
Glucosamine Enhancement of BDNF Expression and Animal Cognitive Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. GLN Enhancement on Animal Cognitive Function
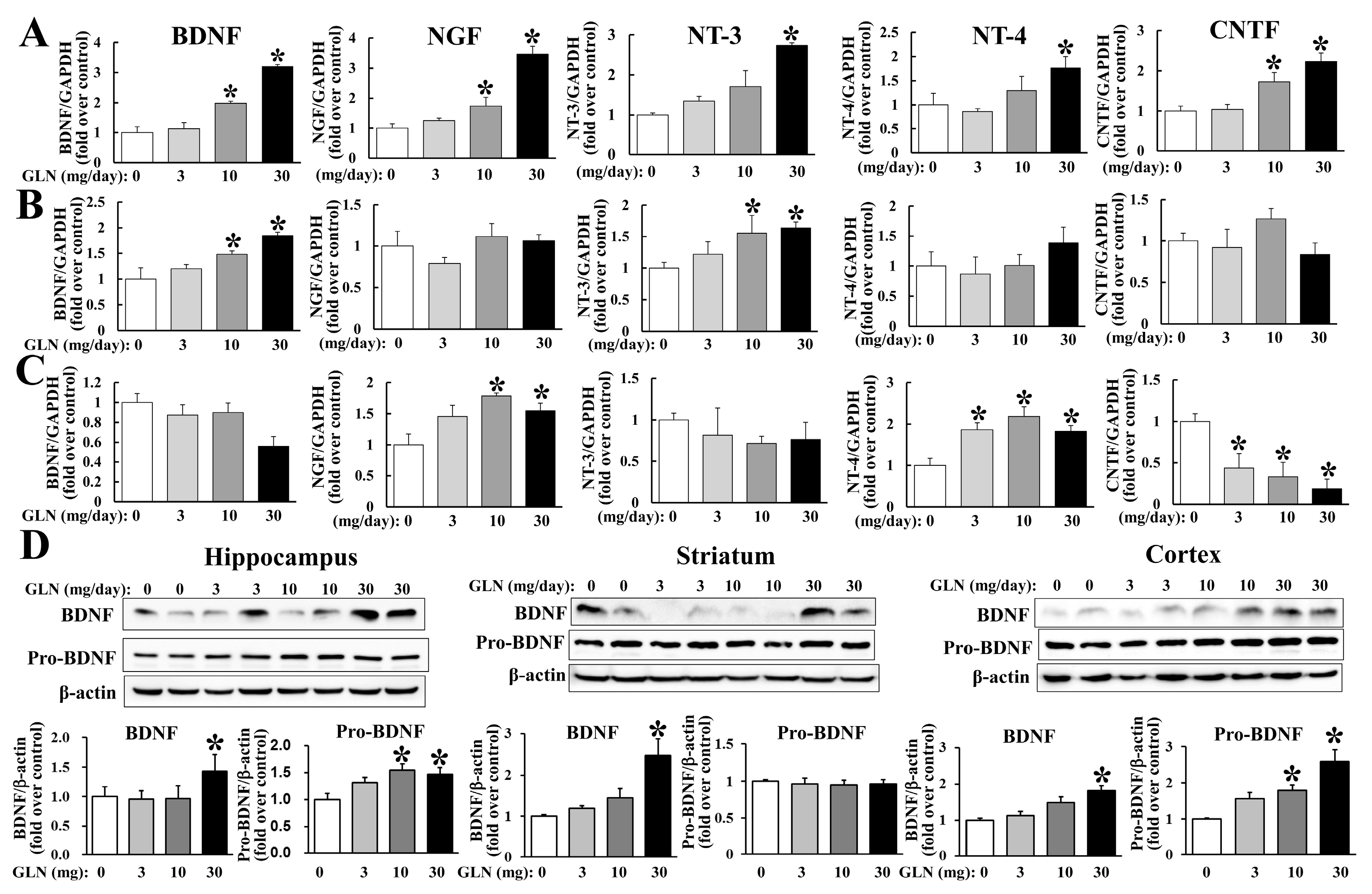
2.2. GLN Induction of the Genes Potentially Associated with the Brain Cognitive Functions
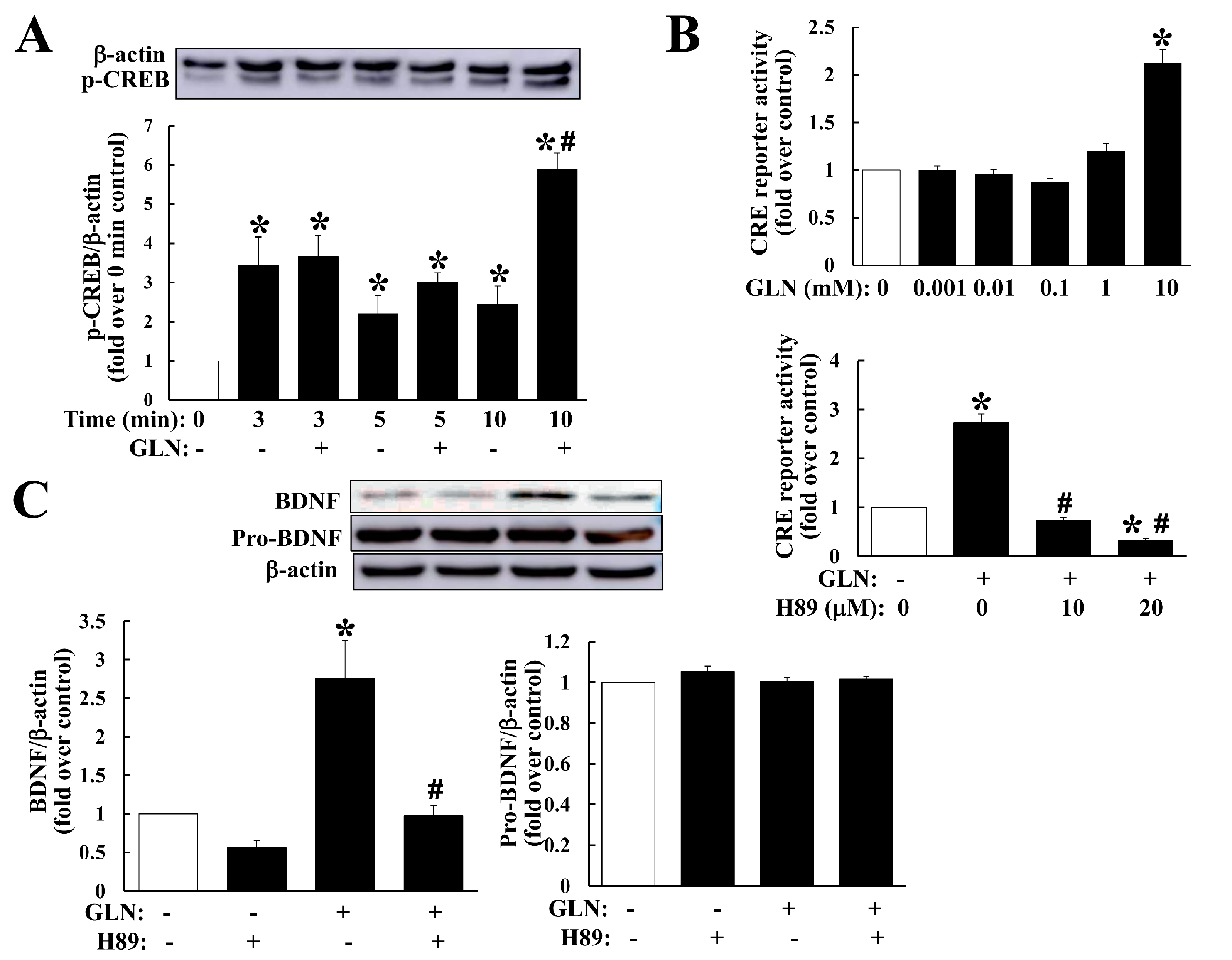
2.3. GLN Impact on Expression of BDNF, CREB5 and PDE4B, and the PKA Signaling in the Hippocampus and HT22 Hippocampal Cells
2.4. Delineation of the GLN Regulation of the cAMP/PKA/CREB Pathway in Relation to BDNF Production
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animal Ethics and Experiments
4.3. Cell Culture
4.4. Determination of Cellular Protein Expression
4.5. Measurement of cAMP by Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Measurement of mRNA Concentration by Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
4.7. Monitoring the CRE Reporter Activity
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yavas, E.; Gonzalez, S.; Fanselow, M.S. Interactions between the hippocampus, prefrontal cortex, and amygdala support complex learning and memory. F1000Research 2019, 8, 1292. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Vogel, E.K.; Lansner, A.; Bergström, F.; Nyberg, L. Neurocognitive Architecture of Working Memory. Neuron 2015, 88, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Zeithamova, D.; Mack, M.L.; Braunlich, K.; Davis, T.; Seger, C.A.; Van Kesteren, M.T.R.; Wutz, A. Brain Mechanisms of Concept Learning. J. Neurosci. 2019, 39, 8259–8266. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, M.; Lucas, G.; Ernfors, P.; Castrén, M.L.; Castren, E. Brain-Derived Neurotrophic Factor and Antidepressant Drugs Have Different But Coordinated Effects on Neuronal Turnover, Proliferation, and Survival in the Adult Dentate Gyrus. J. Neurosci. 2005, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Yi, B.; Wu, C.; Shi, R.; Han, K.; Sheng, H.; Li, B.; Mei, L.; Wang, X.; Huang, Z.; Wu, H. Long-term Administration of Salicylate-induced Changes in BDNF Expression and CREB Phosphorylation in the Auditory Cortex of Rats. Otol. Neurotol. 2018, 39, e173–e180. [Google Scholar] [CrossRef] [Green Version]
- Olsen, C.M.; Liu, Q.-S. Phosphodiesterase 4 inhibitors and drugs of abuse: Current knowledge and therapeutic opportunities. Front. Biol. 2016, 11, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Talarek, S.; Listos, J.; Devi, K.P.; De Oliveira, M.R.; Tewari, D.; Arguelles, S.; Mehrzadi, S.; Hosseinzadeh, A.; D’Onofrio, G.; et al. Phosphodiesterase inhibitors say NO to Alzheimer’s disease. Food Chem. Toxicol. 2019, 134, 110822. [Google Scholar] [CrossRef]
- Blokland, A.; Heckman, P.; Vanmierlo, T.; Schreiber, R.; Paes, D.; Prickaerts, J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol. Sci. 2019, 40, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Akan, I.; Stichelen, S.O.-V.; Bond, M.R.; Hanover, J.A. Nutrient-drivenO-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2017, 144, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Mccarty, M. The neglect of glucosamine as a treatment for osteoarthritis—A personal perspective. Med. Hypotheses 1994, 42, 323–327. [Google Scholar] [CrossRef]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc Modification Protects against Protein Misfolding and Aggregation in Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Hardivillé, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, N. Effects of D-galactosamine and D-glucosamine on retention performance of a brightness discrimination task in rats. Biomed. Biochim. Acta 1985, 44, 611–622. [Google Scholar] [PubMed]
- Setnikar, I.; Rovati, L. Absorption, Distribution, Metabolism and Excretion of Glucosamine Sulfate. Arzneimittelforschung 2011, 51, 699–725. [Google Scholar] [CrossRef]
- Ma, X.; Li, H.; He, Y.; Hao, J. The emerging link between O-GlcNAcylation and neurological disorders. Cell. Mol. Life Sci. 2017, 74, 3667–3686. [Google Scholar] [CrossRef]
- Rocco, M.L.; Soligo, M.; Manni, L.; Aloe, L. Nerve Growth Factor: Early Studies and Recent Clinical Trials. Curr. Neuropharmacol. 2018, 16, 1455–1465. [Google Scholar] [CrossRef]
- Bothwell, M.A. Recent advances in understanding context-dependent mechanisms controlling neurotrophin signaling and function. F1000Research 2019, 8, 1658. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2012, 14, 7–23. [Google Scholar] [CrossRef]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andero, R.; Choi, D.C.; Ressler, K.J. BDNF–TrkB Receptor Regulation of Distributed Adult Neural Plasticity, Memory Formation, and Psychiatric Disorders. Prog. Mol. Biol. Transl. Sci. 2014, 122, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Podda, M.V.; Grassi, C. Role of BDNF Signaling in Memory Enhancement Induced by Transcranial Direct Current Stimulation. Front. Mol. Neurosci. 2018, 12, 427. [Google Scholar] [CrossRef]
- Kawahata, I.; Yoshida, M.; Sun, W.; Nakajima, A.; Lai, Y.; Osaka, N.; Matsuzaki, K.; Yokosuka, A.; Mimaki, Y.; Naganuma, A.; et al. Potent activity of nobiletin-rich Citrus reticulata peel extract to facilitate cAMP/PKA/ERK/CREB signaling associated with learning and memory in cultured hippocampal neurons: identification of the substances responsible for the pharmacological action. J. Neural Transm. 2013, 120, 1397–1409. [Google Scholar] [CrossRef]
- Obrietan, K.; Gao, X.-B.; Pol, A.N.V.D. Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism--a positive feedback circuit in developing neurons. J. Neurophysiol. 2002, 88, 1005–1015. [Google Scholar] [CrossRef]
- Esvald, E.-E.; Tuvikene, J.; Sirp, A.; Patil, S.; Bramham, C.R.; Timmusk, T. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J. Neurosci. 2020, 40, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Jurcovicova, J. Glucose transport in brain—Effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Mardones, L.; Ormazabal, V.; Romo, X.; Jaña, C.; Binder, P.; Peña, E.; Vergara, M.; Zúñiga, F.A. The glucose transporter-2 (GLUT2) is a low affinity dehydroascorbic acid transporter. Biochem. Biophys. Res. Commun. 2011, 410, 7–12. [Google Scholar] [CrossRef]
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002, 524, 199–203. [Google Scholar] [CrossRef]
- Marín-Juez, R.; Rovira, M.; Crespo, D.; Van Der Vaart, M.; Spaink, H.P.; Planas, J.V. GLUT2-Mediated Glucose Uptake and Availability Are Required for Embryonic Brain Development in Zebrafish. Br. J. Pharmacol. 2014, 35, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Nicolosi, R.; Borzelleca, J. Glucosamine effects in humans: A review of effects on glucose metabolism, side effects, safety considerations and efficacy. Food Chem. Toxicol. 2005, 43, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-S.; Yang, M.; Song, M.-S.; Kim, J.-S.; Kim, S.-H.; Kim, J.-C.; Kim, H.; Shin, T.; Wang, H.; Moon, C. Toluene inhibits hippocampal neurogenesis in adult mice. Pharmacol. Biochem. Behav. 2010, 94, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Maher, P.; Schubert, D. Phosphatidylcholine-specific phospholipase C regulates glutamate-induced nerve cell death. Proc. Natl. Acad. Sci. USA 1998, 95, 7748–7753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples and the compounds are not available from the authors. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, L.-Y.; Chao, Y.-M.; Peng, Y.-C.; Lin, H.-C.; Wu, Y.-L. Glucosamine Enhancement of BDNF Expression and Animal Cognitive Function. Molecules 2020, 25, 3667. https://doi.org/10.3390/molecules25163667
Chou L-Y, Chao Y-M, Peng Y-C, Lin H-C, Wu Y-L. Glucosamine Enhancement of BDNF Expression and Animal Cognitive Function. Molecules. 2020; 25(16):3667. https://doi.org/10.3390/molecules25163667
Chicago/Turabian StyleChou, Lien-Yu, Yu-Ming Chao, Yen-Chun Peng, Hui-Ching Lin, and Yuh-Lin Wu. 2020. "Glucosamine Enhancement of BDNF Expression and Animal Cognitive Function" Molecules 25, no. 16: 3667. https://doi.org/10.3390/molecules25163667
APA StyleChou, L.-Y., Chao, Y.-M., Peng, Y.-C., Lin, H.-C., & Wu, Y.-L. (2020). Glucosamine Enhancement of BDNF Expression and Animal Cognitive Function. Molecules, 25(16), 3667. https://doi.org/10.3390/molecules25163667