
Sustainable Access to π-Conjugated Molecular Materials via Direct (Hetero)Arylation Reactions in Water and under Air

 , , and
, , and
Abstract
:
1. Introduction
2. Results
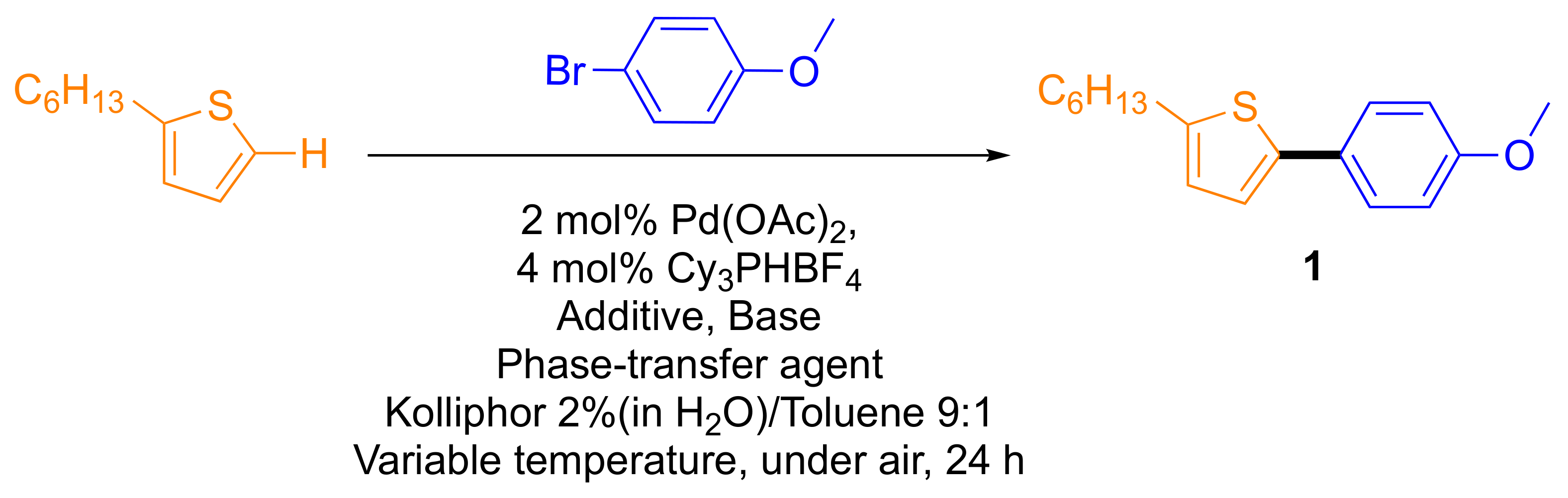
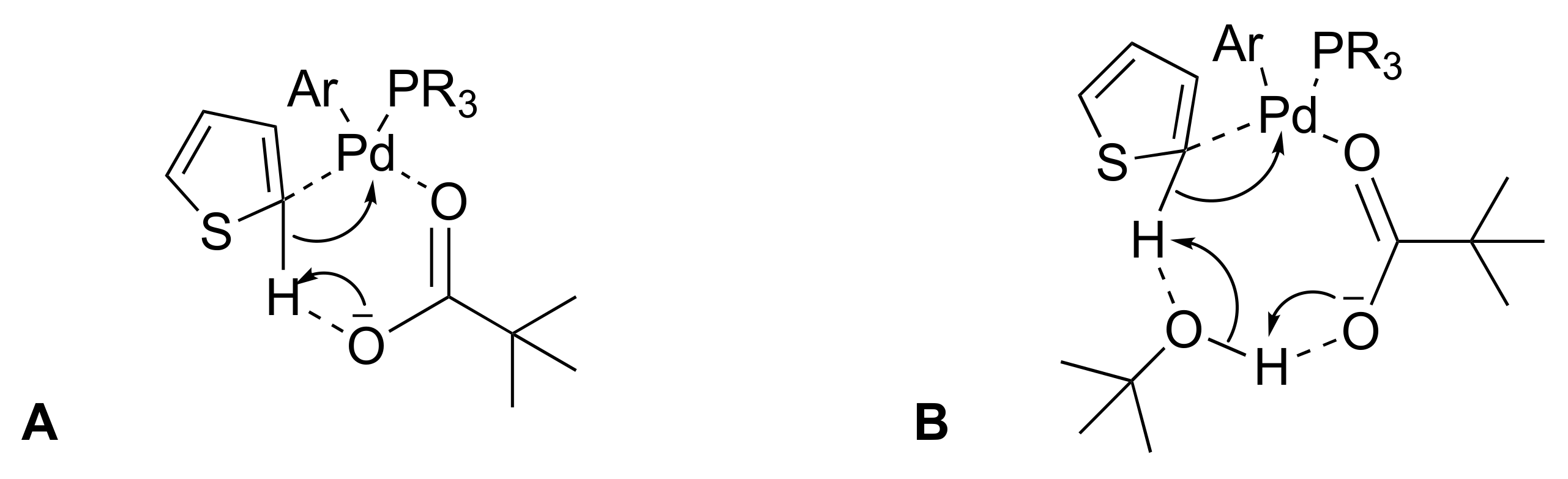
2.1. Optimization of Surfactant Enhanced DHA in Water on a Model Reaction
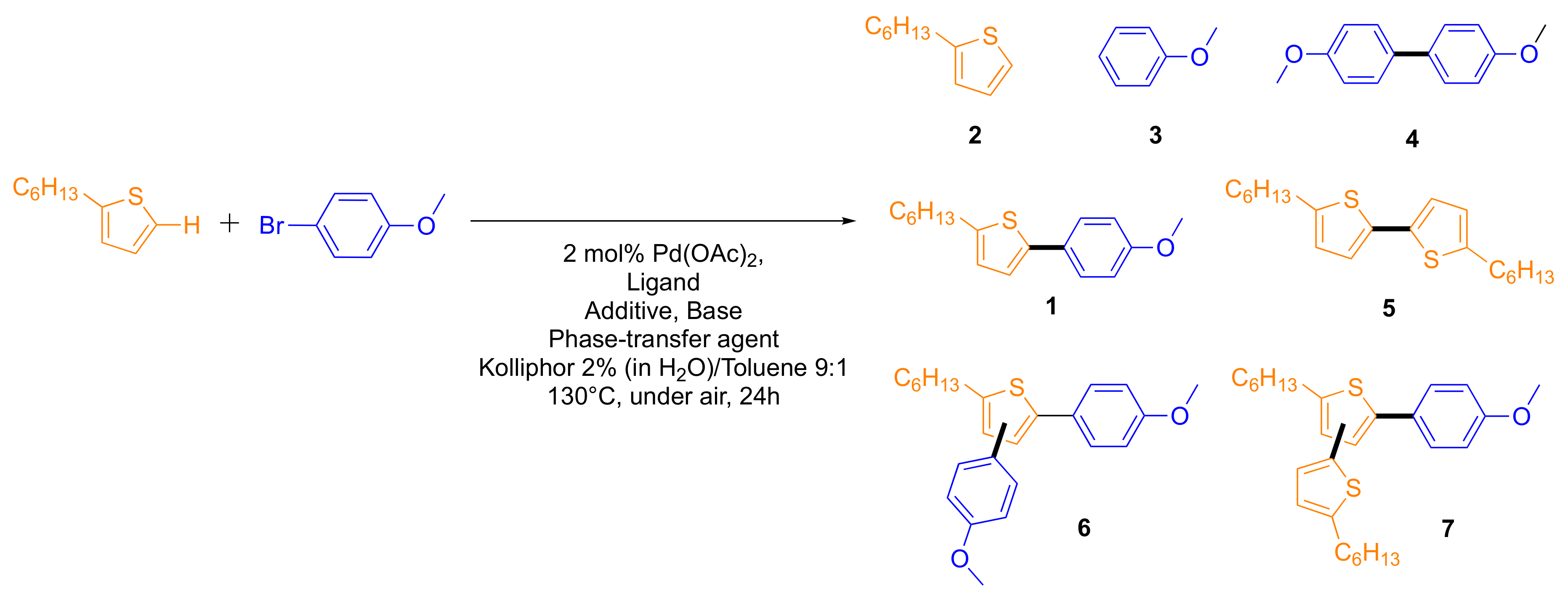
2.2. Scope and Generality of the Method in the Preparation of Conjugated Building Blocks
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clark, J.; Lanzani, G. Organic photonics for communications. Nat. Photonics 2010, 4, 438–446. [Google Scholar] [CrossRef]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-conjugated systems in field-effect transistors: A material odyssey of organic electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, A. π-Conjugated polymers for organic electronics and photovoltaic cell applications. Chem. Mater. 2011, 23, 733–758. [Google Scholar] [CrossRef]
- Sekine, C.; Tsubata, Y.; Yamada, T.; Kitano, M.; Doi, S. Recent progress of high performance polymer OLED and OPV materials for organic printed electronics. Sci. Technol. Adv. Mater. 2014, 15. [Google Scholar] [CrossRef]
- Duan, L.; Hou, L.; Lee, T.W.; Qiao, J.; Zhang, D.; Dong, G.; Wang, L.; Qiu, Y. Solution processable small molecules for organic light-emitting diodes. J. Mater. Chem. 2010, 20, 6392–6407. [Google Scholar] [CrossRef] [Green Version]
- Roncali, J.; Leriche, P.; Blanchard, P. Molecular materials for organic photovoltaics: Small is beautiful. Adv. Mater. 2014, 26, 3821–3838. [Google Scholar] [CrossRef] [Green Version]
- Po, R.; Bernardi, A.; Calabrese, A.; Carbonera, C.; Corso, G.; Pellegrino, A. From lab to fab: How must the polymer solar cell materials design change? An industrial perspective. Energy Environ. Sci. 2014, 7, 925–943. [Google Scholar] [CrossRef]
- Po, R.; Bianchi, G.; Carbonera, C.; Pellegrino, A. “All that glisters is not gold”: An analysis of the synthetic complexity of efficient polymer donors for polymer solar cells. Macromolecules 2015, 48, 453–461. [Google Scholar] [CrossRef]
- Huang, Y.; Elder, D.L.; Kwiram, A.L.; Jenekhe, S.A.; Jen, A.K.Y.; Dalton, L.R.; Luscombe, C.K. Organic Semiconductors at the University of Washington: Advancements in Materials Design and Synthesis and toward Industrial Scale Production. Adv. Mater. 2019, 1904239, 1–23. [Google Scholar] [CrossRef]
- Beach, E.S.; Cui, Z.; Anastas, P.T. Green Chemistry: A design framework for sustainability. Energy Environ. Sci. 2009, 2, 1038–1049. [Google Scholar] [CrossRef]
- Zvezdin, A.; Di Mauro, E.; Rho, D.; Santato, C.; Khalil, M. En route toward sustainable organic electronics. MRS Energy Sustain. 2020, 7, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Irimia-Vladu, M. “Green” electronics: Biodegradable and biocompatible materials and devices for sustainable future. Chem. Soc. Rev. 2014, 43, 588–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Lee, M.Y.; Park, C.H.; Lee, H.R.; Oh, J.H. Toward Environmentally Robust Organic Electronics: Approaches and Applications. Adv. Mater. 2017, 29, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Carsten, B.; He, F.; Son, H.J.; Xu, T.; Yu, L. Stille polycondensation for synthesis of functional materials. Chem. Rev. 2011, 111, 1493–1528. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Rehahn, M.; Wegner, G.; Schlüter, A.D. Suzuki polycondensation: Polyarylenes à la carte. Macromol. Rapid Commun. 2009, 30, 653–687. [Google Scholar] [CrossRef]
- Marrocchi, A.; Facchetti, A.; Lanari, D.; Petrucci, C.; Vaccaro, L. Current methodologies for a sustainable approach to π-conjugated organic semiconductors. Energy Environ. Sci. 2016, 9, 763–786. [Google Scholar] [CrossRef]
- McAfee, S.M.; McCahill, J.S.J.; MacAulay, C.M.; Hendsbee, A.D.; Welch, G.C. Utility of a heterogeneous palladium catalyst for the synthesis of a molecular semiconductor via Stille, Suzuki, and direct heteroarylation cross-coupling reactions. RSC Adv. 2015, 5, 26097–26106. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-metal-catalyzed direct arylation of (hetero)arenes by C–H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- Mercier, L.G.; Leclerc, M. Direct (hetero)arylation: A new tool for polymer chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef]
- Wu, W.; Xin, H.; Ge, C.; Gao, X. Application of direct (hetero)arylation in constructing conjugated small molecules and polymers for organic optoelectronic devices. Tetrahedron Lett. 2017, 58, 175–184. [Google Scholar] [CrossRef]
- Facchetti, A.; Vaccaro, L.; Marrocchi, A. Semiconducting polymers prepared by direct arylation polycondensation. Angew. Chem. Int. Ed. 2012, 51, 3520–3523. [Google Scholar] [CrossRef]
- Suraru, S.L.; Lee, J.A.; Luscombe, C.K. C–H Arylation in the Synthesis of π-Conjugated Polymers. ACS Macro Lett. 2016, 5, 724–729. [Google Scholar] [CrossRef] [Green Version]
- Gorelsky, S.I. Origins of regioselectivity of the palladium-catalyzed (aromatic)CH bond metalation-deprotonation. Coord. Chem. Rev. 2013, 257, 153–164. [Google Scholar] [CrossRef]
- Hendsbee, A.D.; Li, Y. Performance comparisons of polymer semiconductors synthesized by direct (Hetero)Arylation Polymerization (DHAP) and conventional methods for organic thin film transistors and organic photovoltaics. Molecules 2018, 23, 1255. [Google Scholar] [CrossRef] [Green Version]
- Bohra, H.; Wang, M. Direct C–H arylation: A “greener” approach towards facile synthesis of organic semiconducting molecules and polymers. J. Mater. Chem. A 2017, 5, 11550–11571. [Google Scholar] [CrossRef]
- Yu, S.; Liu, F.; Yu, J.; Zhang, S.; Cabanetos, C.; Gao, Y.; Huang, W. Eco-friendly direct (hetero)-arylation polymerization: Scope and limitation. J. Mater. Chem. C 2017, 5, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, J.; Fujie, Y.; Maruyama, K.; Yasuda, T.; Kanbara, T. Suppression of Homocoupling Side Reactions in Direct Arylation Polycondensation for Producing High Performance OPV Materials. Macromolecules 2016, 49, 9388–9395. [Google Scholar] [CrossRef]
- Matsidik, R.; Komber, H.; Luzio, A.; Caironi, M.; Sommer, M. Defect-free Naphthalene Diimide Bithiophene Copolymers with Controlled Molar Mass and High Performance via Direct Arylation Polycondensation. J. Am. Chem. Soc. 2015, 137, 6705–6711. [Google Scholar] [CrossRef]
- Bura, T.; Beaupré, S.; Légaré, M.A.; Quinn, J.; Rochette, E.; Blaskovits, J.T.; Fontaine, F.G.; Pron, A.; Li, Y.; Leclerc, M. Direct heteroarylation polymerization: Guidelines for defect-free conjugated polymers. Chem. Sci. 2017, 8, 3913–3925. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Tatum, W.; Zhang, J.; Scott, C.; Luscombe, C.K.; Marder, S.R.; Blakey, S.B. The Direct Arylation Polymerization (DArP) of Well-Defined Alternating Copolymers Based on 5,6-Dicyano[2,1,3]benzothiadiazole (DCBT). Asian J. Org. Chem. 2018, 7, 1419–1425. [Google Scholar] [CrossRef]
- Gorelsky, S.I. Reactivity and regioselectivity of palladium-catalyzed direct arylation in noncooperative and cooperative processes. Organometallics 2012, 31, 4631–4634. [Google Scholar] [CrossRef]
- Pouliot, J.R.; Grenier, F.; Blaskovits, J.T.; Beaupré, S.; Leclerc, M. Direct (Hetero)arylation Polymerization: Simplicity for Conjugated Polymer Synthesis. Chem. Rev. 2016, 116, 14225–14274. [Google Scholar] [CrossRef] [PubMed]
- Bura, T.; Blaskovits, J.T.; Leclerc, M. Direct (Hetero)arylation Polymerization: Trends and Perspectives. J. Am. Chem. Soc. 2016, 138, 10056–10071. [Google Scholar] [CrossRef] [PubMed]
- Lafrance, M.; Fagnou, K. Palladium-catalyzed benzene arylation: Incorporation of catalytic pivalic acid as a proton shuttle and a key element in catalyst design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [Google Scholar] [CrossRef]
- Rudenko, A.E.; Thompson, B.C. Influence of the carboxylic acid additive structure on the properties of poly(3-hexylthiophene) prepared via direct arylation polymerization (DArP). Macromolecules 2015, 48, 569–575. [Google Scholar] [CrossRef]
- Morin, P.O.; Bura, T.; Sun, B.; Gorelsky, S.I.; Li, Y.; Leclerc, M. Conjugated polymers à la carte from time-controlled direct (hetero)arylation polymerization. ACS Macro Lett. 2015, 4, 21–24. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S. Transitioning organic synthesis from organic solvents to water. What’s your e Factor? Green Chem. 2014, 16, 3660–3679. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S. Transition-metal-catalyzed cross-couplings going green: In water at room temperature. Aldrichim. Acta 2008, 41, 59–72. [Google Scholar]
- Lipshutz, B.H.; Ghorai, S.; Cortes-Clerget, M. The Hydrophobic Effect Applied to Organic Synthesis: Recent Synthetic Chemistry “in Water”. Chem. Eur. J. 2018, 24, 6672–6695. [Google Scholar] [CrossRef]
- Lipshutz, B.H. When Does Organic Chemistry Follow Nature’s Lead and “make the Switch”? J. Org. Chem. 2017, 82, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Mattiello, S.; Rooney, M.; Sanzone, A.; Brazzo, P.; Sassi, M.; Beverina, L. Suzuki-Miyaura Micellar Cross-Coupling in Water, at Room Temperature, and under Aerobic Atmosphere. Org. Lett. 2017, 19, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.; Mattiello, S.; Stara, R.; Sanzone, A.; Brazzo, P.; Sassi, M.; Beverina, L. Suzuki-Miyaura cross-coupling of latent pigments in water/toluene emulsion under aerobic atmosphere. Dye. Pigment. 2018, 149, 893–901. [Google Scholar] [CrossRef]
- Vaghi, L.; Sanzone, A.; Sassi, M.; Pagani, S.; Papagni, A.; Beverina, L. Synthesis of Fluorinated Acridines via Sequential Micellar Buchwald-Hartwig Amination/Cyclization of Aryl Bromides. Synthesizer 2018, 50, 1621–1628. [Google Scholar] [CrossRef]
- Sanzone, A.; Calascibetta, A.; Ghiglietti, E.; Ceriani, C.; Mattioli, G.; Mattiello, S.; Sassi, M.; Beverina, L. Suzuki-Miyaura Micellar One-Pot Synthesis of Symmetrical and Unsymmetrical 4,7-Diaryl-5,6-difluoro-2,1,3-benzothiadiazole Luminescent Derivatives in Water and under Air. J. Org. Chem. 2018, 83, 15029–15042. [Google Scholar] [CrossRef]
- Sanzone, A.; Mattiello, S.; Garavaglia, G.M.; Calascibetta, A.M.; Ceriani, C.; Sassi, M.; Beverina, L. Efficient synthesis of organic semiconductors by Suzuki-Miyaura coupling in an aromatic micellar medium. Green Chem. 2019, 21, 4400–4405. [Google Scholar] [CrossRef]
- Sassi, M.; Mattiello, S.; Beverina, L. Syntheses of Organic Semiconductors in Water. Recent Advancement in the Surfactants Enhanced Green Access to Polyconjugated Molecules. Eur. J. Org. Chem. 2020, 2020. [Google Scholar] [CrossRef]
- Nishikata, T.; Abela, A.R.; Huang, S.; Lipshutz, B.H. Cationic Pd(II)-catalyzed C–H activation/cross-coupling reactions at room temperature: Synthetic and mechanistic studies. Beilstein J. Org. Chem. 2016. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, G.N.; Fiske, S.; Verma, H.; Lokhande, S.K.; Kumar, D. A micellar catalysis strategy applied to the Pd-catalyzed C–H arylation of indoles in water. Green Chem. 2019, 21, 1448–1454. [Google Scholar] [CrossRef]
- Daniels, M.H.; Armand, J.R.; Tan, K.L. Sequential Regioselective C–H Functionalization of Thiophenes. Org. Lett. 2016. [Google Scholar] [CrossRef]
- Grenier, F.; Goudreau, K.; Leclerc, M. Robust Direct (Hetero)arylation Polymerization in Biphasic Conditions. J. Am. Chem. Soc. 2017, 139, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Mainville, M.; Tremblay, V.; Fenniri, M.Z.; Laventure, A.; Farahat, M.E.; Ambrose, R.; Welch, G.C.; Hill, I.G.; Leclerc, M. Water Compatible Direct (Hetero)arylation Polymerization of PPDT2FBT: A Pathway Towards Large-Scale Production of Organic Solar Cells. Asian J. Org. Chem. 2020, 1–9. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Abela, A.R.; Moser, R.; Nishikata, T.; Duplais, C.; Gaston, R.D.; Gadwood, R.C. TPGS-750-M: A Second-Generation Amphiphile for Metal-Catalyzed Cross-Couplings in Water at Room Temperature. J. Org. Chem. 2011, 76, 4379–4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollet, H.; Grubenmann, A. Formulation Technology; Wiley-VCH: Weinheim, Germany, 2001; ISBN 3527612939. [Google Scholar]
- Gabriel, C.M.; Lee, N.R.; Bigorne, F.; Klumphu, P.; Parmentier, M.; Gallou, F.; Lipshutz, B.H. Effects of co-solvents on reactions run under micellar catalysis conditions. Org. Lett. 2017, 19, 194–197. [Google Scholar] [CrossRef]
- Mao, S.; Shi, X.; Soulé, J.F.; Doucet, H. Exploring Green Solvents Associated to Pd/C as Heterogeneous Catalyst for Direct Arylation of Heteroaromatics with Aryl Bromides. Adv. Synth. Catal. 2018, 360, 3306–3317. [Google Scholar] [CrossRef]
- Kuwabara, J.; Yamazaki, K.; Yamagata, T.; Tsuchida, W.; Kanbara, T. The effect of a solvent on direct arylation polycondensation of substituted thiophenes. Polym. Chem. 2015, 6, 891–895. [Google Scholar] [CrossRef] [Green Version]
- Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. Establishment of Broadly Applicable Reaction Conditions for the Palladium-Catalyzed Direct Arylation of Heteroatom-Containing Aromatic Compounds Establishment of Broadly Applicable Reaction Conditions for the Palladium-Catalyzed Direct Arylation of Hetero. J. Org. Chem. 2009, 74, 1826–1834. [Google Scholar] [CrossRef]
- Schipper, D.J.; Fagnou, K. Direct arylation as a synthetic tool for the synthesis of thiophene-based organic electronic materials. Chem. Mater. 2011, 23, 1594–1600. [Google Scholar] [CrossRef]
- Lombeck, F.; Komber, H.; Gorelsky, S.I.; Sommer, M. Identifying homocouplings as critical side reactions in direct arylation polycondensation. ACS Macro Lett. 2014, 3, 819–823. [Google Scholar] [CrossRef]
- Novikov, A.A.; Semenov, A.P.; Monje-Galvan, V.; Kuryakov, V.N.; Klauda, J.B.; Anisimov, M.A. Dual Action of Hydrotropes at the Water/Oil Interface. J. Phys. Chem. C 2017, 121, 16423–16431. [Google Scholar] [CrossRef]
- Bura, T.; Morin, P.O.; Leclerc, M. En Route to Defect-Free Polythiophene Derivatives by Direct Heteroarylation Polymerization. Macromolecules 2015, 48, 5614–5620. [Google Scholar] [CrossRef]
- Mattiello, S.; Monguzzi, A.; Pedrini, J.; Sassi, M.; Villa, C.; Torrente, Y.; Marotta, R.; Meinardi, F.; Beverina, L. Self-Assembled Dual Dye-Doped Nanosized Micelles for High-Contrast Up-Conversion Bioimaging. Adv. Funct. Mater. 2016, 26, 8447–8454. [Google Scholar] [CrossRef]
- He, K.; Li, W.; Tian, H.; Zhang, J.; Yan, D.; Geng, Y.; Wang, F. Asymmetric Conjugated Molecules Based on [1]Benzothieno[3,2-b][1]benzothiophene for High-Mobility Organic Thin-Film Transistors: Influence of Alkyl Chain Length. ACS Appl. Mater. Interfaces 2017, 9, 35427–35436. [Google Scholar] [CrossRef] [PubMed]
- Berlinguette, C.; Bomben, P. Cyclometalated trasition metal dyes. U.S. Patent WO2012155247, 17 April 2012. [Google Scholar]
- Pommerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R.F.; Bässler, H.; Porsch, M.; Daub, J. Efficient Two Layer LEDs on a Polymer Blend Basis. Adv. Mater. 1995, 7, 551–554. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 8–15 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Additive (30 mol%) | Base 1.5 eq | Phase-Transfer Agent (30 mol%) | T (°C) | Conversion to Product (%) |
|---|---|---|---|---|---|
| 1 | PivOH | Na2CO3 | _ | 80 | trace |
| 2 a | PivOK | _ | _ | 80 | trace |
| 3 a | PivONBu4 | _ | _ | 80 | trace |
| 4 | PivOH | Na2CO3 | _ | 130 | 30 |
| 5 | PivOH | Cs2CO3 | _ | 130 | 27 |
| 6 | PivOH | Na2CO3 | Aliquat 336 | 130 | 53 (44 isolated) |
| 7 | PivOH | Na2CO3 | Aliquat HTA-1 | 130 | 55 |
| 8 | PivOH | NaOH | Aliquat HTA-1 | 130 | 48 |
| 9 | PivOH | tBuONa | Aliquat HTA-1 | 130 | 59 |
| 10 | NDA | Na2CO3 | Aliquat HTA-1 | 130 | 68 |
| 11 | NDA | NaOH | Aliquat HTA-1 | 130 | 77 |
| 12 | NDA | tBuONa | Aliquat HTA-1 | 130 | 80 |
| 13 b | NDA | tBuONa | Aliquat HTA-1 | 130 | 88 (86 isolated) |
| 14 b | NDA | tBuONa | - | 130 | 59 |
| Entry | Ligand (4 mol%) | RCOOH (eq) | Base (eq) | Phase-Transfer Agent (eq) | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cy3PHBF4 | NDA | tBuONa | Aliquat HTA-1 | 88% | 3% | 2% | 1% | 2% | 4% | 0% |
| 0.3 | 3 | 0.3 | |||||||||
| 2 | Cy3PHBF4 | NDA | tBuONa | Aliquat HTA-1 | 87% | 3% | 1% | 2% | 2% | 3% | 1% |
| 1 | 3 | 1 | |||||||||
| 3 | tBu3PHBF4 | NDA | tBuONa | Aliquat HTA-1 | 83% | 9% | 3% | 1% | 2% | 2% | 0% |
| 0.3 | 3 | 0.3 | |||||||||
| 4 a | Cy3PHBF4 | NDA | tBuONa | Aliquat HTA-1 | 85% | 3% | 1% | 1% | 4% | 4% | 3% |
| 0.3 | 3 | 0.3 | |||||||||
| 5 a,b | Cy3PHBF4 | NDA | tBuONa | Aliquat HTA-1 | 86% | 4% | 2% | 1% | 2% | 4% | 1% |
| 0.3 | 3 | 0.3 |
| Derivative | λmax (abs) [nm] | λmax (em) [nm] | Stokes Shift [eV] | E1/2 (V) | HOMO (eV) |
|---|---|---|---|---|---|
| 8 | 351 | 392 | 0.37 | 0.38 | −5.18 |
| 9 | 334 | 430 | 0.83 | 0.69 | −5.49 |
| 10 | 364 | 379 | 0.14 | 0.78 | −5.58 |
| 11 | 375 | 430 | 0.43 | 0.64 | −5.44 |
| 12 | 351 | 391 | 0.36 | 0.74 | −5.54 |
| 13 | 401 | 461 | 0.40 | 0.78 | −5.59 |
| 14 | 469 | 603 | 0.58 | −1.78 | −3.00 |
| 15 a | 267 | 341 | 1.00 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calascibetta, A.M.; Mattiello, S.; Sanzone, A.; Facchinetti, I.; Sassi, M.; Beverina, L. Sustainable Access to π-Conjugated Molecular Materials via Direct (Hetero)Arylation Reactions in Water and under Air. Molecules 2020, 25, 3717. https://doi.org/10.3390/molecules25163717
Calascibetta AM, Mattiello S, Sanzone A, Facchinetti I, Sassi M, Beverina L. Sustainable Access to π-Conjugated Molecular Materials via Direct (Hetero)Arylation Reactions in Water and under Air. Molecules. 2020; 25(16):3717. https://doi.org/10.3390/molecules25163717
Chicago/Turabian StyleCalascibetta, Adiel Mauro, Sara Mattiello, Alessandro Sanzone, Irene Facchinetti, Mauro Sassi, and Luca Beverina. 2020. "Sustainable Access to π-Conjugated Molecular Materials via Direct (Hetero)Arylation Reactions in Water and under Air" Molecules 25, no. 16: 3717. https://doi.org/10.3390/molecules25163717