Abstract
A density functional theory (DFT) study of the 1H- and 13C-NMR chemical shifts of the geometric isomers of 18:2 ω-7 conjugated linoleic acid (CLA) and nine model compounds is presented, using five functionals and two basis sets. The results are compared with available experimental data from solution high resolution nuclear magnetic resonance (NMR). The experimental 1H chemical shifts exhibit highly diagnostic resonances due to the olefinic protons of the conjugated double bonds. The “inside” olefinic protons of the conjugated double bonds are deshielded than those of the “outside” protons. Furthermore, in the cis/trans isomers, the signals of the cis bonds are more deshielded than those of the trans bonds. These regularities of the experimental 1H chemical shifts of the olefinic protons of the conjugated double bonds are reproduced very accurately for the lowest energy DFT optimized single conformer, for all functionals and basis sets used. The other low energy conformers have negligible effects on the computational 1H-NMR chemical shifts. We conclude that proton NMR chemical shifts are more discriminating than carbon, and DFT calculations can provide a valuable tool for (i) the accurate prediction of 1H-NMR chemical shifts even with less demanding functionals and basis sets; (ii) the unequivocal identification of geometric isomerism of CLAs that occur in nature, and (iii) to derive high resolution structures in solution.
1. Introduction
High resolution nuclear magnetic resonance (NMR) spectroscopy has become a widely utilized method in structure elucidation and in the qualitative and quantitative analysis of lipid molecules [1,2,3,4,5,6]. NMR spectroscopy, even in mixture analysis, has several advantages compared to common chromatographic analysis methods, since it is non-destructive, no specific standards are necessary for quantification, and chemical shifts and coupling constants are a function of the nucleus and its environment. NMR-based analytical and structural methods in lipid research, however, are limited by overlapping signals in the 1H-NMR spectrum, the low natural abundance of 13C, and the high cost of instrumentation. Nevertheless, high-resolution NMR spectroscopy has become increasingly popular in the study of lipids due to immense advances in high field instrumentation, the use of high sensitivity cryogenic probes, advanced data processing, improved automated methods for assignment, and the great variety of 1D and 2D NMR techniques [1,2,3,4,5,6,7,8,9,10].
Conjugated linoleic acids (CLAs) refer to an important class of polyunsaturated fatty acids that are a mixture of geometric (cis-cis, trans-trans, cis-trans and trans-cis) and positional (6–8 to 13–15) fatty acid isomers that contain conjugated double bonds [11]. CLA isomers occur naturally in foods derived from ruminants and are produced as intermediates of the biohydrogenation of polyunsaturated fatty acids, especially linoleic (cis-9, cis-12) 18:2 and α-linolenic (cis-9, cis-12, cis-15) 18:3 by rumen bacteria [12]. Cis-9, trans-11 CLA (Figure 1) is also called rumenic acid and it accounts for 72–94% of the total CLA in foods from ruminant animals. CLAs have numerous effects with regard to cancer, atherosclerosis, obesity, and immune response [13]. Thus, it has been claimed that the cis-9, trans-11 18:2 CLA isomer, in a range of animal models, inhibits chemically induced cancer [14] and cancer cell proliferation [15], and is implicated in immunomodulation and anti-atherosclerosis effects. On the contrary, there is evidence that the trans-10, cis-12 18:2 CLA isomer may adversely affect insulin sensitivity and result in pro-carcinogenic effects in animal models [16,17].
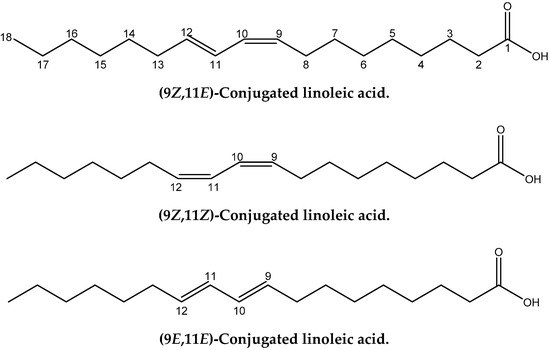
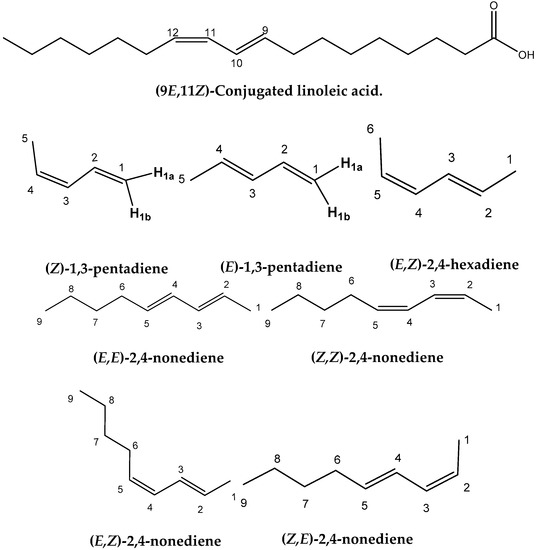
Figure 1.
Chemical structures of the four geometric isomers of the 18:2 ω-7 conjugated linoleic acid (rumenic acid) and the model compounds investigated in the present work.
Conjugated linoleic acids (CLAs) have been extensively investigated using 1H- and 13C-NMR [6,18,19,20,21,22]. NOESY experiments, and the analyses of spin–spin coupling constants were utilized to identify the geometric configurations of the double bonds [23]. The DFT calculations of NMR chemical shifts in conjugated systems, however, are limited to 1H-NMR chemical shifts and 13C chemical shift tensors of all-trans, 13-cis, 11-cis-12-s-cis, 11-cis-12-s-trans, and 9-cis isomers of retinal using the gauge including atomic orbital (GIAO) technique in combination with the B3LYP/6-31G method [24]. It was concluded that the DFT theory provides a valuable tool for the prediction of 13C- NMR properties, even with a small basis set. For the accurate prediction of trends in the experimental 1H-NMR chemical shifts a larger basis set, including polarization functions, was recommended [24].
A significant number of studies have been published that combine experimental NMR chemical shifts with computations [24,25,26,27,28] and investigating high resolution structures in solution [28,29,30,31,32,33,34,35,36]. When we consider the importance of calculations of NMR parameters in structural chemistry, it is surprising that no applications in lipid research have so far been reported. Furthermore, since no X-ray structures of CLAs have so far been published, it would be of interest to utilize the quantum chemical calculations of the chemical shifts of structure elucidation in solution of this important class of natural products. In this paper we discuss: (i) the accuracy of the calculation of 1H- and 13C-NMR chemical shifts performed within the DFT framework using the GIAO [37] technique on several geometric isomers of CLA and model compounds (Figure 1), and (ii) the use of 1H- and 13C- NMR chemical shifts for the unequivocal assignment of geometric isomerism of CLAs and as a tool for investigating high resolution structures in solution.
2. Results and Discussion
2.1. DFT-Calculated vs. Experimental 1H-and 13C-NMR Chemical Shifts of Model Compounds in Solution: Effects of Various Functionals and Basis Sets
2.1.1. 1H-NMR
Figure 2A and Table S1 show calculated, δcalc, (at the GIAO/B3LYP/6-311+G(2d,p) level with CPCM in CHCl3/CH3CN) vs. experimental, δexp, 1H-NMR chemical shifts of (Z)-1,3-pentadiene, (E)-1,3-pentadiene, and (E,Z)-2.4-hexadiene in CDCl3 [38], and (E,E)-2,4-nonediene, (Z,Z)-2,4-nonediene, (E,Z)-2,4-nonediene, and (Z,E)-2,4-nonediene in CD3CN [39]. The optimization of the structures was performed at the B3LYP/6-31+G(d), B3LYP/6-311++G(d,p), APFD/6-31+G(d), APFD/6-311++G(d,p), PBE0/6-31+G(d), PBE0/6-311++G(d,p), M06-2X/6-31+G(d), M06-2X/6-311++G(d,p), ωB97XD/6-31+G(d), and ωB97XD/6-311++G(d,p) level. Excellent linear regression correlation coefficients and standard deviations of δ(1H) were obtained for all functionals and basis sets used, including the B3LYP/6-31+G(d) level (Table S2). This is in agreement with the conclusion of a comprehensive review article that increasing basis set and computational time does not necessarily result in more accurate chemical shifts [26]. In general, slopes that deviate from the ideal value of +1.00 by ≤0.05 and R2 values > 0.995 are indicative of very well performing method. Figure 2B illustrates δcalc vs. δexp 1H-NMR chemical shifts of the olefinic region of Figure 2A Again, excellent linear regression correlation coefficients (≥ 0.968) were obtained with the exception of the calculations at the APFD/6-31+G(d) (R2 = 0.941) and M06-2X/6-31+G(d) (R2 = 0.938) levels (Table S2).
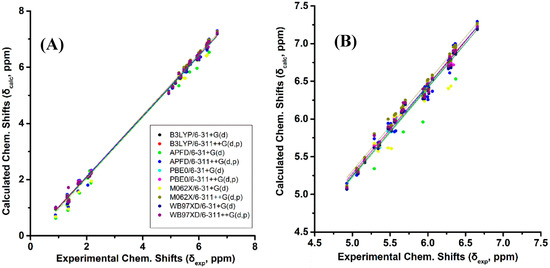
Figure 2.
(A) Calculated, δcalc, 1H-NMR chemical shifts (at the GIAO/B3LYP/6-311+G(2d,p) level of theory with CPCM in CHCl3/CH3CN) vs. experimental, δexp, chemical shifts with energy minimization using various functionals and basis sets for (Z)-1,3-pentadiene, (E)-1,3-pentadiene, (E,Z)-2.4-hexadiene, (E,E)-2,4-nonediene, (Z,Z)-2,4-nonediene, (E,Z)- 2,4-nonediene, and (Z,E)-2,4-nonediene (Figure 1). (Β) Calculated, δcalc, 1H-NMR chemical shifts of the olefinic protons vs. experimental, δexp, chemical shifts of the data of Figure 2A.
The experimental 1H-NMR chemical shifts of the model compounds of Figure 1 exhibit highly diagnostic resonances due to the –CH= protons of the conjugated double bonds in the region of 4.93 to 6.66 ppm. Some of these resonances are clearly shifted to higher frequencies than those of the isolated double bonds, thus illustrating the increased delocalization of conjugated double bonds. The “inside” olefinic protons of the conjugated double bonds are more deshielded than those of the “outside” protons. Thus, the experimental C(4)–H and C(3)–H protons of (E,E)-2,4-nonediene both appear at 6.00 ppm, while those of C(5)–H and C(2)–H appear at 5.56 and 5.57 ppm, respectively. Furthermore, in the cis/trans isomers the signals of the cis bonds are more deshielded than those of the trans bond. Thus, C(3)–H and C(2)–H of (Z,E)-2,4-nonediene appear at 5.96 and 5.36 ppm, respectively, while those of C(4)–H and C(5)–H appear at 6.36 and 5.68 ppm, respectively. In the case of identical bond configuration, the two “inside” protons have very similar chemical shifts as well as the two signals of the “outside” protons.
The above regularities of δexp of the olefinic protons are clearly reproduced for all functionals and basis sets used (Tables S1 and S2) and, thus can be used as criteria of the geometric isomerism of the conjugated system. Figure 3B illustrates that very poor correlation was obtained (R2: 0.454 to 0.657, intercept: 3.168 to 3.544, and slope: 0.457 to 0.528) when δcalc of (E,E)-2,4-nonediene was plotted vs. δexp of (E,Z)-2,4-nonediene. Similarly, a very poor correlation was obtained when δcalc of (E,E)-2,4-nonediene was plotted vs. δexp of (Z,Z)-2,4-nonediene (intercept: 2.626 to 2.906, and slope: 0.551 to 0.616) and when δcalc of (E,Z)-2,4-nonediene was plotted vs. δexp of (Z,E)-2,4-nonediene (intercept: 3.403 to 3.922, and slope: 0.408 to 0.486) (Figures S1 and S2). It can, therefore, be concluded that 1H-NMR chemical shift calculations very accurately reproduce the general trends of the experimental data, and that the minor basis set dependence can result in levels of accuracy which are necessary for high resolution three-dimensional structure determinations (see below).
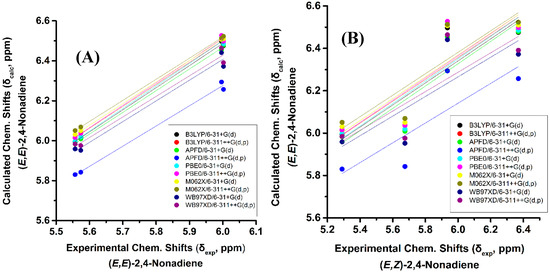
Figure 3.
(A) Calculated, δcalc, of the olefinic protons (at the GIAO/B3LYP/6-311+G(2d,p) level of theory with CPCM in CH3CN) of (E,E)-2,4-nonediene vs. experimental, δexp, olefinic protons in CD3CN of (E,E)-2,4-nonediene with energy minimization using the B3LYP/6-31+G(d), B3LYP/6-311++G(d,p), APFD/6-31+G(d), APFD/6-311G++(d,p), PBE0/6-31+G(d), and PBE0/6-311++G(d,p) methods. (B) Calculated, δcalc, of the olefinic protons (at the GIAO/B3LYP/6-311+G(2d,p) level of theory with CPCM in CH3CN) of (E,E)-2,4-nonediene vs. experimental, δexp, olefinic protons in CD3CN of (E,Z)-2,4-nonediene with energy minimization using the same basis sets and functionals as in (A).
2.1.2. 13C-NMR
Figure S3 and Table S3 show calculated, δcalc, (at the GIAO/B3LYP/6-311+G(2d,p) level with CPCM in CHCl3/CH3CN) vs. experimental, δexp, 13C-NMR chemical shifts of (Z)-1,3-pentadiene, (E)-1,3-pentadiene, and (E,Z)-2,4-hexadiene in CDCl3 [38], and (E,E)-2,4-nonediene, (Z,Z)-2,4-nonediene, (E,Z)-2,4-nonediene, and (Z,E)-2,4-nonediene in CD3CN [39]. The optimization of the structures was performed at the B3LYP/6-31+G(d), B3LYP/6-311++G(d,p), APFD/6-31+G(d), APFD/6-311++G(d,p), PBE0/6-31+G(d), PBE0/6-311++G(d,p), M06-2X/6-31+G(d), M06-2X/6-311++G(d,p), ωB97XD/6-31+G(d), and ωB97XD/6-311++G(d,p) level. Excellent linear regression correlation coefficient (R2: 0.997 to 0.996) was obtained, however, the mean square error (7.876 to 12.742), intercept (2.674 to 4.237), and slope (1.026 to 1.032) differed markedly from the ideal values for all functionals and basis sets used (Table S4). Figure S3B illustrates δcalc vs. δexp13C-NMR chemical shifts of the olefinic region of Figure S3A. Poor linear regression correlation coefficients (R2: 0.873 to 0.911) were obtained for the functional and basis sets used. The mean square error (3.568 to 5.015), intercept (−13.154 to −23.743), and slope (1.160 to 1.242) of δcalc vs. δexp differed markedly from the ideal values, which is a serious deficiency in any predictive scheme (Table S5).
2.2. Effects of Out-of-Plane Deformation of the Conjugated System and Conformation of Substituents on the Calculated 1H- and 13C-NMR Chemical Shifts in Model Compounds
2.2.1. 1H-NMR
Variation of the torsion angle φ(C-1,C-2,C-3,C-4) of the conjugated system of (Z)-1,3-pentadiene in steps of 5° in the range of 180° to 0°, results in a significant increase in the electronic energy (ΔE) (Figure 4B). A second minimum at φ = 31.0° (s-cis conformer) was observed with ΔE = 3.56 kcal·mol−1 (ΔG = 3.40 kcal·mol−1) higher than that of the s-trans conformer. Table S6 shows that the computed 1H-NMR chemical shifts of the two low energy conformers with φ = 31.0° and φ = 180.0°, weighting by the respective Boltzmann factor, are nearly identical with those of the φ = 180.0° conformer. The effect of the population of the φ = 31.0° conformer can thus be neglected. Conjugation of C=C double bonds strengthens the anisotropy effect which can be quantitatively defined by the extent of π-electron delocalization [40]. A more pronounced π-delocalization along the conjugated double bonds results in a stronger overall anisotropy effect (c.f. φ = 0° vs. φ = 180°). The computational data indicate a similar behavior of the “inside” H2 and H3 protons with a pronounced shielding at φ < 90.0° (Figure 4C). The “outside” H1b and H4 protons show a similar characteristic Karplus-like variation with a strong deshielding at φ < 60.0°. Surprisingly, the effect on the olefinic 1H-NMR chemical shifts in the range of 150.0° < φ < 180.0° is very small and thus can be neglected. Interestingly, in this range of torsion angles the “inside” H2 and H3 protons are significantly deshielded than those of the “outside” H1b and H4 protons, in excellent agreement with the experimental data of the minimum energy conformer with φ = 180.0°.
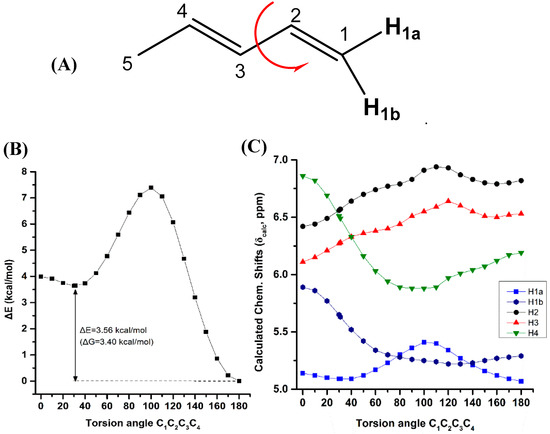
Figure 4.
Effect of variation of the C1C2C3C4 torsion angle of (E)-1,3-pentadiene (A) on the electronic energy ΔΕ (kcal·mol−1) (B), and olefinic 1H-NMR chemical shifts (C) with energy minimization at the B3LYP/6-31+G(d) level. The Gibbs energy difference, ΔG, of the two low energy conformers is also indicated.
Figure 5 illustrates the effect of variation of the C2C3C4C5 torsion angle of (E,E)-2,4-nonediene on the electronic energy ΔΕ(kcal·mol−1), and olefinic 1H chemical shifts with energy minimization at the B3LYP/6-31+G(d) level. As in the case of (Z)-1,3-pentadiene, a second minimum at φ = 31.2° was observed (s-cis conformer) with ΔE = 3.60 kcal·mol−1 (ΔG = 3.41 kcal·mol−1) higher than that of the s-trans conformer (φ = 180.0°). Table S7 shows that the computed 1H-NMR chemical shifts of the two low energy conformers with φ = 31.2° and φ = 180.0°, weighting by the respective Boltzmann factors, are essentially the same with those of the φ = 180.0° conformer. This is due to the negligible population of the higher minimum conformer with φ = 31.2°. The “inside” olefinic protons are significantly more deshielded with respect to those of the “outside” protons for a wide range of torsion angles 60° < φ < 180° (Figure 5C). For <40° a significant cross-over of the chemical shifts is observed which results in the deshielding of the “outside” protons with respect to the “inside” protons, contrary to the experimental data (Table S1).
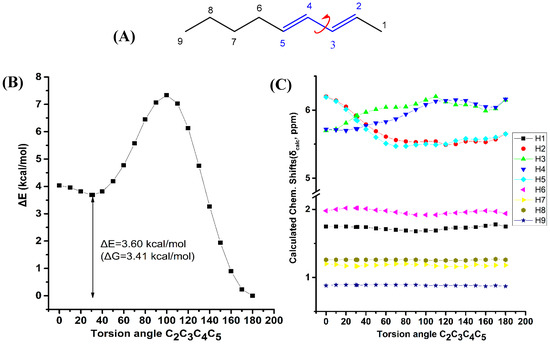
Figure 5.
Effect of variation of the C2C3C4C5 torsion angle of (E,E)-2,4-nonediene (A) on the electronic energy ΔΕ(kcal·mol−1) (B), and the 1H-NMR chemical shifts (C) with energy minimization at the B3LYP/6-31+G(d) level. The Gibbs energy difference, ΔG, of the two low energy conformers is also indicated.
The shielding changes due to variation of the C1C2C3C4 torsion angle of (Z)-1,3-pentadiene (Figure 4) are significantly larger than those of the C2C3C4C5 torsion angle of (E,E)-2,4-nonediene. It can be concluded that although the shielding tendencies remain the same for both compounds, the C(1)-CH3 substitution of (E,E)-2,4-nonediene results in a significant reduction in the shielding ranges.
Figure 6 illustrates the effect of variation of the C4C5C6C7 torsion angle of the (E,E)-2,4-nonediene on the electronic energy ΔΕ, and the 1H-NMR chemical shifts with energy minimization at the B3LYP/6-31+G(d) level. Two low energy conformers with φ = 120.0° and φ = 0.1° were observed with ΔE = 1.02 kcal·mol−1 (ΔG = 1.21 kcal·mol−1). For a wide range of torsion angles 100° < φ < 180°, the “inside” protons are more deshielded than the “outside” protons. The experimental 1H-NMR chemical shifts of the olefinic protons (Table S1) are in excellent agreement with the calculated chemical shifts of the minimum energy conformer with φ = 120.0°. For φ = 0.1°, the chemical shift of the “outside” H5 becomes similar to that of the “inside” H4 proton, contrary to the experimental data. Table S7 shows that the computed 1H-NMR chemical shifts of the low energy conformers, weighting by the respective Boltzmann factor, are essentially the same with those of the φ = 120.0° conformer. In this case, the weights of the higher minimum conformer are not vanishing, but the respective chemical shifts of the φ = 120.0° and φ = 0.1° conformers are not significantly different. A systematic investigation of 1H chemical shifts in alkenes by Abraham et al. [41] showed that the 1H chemical shifts of olefins are influenced by both diamagnetic anisotropy and steric effects of the double bonds. There is a deshielding above the C=C bond at small distances due to the van der Waals term and shielding at large distances due to the bond anisotropy. The plane of the C=C bond there is always a deshielding effect [42]. The olefinic group has significant γ effects on protons three bonds away. This is in agreement with our results of the significant effect of the C4C5C6C7 torsion angle on the computed 1H-NMR chemical shifts of the H7.
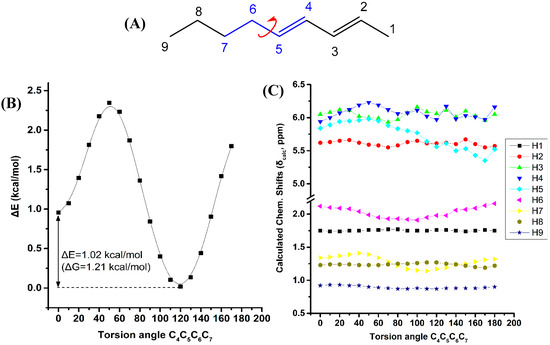
Figure 6.
Effect of variation of the C4C5C6C7 torsion angle of (E,E)-2,4-nonediene (A) on the electronic energy ΔΕ(kcal·mol−1) (B), and the 1H-NMR chemical shifts (C) with energy minimization at the B3LYP/6-31+G(d) level. The Gibbs energy difference, ΔG, of the two low energy conformers is also indicated.
Figure 7 illustrates the effect of variation of the C6C7C8C9 torsion angle of the (E,E)-2,4-nonediene on the electronic energy, ΔΕ, and the 1H-NMR chemical shifts with energy minimization at the B3LYP/6-31+G(d) level. Two minima in the electronic energy were observed at 180.0° and 64.8°, with ΔE = 0.98 kcal·mol−1 (ΔG = 0.87 kcal·mol−1). In this case the weights of the higher minimum conformer are not vanishing, however, the H7 and H8 chemical shifts exhibit a parallel pattern as a function of the C6C7C8C9 torsion angle with very small chemical shift difference. This indicates that δ(H7) and δ(H8) are of minor importance in conformational analysis.
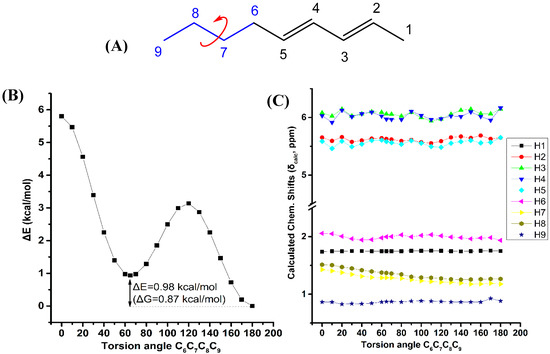
Figure 7.
Effect of variation of the C6C7C8C9 torsion angle of (E,E)-2,4-nonediene (A) on the electronic energy ΔΕ(kcal·mol−1) (B), and the 1H-NMR chemical shifts (C) with energy minimization at the B3LYP/6-31+G(d) level. The Gibbs energy difference, ΔG, of the two low energy conformers is also indicated.
From the above it can be concluded that the experimental 1H-NMR chemical shifts are reproduced very accurately for the lowest energy DFT optimized single conformer. The other low energy conformers have negligible effects on the computational 1H-NMR chemical shifts.
Figure 8A illustrates a plot of NBO bond order of the olefinic C–H bonds vs. δcalc(1H). The resulting good correlation (R2 = 0.903) demonstrate that the NBO bond order is a primary factor which determines δcalc(1H). Similar correlation was obtained when the AIM bond order [43] was plotted vs. δcalc(1H) (Figure 8B). On the contrary, no functional relationship was found for both NBO and AIM charge densities of the olefinic C-H protons vs. δcalc(1H).
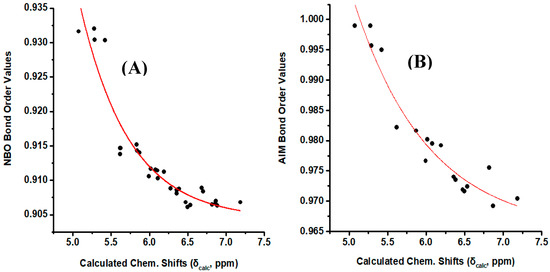
Figure 8.
(A) NBO bond order of the olefinic C–H bonds of the model compounds of Figure 1 vs. calculated, δcalc, 1H-NMR chemical shifts with energy minimization at the B3LYP/6-31+G(d) level. (B) AIM bond order of the olefinic C–H bonds of the model compounds (Z)-1,3-pentadiene, (E)-1,3 pentadiene, (E,Z)-2,4 hexadiene, (E,E)-2,4 nonadiene vs. calculated, δcalc, 1H-NMR chemical shifts with energy minimization at the B3LYP/6-31+G(d) level.
2.2.2. 13C-NMR
Figure S4 illustrates the effect of the out-of-plane deformation of the conjugated system on the 13C chemical shifts of (E)-1,3-pentadiene and (E,E)-2,4-nonediene, respectively, with calculations at the B3LYP/6-31+G(d) level. For the (E,E)-2,4-nonediene, the “inside” carbons C-3 and C-4 indicate a similar dependence with a rather complex cross-over with C-2. Figure S5 illustrates a very significant effect of variation of the C4C5C6C7 torsion angle of (E,E)-2,4-nonediene on the 13C chemical shifts of the olefinic C-4 and the aliphatic C6 and C7 carbons. However, the low energy conformers with φ = 120.0° and 0.1° are characterized with minimum chemical shifts differences. From the above it can be concluded that 13C-NMR chemical shifts are less discriminating than 1H, also considering the statistical data of Tables S2, S4 and S5. Similar conclusions can be drown from the effect of variation of the C6C7C8C9 torsion angle of (E,E)-2,4-nonediene on the 13C chemical shifts (Figure S5).
2.3. DFT-Calculated vs. Experimental 1H-NMR Chemical Shifts: 3D-Structures of Geometric Isomers of 18:2 ω-7 CLA in Solution
The 1H-NMR spectra of the four geometric isomers of 18:2 ω-7 CLA exhibit highly diagnostic resonances due to the –CH= protons of the conjugated double bonds in the region of 6.34 to 5.27 ppm. As in the case of the model compounds of Figure 1, the “inside” olefinic protons of C10 and C11 are more deshielded than those of the olefinic protons of C9 and C12. Furthermore, in the cis/trans isomers the signals of the cis bonds are more deshielded than those of the trans bonds. In the case of identical bond configuration, the two “inside” protons have very similar chemical shifts as well as the two signals of the “outside” protons (Table S8). Figure S6 illustrates the olefinic region of the 1H-NMR spectra of (9Z,11E)-CLA in CDCl3 (ε = 4.81), CD3CN (ε = 38.8) and DMSO-d6 (ε = 46.7) solution. Despite the use of three solvents with significantly different dielectric constants, ε, solvation and hydrogen bonding ability, the olefinic proton chemicals shifts are essentially solvent independent. The carboxylic proton was found to be extremely broad due to the fast proton exchange rate with traces of H2O in the organic solvents. The resulting chemical shifts in DMSO-d6 (δ = 11.96), CD3CN (δ = 8.82), and CDCl3 (δ = 10.50) (Table S9) clearly demonstrate that hydrogen bond between the COOH group and the DMSO-d6 molecule is more efficient than in CD3CN. In CDCl3, a strong deshielding is observed due to the presence of dimeric hydrogen bonded species. However, the chemical shift is smaller than that of the dimeric acetic acid in CDCl3 (δ = 11.51 ppm, concentration 160 mM [44]), due to the equilibrium between dimeric and monomeric species at the concentration used (25 mM).
Table 1 shows ΔG values (kcal·mol−1) and % populations of the various low energy conformers of the geometric isomers of 18:2 ω-7 CLA in the gas phase with energy minimization at the B3LYP/6-31+G(d) and APFD/6-31+G(d) level. According to the most stable conformation of the butyl group in 2,4-nonediene geometric model isomers, a similar conformational behavior is expected for the two alkyl groups which are attached to the conjugated system of linoleic acid. Consequently, a variety of five low energy conformers was obtained. In Figures S7–S11 the structures of five conformers (A), (B), (C), (D), and (E) of the four geometric isomers of 18:2 ω-7 CLA, are presented.

Table 1.
ΔG values (kcal·mol−1) and % populations of the various conformers of 18:2 ω-7 CLA geometric isomers in the gas phase at the B3LYP/6-31+G(d) and APFD/6-31+G(d) level.
Figure S12 shows calculated (at the GIAO/WP04/6-311+G(2d,p) (CPCM, CHCl3) level of theory) vs. experimental 1H-NMR chemical shifts of the four 9,11-conjugated linoleic acid geometric isomers with optimization of the structures at the B3LYP/6-31+G(d), and APFD/6-31+G(d) level of theory. The resulting statistical data of Table S10 demonstrate that computational 1H-NMR chemical shifts reproduce accurately the general trends of the experimental data. Very good correlations were also obtained when a discrete solvation molecule was utilized to model hydrogen bond interaction of the COOH group of the (9Z,11E)-CLA in DMSO and CH3CN solution (Figure S13). The dimeric form in CDCl3 was modeled with (9Z,11E)-CLA complexed with a molecule of CH3COOH to facilitate computations. In all cases, solvation of the COOH group induces minor changes in conformation (Figures S8 and S14), in excellent agreement with the experimental 1H-NMR chemical shift data of Table S9.
A typical workflow that can be used for investigating homologous CLA compounds includes the following steps:
- (i)
- The 1H-NMR spectra are recorded in CDCl3 solution at 298 K, and the experimental chemical shifts, δexp, are determined.
- (ii)
- The 1H-NMR chemical shifts are computed at the GIAO B3LYP/6-311+G(2d,p) level with the CPCM model with energy minimization using the B3LYP/6-311++G(d,p) method.
- (iii)
- A very good linear correlation between experimental NMR chemical shifts, δexp, and calculated δcalc provides strong indication that the computational procedure is working.
3. Materials and Method
3.1. Chemicals
Conjugated (9Z,11E) 18:2 linoleic acid, purity ≥96% (HPLC) was purchased from Fluka Chemie GmbH (Buchs, Switzerland). CDCl3, DMSO-d6, and CD3CN were purchased from Deutero (Kastellaun, Germany).
3.2. NMR
The NMR experiments were performed on a Bruker AV-500 MHz, Bruker (Billerica, MA, USA). Samples of (9Z,11E)-CLA were dissolved in 0.6 mL of deuterated solvent (25 mM) and transferred to 5 mm NMR tubes. Chemical shifts were measured with reference to the residual 1H signal of the incompletely deuterated solvent.
3.3. Computational Methods
The computational study was performed by using the Gaussian 09 with the DFT method [45]. The structures of the model compounds were optimized at the B3LYP/6-31+G(d,p), B3LYP/6-311++G(d,p), APFD/6-31+G(d), APFD/6-311++G(d,p), PBE0/6-31+G(d), PBE0/6-311++G(d,p), M06-2X/6-31+G(d), M06-2X/6-311++G(d,p), ωB97XD/6-31+G(d), and ωB97XD/6-311++G(d,p) level. The structures of the (9Z,11E)-CLA geometric isomers were optimized at the B3LYP/6-31+G(d) and APFD/6-31+G(d) level. The optimized geometries were verified by performing frequency calculation at the same level (zero imaginary frequencies). The computed 1H- and 13C-NMR chemical shifts, at the GIAO/B3LYP/6-311+G(2d,p) level, of the compounds investigated, were referenced with respect to the standard TMS which was optimized at the same level. The scanning of torsional angles was performed by using the redundant coordinates in Gaussian 09.
4. Conclusions
From the calculated data reported herein, it can be concluded that:
- (i)
- Excellent linear correlations can be obtained between DFT-calculated and experimental 1H-NMR chemical for the lowest energy DFT optimized single conformer for various functionals and basis sets, especially at the B3LYP/6-311++G(d,p) level. The other low energy conformers have negligible effects on the computational 1H-NMR chemical shifts.
- (ii)
- The computational 1H-NMR chemical shifts can provide an unequivocal assignment of the geometric isomerism in conjugated systems of biological systems such as CLAs.
- (iii)
- The great sensitivity of 1H-NMR chemical shifts to geometric isomerism and conformation of substituents can provide an excellent method for obtaining high resolution structures in solution.
- (iv)
- The typical workflow for investigating 3D structures in solution includes a few applicable steps.
The present method of DFT calculations of 1H-NMR chemicals shifts in parallel with the use of the computer assisted structure elucidation (CASE) program [46,47] and the development of experimental methods [6,10,48], could be of primary importance in geometric isomer identification and high resolution structural determination not only in isolated CLAs and their primary oxidation products [49], but also in mixture analysis in the emerging field of lipidomics [50,51].
Supplementary Materials
Supplementary Materials are available online.
Author Contributions
T.V., C.O., M.G.S. and P.C.V. performed DFT calculations; T.V., A.P. and E.A. performed NMR experiments and literature search; M.G.S. and I.P.G. conceived and designed the study and contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Funding
I.P.G. received funding from the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project Number: 2050). The APC was found by MDPI.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Gunstone, F.D. High resolution 13C-NMR spectroscopy of lipids. In Advances in Lipid Methodology, 2nd ed.; The Oily Press: Dundee, UK, 1993; pp. 1–68. [Google Scholar]
- Vlahov, G. Application of NMR to the study of olive oils. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 35, 341–357. [Google Scholar] [CrossRef]
- Diehl, B.W.K. High resolution NMR spectroscopy. Eur. J. Lipid Sci. Technol. 2001, 103, 830–834. [Google Scholar] [CrossRef]
- Diehl, B.W.K. Multinuclear high resolution nuclear magnetic resonance spectroscopy. In Lipid Analysis in Oils and Fats; Hamilton, R.J., Ed.; Springer: Boston, MA, USA, 1998; pp. 87–135. [Google Scholar]
- Gunstone, F.D.; Knothe, G.H. Nuclear Magnetic Resonance Spectroscopy of Fatty Acids and Their Derivatives. AOCS Lipid Library. Available online: www.aocs.org (accessed on 12 December 2019).
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.; Tsiafoulis, C.; Gerothanassis, I.P. High resolution NMR spectroscopy as a structural and analytical tool for unsaturated lipids in solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef] [PubMed]
- Spyros, A.; Dais, P. 31P NMR spectroscopy in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 195–207. [Google Scholar] [CrossRef]
- Mannina, L.; Sobolev, A.P.; Viel, S. Liquid state 1H high field NMR in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 66, 1–39. [Google Scholar] [CrossRef]
- Μartínez-Yusta, A.; Goicoechea, E.; Guillén, M.D. A review of thermo-oxidative degradation of food lipids studied by 1H NMR spectroscopy: Influence of degradative conditions and food lipid nature. Compr. Rev. Food Sci. Food Saf. 2014, 13, 838–859. [Google Scholar] [CrossRef]
- Spyros, A.; Dais, P. NMR Spectroscopy in Food Analysis; RSC: Cambridge, UK, 2012. [Google Scholar]
- Sebedio, J.L.; Christie, W.W.; Adolf, R.O. Advances in Conjugated Linoleic Acid Research; American Oil Chemists’ Society: Champaign, IL, USA, 2003; Volume 2. [Google Scholar]
- Shingfield, K.J.; Wallace, R.J. Synthesis of conjugated linoleic acid in ruminants and humans. In Conjugated Linoleic Acids and Conjugated Vegetable Oils; Sels, B., Philippaerts, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2014; pp. 1–65. [Google Scholar]
- Dilzer, A.; Park, Y. Implication of conjugated linoleic acid (CLA) in human health. Crit. Rev. Food Sci. Nutr. 2012, 52, 488–513. [Google Scholar] [CrossRef]
- Ha, Y.L.; Grimm, N.K.; Pariza, M.W. Anticarcinogens from fried ground beef: Heat-altered derivatives of linoleic acid. Carcinogenesis 1987, 8, 1881–1887. [Google Scholar] [CrossRef]
- Ochoa, J.J.; Farquharson, A.J.; Grant, I.; Moffat, L.E.; Heys, S.D.; Wahle, K.W. Conjugated linoleic acids (CLAs) decrease prostate cancer cells proliferation: Different molecular mechanisms for cis-9, trans-11 and trans-10, cis-12 isomers. Carcinogenesis 2004, 25, 1185–1191. [Google Scholar] [CrossRef]
- Wahle, K.W.; Heys, S.D.; Rotondo, D. Conjugated linoleic acids: Are they beneficial or detrimental to health? Prog. Lipid Res. 2004, 43, 553–587. [Google Scholar] [CrossRef]
- Tricon, S.; Burdge, G.C.; Williams, C.M.; Calder, P.C.; Yaqoob, P. The effects of conjugated linoleic acid on human health-related outcomes. Proc. Nutr. Soc. 2005, 64, 171–182. [Google Scholar] [CrossRef]
- Davis, A.L.; Mc Neill, G.P.; Caswell, D.C. Analysis of conjugated linoleic acid isomers by 13C NMR spectroscopy. Chem. Phys. Lipids 1999, 97, 155–156. [Google Scholar] [CrossRef]
- Kellersmann, C.; Steinhart, H.; Francke, W. Syntheses of conjugated octadecadienoic acids. Lipids 2006, 41, 777–788. [Google Scholar] [CrossRef]
- Tsiafoulis, C.G.; Skarlas, T.; Tzamaloukas, O.; Miltiadou, D.; Gerothanassis, I.P. Direct nuclear magnetic resonance identification and quantification of geometric isomers of conjugated linoleic acid in milk lipid fraction without derivatization steps: Overcoming sensitivity and resolution barriers. Anal. Chim. Acta 2014, 821, 62–71. [Google Scholar] [CrossRef]
- Tsiafoulis, C.G.; Papaemmanouil, C.; Alivertis, D.; Tzamaloukas, O.; Miltiadou, D.; Balayssac, S.; Malet-Martino, M.; Gerothanassis, I.P. NMR-based metabolomics of the lipid fraction of organic and conventional bovine milk. Molecules 2019, 24, 1067. [Google Scholar] [CrossRef]
- Symeou, S.; Tsiafoulis, C.G.; Gerothanassis, I.P.; Miltiadou, D.; Tzamaloukas, O. Nuclear magnetic resonance screening of changes in fatty acid and cholesterol content of ovine milk induced by ensiled olive cake inclusion in Chios sheep diets. Small Rumin. Res. 2019, 177, 111–116. [Google Scholar] [CrossRef]
- Kishino, S.; Ogawa, J.; Ando, A.; Iwashita, T.; Fujita, T.; Kawashima, H.; Shimizu, S. Structural analysis of conjugated linoleic acid produced by Lactobacillus plantarum, and factors affecting isomer production. Biosci. Biotechnol. Biochem. 2003, 67, 179–182. [Google Scholar] [CrossRef]
- Touw, S.I.E.; de Groot, H.J.M.; Buda, F. DFT calculations of the 1H NMR chemical shifts and 13C chemical shifts tensors of retinal isomers. J. Mol. Struct. 2004, 711, 141–147. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem. 2009, 74, 4597–4607. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Siskos, M.G.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Investigation of solute–solvent interactions in phenol compounds: Accurate ab initio calculations of solvent effects on 1H NMR chemical shifts. Org. Biomol. Chem. 2013, 11, 7400–7411. [Google Scholar] [CrossRef]
- Tarazona, G.; Benedit, G.; Fernández, R.; Pérez, M.; Rodriguez, J.; Jiménez, C.; Cuevas, C. Can stereoclusters separated by two methylene groups be related by DFT studies? The case of the cytotoxic meroditerpeneshalioxepines. J. Nat. Prod. 2018, 81, 343–348. [Google Scholar] [CrossRef]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H…O and O–H…O proton chemical shifts: Towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances. Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Tzakos, A.G.; Gerothanassis, I.P. 1H NMR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations–The challenge of accurate positions of labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen atomic positions of O–H…O hydrogen bonds in solution and in the solid state: The synergy of quantum chemical calculations with 1H-NMR chemical shifts and X-ray diffraction methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Refinement of labile hydrogen positions based on DFT calculations of 1H NMR chemical shifts: Comparison with X-ray and neutron diffraction methods. Org. Biomol. Chem. 2017, 15, 4655–4666. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. DFT-calculated structures based on 1H NMR chemical shifts in solution vs. structures solved by single-crystal X-ray and crystalline-sponge methods: Assessing specific sources of discrepancies. Tetrahedron 2018, 74, 4728–4737. [Google Scholar] [CrossRef]
- Torralba, M.P.; Sanz, D.; Claramunt, R.M.; Alkorta, I.; Dardonville, C.; Elguero, J. The structure of fosfomycin salts in solution and in the solid state by nuclear magnetic resonance spectroscopy and DFT calculations. Tetrahedron 2018, 74, 3937–3942. [Google Scholar] [CrossRef]
- Mari, S.H.; Varras, P.C.; Wahab, A.-T.; Choudhary, I.M.; Siskos, M.G.; Gerothanassis, I.P. Solvent-dependent structures of natural products based on the combined use of DFT calculations and 1H-NMR chemical shifts. Molecules 2019, 24, 2290. [Google Scholar] [CrossRef]
- Siskos, M.G.; Varras, P.C.; Gerothanassis, I.P. DFT calculations of O-H....O 1H NMR chemical shifts in investigating enol – enol tautomeric equilibria: Probing the impacts of intramolecular hydrogen bonding vs. stereoelectronic interactions. Tetrahedron 2020, 76. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Saito, T.; Yamaji, T.; Hayamizu, K.; Yanagisawa, M.; Yamamoto, O. Spectral Database for Organic Compounds (SDBS). Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi (accessed on 12 December 2019).
- Ruychev, V.Y. Regioselectivity of the Photoisomerization of Acyclic 1,3-Dienes and Stable Trans Cycloheptenes. Ph.D. Thesis, Auburn University, Auburn, AL, USA, 2012. [Google Scholar]
- Klod, S.; Kleinpeter, E. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes-application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2 2001, 10, 1893–1898. [Google Scholar]
- Abraham, R.J.; Canton, M.; Griffiths, L. Proton chemical shifts in NMR: Part 17. Chemical shifts in alkenes and anisotropic and steric effects of the double bond. Magn. Reson. Chem. 2001, 39, 421–431. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Ab initio hybrid DFT–GIAO calculations of the shielding produced by carbon–carbon bonds and aromatic rings in 1H NMR spectroscopy. New J. Chem. 1998, 22, 381–385. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; Wiley: Chichester, UK, 2008. [Google Scholar]
- Abraham, R.J.; Byrne, J.J.; Griffiths, L.; Perez, M. 1H chemical shifts in NMR: Part 23, the effect of dimethyl sulphoxide versus chloroform solvent on 1H chemical shifts. Magn. Reson. Chem. 2006, 44, 491–509. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 0.9, Revision. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Williams, A.J.; Elyashberg, M.E.; Blinov, K.A.; Lankin, D.C.; Martin, G.E.; Reynolds, W.F.; Porco, J.A., Jr.; Singleton, C.A.; Su, S. Applying computer-assisted structure elucidation algorithms for the purpose of structure validation: Revisiting the NMR assignments of hexacyclinol. J. Nat. Prod. 2008, 71, 581–588. [Google Scholar] [CrossRef]
- Burns, D.C.; Mazzola, E.P.; Reynolds, W.F. The role of computer-assisted structure elucidation (CASE) programs in the structure elucidation of complex natural products. Nat. Prod. Rep. 2019, 36, 919–933. [Google Scholar] [CrossRef]
- Dais, P.; Misiaka, M.; Hatzakis, E. Analysis of marine dietary supplements using NMR spectroscopy. Anal. Methods 2015, 7, 5226–5238. [Google Scholar] [CrossRef]
- Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Gerothanassis, I.P. 1H-13C HMBC NMR experiments as a structural and analytical tool for the characterization of elusive trans/cis hydroperoxide isomers from oxidized unsaturated fatty acids in solution. Magn. Reson. Chem. 2019, 57, S67–S74. [Google Scholar] [CrossRef]
- Boccia, A.C.; Cusano, E.; Scano, P.; Consonni, R. NMR lipid profile of milk from alpine goats with supplemented hempseed and linseed diets. Molecules 2020, 25, 1491. [Google Scholar] [CrossRef]
- Wang, C.; Timári, I.; Zhang, B.; Li, D.-W.; Leggett, A.; Amer, A.O.; Bruschweiler-Li, L.; Kopec, R.E.; Brüschweiler, R. COLMAR lipids web server and ultrahigh-resolution methods for 2D NMR- and MS-based lipidomics. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
Sample Availability: Samples of the four geometric isomers of 18:2 ω-7 conjugated linoleic acid (CLA) are available from the authors. |









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).