
New Deoxyribozymes for the Native Ligation of RNA


Abstract
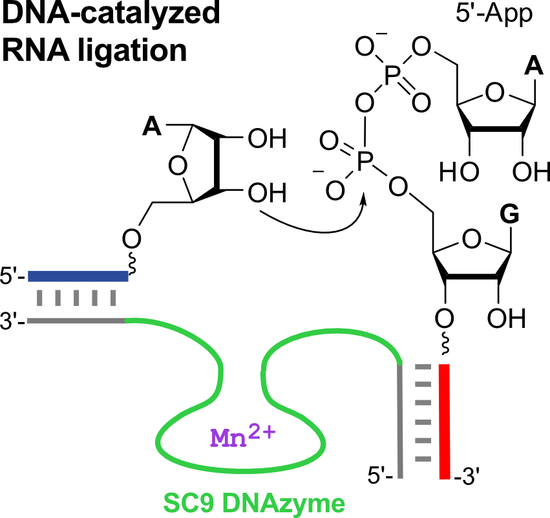
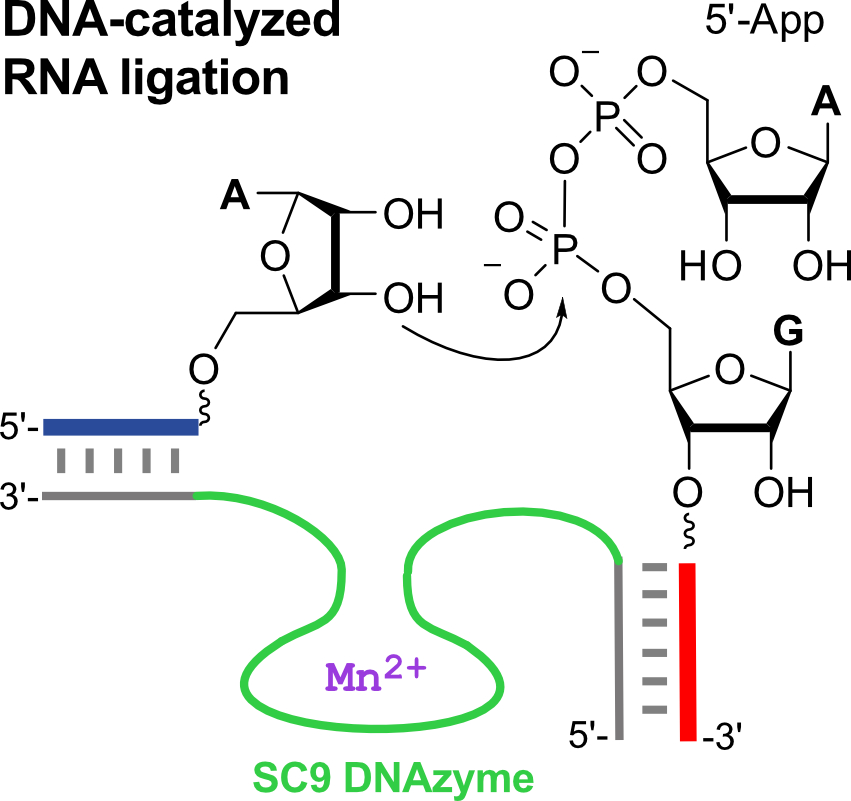
:
1. Introduction
2. Results and Discussion
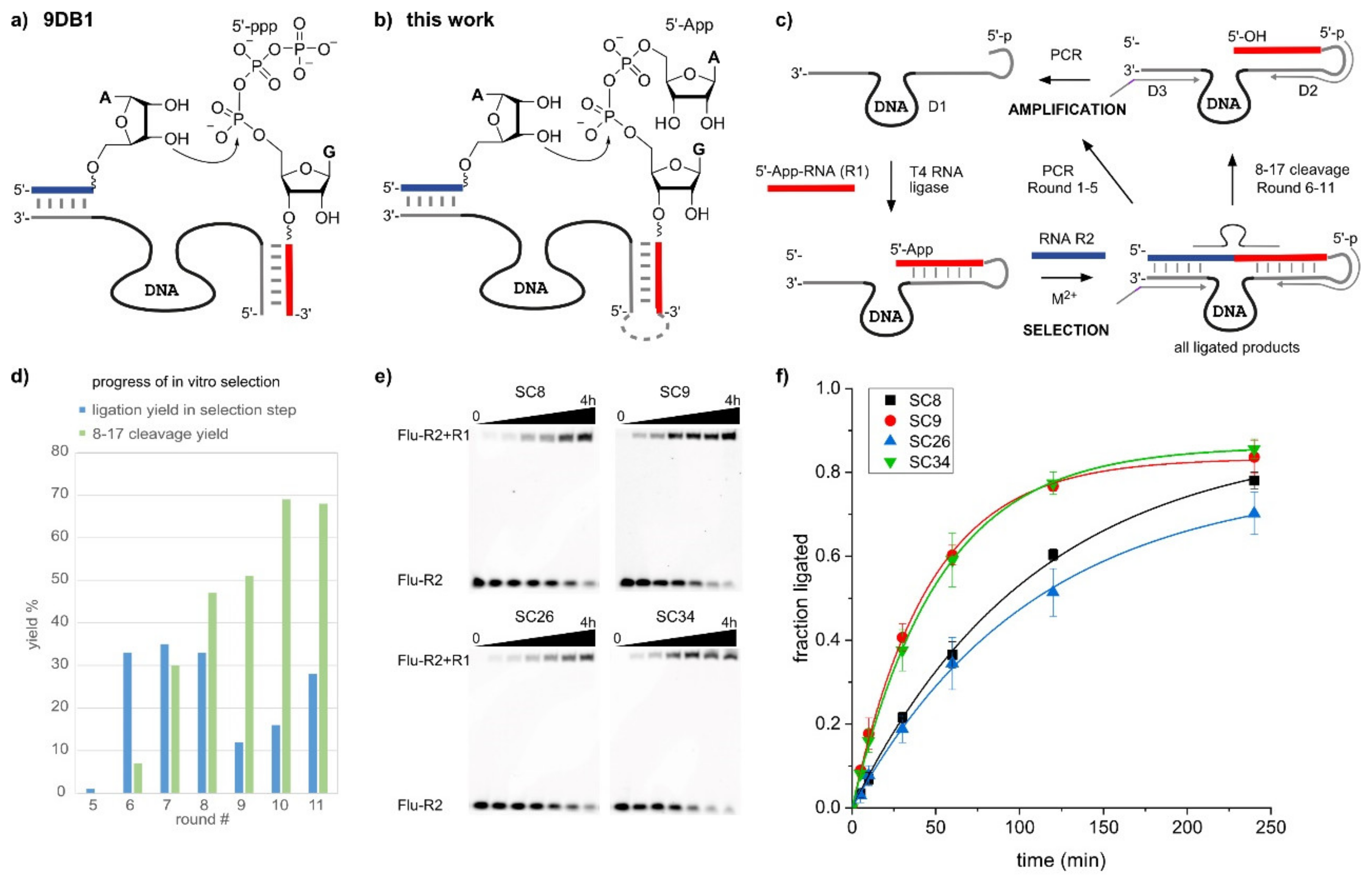
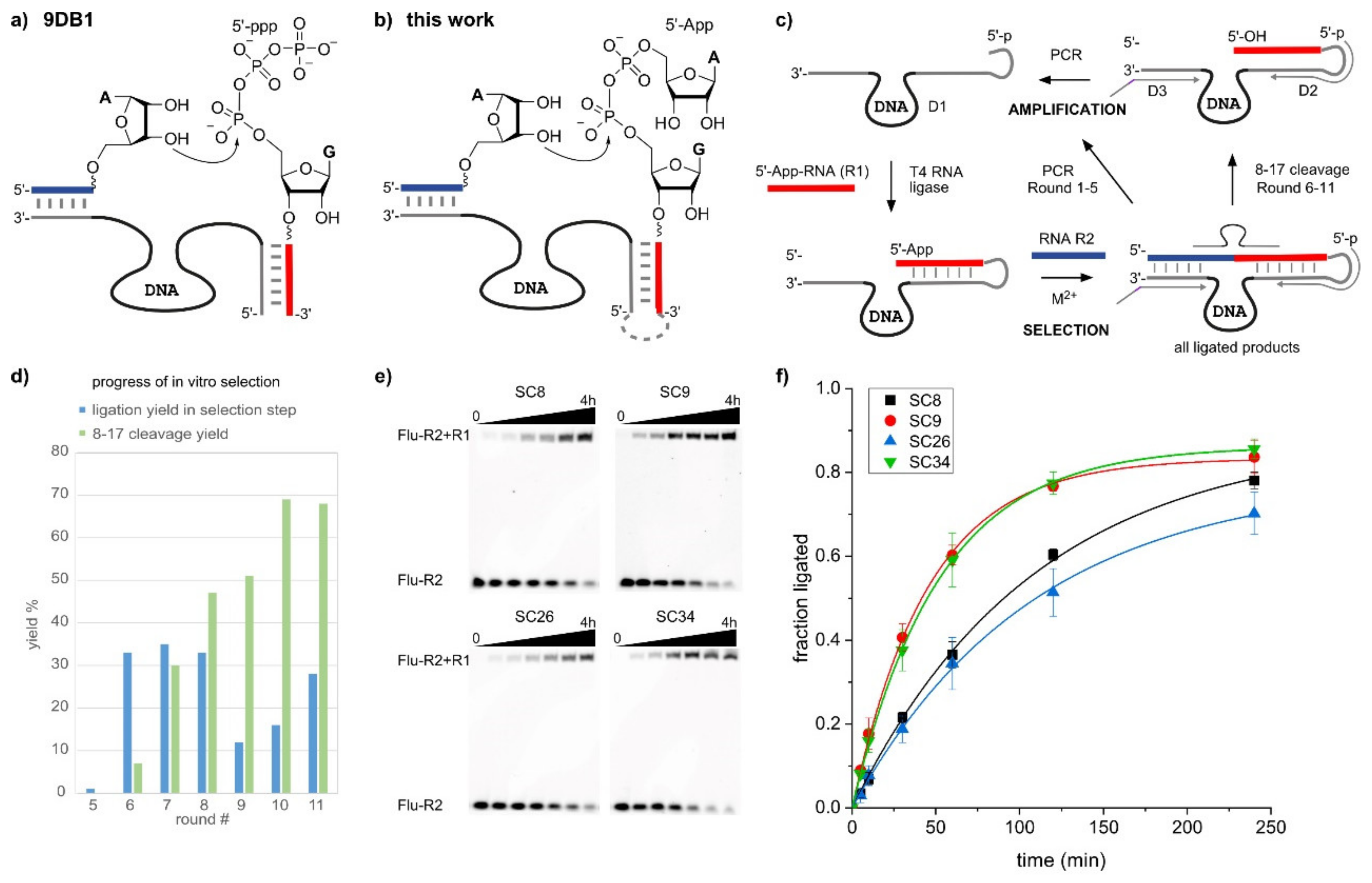
2.1. In Vitro Selection of RNA-Ligating Deoxyribozymes
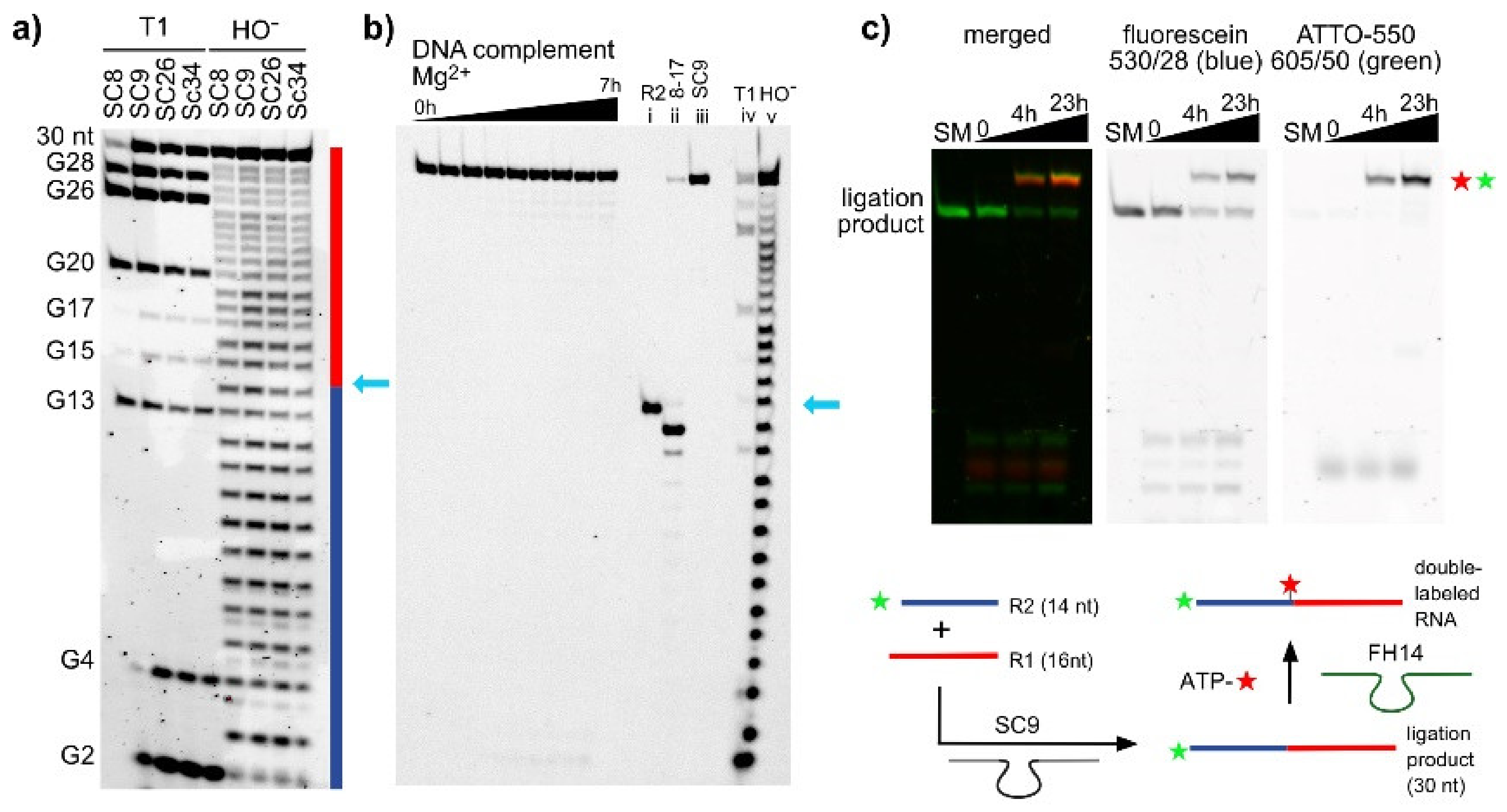
2.2. The SC DNA Enzymes Synthesize Native 3′-5′-Linked RNA
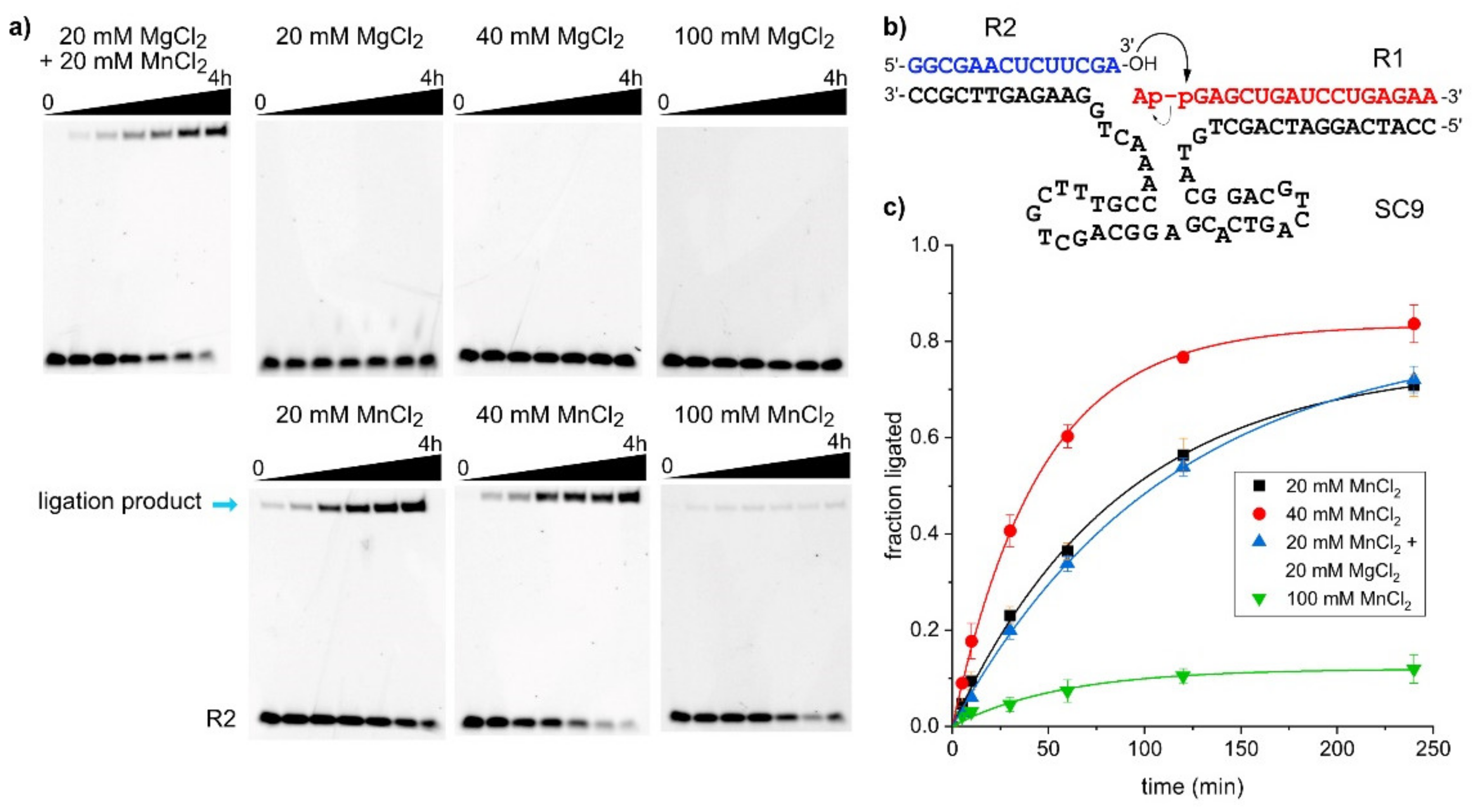
2.3. The SC9 Deoxyribozyme Requires Mn2+ as a Cofactor
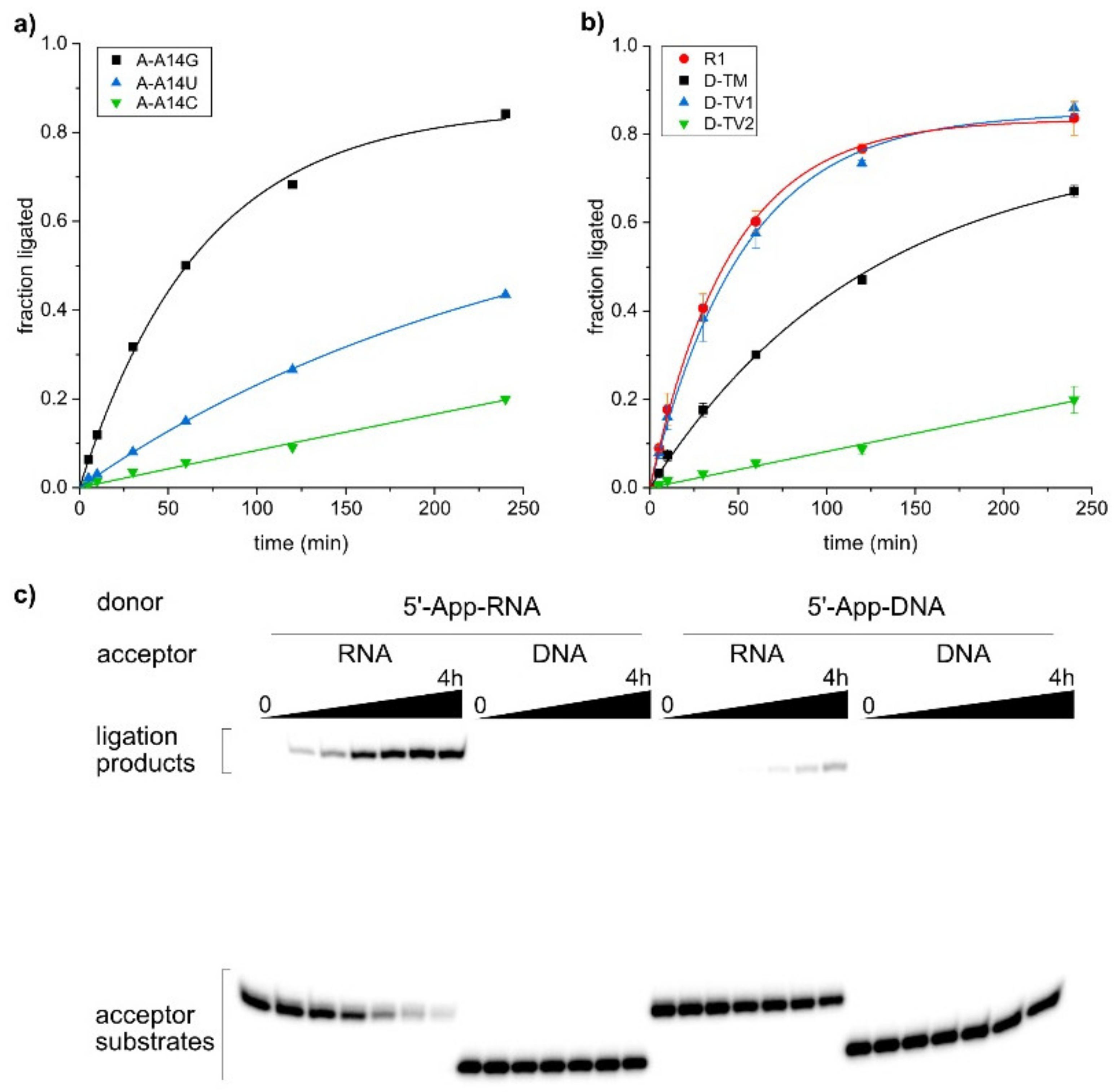
2.4. The SC9 Deoxyribozyme Has a Broad RNA Substrate Scope
3. Conclusions
4. Materials and Methods
4.1. General Information and Sequences of DNAs and RNAs Used in This Study
4.2. Synthesis of Donor and Acceptor Substrates
4.3. In Vitro Selection
4.3.1. Preparation of the Starting Pool
4.3.2. Selection Step
4.3.3. PCR Amplification of the Enriched Pool
4.3.4. Cloning and Sequencing of the Enriched Pool
4.4. Characterization of the Deoxyriboyzmes
4.4.1. Kinetic Analysis of Intermolecular Ligation Activity (Reaction in trans)
4.4.2. Characterization of the Ligation Products
RNase T1 Digestion
Alkaline Hydrolysis
Cleavage by 8-17 Deoxyribozyme
MgCl2 Cleavage in RNA-DNA Duplex
In Situ Labeling of the Ligation Product by FH14 Ribozyme Using N6-(6-amino)hexyl-ATP
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Müller, S.; Appel, B.; Balke, D.; Hieronymus, R.; Nübel, C. Thirty-five years of research into ribozymes and nucleic acid catalysis: Where do we stand today? F1000Res 2016, 5, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, S.K. Catalytic DNA: Scope, applications, and biochemistry of deoxyribozymes. Trends Biochem. Sci. 2016, 41, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. DNA catalysis: The chemical repertoire of DNAzymes. Molecules 2015, 20, 20777–20804. [Google Scholar] [CrossRef] [Green Version]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Schlosser, K.; Li, Y. A versatile endoribonuclease mimic made of DNA: Characteristics and applications of the 8-17 RNA-cleaving DNAzyme. ChemBioChem 2010, 11, 866–879. [Google Scholar] [CrossRef]
- Schlosser, K.; Li, Y. Biologically inspired synthetic enzymes made from DNA. Chem. Biol. 2009, 16, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, J. Catalytic nucleic acids: Biochemistry, chemical biology, biosensors, and nanotechnology. iScience 2020, 23, 100815. [Google Scholar] [CrossRef] [Green Version]
- Silverman, S.K. Deoxyribozymes: Selection design and serendipity in the development of DNA catalysts. Acc. Chem. Res. 2009, 42, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Purtha, W.E.; Coppins, R.L.; Smalley, M.K.; Silverman, S.K. General deoxyribozyme-catalyzed synthesis of native 3′-5′ RNA linkages. J. Am. Chem. Soc. 2005, 127, 13124–13125. [Google Scholar] [CrossRef] [Green Version]
- Büttner, L.; Seikowski, J.; Wawrzyniak, K.; Ochmann, A.; Höbartner, C. Synthesis of spin-labeled riboswitch RNAs using convertible nucleosides and DNA-catalyzed RNA ligation. Bioorg. Med. Chem. 2013, 21, 6171–6180. [Google Scholar] [CrossRef]
- Martin, L.L.; Unrau, P.J.; Müller, U.F. RNA synthesis by in vitro selected ribozymes for recreating an RNA world. Life 2015, 5, 247–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachowius, F.; Attwater, J.; Holliger, P. Nucleic acids: Function and potential for abiogenesis. Q. Rev. Biophys. 2017, 50, e4. [Google Scholar] [CrossRef] [PubMed]
- Wachowius, F.; Höbartner, C. Probing essential nucleobase functional groups in aptamers and deoxyribozymes by nucleotide analogue interference mapping of DNA. J. Am. Chem. Soc. 2011, 133, 14888–14891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachowius, F.; Javadi-Zarnaghi, F.; Höbartner, C. Combinatorial mutation interference analysis reveals functional nucleotides required for DNA catalysis. Angew. Chem. Int. Ed. 2010, 49, 8504–8508. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Salvatierra, A.; Wawrzyniak-Turek, K.; Steuerwald, U.; Höbartner, C.; Pena, V. Crystal structure of a DNA catalyst. Nature 2016, 529, 231–234. [Google Scholar] [CrossRef]
- Höbartner, C. How DNA catalyses RNA ligation. Nat. Catal. 2019, 2, 483–484. [Google Scholar] [CrossRef]
- Aranda, J.; Terrazas, M.; Gomez, H.; Villegas, N.; Orozco, M. An artificial DNAzyme RNA ligase shows a reaction mechanism resembling that of cellular polymerases. Nat. Catal. 2019, 2, 544–552. [Google Scholar] [CrossRef]
- Mattioli, E.J.; Bottoni, A.; Calvaresi, M. DNAzymes at work: A DFT computational investigation on the mechanism of 9DB1. J. Chem. Inf. Model. 2019, 59, 1547–1553. [Google Scholar] [CrossRef]
- Wawrzyniak-Turek, K.; Höbartner, C. Deoxyribozyme-mediated ligation for incorporating EPR spin labels and reporter groups into RNA. Methods Enzymol. 2014, 549, 85–104. [Google Scholar] [CrossRef]
- Goldeck, M.; Tuschl, T.; Hartmann, G.; Ludwig, J. Efficient solid-phase synthesis of pppRNA by using product-specific labeling. Angew. Chem. Int. Ed. 2014, 53, 4694–4698. [Google Scholar] [CrossRef]
- Sarac, I.; Meier, C. Efficient automated solid-phase synthesis of DNA and RNA 5′-triphosphates. Chem. Eur. J. 2015, 21, 16421–16426. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.E.; Müller, U.F. A ribozyme that triphosphorylates RNA 5′-hydroxyl groups. Nucleic Acids Res. 2014, 42, 4767–4778. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.K.; Wang, L.K.; Lima, C.D.; Shuman, S. Structure and mechanism of RNA ligase. Structure 2004, 12, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Silverman, S.K. Efficient RNA 5′-adenylation by T4 DNA ligase to facilitate practical applications. RNA 2006, 12, 1142–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkubo, A.; Tago, N.; Yokouchi, A.; Nishino, Y.; Yamada, K.; Tsunoda, H.; Seio, K.; Sekine, M. Synthesis of 5′-terminal capped oligonucleotides using O-N phosphoryl migration of phosphoramidite derivatives. Org. Lett. 2012, 14, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Zelin, E.; Wang, Y.; Silverman, S.K. Adenosine is inherently favored as the branch-site RNA nucleotide in a structural context that resembles natural RNA splicing. Biochemistry 2006, 45, 2767–2771. [Google Scholar] [CrossRef] [Green Version]
- Zelin, E.; Silverman, S.K. Allosteric control of ribozyme catalysis by using DNA constraints. ChemBioChem 2007, 8, 1907–1911. [Google Scholar] [CrossRef]
- Wanat, P.; Walczak, S.; Wojtczak, B.A.; Nowakowska, M.; Jemielity, J.; Kowalska, J. Ethynyl, 2-propynyl, and 3-butynyl C-phosphonate analogues of nucleoside di- and triphosphates: Synthesis and reactivity in CuAAC. Org. Lett. 2015, 17, 3062–3065. [Google Scholar] [CrossRef]
- Flynn-Charlebois, A.; Wang, Y.; Prior, T.K.; Rashid, I.; Hoadley, K.A.; Coppins, R.L.; Wolf, A.C.; Silverman, S.K. Deoxyribozymes with 2′-5′ RNA ligase activity. J. Am. Chem. Soc. 2003, 125, 2444–2454. [Google Scholar] [CrossRef]
- Ghaem Maghami, M.; Dey, S.; Lenz, A.K.; Höbartner, C. Repurposing antiviral drugs for orthogonal RNA-catalyzed labeling of RNA. Angew. Chem. Int. Ed. 2020, 59, 9335–9339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaem Maghami, M.; Scheitl, C.P.M.; Höbartner, C. Direct in vitro selection of trans-acting ribozymes for posttranscriptional, site-specific, and covalent fluorescent labeling of RNA. J. Am. Chem. Soc. 2019, 141, 19546–19549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torabi, S.F.; Wu, P.; McGhee, C.E.; Chen, L.; Hwang, K.; Zheng, N.; Cheng, J.; Lu, Y. In vitro selection of a sodium-specific DNAzyme and its application in intracellular sensing. Proc. Natl. Acad. Sci. USA 2015, 112, 5903–5908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Kartik, S.; Liu, B.; Liu, J. From general base to general acid catalysis in a sodium-specific DNAzyme by a guanine-to-adenine mutation. Nucleic Acids Res. 2019, 47, 8154–8162. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Saran, R.; Chen, Q.; Ding, J.; Liu, J. A new Na+-dependent RNA-cleaving DNAzyme with over 1000-fold rate acceleration by ethanol. ChemBioChem 2016, 17, 159–163. [Google Scholar] [CrossRef]
- Zhou, W.; Saran, R.; Ding, J.; Liu, J. Two completely different mechanisms for highly specific Na+ recognition by DNAzymes. ChemBioChem 2017, 18, 1828–1835. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, E.; Lam, C.H.; Perrin, D.M. A densely modified M2+-independent DNAzyme that cleaves RNA efficiently with multiple catalytic turnover. Chem. Sci. 2018, 9, 1813–1821. [Google Scholar] [CrossRef]
- Renders, M.; Miller, E.; Hollenstein, M.; Perrin, D. A method for selecting modified DNAzymes without the use of modified DNA as a template in PCR. Chem. Commun. 2015, 51, 1360–1362. [Google Scholar] [CrossRef]
- Schlosser, K.; Gu, J.; Sule, L.; Li, Y. Sequence-function relationships provide new insight into the cleavage site selectivity of the 8-17 RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2008, 36, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Höbartner, C.; Pradeepkumar, P.I. DNA catalysts for synthetic applications in biomolecular chemistry. In New Strategies in Chemical Synthesis and Catalysis; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2012; Chapter 6; pp. 129–155. [Google Scholar]
- Lee, C.S.; Mui, T.P.; Silverman, S.K. Improved deoxyribozymes for synthesis of covalently branched DNA and RNA. Nucleic Acids Res. 2011, 39, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkumar, P.I.; Höbartner, C.; Baum, D.A.; Silverman, S.K. DNA-catalyzed formation of nucleopeptide linkages. Angew. Chem. Int. Ed. 2008, 47, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Höbartner, C.; Silverman, S.K. Engineering a selective small-molecule substrate binding site into a deoxyribozyme. Angew. Chem. Int. Ed. 2007, 46, 7420–7424. [Google Scholar] [CrossRef] [PubMed]
- Turriani, E.; Höbartner, C.; Jovin, T.M. Mg2+-dependent conformational changes and product release during DNA-catalyzed RNA ligation monitored by bimane fluorescence. Nucleic Acids Res. 2015, 43, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, B.; Höbartner, C. Combinatorial nucleoside-deletion-scanning mutagenesis of functional DNA. Angew. Chem. Int. Ed. 2013, 52, 2995–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Y.; Breaker, R.R. Capping DNA with DNA. Biochemistry 2000, 39, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Walton, T.; DasGupta, S.; Duzdevich, D.; Oh, S.S.; Szostak, J.W. In vitro selection of ribozyme ligases that use prebiotically plausible 2-aminoimidazole-activated substrates. Proc. Natl. Acad. Sci. USA 2020, 117, 5741–5748. [Google Scholar] [CrossRef] [Green Version]
- Höbartner, C.; Kreutz, C.; Flecker, E.; Ottenschläger, E.; Pils, W.; Grubmayr, K.; Micura, R. The synthesis of 2′-O-[(triisopropylsilyl)oxy] methyl (TOM) phosphoramidites of methylated ribonucleosides (m1G, m2G, m22G, m1 I, m3U, m4C, m6A, m62A) for use in automated RNA solid-phase synthesis. Monatsh. Chem. 2003, 134, 851–873. [Google Scholar] [CrossRef]
- Rio, D.C. Expression and purification of active recombinant T7 RNA polymerase from E. coli. Cold Spring Harb. Protoc. 2013, 1094–1098. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNAzyme | Core Sequence | Number of Clones | kobs (min−1) 1 |
|---|---|---|---|
| SC8 | GCGGGCCCCAATTCATTGGCTTAACTACGAGGACGTCCAG | 1 | 0.0092 ± 0.0004 |
| SC9 | GTACGGACGTCAGTCACGAGGCAGCTGCTTTGCCAAACTG | 8 | 0.0219 ± 0.0006 |
| SC26 | ACCGTCACAAAAGACTGTAGTTAATACCACTGCAGGTCGT | 5 | 0.0093 ± 0.0004 |
| SC34 | GCACAGGGGTATAATTGCCTCGTACAACTTTATGTCCGAA | 1 | 0.0193 ± 0.0003 |
| Name | 5′-Sequence-3′ |
|---|---|
| D1 DNA Pool | pCCATCAGGATCAGCT-N40-GAAGAGTTCGCCGp |
| D2 Fwd primer | pCCATCAGGATCAGCT |
| D3 Rev primer | ACCACCAACAACA-X(spacer18)-ATGCTCGGCGAACTCTTC |
| D4 8-17 | GATGGTTCAGGATCAGCTTCCGAGCCGGACGACGAAGAGTTCGCC |
| D5 Splint | CAGGATCAGCTCTCGAAGAGTTC |
| SC8 | CCATCAGGATCAGCTGCGGGCCCCAATTCATTGGCTTAACTACGAGGACGTCCAGGAAGAGTTCGCC |
| SC9 | CCATCAGGATCAGCTGTACGGACGTCAGTCACGAGGCAGCTGCTTTGCCAAACTGGAAGAGTTCGCC |
| SC26 | CCATCAGGATCAGCTACCGTCACAAAAGACTGTAGTTAATACCACTGCAGGTCGTGAAGAGTTCGCC |
| SC34 | CCATCAGGATCAGCTGCACAGGGGTATAATTGCCTCGTACAACTTTATGTCCGAAGAAGAGTTCGCC |
| SC9-D-TM | CCACTGAAGCTGATCGTACGGACGTCAGTCACGAGGCAGCTGCTTTGCCAAACTGGAAGAGTTCGCC |
| SC9-D-TV1 | CCAAGTCCTAGTCGAGTACGGACGTCAGTCACGAGGCAGCTGCTTTGCCAAACTGGAAGAGTTCGCC |
| SC9-D-TV2 | CCAGACTTCGACTAGGTACGGACGTCAGTCACGAGGCAGCTGCTTTGCCAAACTGGAAGAGTTCGCC |
| 9DB1 | CAGGATCAGCTGGATCATACGGTCGGAGGGGTTTGCCGTTTACGAAGAGTTCGC |
| Donor DNA | GAGCTGATCCTGAGAA |
| Acceptor DNA | GGCGAACTCTTCGA |
| R1 Donor | GAGCUGAUCCUGAGAA |
| R2 Acceptor | GGCGAACUCUUCGA |
| D-TM | GGAUCAGCUUCAGGAA |
| D-TV1 | GUCGACUAGGACUGAA |
| D-TV2 | GCUAGUCGAAGUCGAA |
| D-G1A | AAGCUGAUCCUGAGAA |
| D-G1U | UAGCUGAUCCUGAGAA |
| D-G1C | CAGCUGAUCCUGAGAA |
| A-A14G | GGCGAACUCUUCGG |
| A-A14C | GGCGAACUCUUCGC |
| A-A14U | GGCGAACUCUUCGU |
| FH14 | GGAUCAGCUCCACGCUGAGUAGACAUACUUGCAAACGCUGCAAACAUAGACGAAGAGUUC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheitl, C.P.M.; Lange, S.; Höbartner, C. New Deoxyribozymes for the Native Ligation of RNA. Molecules 2020, 25, 3650. https://doi.org/10.3390/molecules25163650
Scheitl CPM, Lange S, Höbartner C. New Deoxyribozymes for the Native Ligation of RNA. Molecules. 2020; 25(16):3650. https://doi.org/10.3390/molecules25163650
Chicago/Turabian StyleScheitl, Carolin P. M., Sandra Lange, and Claudia Höbartner. 2020. "New Deoxyribozymes for the Native Ligation of RNA" Molecules 25, no. 16: 3650. https://doi.org/10.3390/molecules25163650
APA StyleScheitl, C. P. M., Lange, S., & Höbartner, C. (2020). New Deoxyribozymes for the Native Ligation of RNA. Molecules, 25(16), 3650. https://doi.org/10.3390/molecules25163650