A.M.R.; Renili, F.; Zacchini, S. Ethylene Polymerization by Niobium(V) N,N-Dialkylcarbamates Activated with Aluminum Co-catalysts. Organometallics 2011, 30, 1682–1688. [Google Scholar] [CrossRef]
- Domide, D.; Hübner, O.; Behrens, S.; Walter, O.; Wadepohl, H.; Kaifer, E.; Himmel, H.J. Synthesis and reactivity of a new oxidation-labile heterobimetallic Mn 6Zn2 carbamate cluster and precursor to nanosized magnetic oxide particles. Eur. J. Inorg. Chem. 2011, 3, 1387–1394. [Google Scholar] [CrossRef]
- Domide, D.; Walter, O.; Behrens, S.; Kaifer, E.; Himmel, H.-J. Synthesis of Heterobimetallic Zn/Co Carbamates: Single-Source Precursors of Nanosized Magnetic Oxides Under Mild Conditions. Eur. J. Inorg. Chem. 2011, 2011, 860–867. [Google Scholar] [CrossRef]
- Haywood, P.F.; Hill, M.R.; Roberts, N.K.; Craig, D.C.; Russell, J.J.; Lamb, R.N. Synthesis and Isomerisation Reactions of Tetranuclear and Octanuclear (Carbamato)zinc Complexes. Eur. J. Inorg. Chem. 2008, 12, 2024–2032. [Google Scholar] [CrossRef]
- Buhro, W.E.; Chisholm, M.H.; Martin, J.D.; Huffman, J.C.; Folting, K.; Streib, W.E. Reactions involving carbon dioxide and mixed amido-phosphido dinuclear compounds. M2(NMe2)4(PR2)2(M.tplbond.M), where M = Mo and W. Comparative study of the insertion of carbon dioxide into metal-nitr. J. Am. Chem. Soc. 1989, 111, 8149–8156. [Google Scholar] [CrossRef]
- Ito, H.; Ito, T. Dialkylcarbamato Complexes of Ni(II), Zn(II), and Cd(II)–Tetraazacycloalkanes Obtained from CO2-Uptake, and X-Ray Structure of (Diethylcarbamato)((7RS,14RS)5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraaza-cyclotetradecane)nickel(II) Perchlorate. Bull. Chem. Soc. Jpn. 1985, 58, 1755–1760. [Google Scholar] [CrossRef]
- Agostinelli, E.; Belli Dell’Amico, D.; Calderazzo, F.; Fiorani, D.; Pelizzi, G. Synthesis, properties and crystal and molecular strucrture of Cu2(O2CNEt2)4 2NHEt2. Gazz. Chim. Ital. 1988, 188, 729–740. [Google Scholar]
- Belli, D.; Amico, D.; Boschi, D.; Calderazzo, F.; Ianelli, S.; Labella, L.; Marchetti, F.; Pelizzi, G.; Guy, E.; Quadrelli, F. N,N-Dialkylcarbamato m-oxo derivatives of iron (III). Inorg. Chim. Acta. 2000, 302, 882–891. [Google Scholar] [CrossRef]
- Belli, D.; Amico, D.; Calderazzo, F.; Giurlani, U.; Pelizzi, G. Synthesis and Reactivity of N,N-Dialkylcarbamato Complexes of Titanium (III) and Vanadium (III). Crystal and Molecular Structure of an Anionic Dimeric Titanium (III) Derivative. Chem. Ber. 1987, 6, 955–964. [Google Scholar]
- Shawn McCowan, C.; Tyler Caudle, M. Evidence for unimolecular CO2 elimination in C–N bond metathesis reactions of basic carbamatozinc complexes Zn4O(O2CAm)6 (Am = N-diethylamino, N-piperidyl, N-pyrrolidyl). Dalton Trans. 2005, 6, 238–246. [Google Scholar] [CrossRef]
- Bresciani, G.; Marchetti, F.; Rizzi, G.; Gabbani, A.; Pineider, F.; Pampaloni, G. Metal N,N-dialkylcarbamates as easily available catalytic precursors for the carbon dioxide/propylene oxide coupling under ambient conditions. J. CO2 Util. 2018, 28, 168–173. [Google Scholar] [CrossRef]
- Buryi, D.S.; Levashov, A.S. The Reaction of Tin Tetracarbamates with Organyl Chlorosilanes: A Novel Synthetic Route Towards O-Silylurethanes. Russ. J. Gen. Chem. 2019, 89, 924–928. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Calderazzo, F.; Costa, L.; Franchi, E.; Gini, L.; Labella, L.; Marchetti, F. Octanuclear μ-oxo N,N-diethylcarbamato derivatives of titanium(IV) obtained by a high-yielding hydrolytic process. J. Mol. Struct. 2008, 890, 295–297. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F.; Labella, L.; Marchetti, F. Triphenylphosphine-substituted N,N-di-iso-propylcarbamate of ruthenium(II) and its reactions with carbon monoxide. J. Organomet. Chem. 2000, 596, 144–151. [Google Scholar] [CrossRef]
- Komarov, N.V.; Ryzhkova, N.A.; Andreev, A.A. Synthesis of alkoxystannanes by reactions of O-(organylstannyl) carbamates with alcohols. Russ. Chem. Bull. 2004, 53, 936–938. [Google Scholar] [CrossRef]
- Armelao, L.; Belli Dell’Amico, D.; Bottaro, G.; Falvo, P.; Labella, L.; Marchetti, F.; Parisi, D.; Samaritani, S. From lanthanide chlorides to lanthanide pentafluorophenolates via lanthanide N,N-dialkylcarbamates. Polyhedron 2015, 85, 770–776. [Google Scholar] [CrossRef]
- Levashov, A.S.; Andreev, A.A.; Buryi, D.S.; Konshin, V.V. A reaction of tin tetra(N,N-diethylcarbamate) with phenylacetylene as a new route to tetra(phenylethynyl)tin. Russ. Chem. Bull. 2014, 63, 775–776. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Di Giacomo, A.; Falchi, L.; Labella, L.; Marelli, M.; Evangelisti, C.; Lezzerini, M.; Marchetti, F.; Samaritani, S. A convenient preparation of La2CuO4 from molecular precursors. Polyhedron 2017, 123, 33–38. [Google Scholar] [CrossRef]
- Domide, D.; Neuhäuser, C.; Kaifer, E.; Wadepohl, H.; Himmel, H.J. Synthesis of trinuclear, dinuclear and mononuclear carbamato-zinc complexes from tetranuclear precursors: A top-down synthetic approach to new carbamates. Eur. J. Inorg. Chem. 2009, 14, 2170-0178. [Google Scholar] [CrossRef]
- Neuhäuser, C.; Domide, D.; Mautz, J.; Kaifer, E.; Himmel, H.J. Electron density controlled carbamate ligand binding mode: Towards a better understanding of metalloenzyme activity. Dalton Trans. 2008, 4, 1821–1824. [Google Scholar] [CrossRef]
- Deacon, G. Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coord. Chem. Rev. 1980, 33, 227–250. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; ISBN 9780470405840. [Google Scholar]
- Jabri, E.; Carr, M.; Hausinger, R.; Karplus, P. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef]
- Benning, M.M.; Hong, S.-B.; Raushel, F.M.; Holden, H.M. The Binding of Substrate Analogs to Phosphotriesterase. J. Biol. Chem. 2000, 275, 30556–30560. [Google Scholar] [CrossRef]
- Thoden, J.B.; Phillips, G.N.; Neal, T.M.; Raushel, F.M.; Holden, H.M. Molecular Structure of Dihydroorotase: A Paradigm for Catalysis through the Use of a Binuclear Metal Center. Biochemistry 2001, 40, 6989–6997. [Google Scholar] [CrossRef]
- Abendroth, J.; Niefind, K.; May, O.; Siemann, M.; Syldatk, C.; Schomburg, D. The Structure of L-Hydantoinase from Arthobacter aurescens Leads to an Understanding of Dihydropyrimidinase Substrate and Enantio Specificity. Biochemistry 2002, 41, 8589–8597. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Repo, T. Antibiotics from Carbon Dioxide: Sustainable Pathways to Pharmaceutically Relevant Cyclic Carbamates. Eur. J. Org. Chem. 2019, 6, 1180–1188. [Google Scholar] [CrossRef]
- Lamb, K.J.; Ingram, I.D.V.; North, M.; Sengoden, M. Valorization of Carbon Dioxide into Oxazolidinones by Reaction with Aziridines. Curr. Green Chem. 2019, 6, 32–43. [Google Scholar] [CrossRef]
- Carminati, D.; Gallo, E.; Damiano, C.; Caselli, A.; Intrieri, D. Ruthenium Porphyrin Catalyzed Synthesis of Oxazolidin-2-ones by Cycloaddition of CO2 to Aziridines. Eur. J. Inorg. Chem. 2018, 48, 5258–5262. [Google Scholar] [CrossRef]
- Arshadi, S.; Banaei, A.; Ebrahimiasl, S.; Monfared, A.; Vessally, E. Solvent-free incorporation of CO2 into 2-oxazolidinones: A review. RSC Adv. 2019, 9, 19465–19482. [Google Scholar] [CrossRef]
- Ghosh, S.; Khan, T.S.; Ghosh, A.; Chowdhury, A.H.; Haider, M.A.; Khan, A.; Islam, S.M. Utility of Silver Nanoparticles Embedded Covalent Organic Frameworks as Recyclable Catalysts for the Sustainable Synthesis of Cyclic Carbamates and 2-Oxazolidinones via Atmospheric Cyclizative CO2 Capture. ACS Sustain. Chem. Eng. 2020, 8, 5495–5513. [Google Scholar] [CrossRef]
- Li, J.-Y.; Song, Q.-W.; Zhang, K.; Liu, P. Catalytic Conversion of Carbon Dioxide through C-N Bond Formation. Molecules 2019, 24, 182. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Xia, S.; Song, Z.; Xu, H.; Shi, Y.; He, L.; Cheng, P.; Zhao, B. Highly Efficient Conversion of Propargylic Amines and CO2 Catalyzed by Noble-Metal-Free [Zn116] Nanocages. Angew. Chem. Int. Ed. 2020, 59, 8586–8593. [Google Scholar] [CrossRef]
- Sengoden, M.; North, M.; Whitwood, A. Synthesis of Oxazolidinones using Carbon Dioxide as a C-1 Building Block and an Aluminium-based Catalyst. Chem. Sus. Chem. 2019, 12, 3296–3303. [Google Scholar] [CrossRef]
- Adhikari, D.; Miller, A.W.; Baik, M.H.; Nguyen, S.T. Intramolecular ring-opening from a CO2-derived nucleophile as the origin of selectivity for 5-substituted oxazolidinone from the (salen)Cr-catalyzed [aziridine + CO2] coupling. Chem. Sci. 2015, 6, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xie, S.L.; Gao, X.T.; Zhang, R.; Wang, C.H.; Yin, G.Q.; Zhou, J. Activation of (salen)CoI complex by phosphorane for carbon dioxide transformation at ambient temperature and pressure. Green Chem. 2017, 19, 3908–3915. [Google Scholar] [CrossRef]
- Wang, X.; Gao, W.Y.; Niu, Z.; Wojtas, L.; Perman, J.A.; Chen, Y.S.; Li, Z.; Aguila, B.; Ma, S. A metal-metalloporphyrin framework based on an octatopic porphyrin ligand for chemical fixation of CO2 with aziridines. Chem. Commun. 2018, 54, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, R.; Yang, Z.; Zhou, X.; Ji, H. Imidazolium-based ionic liquid decorated zinc porphyrin catalyst for converting CO2 into five-membered heterocyclic molecules. Sustain. Energy Fuels 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Kayaki, Y.; Mori, N.; Ikariya, T. Palladium-catalyzed carboxylative cyclization of a-allenyl amines in dense carbon dioxide. Tetrahedron Lett. 2009, 47, 6491–6493. [Google Scholar] [CrossRef]
- Foo, S.W.; Takada, Y.; Yamazaki, Y.; Saito, S. Dehydrative synthesis of chiral oxazolidinones catalyzed by alkali metal carbonates under low pressure of CO2. Tetrahedron Lett. 2013, 54, 4717–4720. [Google Scholar] [CrossRef]
- Takada, Y.; Foo, S.W.; Yamazaki, Y.; Saito, S. Catalytic fluoride triggers dehydrative oxazolidinone synthesis from CO2. RSC Adv. 2014, 4, 50851–50857. [Google Scholar] [CrossRef]
- Tominaga, K.; Sasaki, Y. Synthesis of 2-Oxazolidinones from CO2 and 1,2-Aminoalcohols Catalyzed by n-Bu2SnO. Synlett 2002, 02, 0307–0309. [Google Scholar] [CrossRef]
- Xiong, W.; Qi, C.; Guo, T.; Zhang, M.; Chen, K.; Jiang, H. A copper-catalyzed oxidative coupling reaction of arylboronic acids, amines and carbon dioxide using molecular oxygen as the oxidant. Green Chem. 2017, 19, 1642–1645. [Google Scholar] [CrossRef]
- Wang, L.; Qi, C.; Cheng, R.; Liu, H.; Xiong, W.; Jiang, H. Direct Access to Trifluoromethyl-Substituted Carbamates from Carbon Dioxide via Copper-Catalyzed Cascade Cyclization of Enynes. Org. Lett. 2019, 21, 7386–7389. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G. Iron(III) N,N-Dialkylcarbamate-Catalyzed Formation of Cyclic Carbonates from CO2 and Epoxides under Ambient Conditions by Dynamic CO2 Trapping as Carbamato Ligands. Chem. Sus. Chem. 2018, 11, 2737–2743. [Google Scholar] [CrossRef]
- Bresciani, G.; Marchetti, F.; Pampaloni, G. Carboxylation of terminal alkynes promoted by silver carbamate at ambient pressure. New J. Chem. 2019, 43, 10821–10825. [Google Scholar] [CrossRef]
- Bresciani, G.; Bortoluzzi, M.; Ghelarducci, C.; Marchetti, F.; Pampaloni, G. Synthesis of α-Alkylidene Cyclic Carbonates via CO2 Fixation Promoted by Easily Available Silver Compounds Under Ambient Conditions. (submitted).
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Galletti, A.M.R.; Pampaloni, G.; D’Alessio, A.; Patil, Y.; Renili, F.; Giaiacopi, S. Novel highly active niobium catalysts for ring opening metathesis polymerization of norbornene. Macromol. Rapid Commun. 2009, 30, 1762–1768. [Google Scholar] [CrossRef]
- Hayatifar, M.; Pampaloni, G.; Bernazzani, L.; Capacchione, C.; Kissin, Y.V.; Raspolli Galletti, A.M. A new post-metallocene catalyst for alkene polymerization: Copolymerization of ethylene and 1-hexene with titanium complexes bearing N,N-dialkylcarbamato ligands. Polym. Int. 2014, 63, 560–567. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Pinzino, C.; Renili, F.; Repo, T.; Vuorinen, S. Ring opening polymerization of rac-lactide by group 4 tetracarbamato complexes: Activation, propagation and role of the metal. Dalton Trans. 2013, 42, 2792–2802. [Google Scholar] [CrossRef]
- Tsoglin, E.; Chechik, H.; Karseboom, G.; Chinkov, N.; Stanger, A.; Marek, I. Stereoselective synthesis of metalated cyclobutyl derivatives. Adv. Synth. Catal. 2009, 351, 1005–1008. [Google Scholar] [CrossRef]
- Jašíková, L.; Hanikýřová, E.; Škríba, A.; Jašík, J.; Roithová, J. Metal-assisted lossen rearrangement. J. Org. Chem. 2012, 77, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.R.; Jones, A.W.; Russell, J.J.; Roberts, N.K.; Lamb, R.N. Dialkylcarbamato magnesium cluster complexes: Precursors to the single-source chemical vapour deposition of high quality MgO thin films. J. Mater. Chem. 2004, 14, 3198–3202. [Google Scholar] [CrossRef]
- Duce, C.; Spepi, A.; Pampaloni, G.; Piccinelli, F.; Tiné, M.R. Thermal decomposition of metal N,N-dialkylcarbamates. J. Therm. Anal. Calorim. 2016, 123, 1563–1569. [Google Scholar] [CrossRef]
- Duan, X.; Tran, N.H.; Roberts, N.K.; Lamb, R.N. Single-source chemical vapor deposition of clean oriented Al2O3 thin films. Thin Solid Films 2009, 517, 6726–6730. [Google Scholar] [CrossRef]
- Deng, H.; Gong, B.; Petrella, A.J.; Russell, J.J.; Lamb, R.N. Characterization of the ZnO thin film prepared by single source chemical vapor deposition under low vacuum condition. Sci. China, Ser. E Technol. Sci. 2003, 46, 6–11. [Google Scholar] [CrossRef]
- Petrella, A.J.; Deng, H.; Roberts, N.K.; Lamb, R.N. Single-Source Chemical Vapor Deposition Growth of ZnO Thin Films Using Zn4O(CO2NEt2)6. Chem. Mater. 2002, 14, 4339–4342. [Google Scholar] [CrossRef]
- Deng, H.; Clausi, D.A. Unsupervised segmentation of synthetic aperture radar sea ice imagery using MRF models. In Proceedings of the First Canadian Conference on Computer and Robot Vision, London, ON, Canada, 17–19 May 2004; Volume 458, pp. 43–50. [Google Scholar] [CrossRef]
- Lee, E.; Russell, J.J.; Lamb, R.N. Nanostructural properties of zinc oxide thin films grown on non-planar substrates. Surf. Interface Anal. 2006, 38, 51–55. [Google Scholar] [CrossRef]
- Hill, M.R.; Russell, J.J.; Lamb, R.N. High-quality ZnxMg1-xO thin films deposited from a single molecular source. Intimate mixing as a means to improved film properties. Chem. Mater. 2008, 20, 2461–2467. [Google Scholar] [CrossRef]
- Trott, G.; Garden, J.A.; Williams, C.K. Heterodinuclear zinc and magnesium catalysts for epoxide/CO2 ring opening copolymerizations. Chem. Sci. 2019, 10, 4618–4627. [Google Scholar] [CrossRef]
- Duan, X.F.; Tran, N.H.; Roberts, N.K.; Lamb, R.N. Solvothermal approach for low temperature deposition of aluminium oxide thin films. Thin Solid Films 2010, 518, 4290–4293. [Google Scholar] [CrossRef]
- Sgrolli, N.; Imlyhen, N.; Volkman, J.; Raspolli-Galletti, A.M.; Serp, P. Copper-based magnetic catalysts for alkyne oxidative homocoupling reactions. Mol. Catal. 2017, 438, 143–151. [Google Scholar] [CrossRef]
- Kim, K.A.; Cha, J.R.; Gong, M.S.; Kim, J.G. Preparation of ZnO2 nanoparticles using organometallic zinc(II) isobutylcarbamate in organic solvent. Bull. Korean Chem. Soc. 2014, 35, 431–435. [Google Scholar] [CrossRef]
- Kwak, W.G.; Cha, J.R.; Gong, M.S. Surface modification of polyester fibers by thermal reduction with silver carbamate complexes. Fibers Polym. 2016, 17, 1146–1153. [Google Scholar] [CrossRef]
- Park, M.S.; Lim, T.H.; Jeon, Y.M.; Kim, J.G.; Gong, M.S.; Joo, S.W. Preparation of new polyelectrolyte/silver nanocomposites and their humidity-sensitive properties. Macromol. Res. 2008, 16, 308–313. [Google Scholar] [CrossRef]
- Park, M.S.; Lim, T.H.; Jeon, Y.M.; Kim, J.G.; Joo, S.W.; Gong, M.S. Humidity sensitive properties of copoly(TEAMPS/VP)/silver nanocomposite films. Sensors Actuators B Chem. 2008, 133, 166–173. [Google Scholar] [CrossRef]
- Park, H.S.; Gong, M.S. Facile preparation of nanosilver-decorated MWNTs using silver carbamate complex and their polymer composites. Bull. Korean Chem. Soc. 2012, 33, 483–488. [Google Scholar] [CrossRef][Green Version]
- Park, H.S.; Park, H.S.; Gong, M.S. Preparation of silver nanocolloids using silver alkylcarbamate complex in organic medium with PVP stabilizer. Bull. Korean Chem. Soc. 2010, 31, 2575–2580. [Google Scholar] [CrossRef][Green Version]
- Jeon, Y.-M.; Gong, M.-S.; Cho, H.-N. Preparation of silver/PMMA beads via the in sito reduction of a silver alkylcarbamate complex. Macromol. Res. 2009, 17, 2–4. [Google Scholar] [CrossRef]
- Kim, K.A.; Cha, J.R.; Gong, M.S. Facile preparation of silver nanoparticles and application to silver coating using latent reductant from a silver carbamate complex. Bull. Korean Chem. Soc. 2013, 34, 505–509. [Google Scholar] [CrossRef][Green Version]
- Yun, S.W.; Cha, J.R.; Gong, M.S. Easy preparation of nanosilver-decorated graphene using silver carbamate by microwave irradiation and their properties. Bull. Korean Chem. Soc. 2014, 35, 2251–2256. [Google Scholar] [CrossRef][Green Version]
- Kwak, W.G.; Oh, M.H.; Gong, M.S. Preparation of silver-coated cotton fabrics using silver carbamate via thermal reduction and their properties. Carbohydr. Polym. 2015, 115, 317–324. [Google Scholar] [CrossRef]
- Kwak, W.G.; Oh, M.H.; Son, S.Y.; Gong, M.S. Silver loading on poly(ethylene terephthalate) fabrics using silver carbamate via thermal reduction. Macromol. Res. 2015, 23, 509–517. [Google Scholar] [CrossRef]
- Ahn, H.Y.; Cha, J.R.; Gong, M.S. Preparation of sintered silver nanosheets by coating technique using silver carbamate complex. Mater. Chem. Phys. 2015, 153, 390–395. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Wang, X.; Zeng, X. Electrically conductive thick film made from silver alkylcarbamates. J. Electron. Mater. 2010, 39, 2267–2273. [Google Scholar] [CrossRef]
- Kim, K.Y.; Gong, M.S.; Park, C.K. Preparation of highly stabilized silver nanopowders by the thermal reduction and their properties. Bull. Korean Chem. Soc. 2012, 33, 3987–3992. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, M.; Zeng, X. Electrical conductivity of thick films made from silver methylcarbamate paste. J. Electron. Mater. 2013, 42, 2990–2997. [Google Scholar] [CrossRef]
- Abis, L.; Armelao, L.; Belli Dell’Amico, D.; Calderazzo, F.; Garbassi, F.; Merigo, A.; Quadrelli, E.A. Gold molecular precursors and gold–silica interactions. J. Chem. Soc. Dalt. Trans. 2001, 18, 2704–2709. [Google Scholar] [CrossRef]
- Abis, L.; Calderazzo, F.; Maichle-Mössmer, C.; Pampaloni, G.; Strähle, J.; Tripepi, G. Cyclopentadienyl–diethylcarbamato derivatives of zirconium(IV) and hafnium(IV), [M(C5H5)(O2CNEt2)3]: Synthesis and use as precursors for chemical implantation on a silica surface. J. Chem. Soc. Dalt. Trans. 1998, 5, 841–846. [Google Scholar] [CrossRef]
- Abis, L.; Dell’amico, D.B.; Busetto, C.; Calderazzo, F.; Caminiti, R.; Ciofi, C.; Garbassi, F.; Masciarelli, G. N,N-Dialkylcarbamato complexes as precursors for the chemical implantation of metal cations on a silica support. J. Mater. Chem. 1998, 8, 751–759. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Bertagnolli, H.; Calderazzo, F.; D’Arienzo, M.; Gross, S.; Labella, L.; Rancan, M.; Scotti, R.; Smarsly, B.M.; Supplit, R.; et al. Nanostructured Copper Oxide on Silica-Zirconia Mixed Oxides by Chemical Implantation. Chem. -Eur. J. 2009, 15, 4931–4943. [Google Scholar] [CrossRef]
- Armelao, L.; Dell’Amico, D.B.; Bellucci, L.; Bottaro, G.; Di Bari, L.; Labella, L.; Marchetti, F.; Samaritani, S.; Zinna, F. Circularly Polarized Luminescence of Silica-Grafted Europium Chiral Derivatives Prepared through a Sequential Functionalization. Inorg. Chem. 2017, 56, 7010–7018. [Google Scholar] [CrossRef] [PubMed]
- Dolci, S.; Domenici, V.; Duce, C.; Tiné, M.R.; Ierardi, V.; Valbusa, U.; Jaglicic, Z.; Boni, A.; Gemmi, M.; Pampaloni, G. Ultrasmall superparamagnetic iron oxide nanoparticles with titanium-N,N-dialkylcarbamato coating. Mater. Res. Express 2014, 1, 035401. [Google Scholar] [CrossRef]
- Abis, L.; Dell’ Amico, D.B.; Busetto, C.; Calderazzo, F.; Garbassi, F.; Tomei, A. N,N-Dialkylcarbamato complexes as precursors for the chemical implantation of metal cations on a silica support. Part 3 Palladium. J. Mater. Chem. 1998, 8, 2855–2861. [Google Scholar] [CrossRef]
- Awuah, S.G.; Zheng, Y.R.; Bruno, P.M.; Hemann, M.T.; Lippard, S.J. A Pt(IV) Pro-drug Preferentially Targets Indoleamine-2,3-dioxygenase, Providing Enhanced Ovarian Cancer Immuno-Chemotherapy. J. Am. Chem. Soc. 2015, 137, 14854–14857. [Google Scholar] [CrossRef]
- Zhang, G.; Zhu, Y.; Wang, Y.; Wei, D.; Wu, Y.; Zheng, L.; Bai, H.; Xiao, H.; Zhang, Z. PH/redox sensitive nanoparticles with platinum(iv) prodrugs and doxorubicin enhance chemotherapy in ovarian cancer. RSC Adv. 2019, 9, 20513–20517. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, Y.; Lin, X.; Ma, W.; Chen, G.; Li, W.; Wang, X.; Yu, Z. Platinum(iv) prodrugs with long lipid chains for drug delivery and overcoming cisplatin resistance. Chem. Commun. 2018, 54, 5369–5372. [Google Scholar] [CrossRef]
- Sommerfeld, N.S.; Strohhofer, D.; Cseh, K.; Theiner, S.; Jakupec, M.A.; Koellensperger, G.; Galanski, M.; Keppler, B.K. Platinum(IV) Complexes Featuring Axial Michael Acceptor Ligands—Synthesis, Characterization, and Cytotoxicity. Eur. J. Inorg. Chem. 2017, 34, 4049–4054. [Google Scholar] [CrossRef]
- Stilgenbauer, M.; Jayawardhana, A.M.D.S.; Datta, P.; Yue, Z.; Gray, M.; Nielsen, F.; Bowers, D.J.; Xiao, H.; Zheng, Y.-R. A spermine-conjugated lipophilic Pt(IV) prodrug designed to eliminate cancer stem cells in ovarian cancer. Chem. Commun. 2019, 55, 6106–6109. [Google Scholar] [CrossRef]
- Mayr, J.; Hager, S.; Koblmüller, B.; Klose, M.H.M.; Holste, K.; Fischer, B.; Pelivan, K.; Berger, W.; Heffeter, P.; Kowol, C.R.; et al. EGFR-targeting peptide-coupled platinum(IV) complexes. JBIC J. Biol. Inorg. Chem. 2017, 22, 591–603. [Google Scholar] [CrossRef]
- Conibear, A.C.; Hager, S.; Mayr, J.; Klose, M.H.M.; Keppler, B.K.; Kowol, C.R.; Heffeter, P.; Becker, C.F.W. Multifunctional α v β 6 Integrin-Specific Peptide–Pt(IV) Conjugates for Cancer Cell Targeting. Bioconjug. Chem. 2017, 28, 2429–2439. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.N.; Zhang, Y.W.; He, L.N.; Yu, B.; Song, Q.W.; Lang, X.D. Equimolar carbon absorption by potassium phthalimide and in situ catalytic conversion under mild conditions. Chem. Sus. Chem. 2014, 7, 1484–1489. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Buchard, A.; Wells, S.A.; Fang, Y.; Torrente-Murciano, L.; Nearchou, A.; Dong, Z.; White, T.J.; Sartbaeva, A.; Ting, V.P. Mechanism of CO2 capture in nanostructured sodium amide encapsulated in porous silica. Surf. Coatings Technol. 2018, 350, 227–233. [Google Scholar] [CrossRef]
- Assaf, K.I.; Qaroush, A.K.; Mustafa, F.M.; Alsoubani, F.; Pehl, T.M.; Troll, C.; Rieger, B.; Eftaiha, A.F. Biomaterials for CO2 harvesting: From regulatory functions to wet scrubbing applications. ACS Omega 2019, 4, 11532–11539. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.M.; Mason, J.A.; Kong, X.; Bloch, E.D.; Gygi, D.; Dani, A.; Crocellà, V.; Giordanino, F.; Odoh, S.O.; Drisdell, W.S.; et al. Cooperative insertion of CO2 in diamine-appended metal-organic frameworks. Nature 2015, 519, 303–308. [Google Scholar] [CrossRef]
- Kim, E.J.; Siegelman, R.L.; Jiang, H.Z.H.; Forse, A.C.; Lee, J.-H.; Martell, J.D.; Milner, P.J.; Falkowski, J.M.; Neaton, J.B.; Reimer, J.A.; et al. Cooperative carbon capture and steam regeneration with tetraamine-appended metal-organic frameworks. Science 2020, 369, 392–396. [Google Scholar] [CrossRef]
- An, J.P.; Fiorella, R.J.; Geib, S.L.; Rosi, N. Synthesis, Structure, Assembly, and Modulation of the CO2 Adsorption Properties of a Zinc-Adeninate Macrocycle. J. Am. Chem. Soc. 2009, 131, 8401–8403. [Google Scholar] [CrossRef]
- McDonald, T.M.; Lee, W.R.; Mason, J.A.; Wiers, B.M.; Hong, C.S.; Long, J.R. Capture of Carbon Dioxide from Air and Flue Gas in the Alkylamine-Appended Metal–Organic Framework mmen-Mg2(dobpdc). J. Am. Chem. Soc. 2012, 134, 7056–7065. [Google Scholar] [CrossRef]
- Siegelman, R.L.; McDonald, T.M.; Gonzalez, M.I.; Martell, J.D.; Milner, P.J.; Mason, J.A.; Berger, A.H.; Bhown, A.S.; Long, J.R. Controlling Cooperative CO2 Adsorption in Diamine-Appended Mg2(dobpdc) Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 10526–10538. [Google Scholar] [CrossRef]
- Milner, P.J.; Siegelman, R.L.; Forse, A.C.; Gonzalez, M.I.; Runčevski, T.; Martell, J.D.; Reimer, J.A.; Long, J.R. A Diaminopropane-Appended Metal–Organic Framework Enabling Efficient CO2 Capture from Coal Flue Gas via a Mixed Adsorption Mechanism. J. Am. Chem. Soc. 2017, 139, 13541–13553. [Google Scholar] [CrossRef]
- Martell, J.D.; Porter-Zasada, L.B.; Forse, A.C.; Siegelman, R.L.; Gonzalez, M.I.; Oktawiec, J.; Runčevski, T.; Xu, J.; Srebro-Hooper, M.; Milner, P.J.; et al. Enantioselective Recognition of Ammonium Carbamates in a Chiral Metal–Organic Framework. J. Am. Chem. Soc. 2017, 139, 16000–16012. [Google Scholar] [CrossRef]
- Hellström, A.K.; Oskarsson, H.; Bordes, R. Formation, physicochemical and interfacial study of carbamate surfactants. J. Colloid Interface Sci. 2018, 511, 84–91. [Google Scholar] [CrossRef]
- Hellström, A.-K.; Bordes, R. Reversible flocculation of nanoparticles by a carbamate surfactant. J. Colloid Interface Sci. 2019, 536, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, L.; Bresciani, G.; Chiappe, C.; Marchetti, F.; Pampaloni, G.; Pomelli, C.S. Modifying bis(triflimide) ionic liquids by dissolving early transition metal carbamates. Phys. Chem. Chem. Phys. 2018, 20, 5057–5066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, P.-P.; Li, L.-F.; Hu, Y.-Y.; Mei, X.-L. A tridecanuclear {ZnGd12} nanoscopic cluster exhibiting large magnetocaloric effect. Inorg. Chim. Acta. 2020, 499, 119170. [Google Scholar] [CrossRef]
Sample Availability: Samples of several compounds cited in this work are available from the authors. |















































{kind=link}
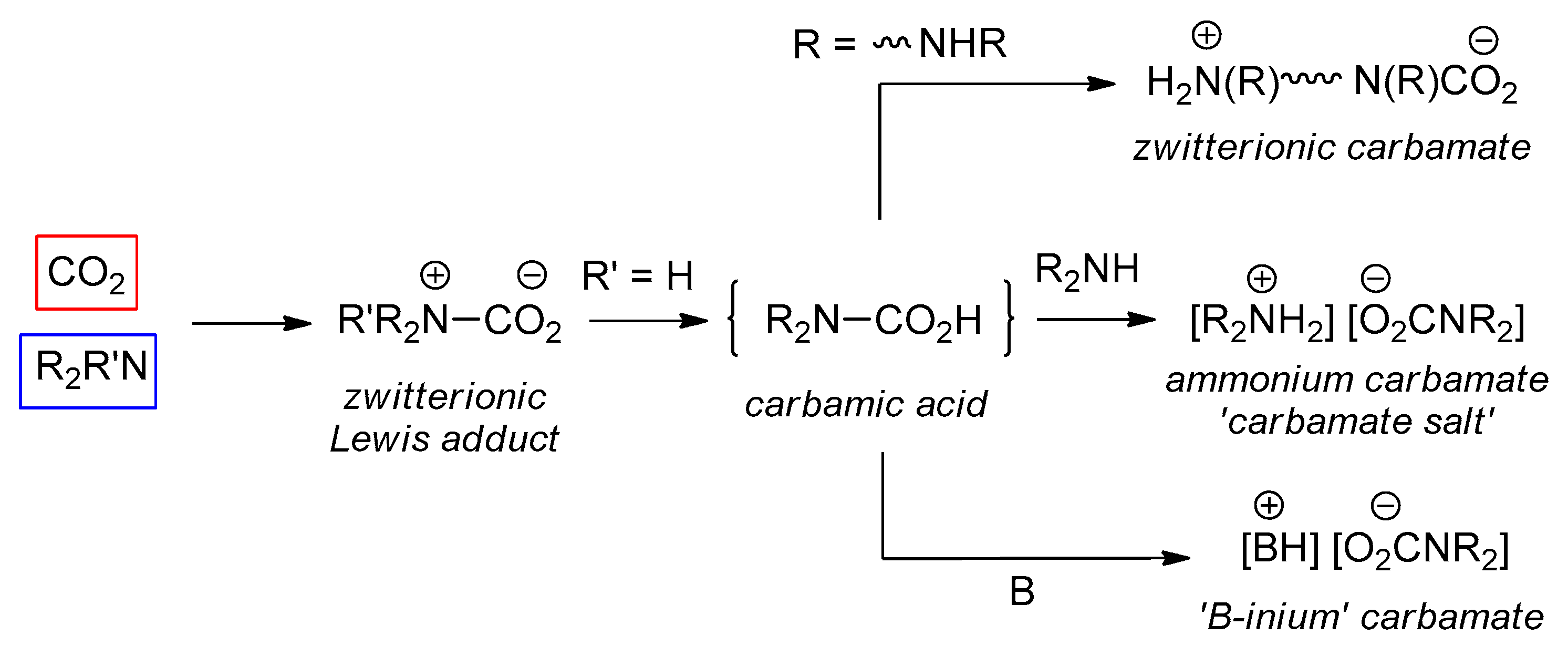{kind=link}
{kind=link}
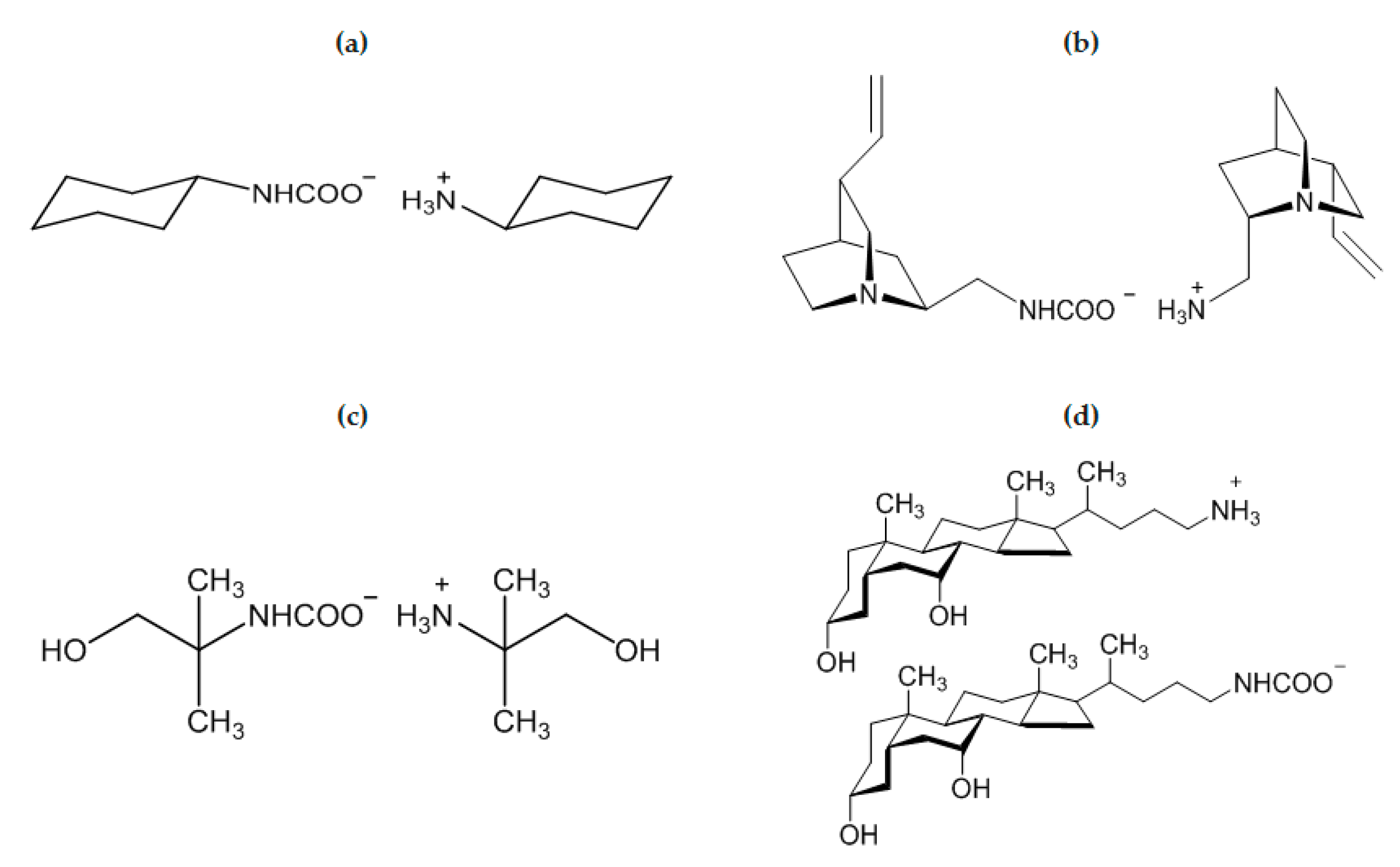{kind=link}
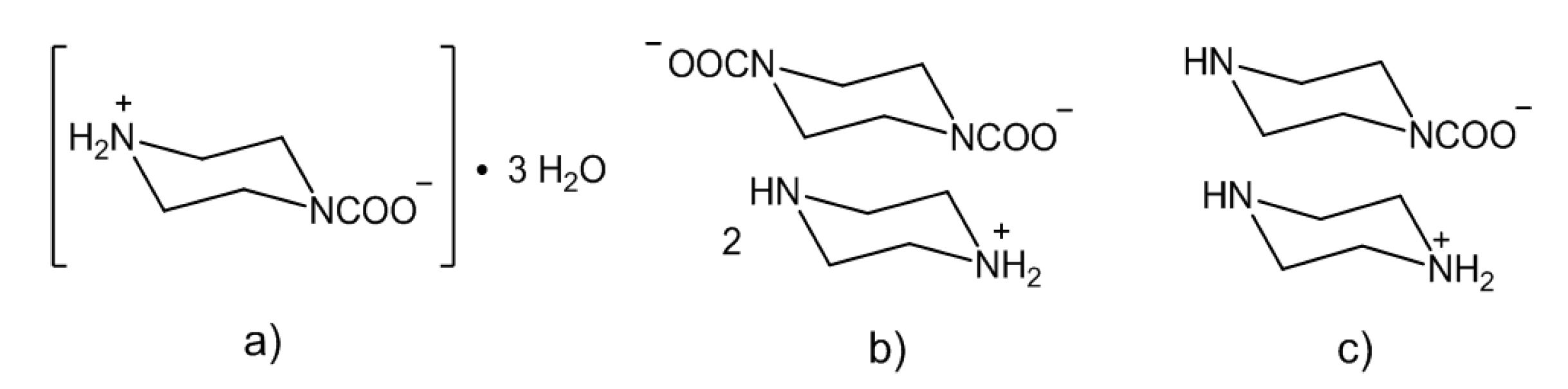{kind=link}
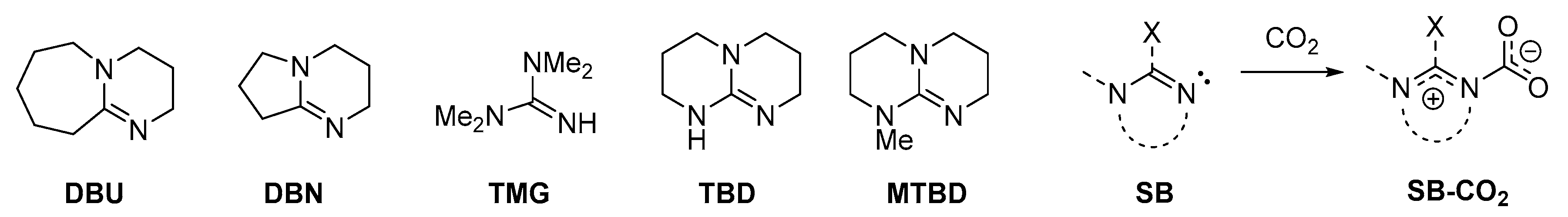{kind=link}
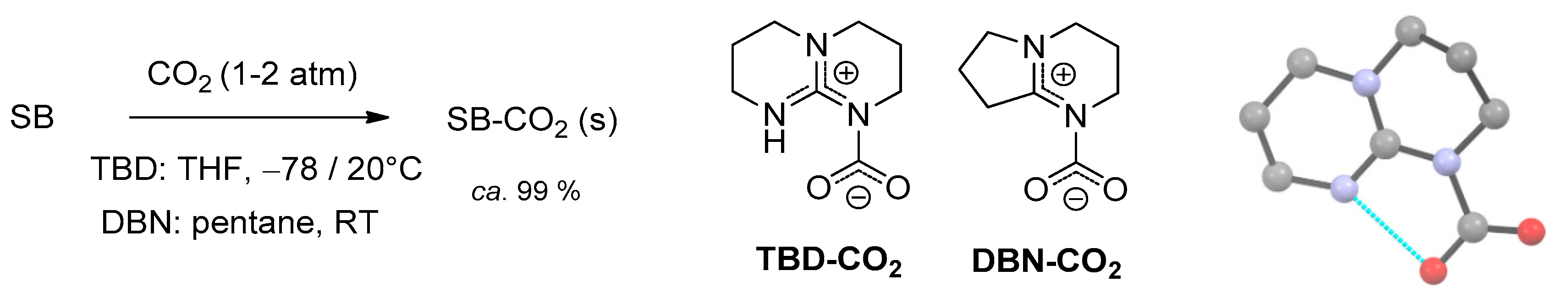{kind=link}
{kind=link}
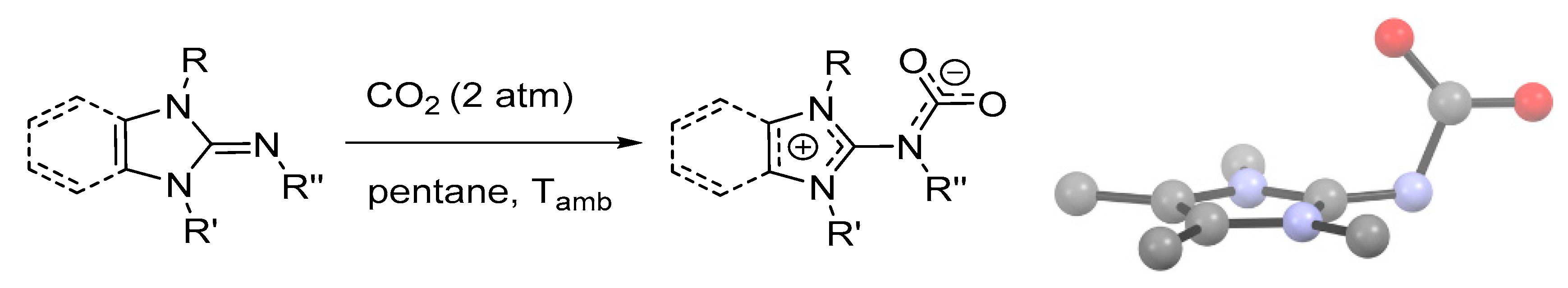{kind=link}
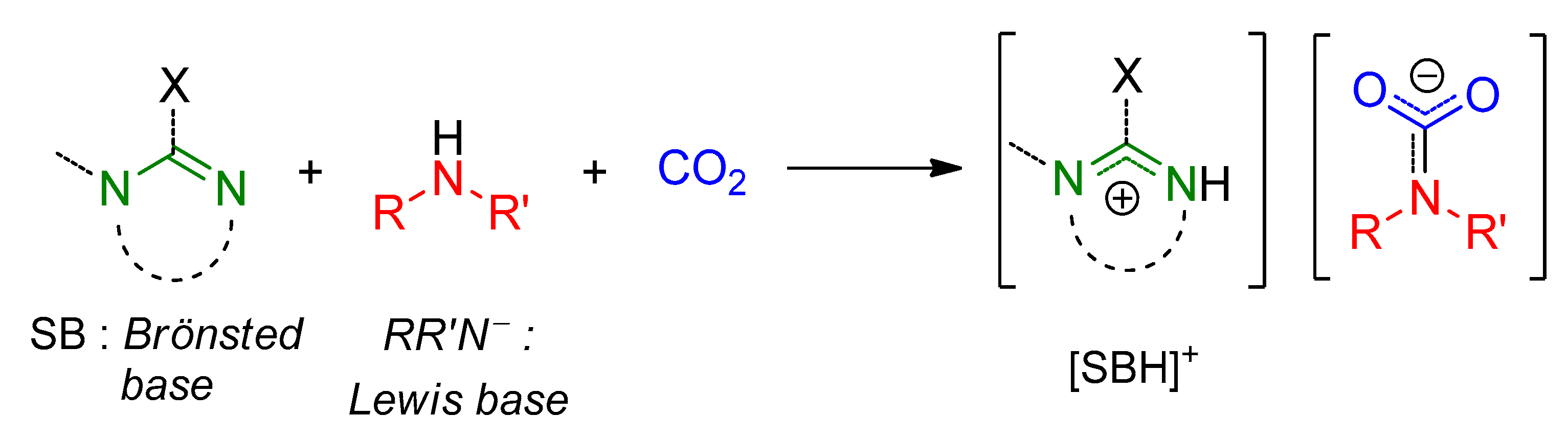{kind=link}
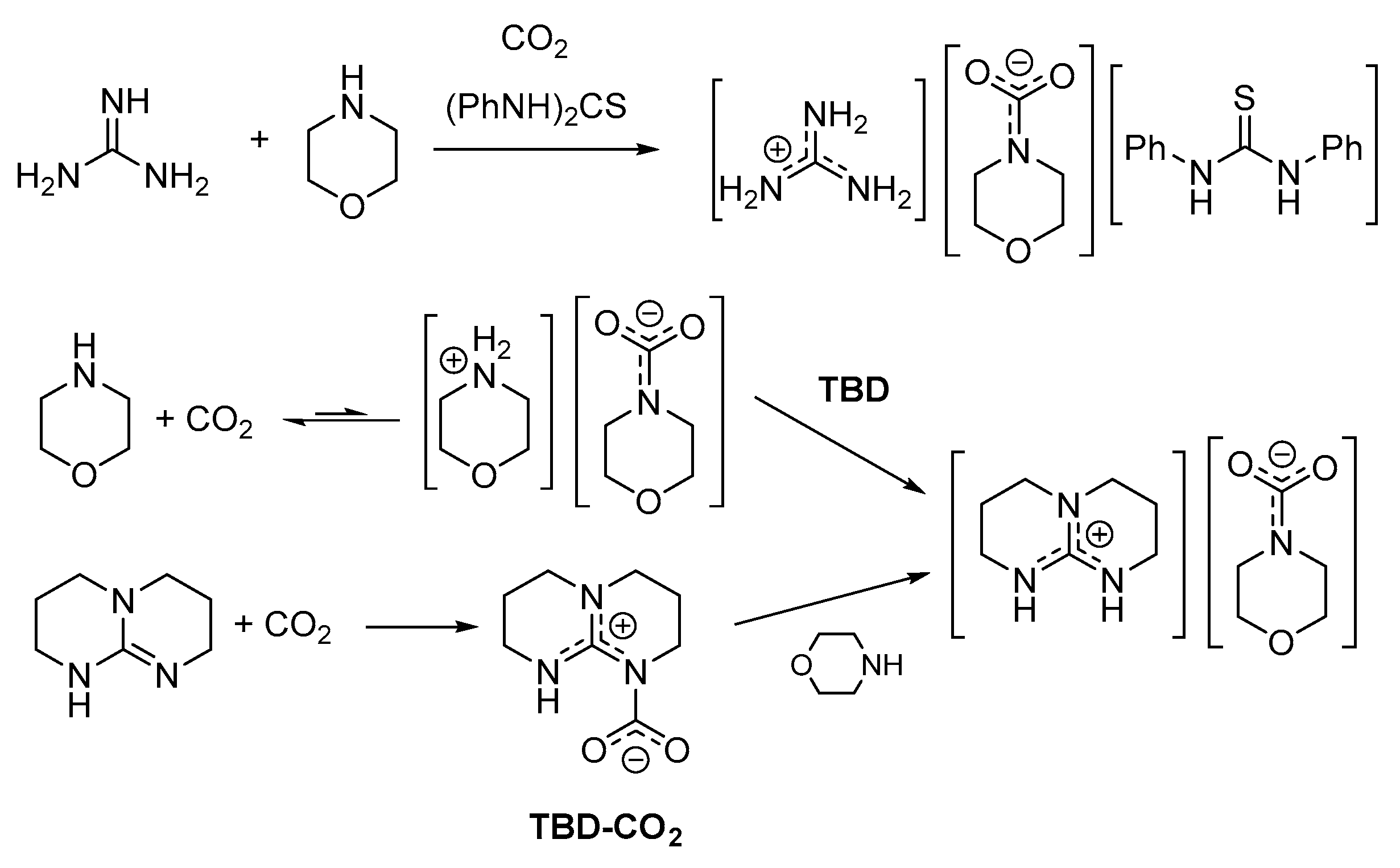{kind=link}
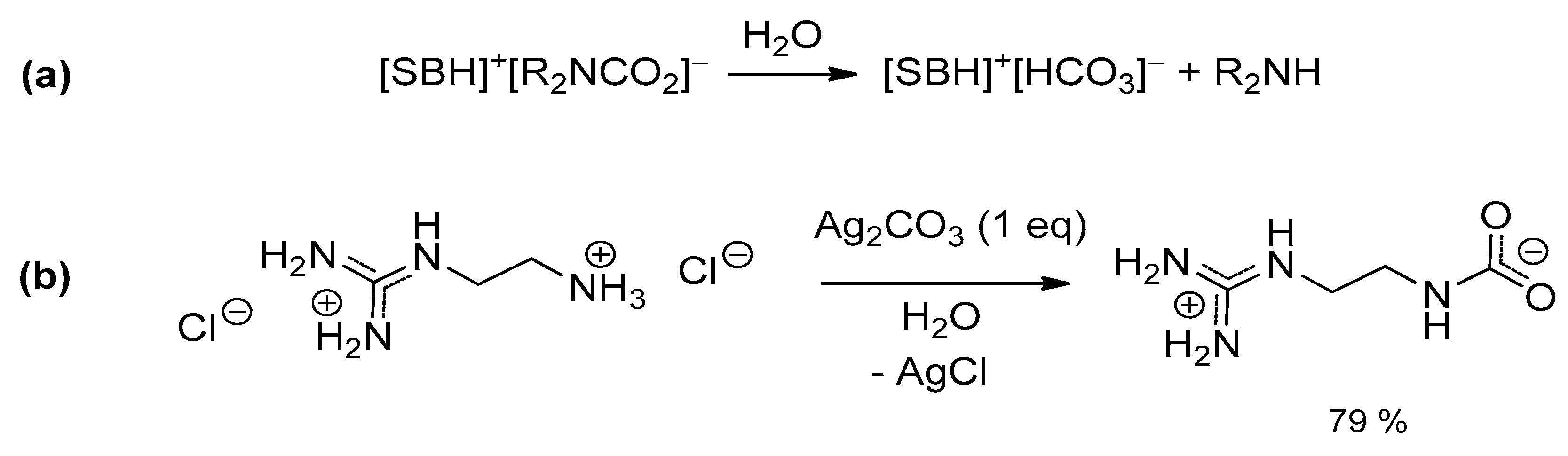{kind=link}
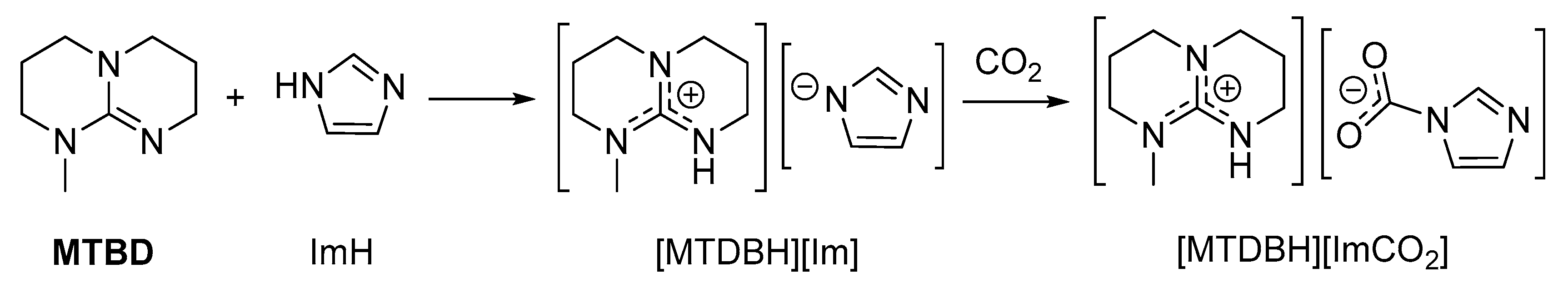{kind=link}
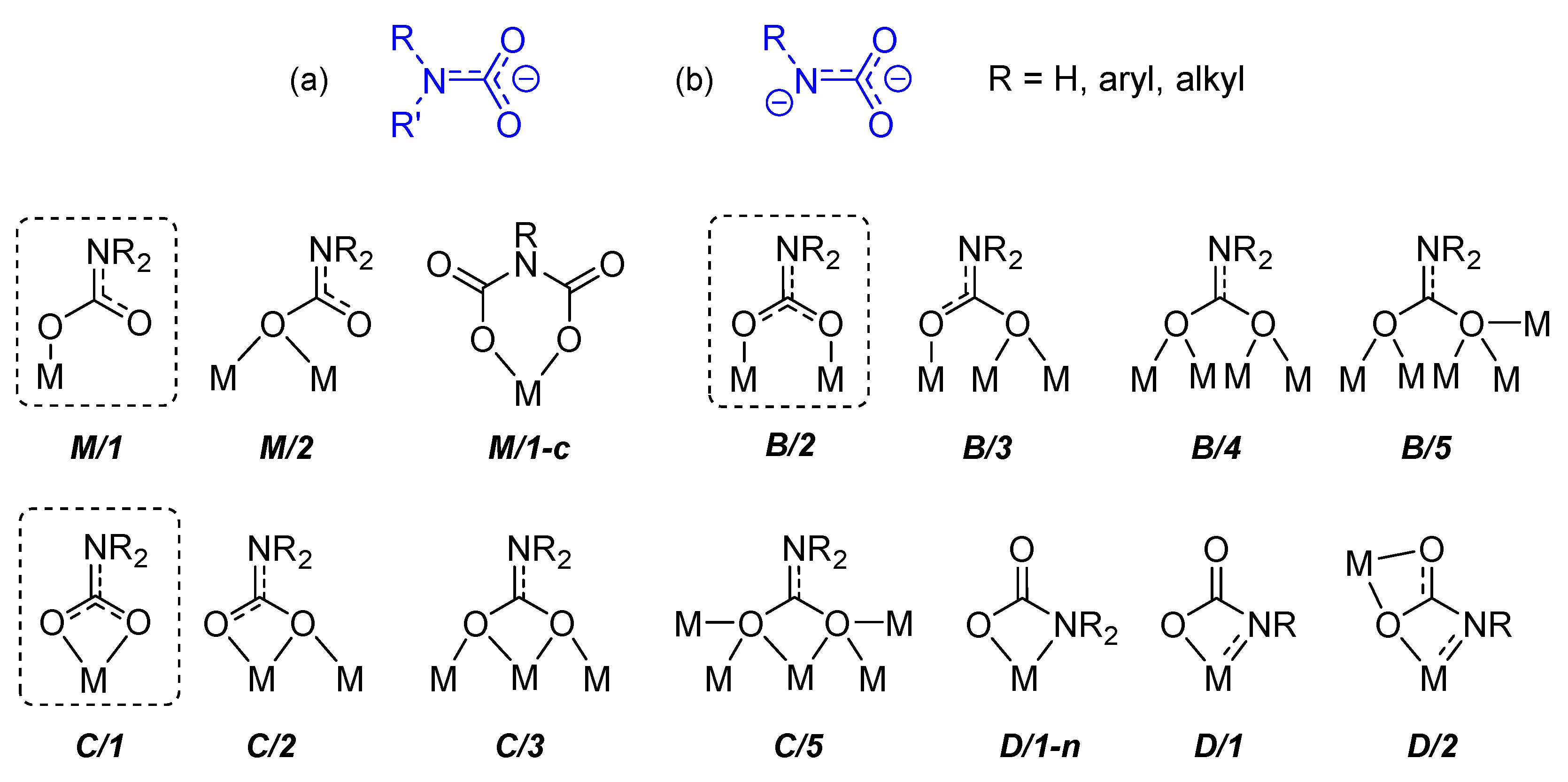{kind=link}
{kind=link}
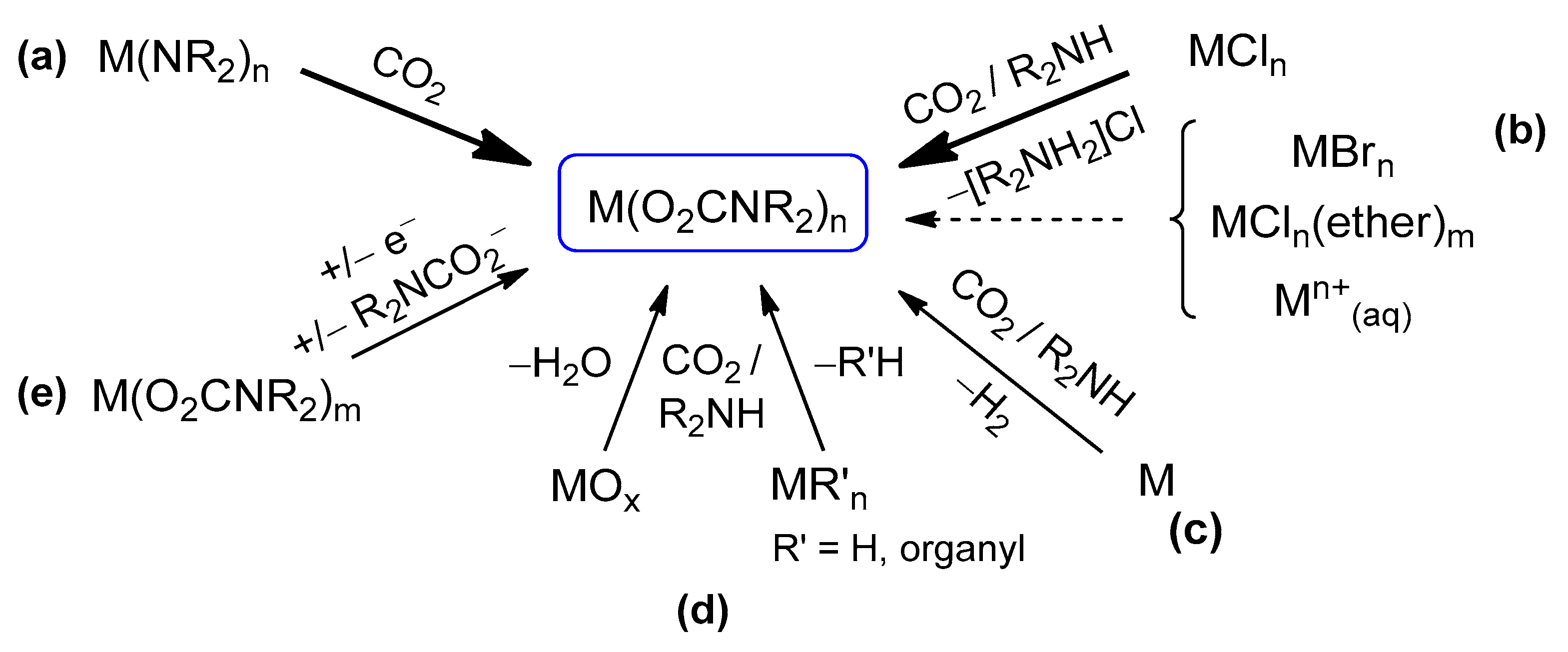{kind=link}
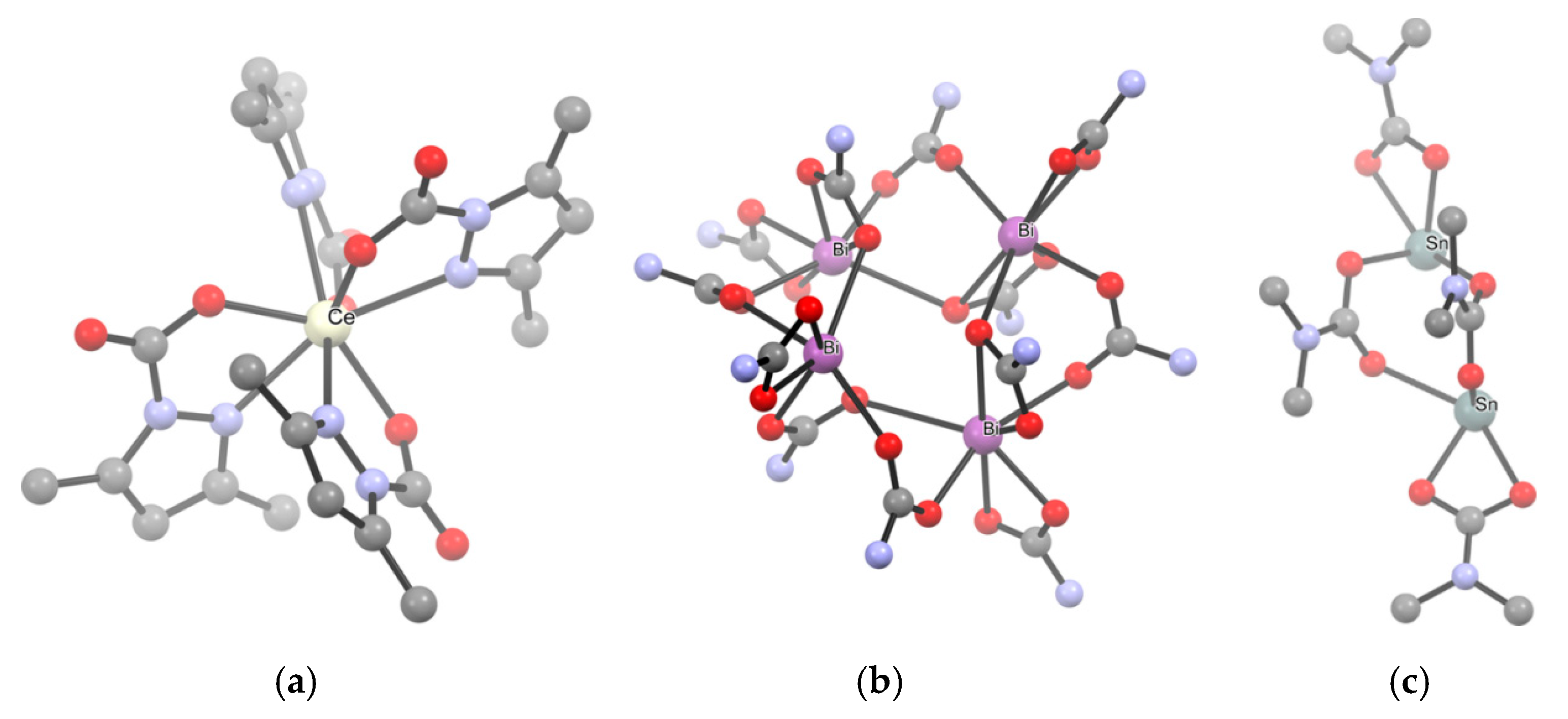{kind=link}
{kind=link}
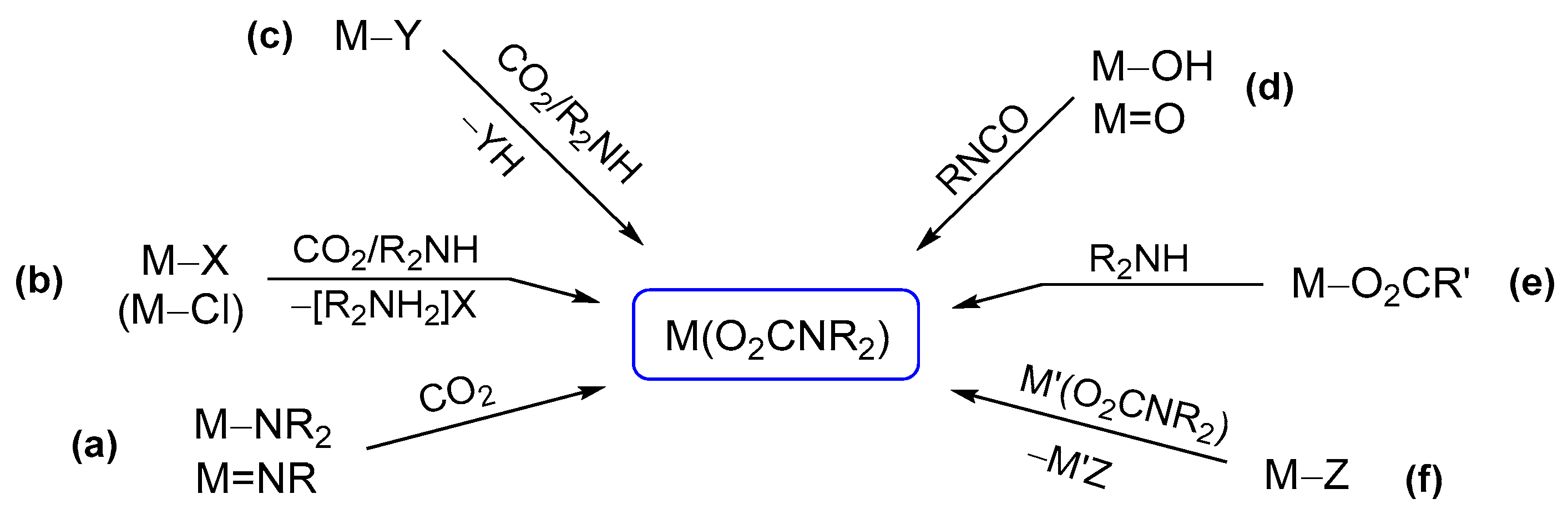{kind=link}
{kind=link}
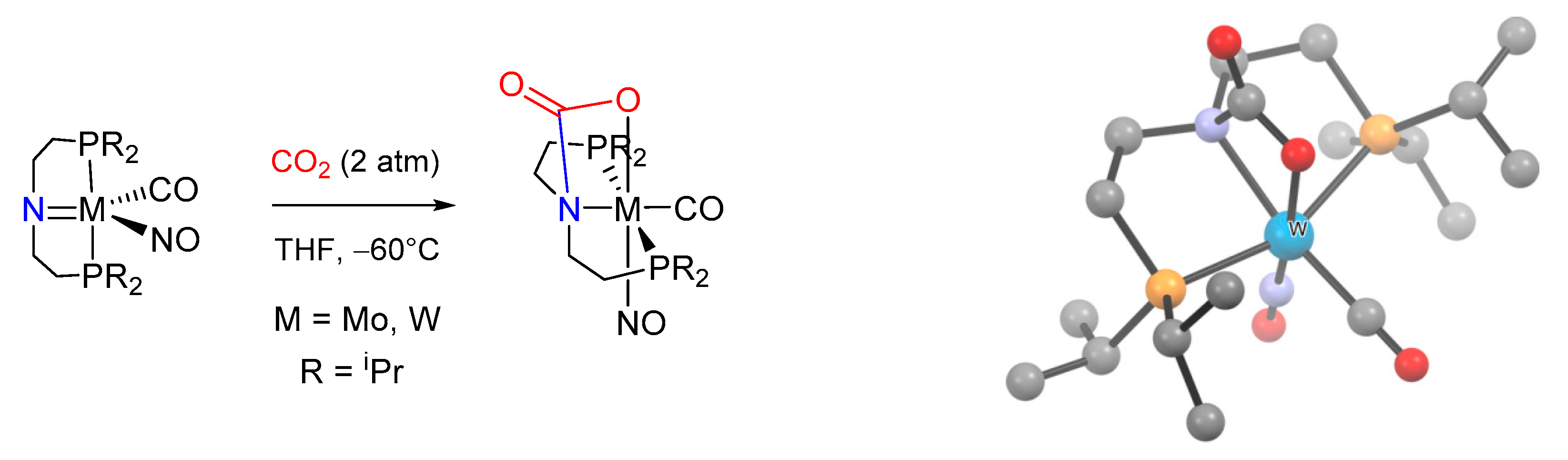{kind=link}
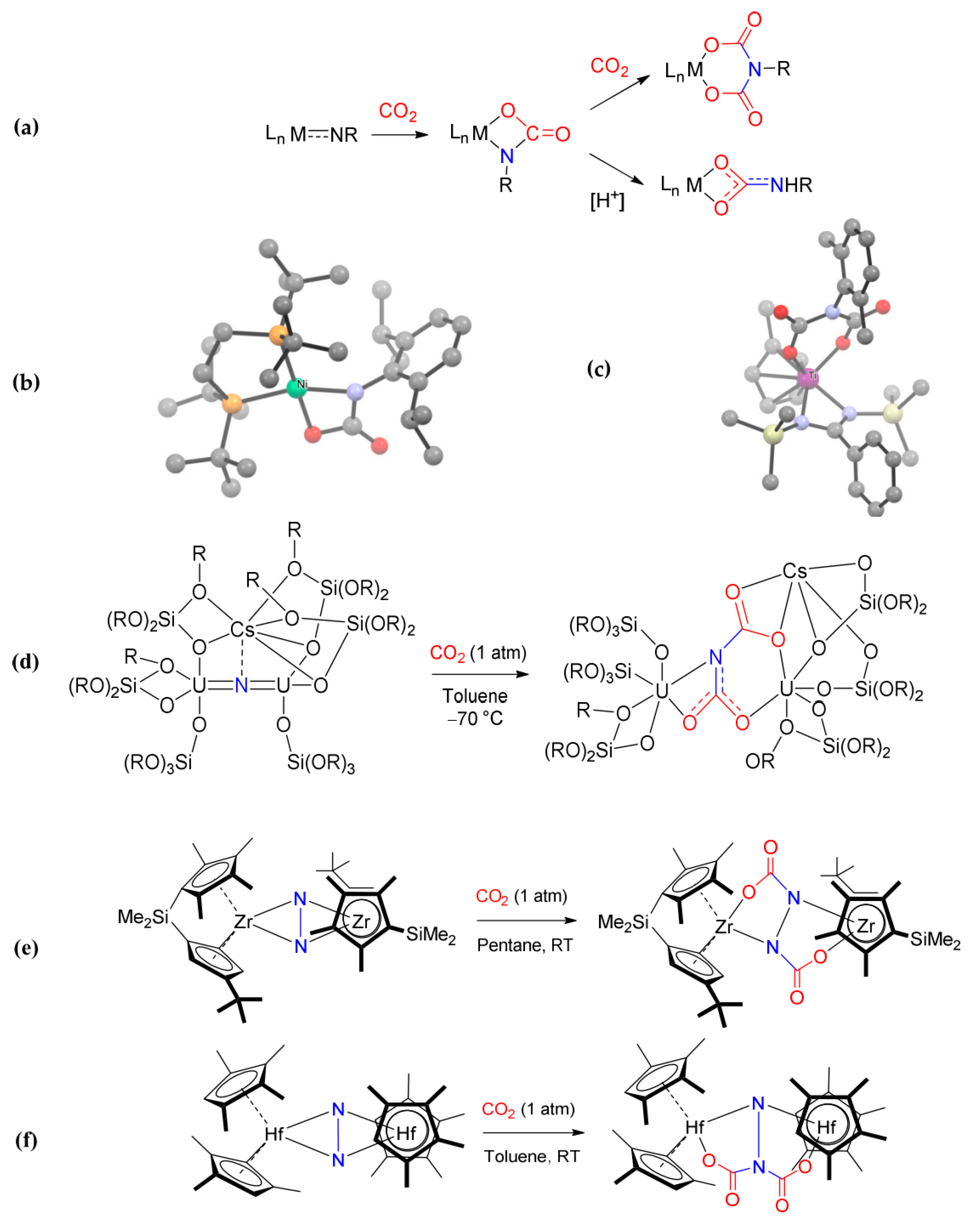{kind=link}
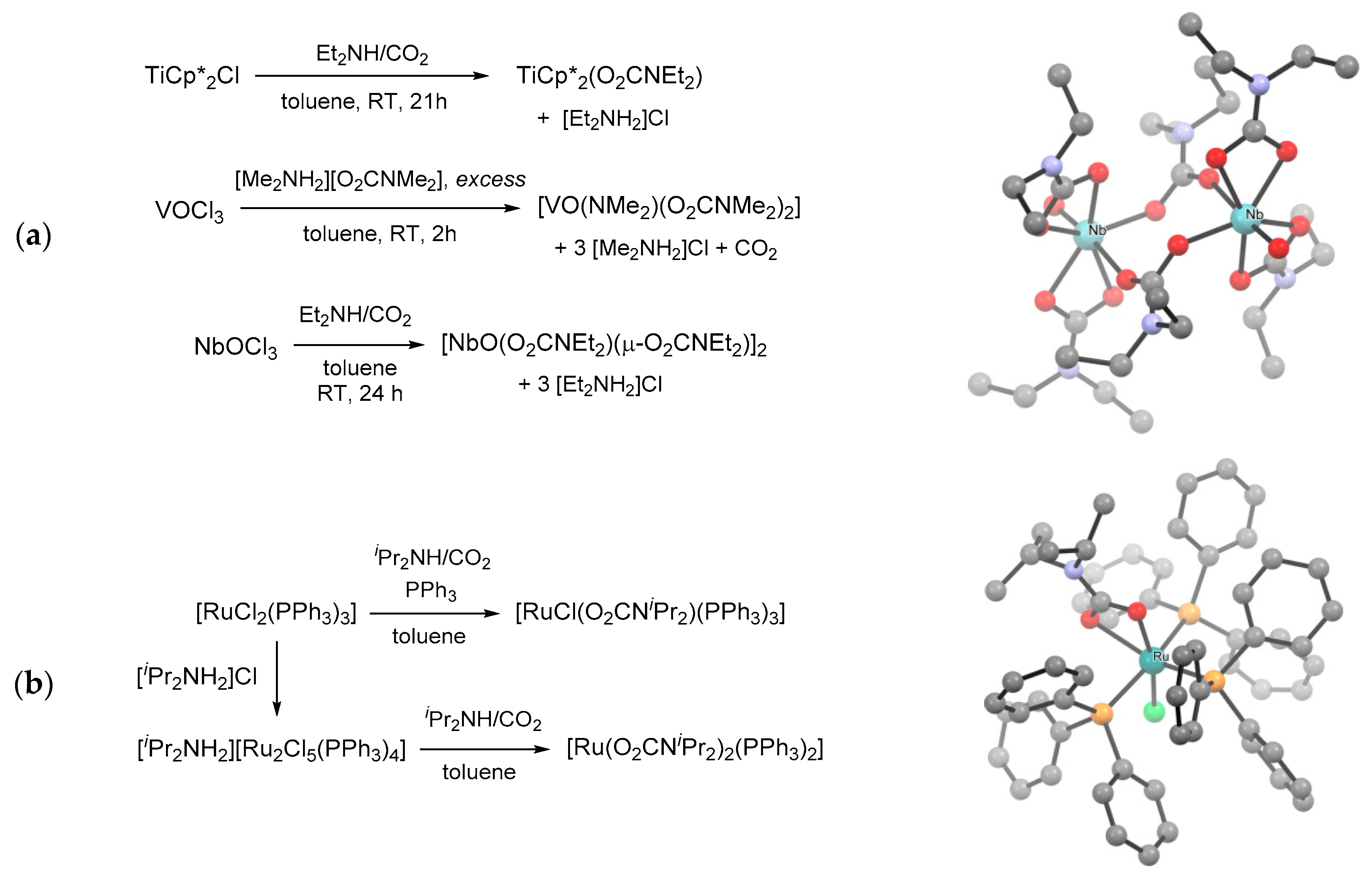{kind=link}
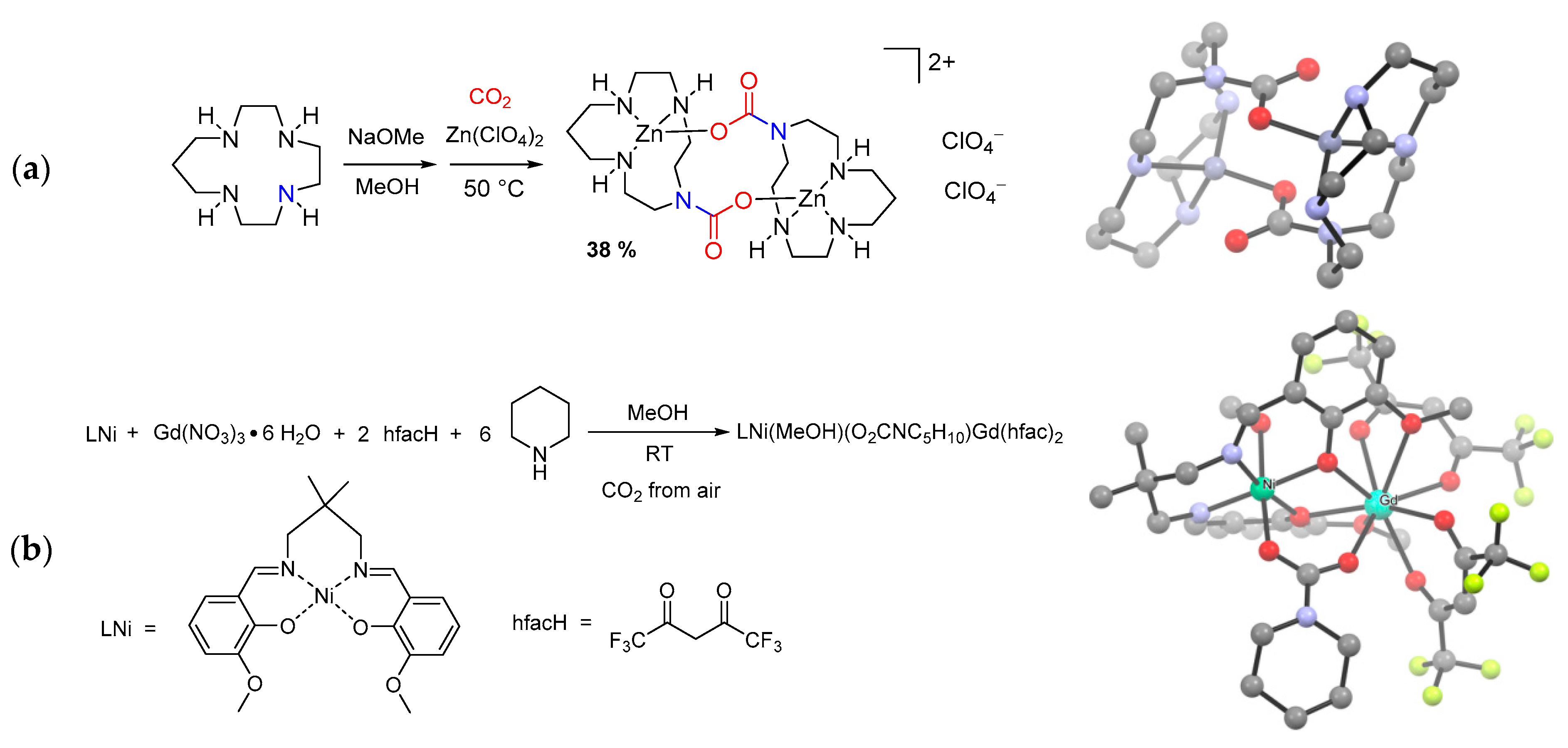{kind=link}
{kind=link}
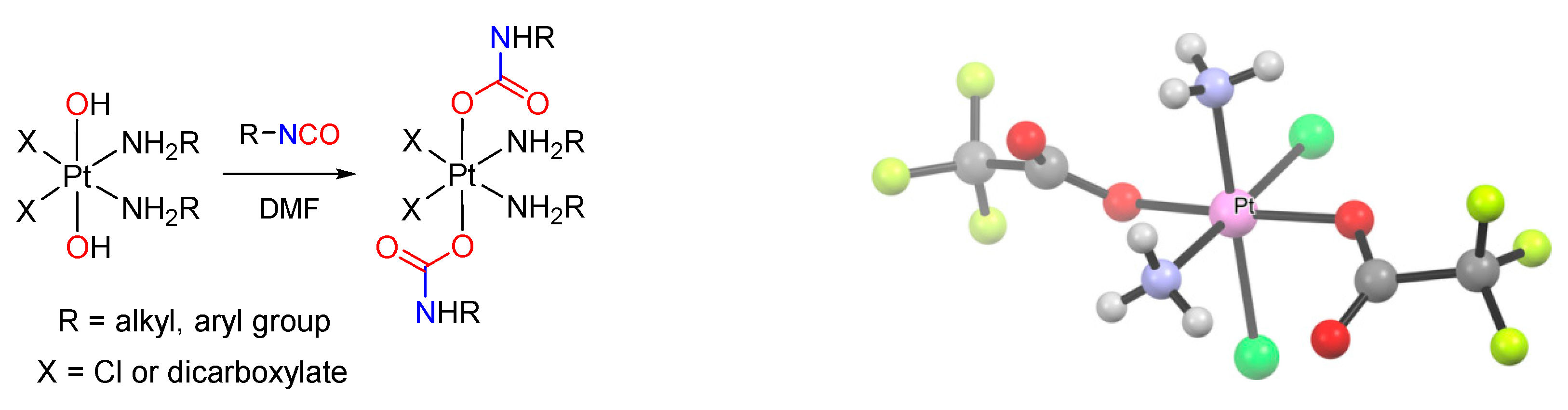{kind=link}
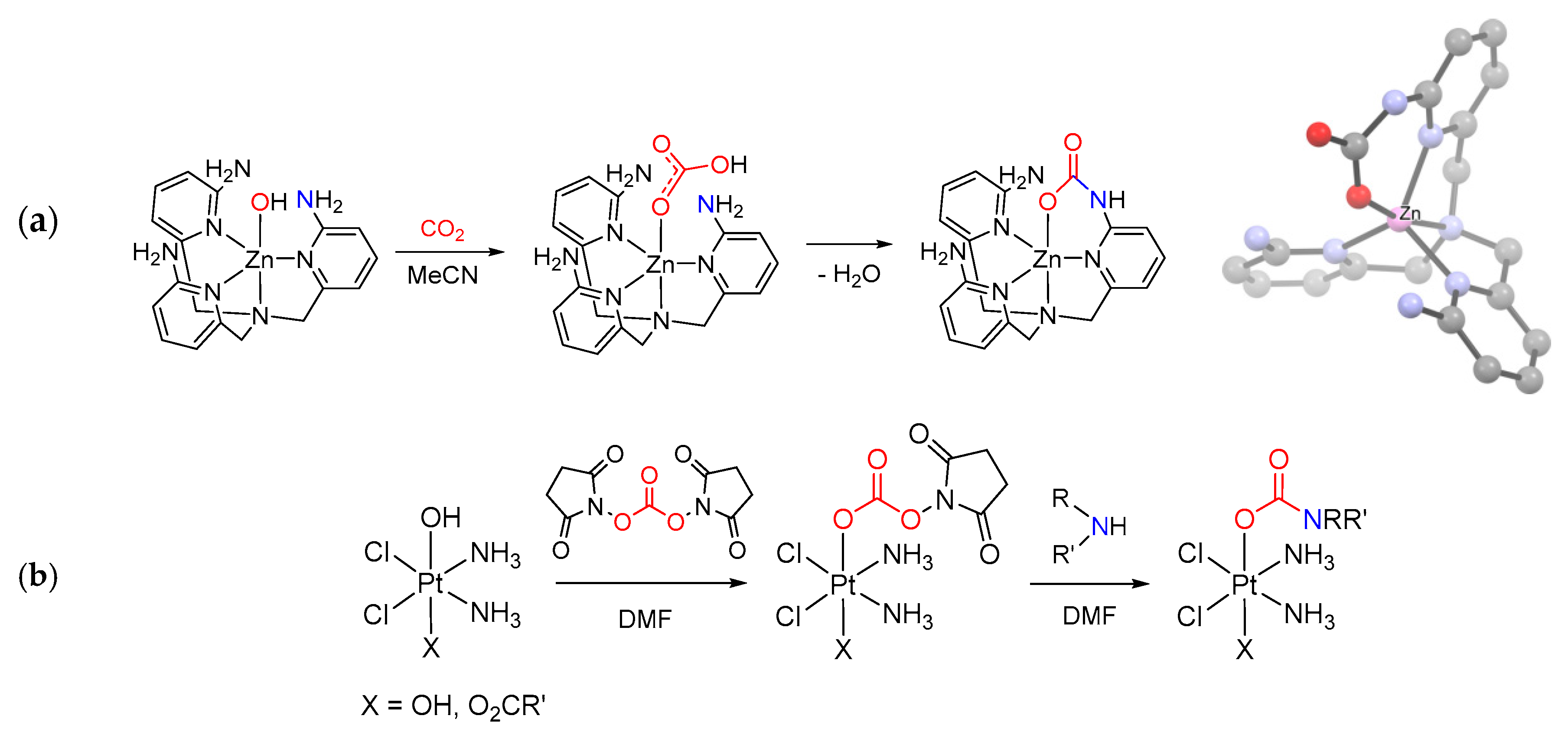{kind=link}
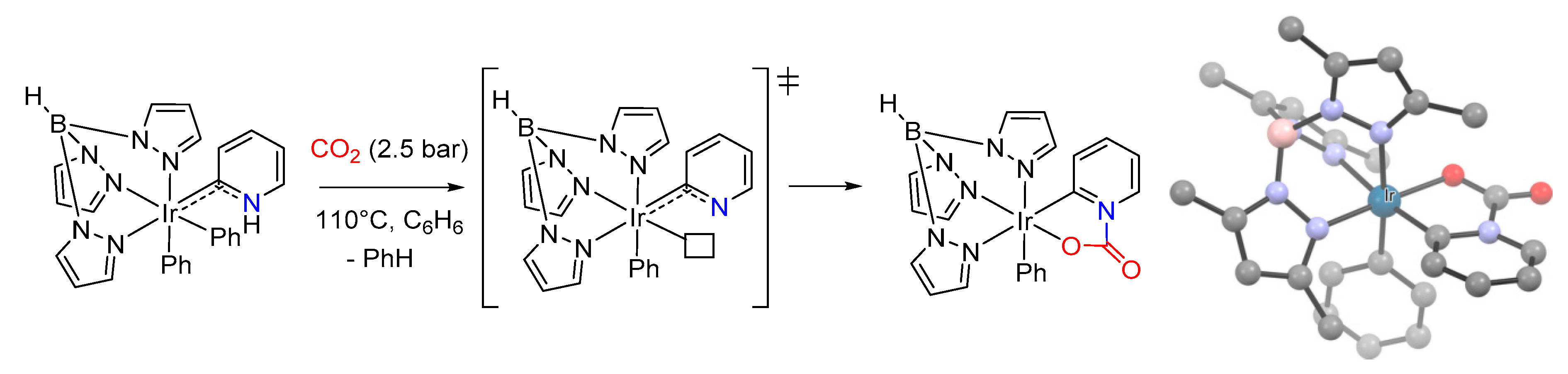{kind=link}
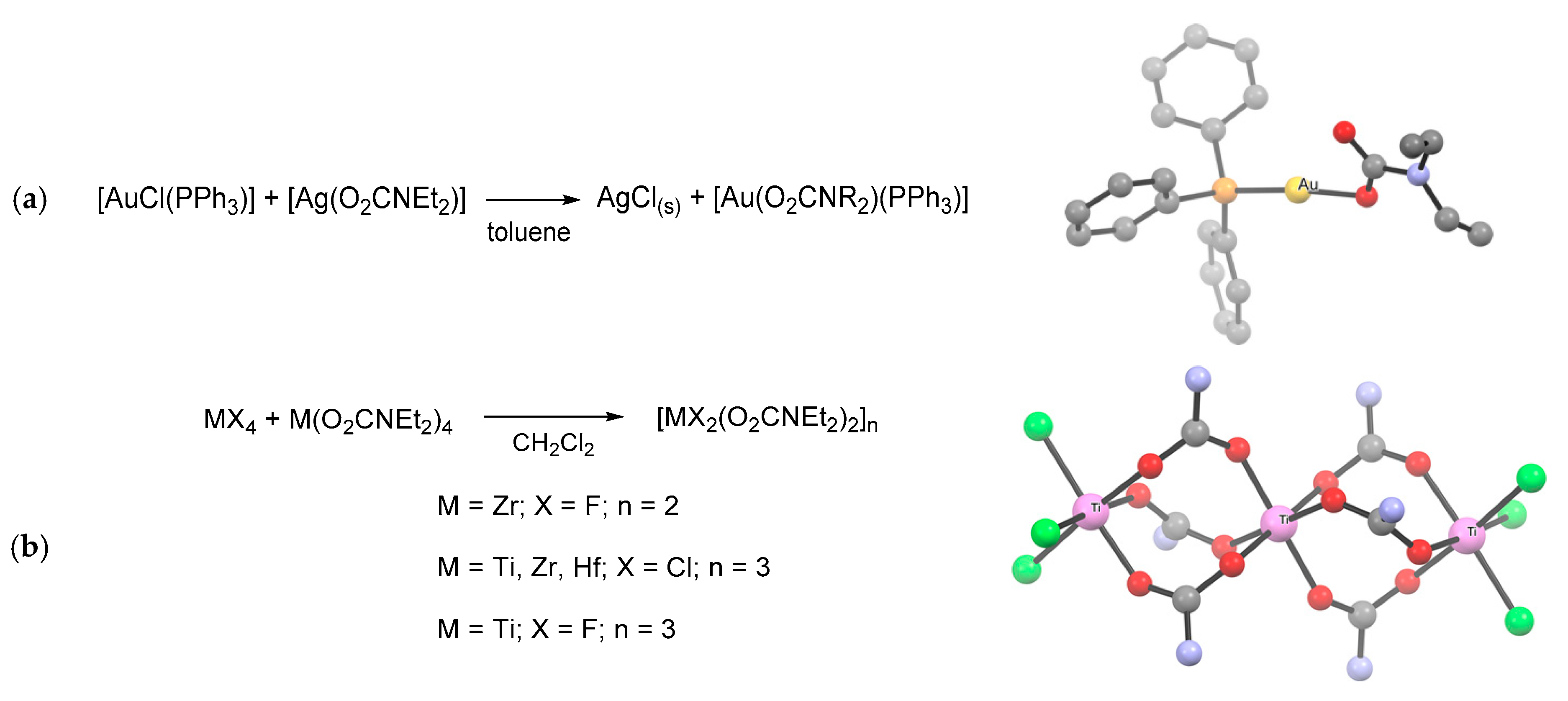{kind=link}
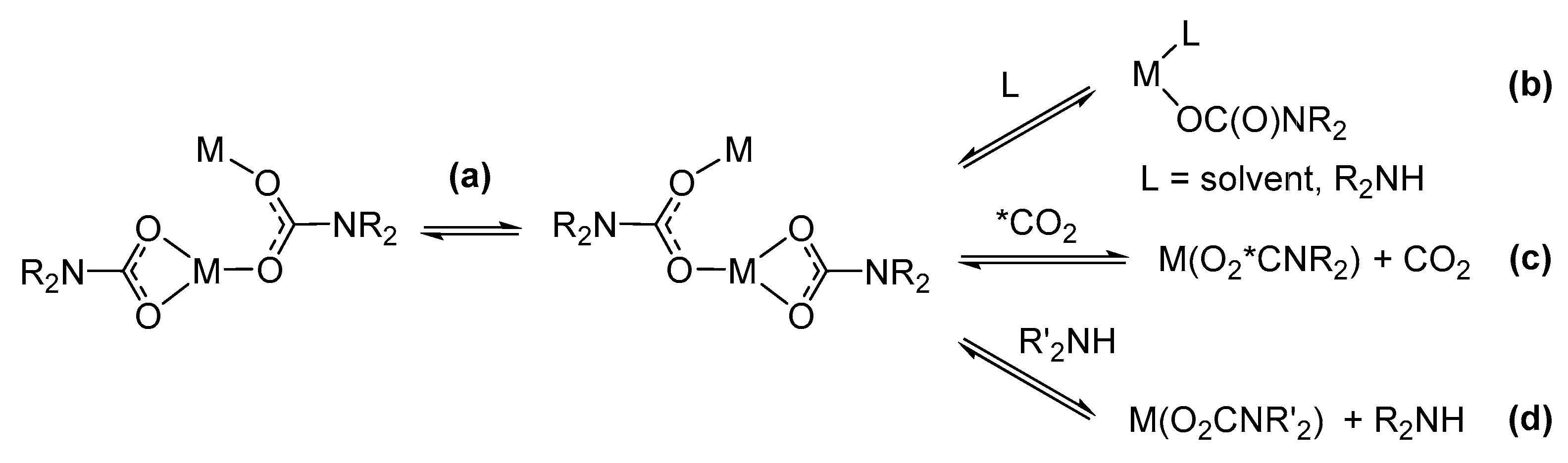{kind=link}
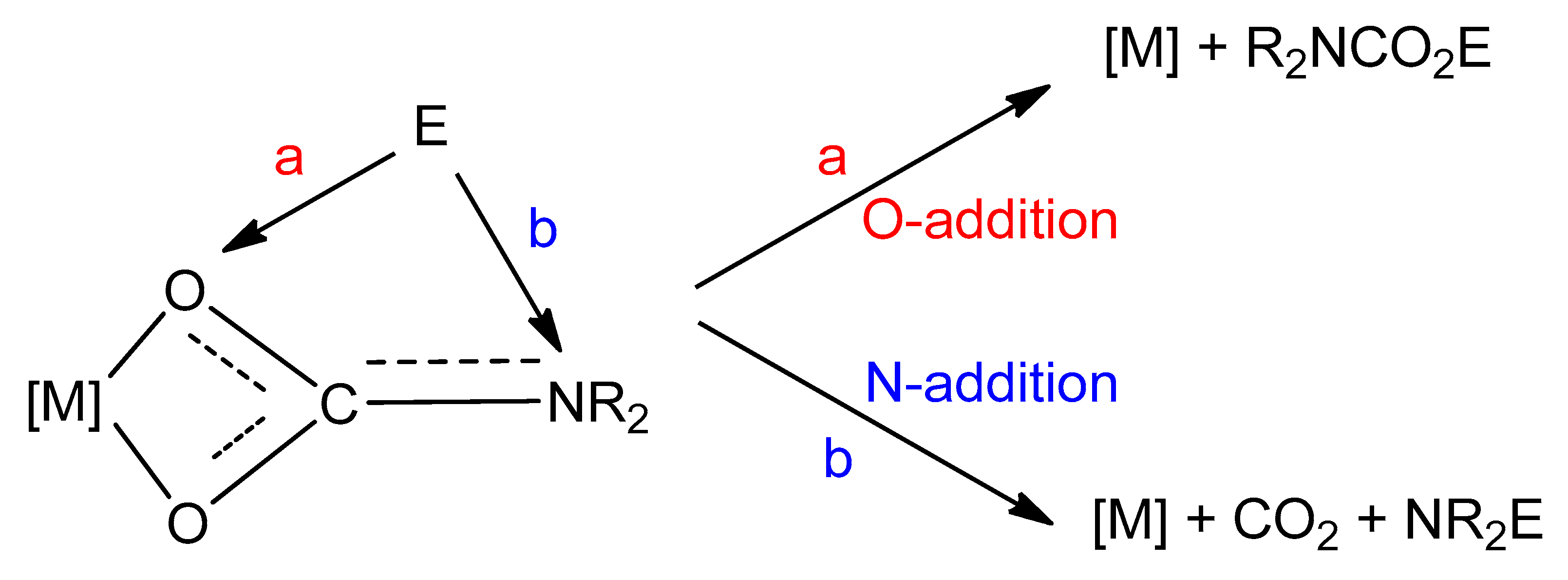{kind=link}
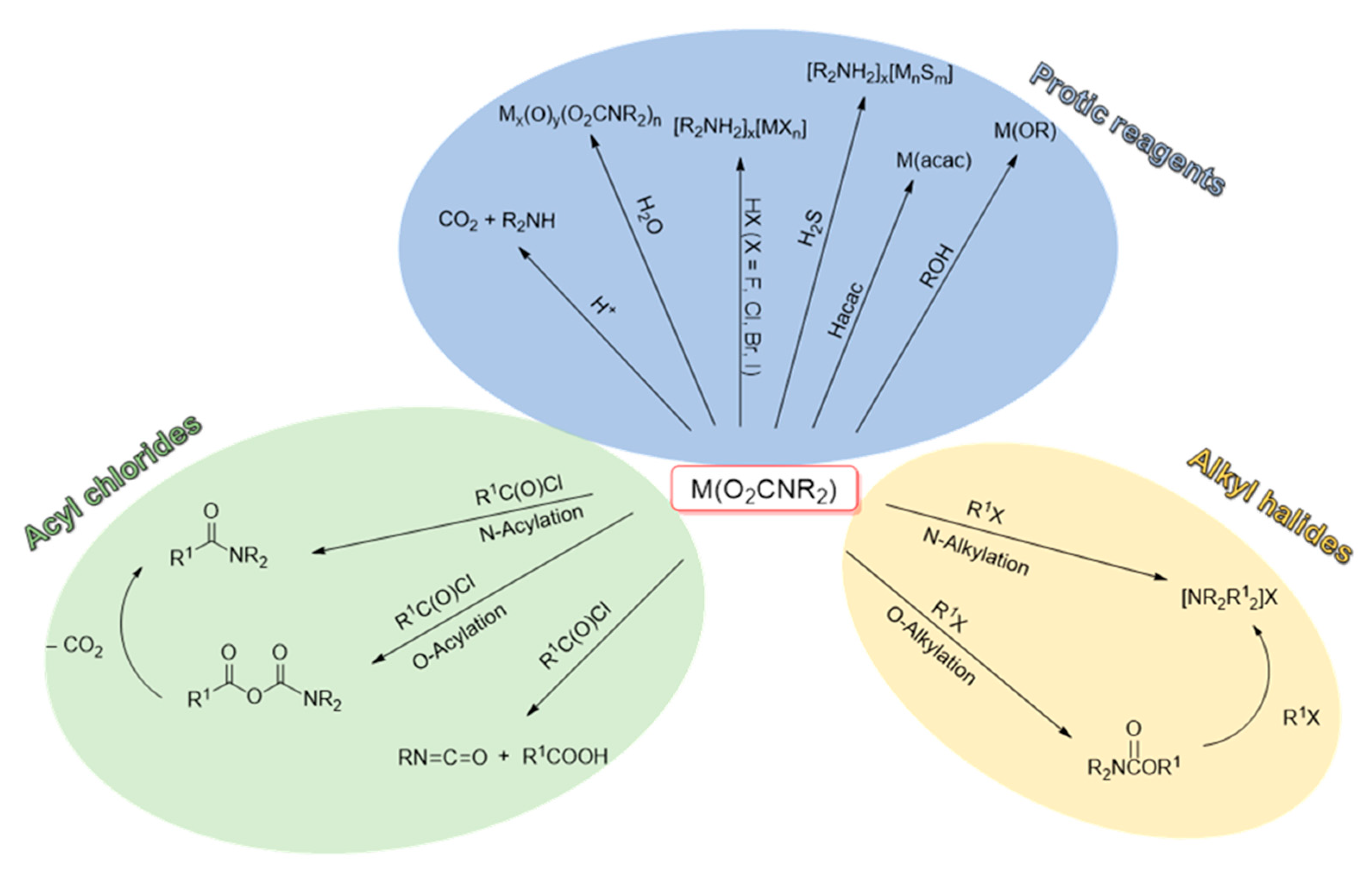{kind=link}
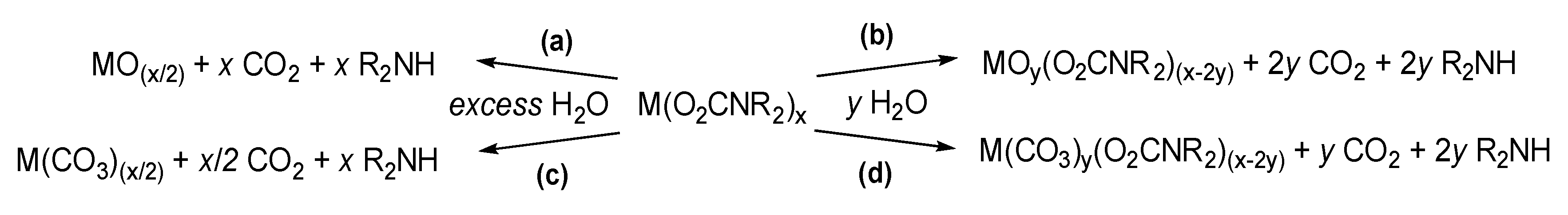{kind=link}
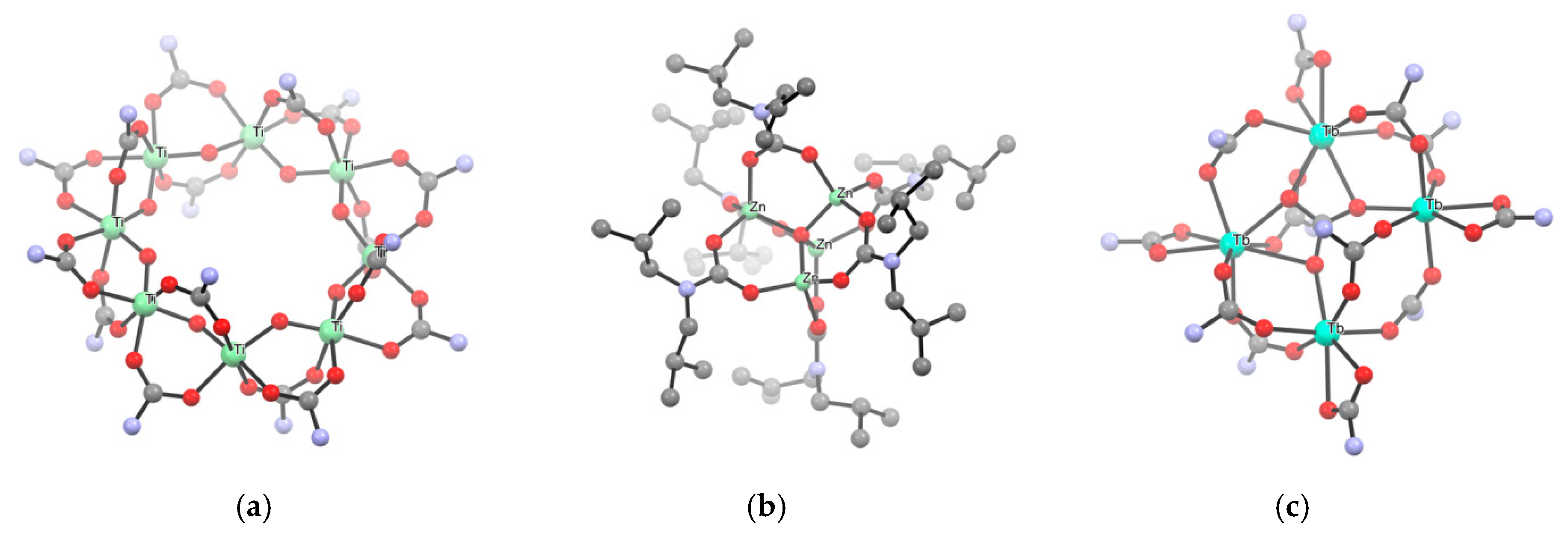{kind=link}
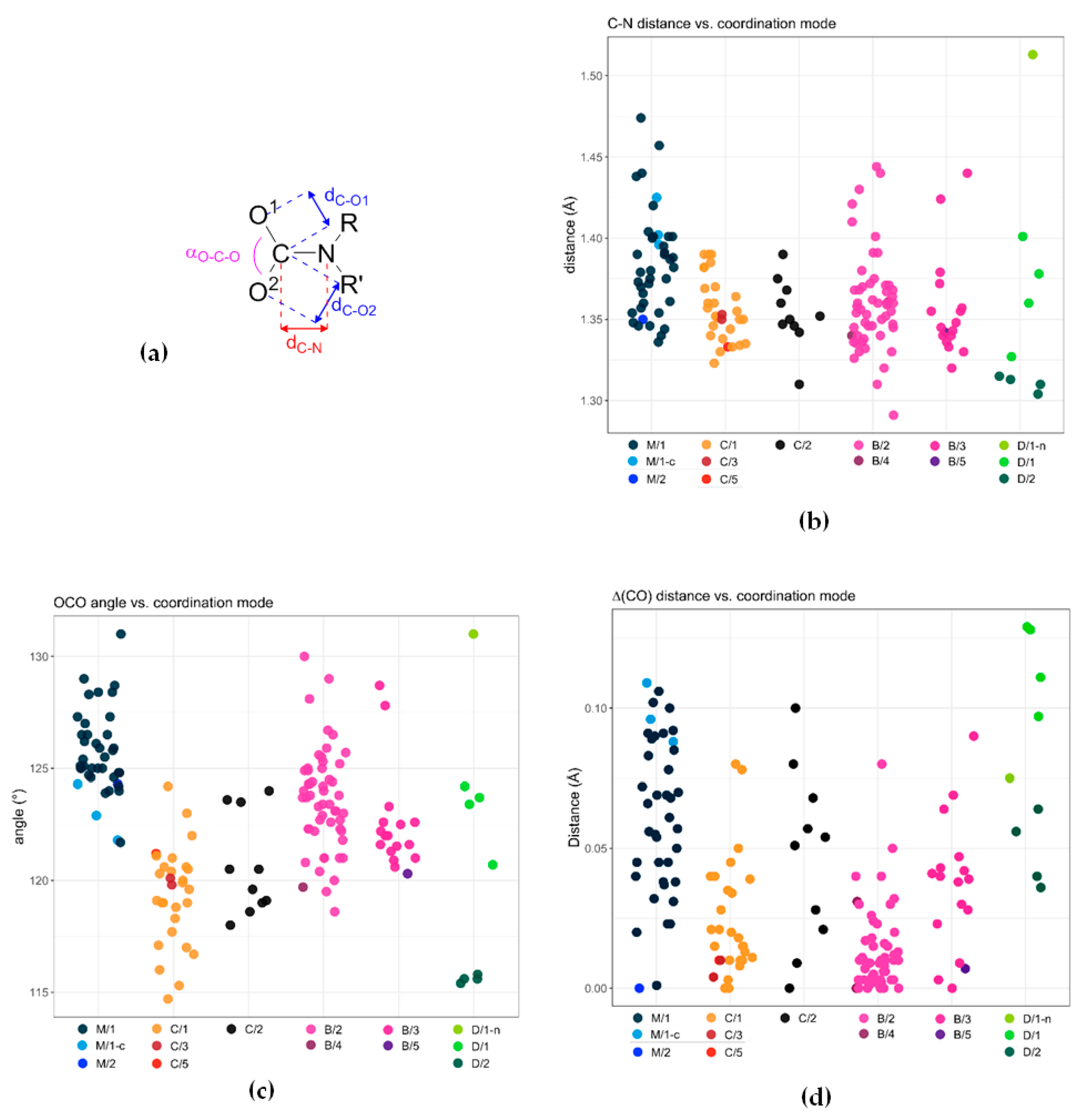{kind=link}
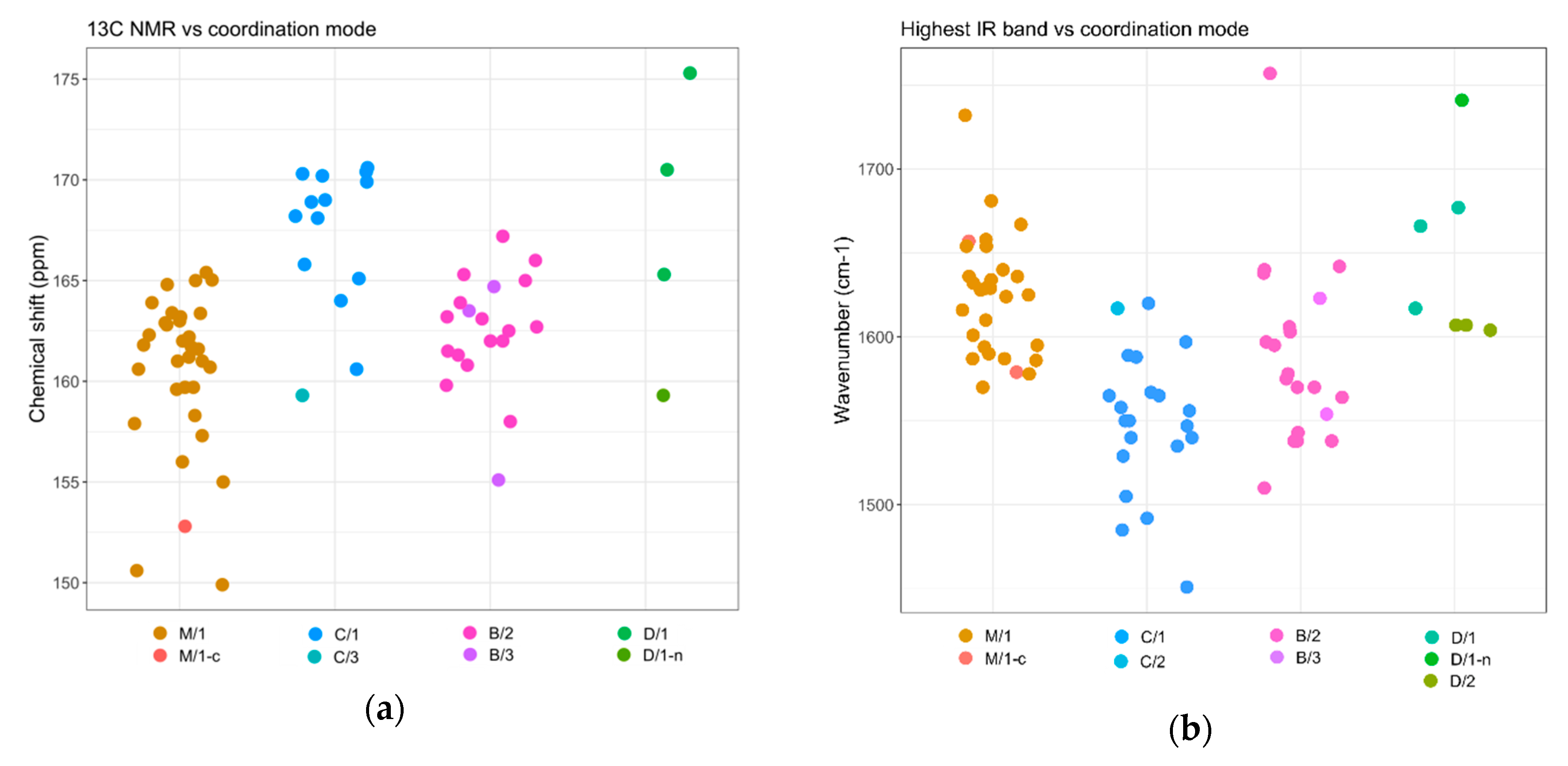{kind=link}
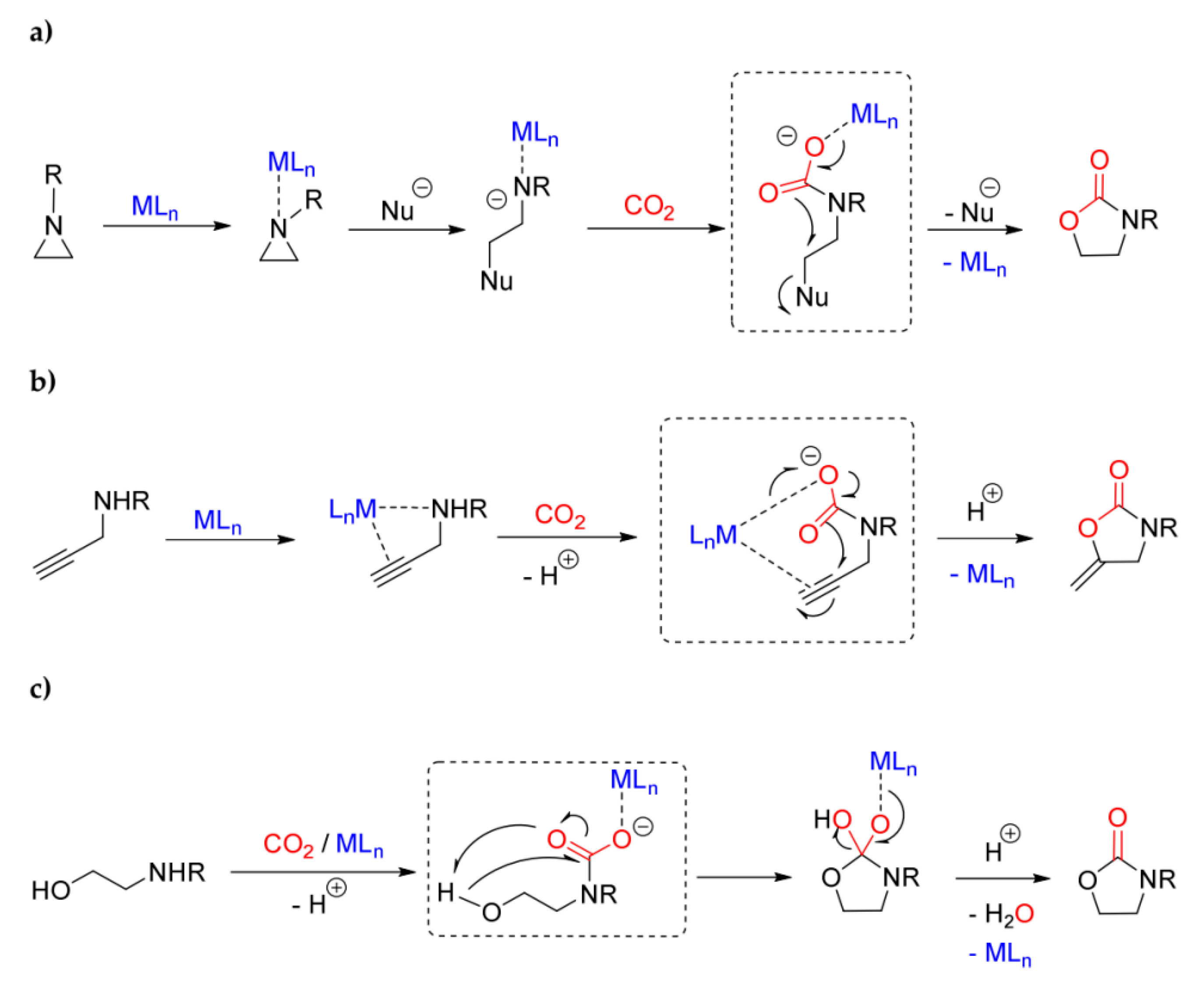{kind=link}
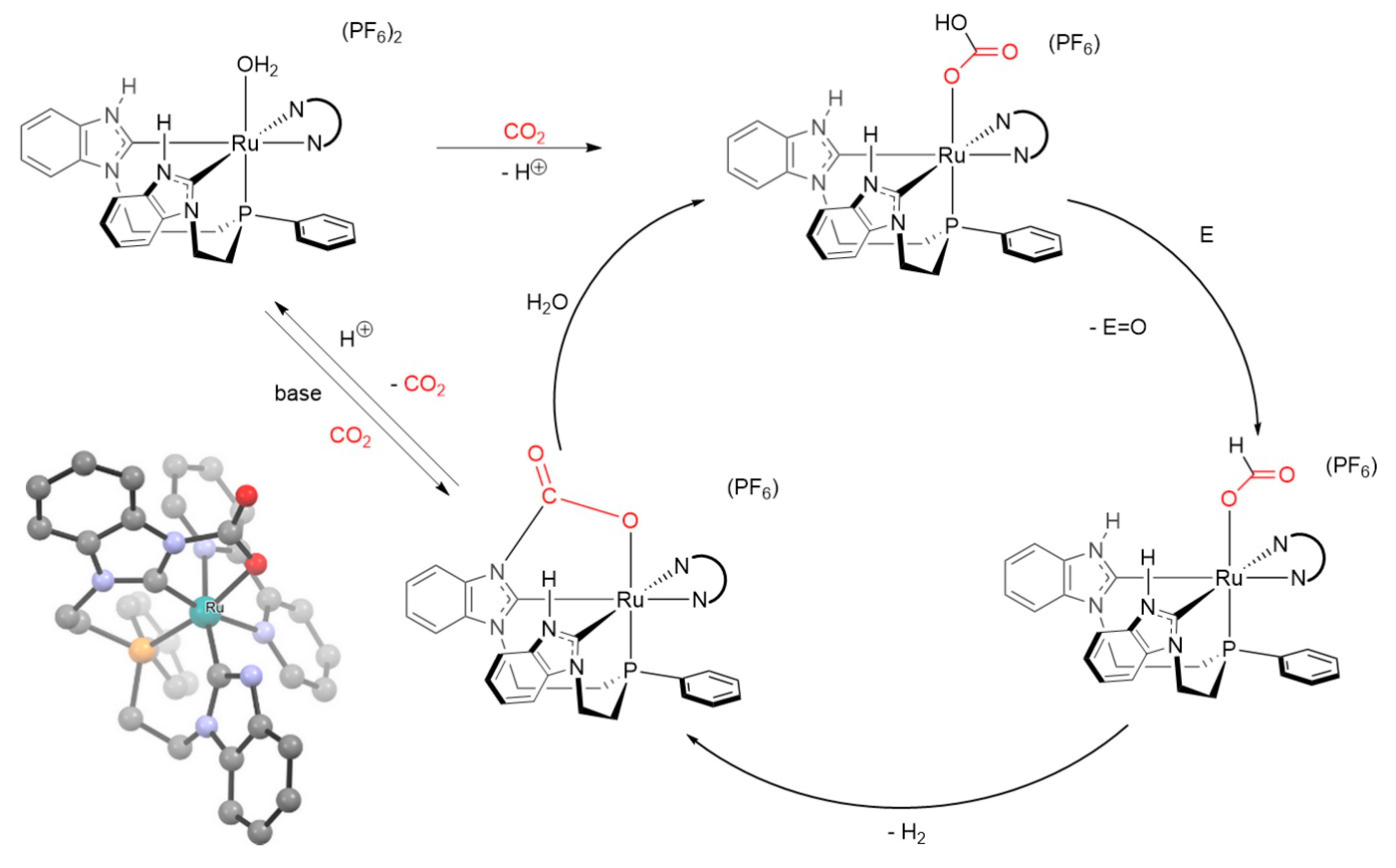{kind=link}
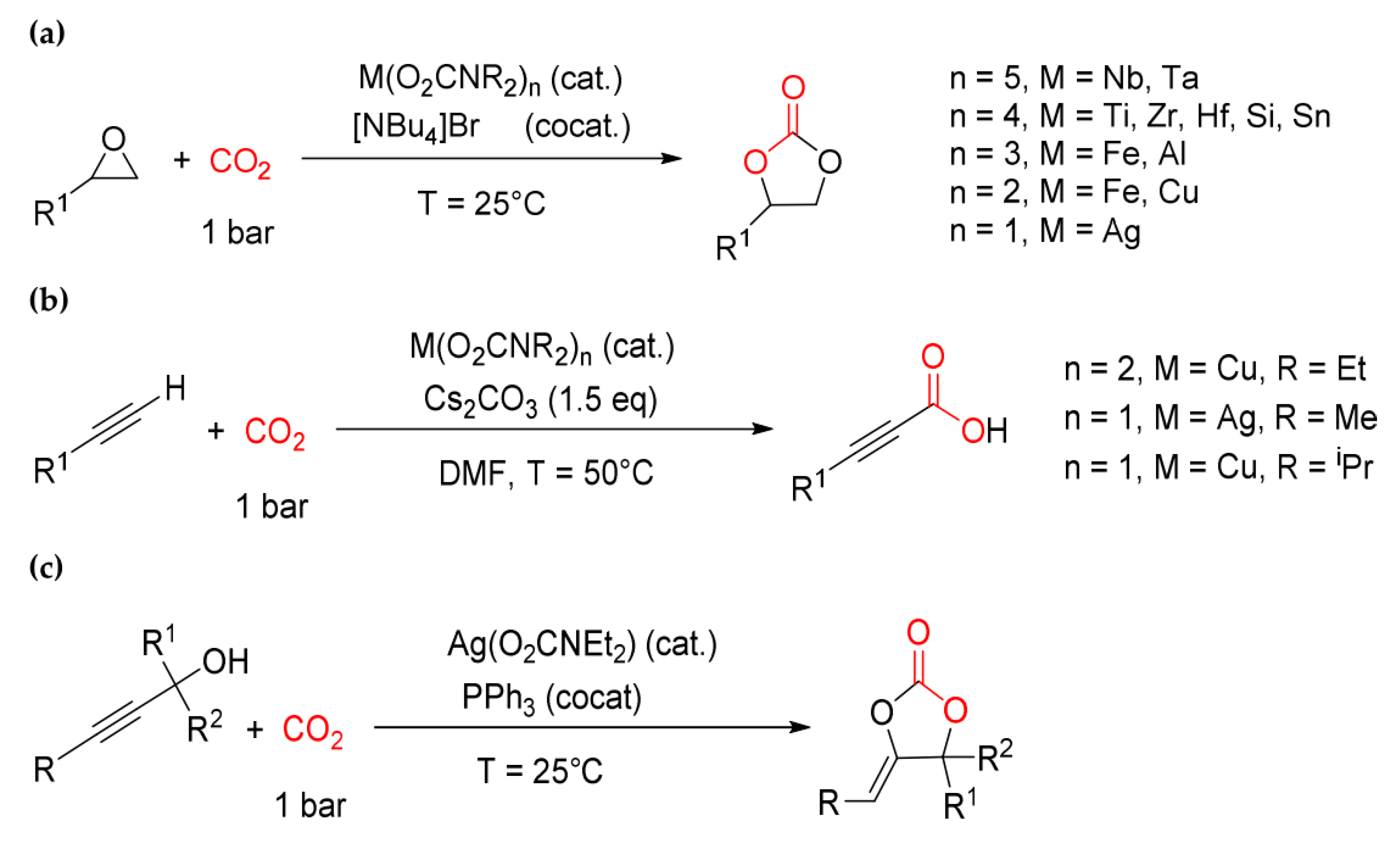{kind=link}
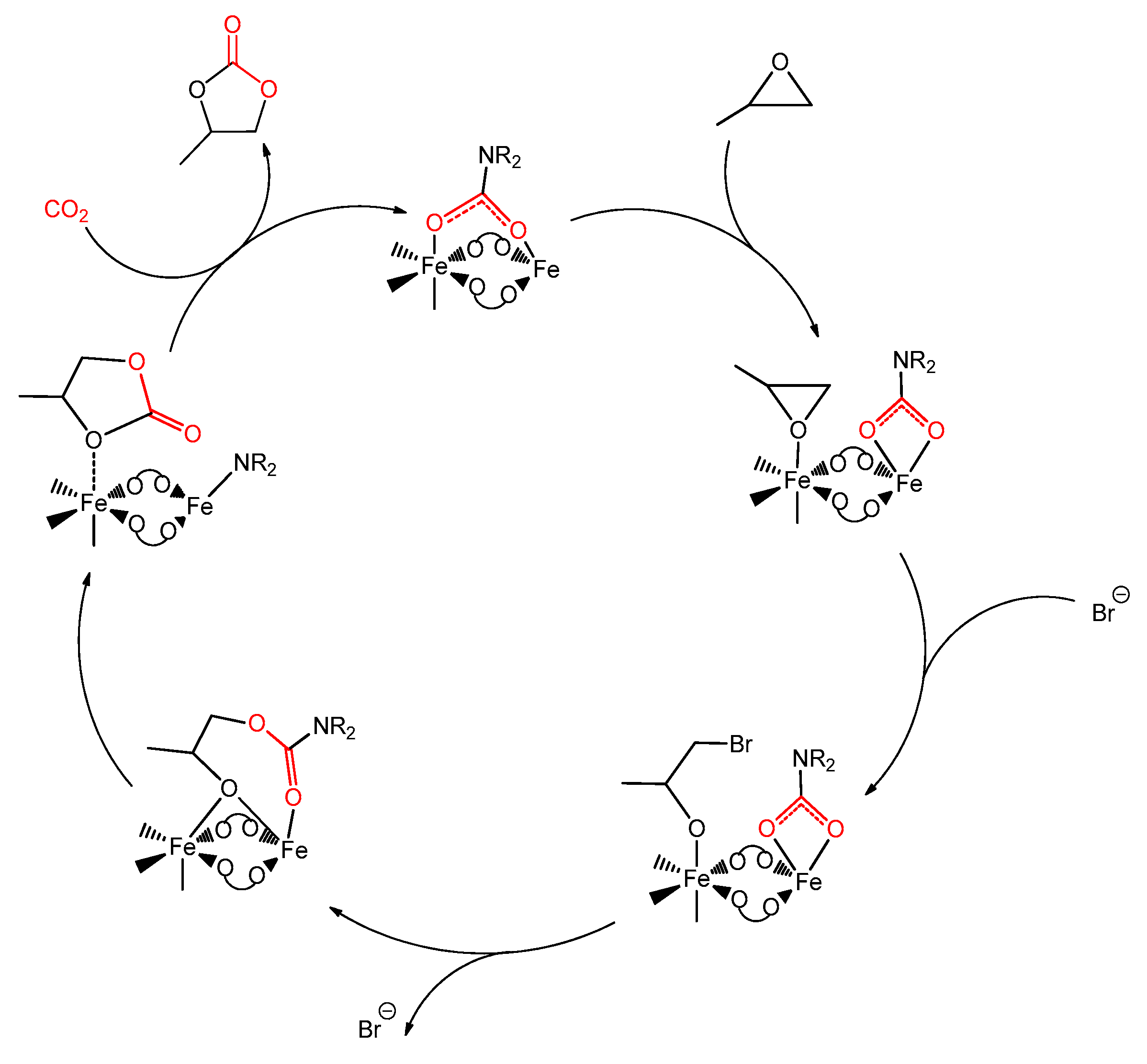{kind=link}
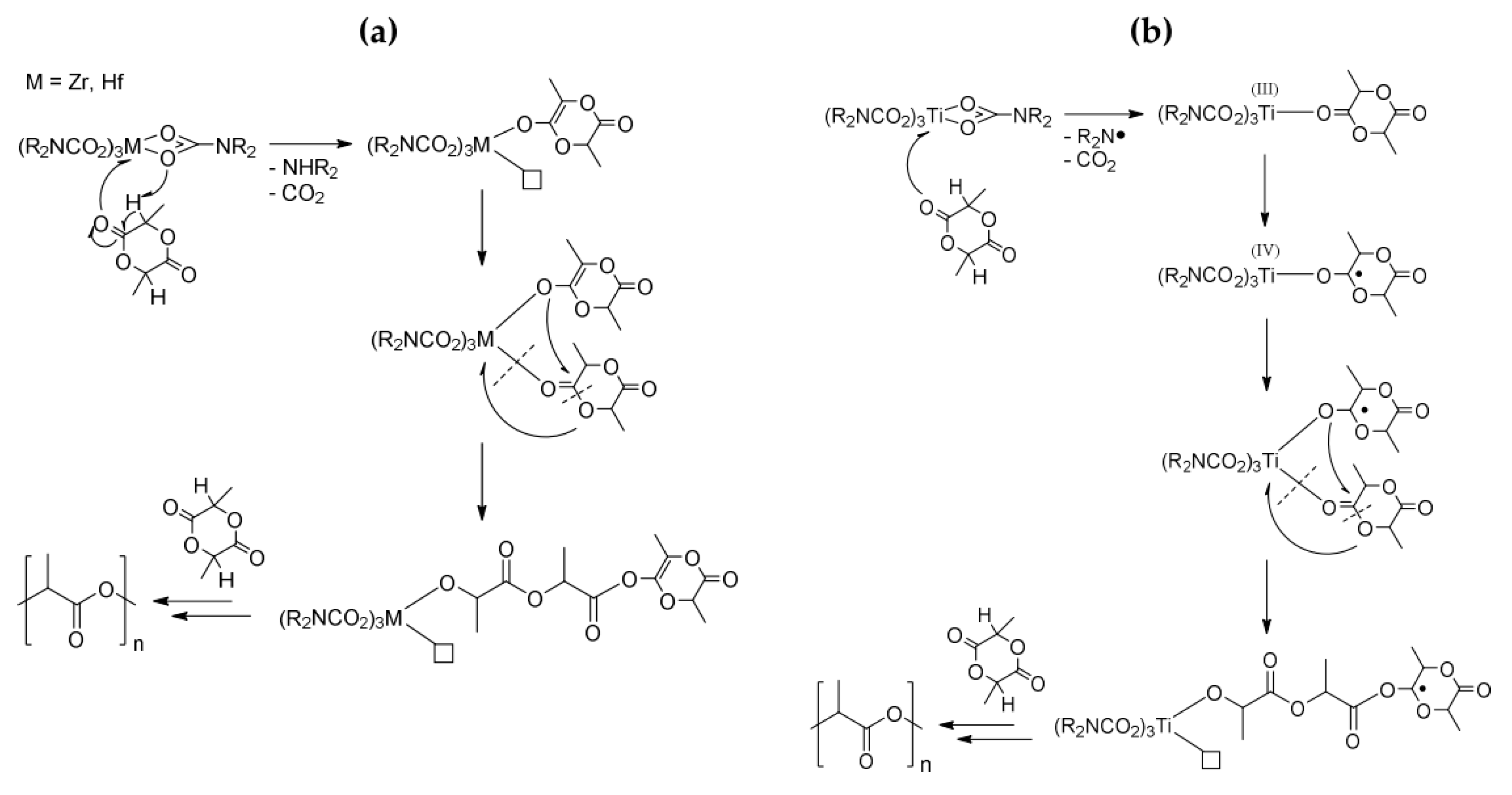{kind=link}
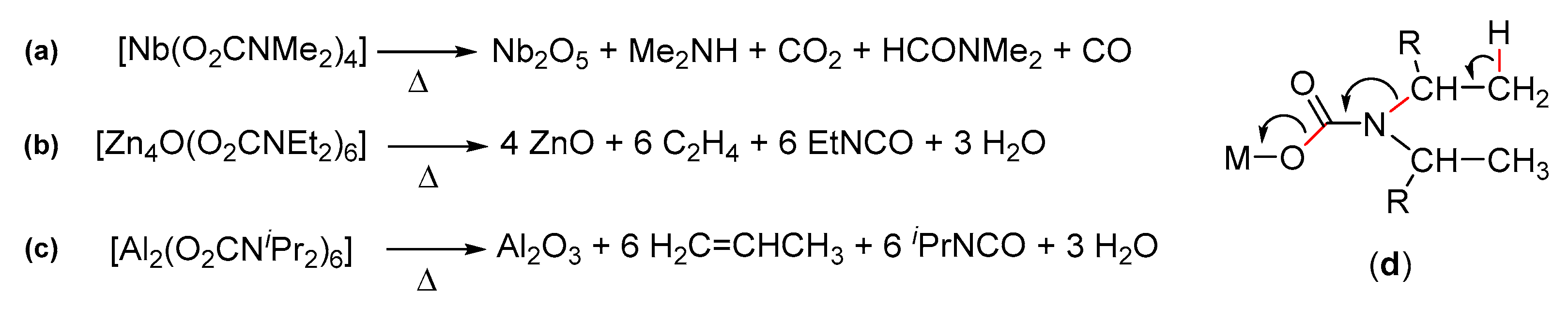{kind=link}
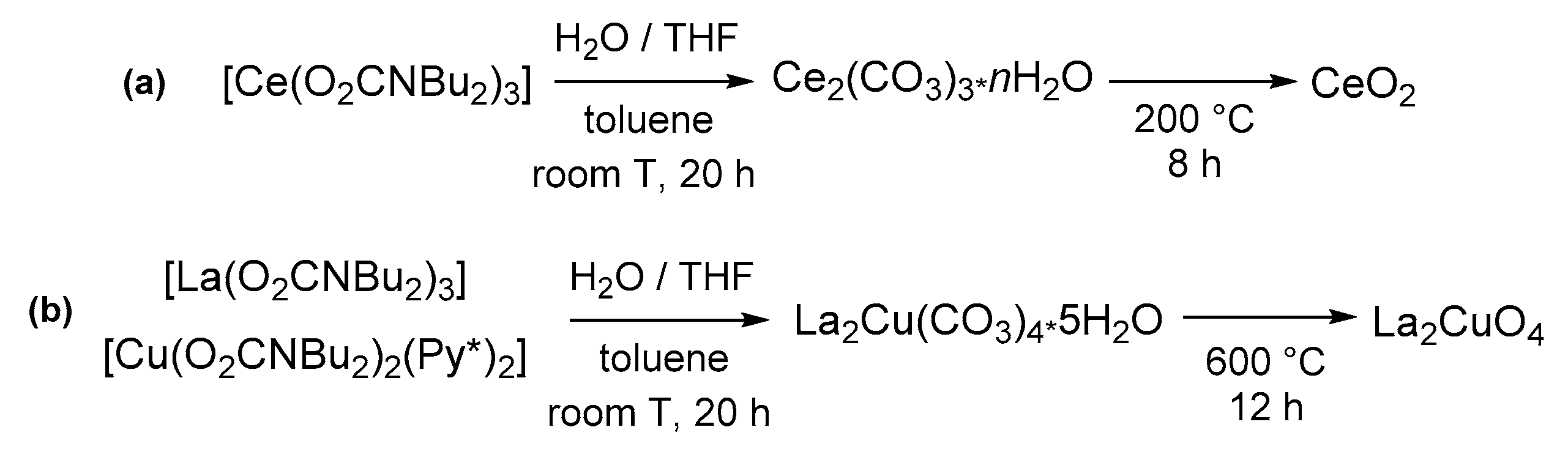{kind=link}
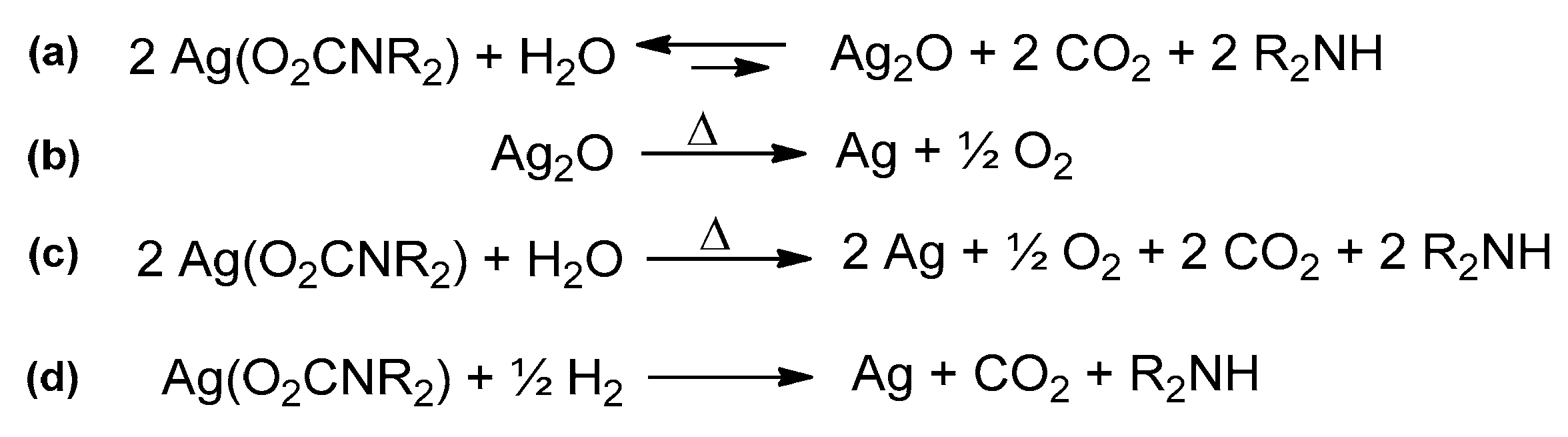{kind=link}
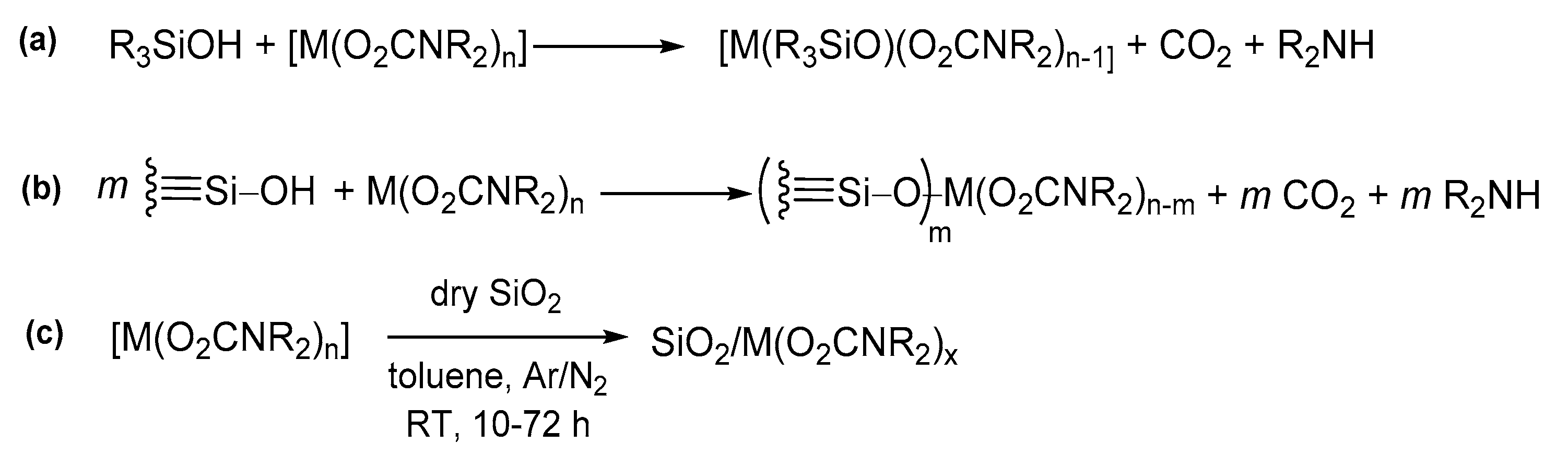{kind=link}
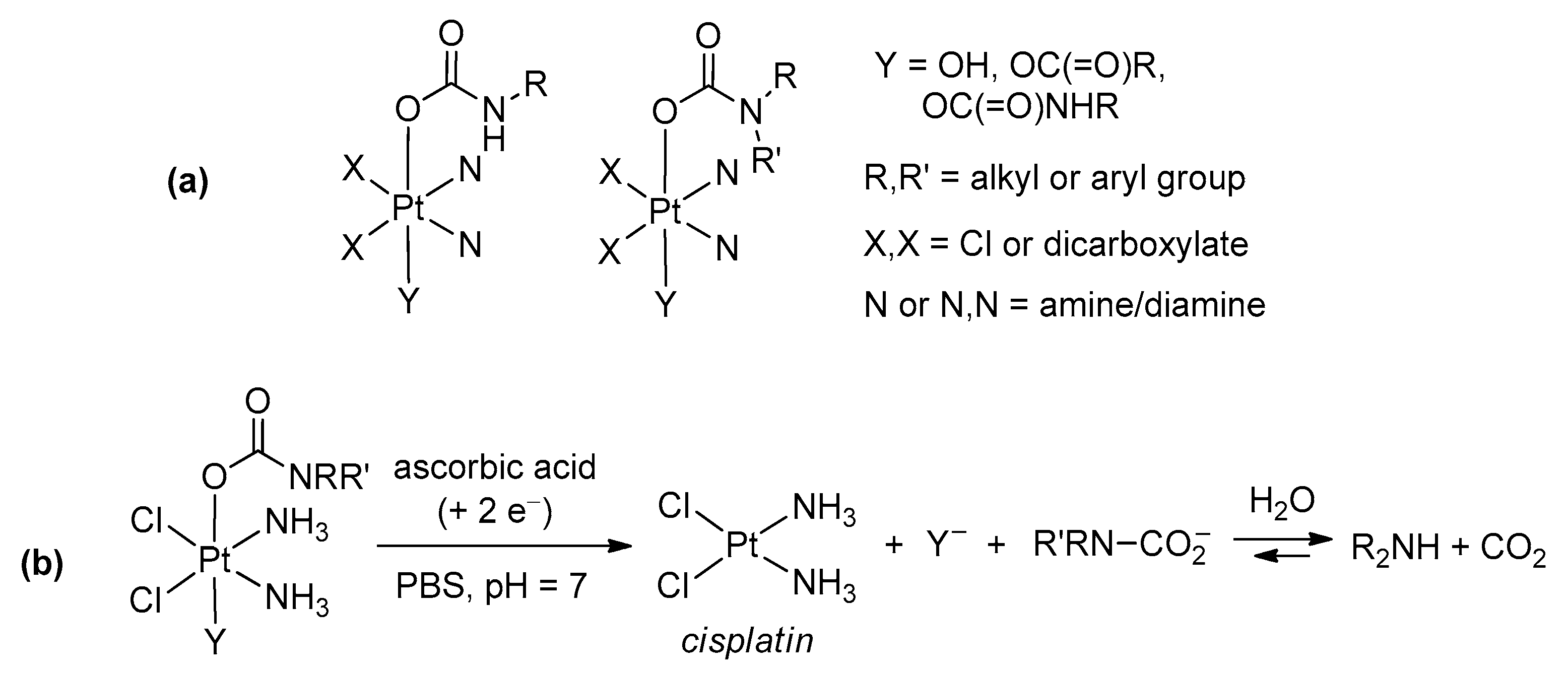{kind=link}
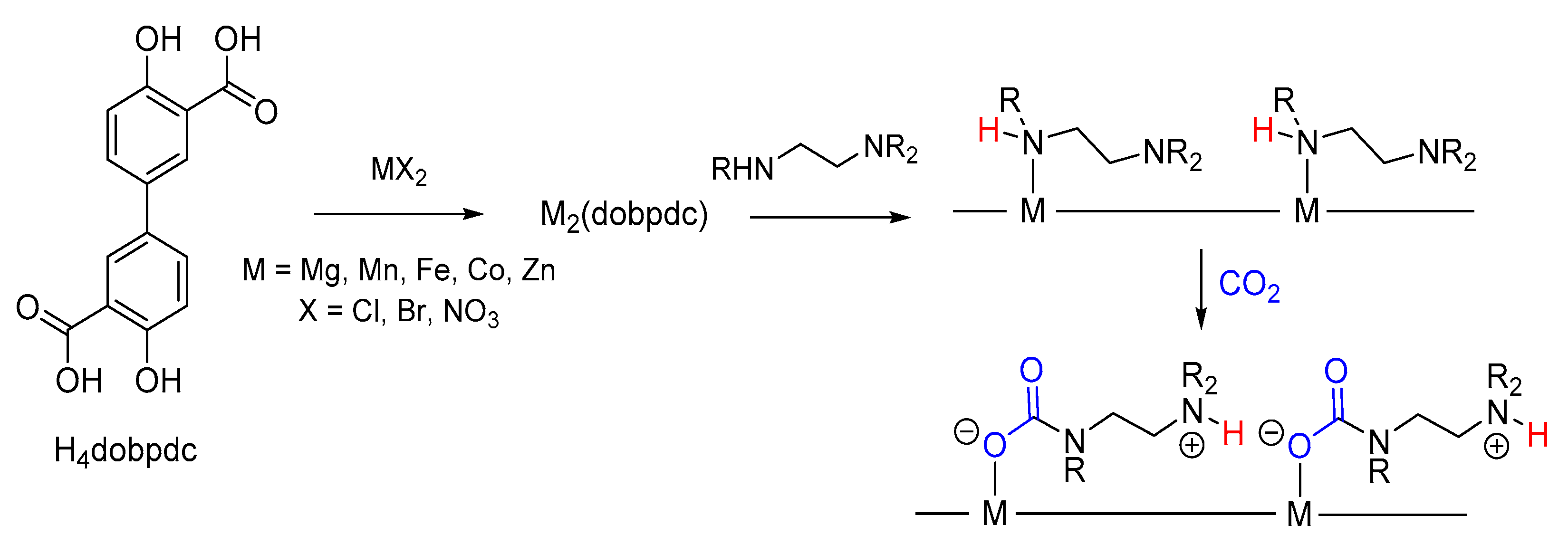{kind=link}
| Amine | pKb [a] | KCBM | KHYD | Ref. |
|---|---|---|---|---|
| NH3 | 4.76 | 2.3 × 103 | 4.4 × 10−1 | [42] |
| NH2Me | 3.38 | 4.0 × 106 | 6.0 × 10−3 | [42] |
| NHMe2 | 3.22 | 1.6 × 106 | 2.2 × 10−2 | [42] |
| NH2Et | 3.19 | 2.0 × 106 | 1.8 × 10−2 | [42] |
| NHEt2 | 3.51 | 7.4 × 104 | 2.4 × 10−1 | [42] |
| NH2iPr | 3.37 | 3.6 × 105 | 6.3 × 10−2 | [42] |
| NHiPr2 | 3.17 | None | - | [42] |
| NH2sBu | 3.44 | 3.8 × 105 | 4.9 × 10−2 | [42] |
| NHsBu2 | - | None | - | [42] |
| NH2Ph | 9.30 | 8.1 × 10−3 | 3.6 | [42] |
| Piperidine | 2.95 | 7.9 × 105 | 8.1 × 10−2 | [42] |
| 3-MPD | 3.12 | 6.9 × 106 | 6.2 × 10−3 | [58] |
| 4-MPD | 3.06 | 5.9 × 106 | 8.3 × 10−3 | [58] |
| Pyrrolidine | 3.16 | 1.9 × 107 | 2.0 × 10−3 | [54,55] |
| MEA | 4.42 | 6.0 × 104 | 1.9 × 10−2 | [42] |
| DEA | 4.98 | 2.1 × 103 | 1.5 × 10−1 | [42] |
| 1-AP [b] | 4.75 | 9.6 × 103 | 1.1 × 10−1 | [54] |
| 2-AP [b] | 4.75 | 4.0 × 103 | 2.5 × 10−1 | [52] |
| MPA [b] | 4.18 | 2.5 × 105 | 1.5 × 10−2 | [52] |
| AMP [b] | 4.73 | None | - | [52] |
| 4-PIPDM [b] | 3.71 | 2.7 × 105 | 4.1 × 10−2 | [54,55] |
| 4-PIPDE [b] | 3.65 | 3.0 × 105 | 4.2 × 10−2 | [55] |
| Morpholine [b] | 5.78 | 1.4 × 103 | 6.8 × 10−2 | [54,55] |
| Thiomorpholine [b] | 5.57 | 9.3 × 102 | 1.6 × 10−1 | [54,55] |
| Piperazine | 4.50 | 5.5 × 104 | 3.2 × 10−2 | [53] |
| 8.67 | 2.6 × 10−1 | 4.6 × 10−1 | ||
| MPIPZ | 4.96 | 5.1 × 103 | 1.2 × 10−1 | [54] |
| 4-AMTHP [b] | 4.37 | 1.9 × 105 | 1.3 × 10−2 | [56] |
| Taurine [b],[c] | 5.19 | 7.1 × 103 | 5.1 × 10−2 | [57] |
| Amino acids | ||||
| Glycine[d] | 4.49 | 4.4 × 104 | 4.2 × 10−2 | [59] |
| Sarcosine [b],[d] | 4.22 | 3.3 × 104 | 1.0 × 10−1 | [60] |
| α-Alanine [d] | 4.40 | 1.8 × 104 | 1.1 × 10−1 | [61] |
| β-Alanine [d] | 3.86 | 1.9 × 105 | 3.1 × 10−2 | [61] |
| Proline [c] | 3.57 | 5.4 × 105 | 2.8 × 10−2 | [62] |
| Lysine [b],[c] | 4.44 [e] | 1.7 × 104 | 1.2 × 10−1 | [63] |
| 3.24 [f] | 6.3 × 104 | 5.1 × 10−1 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bresciani, G.; Biancalana, L.; Pampaloni, G.; Marchetti, F. Recent Advances in the Chemistry of Metal Carbamates. Molecules 2020, 25, 3603. https://doi.org/10.3390/molecules25163603
Bresciani G, Biancalana L, Pampaloni G, Marchetti F. Recent Advances in the Chemistry of Metal Carbamates. Molecules. 2020; 25(16):3603. https://doi.org/10.3390/molecules25163603
Chicago/Turabian StyleBresciani, Giulio, Lorenzo Biancalana, Guido Pampaloni, and Fabio Marchetti. 2020. "Recent Advances in the Chemistry of Metal Carbamates" Molecules 25, no. 16: 3603. https://doi.org/10.3390/molecules25163603
APA StyleBresciani, G., Biancalana, L., Pampaloni, G., & Marchetti, F. (2020). Recent Advances in the Chemistry of Metal Carbamates. Molecules, 25(16), 3603. https://doi.org/10.3390/molecules25163603