
Synthesis and Characterization of Cholesteryl Conjugated Lysozyme (CHLysozyme)



Abstract
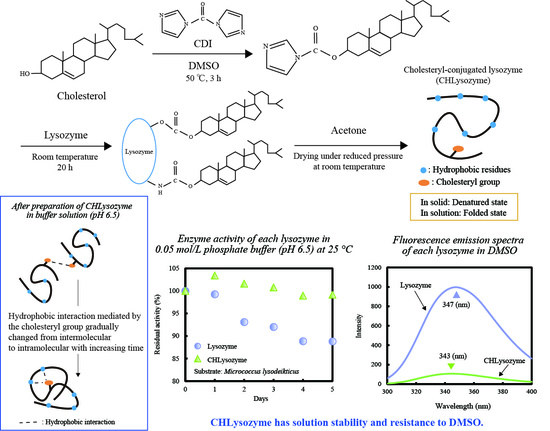
1. Introduction
2. Results and Discussion
2.1. Confirmation of Conjugating the Cholesterol to Lysozyme by NMR Spectroscopy
2.2. Evaluation of Protein State and Hydrophobic Residues in CHLysozyme by DSC
2.3. Evaluation of the Secondary and Tertiary Structure of CHLysozyme by CD Measurement
2.4. Evaluation of Particle Size for CHLysozyme
2.5. Investigation of the Environment Around Aromatic Amino Acid Residues of CHLysozyme by Fluorescence Measurement
2.6. Study of the Hydrophobic Region of CHLysozyme by Fluorescene Measurement with a Hydrophobic Probe
2.7. Enzyme Activity of CHLysozyme
2.7.1. Relative Activity and Enzyme Kinetic Constants of CHLysozyme
2.7.2. Change of Relative and Residual Activity over Time
2.8. Resistance to DMSO of CHLysozyme
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Cholesteryl Conjugated Lysozyme (CHLysozyme)
3.3. Confirmation of Conjugating the Cholesterol to Lysozyme by NMR Spectroscopy
3.4. Evaluation of Protein State and Hydrophobic Residues in CHLysozyme by DSC
3.5. Evaluation of the Secondary and Tertiary Structures of CHLysozyme by CD Measurement
3.6. Evaluation of Particle Size for CHLysozyme
3.7. Investigation of the Environment Around Aromatic Amino Acid Residues of CHLysozyme by Fluorescence Measurement
3.8. Study of the Hydrophobic Region of CHLysozyme by Fluorescence Measurement with a Hydrophobic Probe
3.9. Enzyme Activity of CHLysozyme
3.9.1. Relative Activity and Enzyme Kinetic Constants of CHLysozyme
Relative Activity of CHLysozyme to Lysozyme
Enzyme Kinetic Constants of CHLysozyme
3.9.2. Change of Relative and Residual Activity over Time
3.10. Resistance to DMSO of CHLysozyme
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lutz, S.; Iamurri, S.M. Protein Engineering: Past, Present, and Future. Methods Mol. Biol. 2018, 1685, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T. What are the Limitations of Enzymes in Synthetic Organic Chemistry? Chem. Rec. 2016, 16, 2449–2459. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006, 13, 399–409. [Google Scholar] [CrossRef]
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci 2015, 16, 25831–25864. [Google Scholar] [CrossRef]
- Awwad, S.; Ginn, C.; Brocchini, S. 2-The case for protein PEGylation. In Engineering of Biomaterials for Drug Delivery Systems; Parambath, A., Ed.; Woodhead Publishing: Cambridge, England, 2018; pp. 27–49. [Google Scholar] [CrossRef]
- Lawrence, P.B.; Price, J.L. How PEGylation influences protein conformational stability. Curr. Opin. Chem. Biol. 2016, 34, 88–94. [Google Scholar] [CrossRef]
- Ginn, C.; Khalili, H.; Lever, R.; Brocchini, S. PEGylation and its impact on the design of new protein-based medicines. Future Med. Chem. 2014, 6, 1829–1846. [Google Scholar] [CrossRef]
- Nakamura, S.; Kato, A. Multi-functional biopolymer prepared by covalent attachment of galactomannan to egg-white proteins through naturally occurring Maillard reaction. Nahrung/Food 2000, 44, 201–206. [Google Scholar] [CrossRef]
- Nakamura, S.; Ban, M.; Kato, A. Preparation of bioactive and surface functional oligomannosyl neoglycoprotein using extracellular pH-sensitive glycosylation of mutant lysozyme having N-linked signal sequence in yeast. Bioconjug. Chem. 2006, 17, 1170–1177. [Google Scholar] [CrossRef]
- Gaber, M.; Mabrouk, M.T.; Freag, M.S.; Khiste, S.K.; Fang, J.Y.; Elkhodairy, K.A.; Elzoghby, A.O. Protein-polysaccharide nanohybrids: Hybridization techniques and drug delivery applications. Eur. J. Pharm. Biopharm. 2018, 133, 42–62. [Google Scholar] [CrossRef]
- Gershkovich, A.A.; Verevka, S.V.; Kibirev, V.K.; Demchenko, A.P. Solid-state conjugation of proteins with hydrophobic compounds in non-denaturing conditions. I. Acylation of proteins by dansyl proline using a polymeric N-hydroxysuccinimide ester. J. Biochem. Biophys. Methods 2000, 45, 183–191. [Google Scholar] [CrossRef]
- Lillo, A.M.; Lopez, C.L.; Rajale, T.; Yen, H.J.; Magurudeniya, H.D.; Phipps, M.L.; Balog, E.R.M.; Sanchez, T.C.; Iyer, S.; Wang, H.L.; et al. Conjugation of Amphiphilic Proteins to Hydrophobic Ligands in Organic Solvent. Bioconjug. Chem. 2018, 29, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, K.; Deguchi, S.; Moriguchi, N.; Yamaguchi, S.; Sunamoto, J. Self-aggregates of hydrophobized polysaccharides in water. Formation and characteristics of nanoparticles. Macromolecules 1993, 26, 3062–3068. [Google Scholar] [CrossRef]
- Akiyoshi, K.; Ueminami, A.; Kurumada, S.; Nomura, Y. Self-Association of Cholesteryl-Bearing Poly(l-lysine) in Water and Control of Its Secondary Structure by Host−Guest Interaction with Cyclodextrin. Macromolecules 2000, 33, 6752–6756. [Google Scholar] [CrossRef]
- Tahara, Y.; Sakiyama, M.; Takeda, S.; Nishimura, T.; Mukai, S.A.; Sawada, S.I.; Sasaki, Y.; Akiyoshi, K. Self-Assembled Nanogels of Cholesterol-Bearing Hydroxypropyl Cellulose: A Thermoresponsive Building Block for Nanogel Tectonic Materials. Langmuir 2016, 32, 12283–12289. [Google Scholar] [CrossRef]
- Sawada, S.I.; Yukawa, H.; Takeda, S.; Sasaki, Y.; Akiyoshi, K. Self-assembled nanogel of cholesterol-bearing xyloglucan as a drug delivery nanocarrier. J. Biomater. Sci. Polym. Ed. 2017, 28, 1183–1198. [Google Scholar] [CrossRef]
- Nomura, Y.; Sasaki, Y.; Takagi, M.; Narita, T.; Aoyama, Y.; Akiyoshi, K. Thermoresponsive controlled association of protein with a dynamic nanogel of hydrophobized polysaccharide and cyclodextrin: Heat shock protein-like activity of artificial molecular chaperone. Biomacromolecules 2005, 6, 447–452. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Mukai, S.A.; Sasaki, Y.; Akiyoshi, K. Nanogel Tectonics for Tissue Engineering: Protein Delivery Systems with Nanogel Chaperones. Adv. Healthc. Mater. 2018, 7, 1800729. [Google Scholar] [CrossRef]
- Kobayashi, H.; Katakura, O.; Morimoto, N.; Akiyoshi, K.; Kasugai, S. Effects of cholesterol-bearing pullulan (CHP)-nanogels in combination with prostaglandin E1 on wound healing. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 91, 55–60. [Google Scholar] [CrossRef]
- Morimoto, N.; Yamazaki, M.; Tamada, J.; Akiyoshi, K. Polysaccharide-hair cationic polypeptide nanogels: Self-assembly and enzymatic polymerization of amylose primer-modified cholesteryl poly(L-lysine). Langmuir 2013, 29, 7509–7514. [Google Scholar] [CrossRef]
- Malleshappa Gowder, S.; Chatterjee, J.; Chaudhuri, T.; Paul, K. Prediction and analysis of surface hydrophobic residues in tertiary structure of proteins. Sci. World J. 2014, 2014, 971258. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.S.; Subbiah, S.; Levitt, M. Recognizing native folds by the arrangement of hydrophobic and polar residues. J. Mol. Biol. 1995, 252, 709–720. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.; Hoogeveen, A.; Abeln, S. The hydrophobic temperature dependence of amino acids directly calculated from protein structures. PLoS Comput. Biol. 2015, 11, e1004277. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, Y.; Srebnik, S. Conformational changes of globular proteins upon adsorption on a hydrophobic surface. Phys. Chem. Chem. Phys. 2014, 16, 11698–11707. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Das, U.; Hariprasad, G.; Ethayathulla, A.S.; Manral, P.; Das, T.K.; Pasha, S.; Mann, A.; Ganguli, M.; Verma, A.K.; Bhat, R.; et al. Inhibition of protein aggregation: Supramolecular assemblies of arginine hold the key. PLoS ONE 2007, 2, e1176. [Google Scholar] [CrossRef]
- Kotarek, J.; Stuart, C.; De Paoli, S.H.; Simak, J.; Lin, T.L.; Gao, Y.; Ovanesov, M.; Liang, Y.; Scott, D.; Brown, J.; et al. Subvisible Particle Content, Formulation, and Dose of an Erythropoietin Peptide Mimetic Product Are Associated With Severe Adverse Postmarketing Events. J. Pharm. Sci. 2016, 105, 1023–1027. [Google Scholar] [CrossRef]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef]
- Mine, Y.; Ma, F.; Lauriau, S. Antimicrobial peptides released by enzymatic hydrolysis of hen egg white lysozyme. J. Agric. Food Chem. 2004, 52, 1088–1094. [Google Scholar] [CrossRef]
- Proctor, V.A.; Cunningham, F.E. The chemistry of lysozyme and its use as a food preservative and a pharmaceutical. Crit. Rev. Food Sci. Nutr. 1988, 26, 359–395. [Google Scholar] [CrossRef]
- Freitas Dda, S.; Abrahao-Neto, J. Biochemical and biophysical characterization of lysozyme modified by PEGylation. Int. J. Pharm. 2010, 392, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Nodake, Y.; Yamasaki, N. Some properties of a macromolecular conjugate of lysozyme prepared by modification with a monomethoxypolyethylene glycol derivative. Biosci. Biotechnol. Biochem. 2000, 64, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Briand, V.; Sharma, N.; Ahn, S.-k.; Kasi, R. Polymers Comprising Cholesterol: Synthesis, Self-Assembly, and Applications. Materials 2009, 2, 636–660. [Google Scholar] [CrossRef]
- Yeagle, P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Yeagle, P.L. Modulation of membrane function by cholesterol. Biochimie 1991, 73, 1303–1310. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Grimsey, I.M.; Forbes, R.T. Mapping the solid-state properties of crystalline lysozyme during pharmaceutical unit-operations. J. Pharm. Biomed. Anal. 2015, 114, 176–183. [Google Scholar] [CrossRef]
- Gorinstein, S.; Zemser, M.; Friedman, M.; Chang, S.M. Simultaneous differential scanning calorimetry, X-ray diffraction and FTIR spectrometry in studies of ovalbumin denaturation. Int. J. Pept. Protein Res. 1995, 45, 248–256. [Google Scholar] [CrossRef]
- Pikal, M.J.; Rigsbee, D.; Roy, M.L. Solid state stability of proteins III: Calorimetric (DSC) and spectroscopic (FTIR) characterization of thermal denaturation in freeze dried human growth hormone (hGH). J. Pharm. Sci. 2008, 97, 5122–5131. [Google Scholar] [CrossRef]
- Makhatadze, G.I.; Privalov, P.L. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: Hydration effect. J. Mol. Biol. 1990, 213, 375–384. [Google Scholar] [CrossRef]
- Privalov, P.L.; Makhatadze, G.I. Heat capacity of proteins. II. Partial molar heat capacity of the unfolded polypeptide chain of proteins: Protein unfolding effects. J. Mol. Biol. 1990, 213, 385–391. [Google Scholar] [CrossRef]
- Cinelli, S.; De Francesco, A.; Onori, G.; Paciaroni, A. Thermal stability and internal dynamics of lysozyme as affected by hydration. Phys. Chem. Chem. Phys. 2004, 6, 3591–3595. [Google Scholar] [CrossRef]
- Bye, J.W.; Falconer, R.J. Thermal stability of lysozyme as a function of ion concentration: A reappraisal of the relationship between the Hofmeister series and protein stability. Protein Sci. 2013, 22, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef]
- Yampolskaya, G.P.; Tarasevich, B.N.; Elenskii, A.A. Secondary Structure of Globular Proteins in Adsorption Layers at the Solution-Air Interface by the Data of Fourier Transform IR Spectroscopy. Colloid J. 2005, 67, 385–391. [Google Scholar] [CrossRef]
- Yamamoto, T.; Fukui, N.; Hori, A.; Matsui, Y. Circular dichroism and fluorescence spectroscopy studies of the effect of cyclodextrins on the thermal stability of chicken egg white lysozyme in aqueous solution. J. Mol. Struct. 2006, 782, 60–66. [Google Scholar] [CrossRef]
- Loftsson, T.; Jarho, P.; Masson, M.; Jarvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef]
- Christoforides, E.; Papaioannou, A.; Bethanis, K. Crystal structure of the inclusion complex of cholesterol in beta-cyclodextrin and molecular dynamics studies. Beilstein J. Org. Chem. 2018, 14, 838–848. [Google Scholar] [CrossRef]
- Sawada, S.-I.; Sasaki, Y.; Nomura, Y.; Akiyoshi, K. Cyclodextrin-responsive nanogel as an artificial chaperone for horseradish peroxidase. Colloid Polym. Sci. 2010, 289, 685–691. [Google Scholar] [CrossRef]
- Teale, F.W.; Weber, G. Ultraviolet fluorescence of the aromatic amino acids. Biochem. J. 1957, 65, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Ghisaidoobe, A.B.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Forster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Protein Fluorescence. In Principles of Fluorescence Spectroscopy; Lakowicz, J.R., Ed.; Springer US: Boston, MA, USA, 2006; pp. 529–575. [Google Scholar] [CrossRef]
- Kokufuta, E.; Watanabe, N.; Nakamura, I. On the function of the polyion complex of methemoglobin and polyelectrolytes as cyanide ion exchanger. J. Appl. Polym. Sci. 1981, 26, 2601–2612. [Google Scholar] [CrossRef]
- Fitter, J.; Haber-Pohlmeier, S. Structural stability and unfolding properties of thermostable bacterial alpha-amylases: A comparative study of homologous enzymes. Biochemistry 2004, 43, 9589–9599. [Google Scholar] [CrossRef]
- Duy, C.; Fitter, J. How aggregation and conformational scrambling of unfolded states govern fluorescence emission spectra. Biophys. J. 2006, 90, 3704–3711. [Google Scholar] [CrossRef]
- Kirk, W.R.; Kurian, E.; Prendergast, F.G. Characterization of the sources of protein-ligand affinity: 1-sulfonato-8-(1′)anilinonaphthalene binding to intestinal fatty acid binding protein. Biophys. J. 1996, 70, 69–83. [Google Scholar] [CrossRef]
- Pastukhov, A.V.; Ropson, I.J. Fluorescent dyes as probes to study lipid-binding proteins. Proteins 2003, 53, 607–615. [Google Scholar] [CrossRef]
- Izutsu, K.I. Applications of Freezing and Freeze-Drying in Pharmaceutical Formulations. Adv. Exp. Med. Biol. 2018, 1081, 371–383. [Google Scholar] [CrossRef]
- Gasymov, O.K.; Glasgow, B.J. ANS fluorescence: Potential to augment the identification of the external binding sites of proteins. Biochim. Biophys. Acta 2007, 1774, 403–411. [Google Scholar] [CrossRef]
- Prasad, A.L.; Litwack, G. Measurement of the Lytic Activity of Lysozymes (Muramidases). Anal. Biochem. 1963, 6, 328–334. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Balaram, P. Effects of organic solvents on protein structures: Observation of a structured helical core in hen egg-white lysozyme in aqueous dimethylsulfoxide. Proteins 1997, 29, 492–507. [Google Scholar] [CrossRef]
- Evans, P.A.; Topping, K.D.; Woolfson, D.N.; Dobson, C.M. Hydrophobic clustering in nonnative states of a protein: Interpretation of chemical shifts in NMR spectra of denatured states of lysozyme. Proteins 1991, 9, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Mantsch, H.H. Beware of proteins in DMSO. Biochim. Biophys. Acta 1991, 1078, 231–235. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Aoyama, T.; Ogawa, O.; Tabata, Y. Liver targeting of plasmid DNA by pullulan conjugation based on metal coordination. J. Control. Release 2002, 83, 287–302. [Google Scholar] [CrossRef]
- Koner, A.L.; Krndija, D.; Hou, Q.; Sherratt, D.J.; Howarth, M. Hydroxy-terminated conjugated polymer nanoparticles have near-unity bright fraction and reveal cholesterol-dependence of IGF1R nanodomains. ACS Nano 2013, 7, 1137–1144. [Google Scholar] [CrossRef]
- Varshosaz, J.; Hassanzadeh, F.; Sadeghi Aliabadi, H.; Nayebsadrian, M.; Banitalebi, M.; Rostami, M. Synthesis and characterization of folate-targeted dextran/retinoic acid micelles for doxorubicin delivery in acute leukemia. Biomed. Res. Int. 2014, 2014, 525684. [Google Scholar] [CrossRef]
- Hussain, M.A.; Abbas, K.; Lodhi, B.A.; Sher, M.; Ali, M.; Tahir, M.N.; Tremel, W.; Iqbal, S. Fabrication, characterization, thermal stability and nanoassemblies of novel pullulan-aspirin conjugates. Arab. J. Chem. 2017, 10, S1597–S1603. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Conc. (mg/mL) | Particle Size (nm) |
|---|---|---|
| Lysozyme | 0.2 | N. D. 1 |
| 1.0 | 24 | |
| 2.0 | 75 | |
| CHLysozyme | 0.2 | 119 |
| 1.0 | 238 | |
| 2.0 | 1194 |
| Sample 1 | Km (mg/mL) | Vmax (sec−1) |
|---|---|---|
| Lysozyme | 1.20 | 0.07 |
| CHLysozyme | 0.25 | 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsura, S.; Furuishi, T.; Ueda, H.; Yonemochi, E. Synthesis and Characterization of Cholesteryl Conjugated Lysozyme (CHLysozyme). Molecules 2020, 25, 3704. https://doi.org/10.3390/molecules25163704
Katsura S, Furuishi T, Ueda H, Yonemochi E. Synthesis and Characterization of Cholesteryl Conjugated Lysozyme (CHLysozyme). Molecules. 2020; 25(16):3704. https://doi.org/10.3390/molecules25163704
Chicago/Turabian StyleKatsura, Shinji, Takayuki Furuishi, Haruhisa Ueda, and Etsuo Yonemochi. 2020. "Synthesis and Characterization of Cholesteryl Conjugated Lysozyme (CHLysozyme)" Molecules 25, no. 16: 3704. https://doi.org/10.3390/molecules25163704
APA StyleKatsura, S., Furuishi, T., Ueda, H., & Yonemochi, E. (2020). Synthesis and Characterization of Cholesteryl Conjugated Lysozyme (CHLysozyme). Molecules, 25(16), 3704. https://doi.org/10.3390/molecules25163704