
Compensatory Neuroprotective Response of Thioredoxin Reductase against Oxidative-Nitrosative Stress Induced by Experimental Autoimmune Encephalomyelitis in Rats: Modulation by Theta Burst Stimulation


Abstract
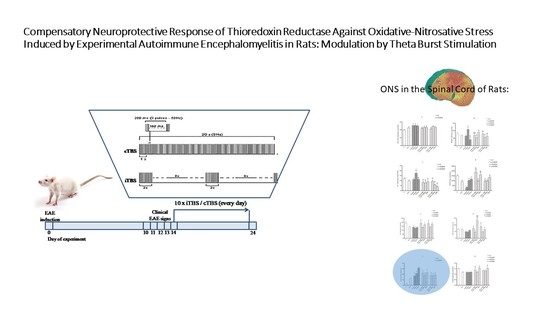
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Procedure
2.2. Clinical Evaluation
2.3. Tissue Homogenates and Biochemical Analyses
2.4. Statistical Analysis
3. Results
3.1. Clinical Observation
3.2. Oxidative Stress Biochemical Analyses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval
Abbreviations
| CFA | Complete Freund’s Adjuvant |
| CNS | central nervous system |
| cTBS | continuous theta burst stimulation |
| cTBSsh | sham stimulated EAE rats, exposed to the sound artifacts of cTBS |
| dpi | postimmunization days |
| EAE | experimental autoimmune encephalomyelitis |
| iTBS | intermittent theta burst stimulation |
| iTBSsh | sham stimulated EAE rats, exposed to the sound artifacts of iTBS |
| MS | multiple sclerosis |
| TBS | theta burst stimulation |
References
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Jovanovic, M.; Ninkovic, M.; Stevanovic, I.; Curcic, M.; Vujanovic, D.; Djurdjevic, D. Intrastriatal pre-treatment with L-NAME protects rats from diquat neurotoxcity. Ann. Agric. Environ. Med. 2012, 19, 666–672. [Google Scholar] [PubMed]
- Djukic, M.M.; Jovanovic, M.D.; Ninkovic, M.; Stevanovic, I.; Ilic, K.; Curcic, M.; Vekic, J. Protective role of glutathione reductase in paraquat induced neurotoxicity. Chem. Biol. Interact. 2012, 199, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, A.; Murthy, M.; Mirault, M.-E. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell. Mol. Life Sci. CMLS 2005, 62, 1063–1080. [Google Scholar] [CrossRef]
- Masutani, H.; Bai, J.; Kim, Y.-C.; Yodoi, J. Thioredoxin as a neurotrophic cofactor and an important regulator of neuroprotection. Mol. Neurobiol. 2004, 29, 229–242. [Google Scholar] [CrossRef]
- Murthy, C.R.; Bender, A.S.; Dombro, R.S.; Bai, G.; Norenberg, M.D. Elevation of glutathione levels by ammonium ions in primary cultures of rat astrocytes. Neurochem. Int. 2000, 37, 255–268. [Google Scholar] [CrossRef]
- Węgrzynowicz, M.; Hilgier, W.; Dybel, A.; Oja, S.S.; Saransaari, P.; Albrecht, J. Upregulation of cerebral cortical glutathione synthesis by ammonia in vivo and in cultured glial cells: The role of cystine uptake. Neurochem. Int. 2007, 50, 883–889. [Google Scholar] [CrossRef]
- Hilgier, W.; Węgrzynowicz, M.; Ruszkiewicz, J.; Oja, S.S.; Saransaari, P.; Albrecht, J. Direct exposure to ammonia and hyperammonemia increase the extracellular accumulation and degradation of astroglia-derived glutathione in the rat prefrontal cortex. Toxicol. Sci. 2010, 117, 163–168. [Google Scholar] [CrossRef]
- De Nuccio, C.; Bernardo, A.; Cruciani, C.; De Simone, R.; Visentin, S.; Minghetti, L. Peroxisome proliferator activated receptor-γ agonists protect oligodendrocyte progenitors against tumor necrosis factor-alpha-induced damage: Effects on mitochondrial functions and differentiation. Exp. Neurol. 2015, 271, 506–514. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, I.; Mančić, B.; Ilić, T.; Milosavljević, P.; Lavrnja, I.; Stojanović, I.; Ninković, M. Theta burst stimulation influence the expression of BDNF in the spinal cord on the experimental autoimmune encephalomyelitis. Folia Neuropathol. 2019, 57, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Ugawa, Y. Basic mechanisms of TMS. J. Clin. Neurophysiol. 2002, 19, 322–343. [Google Scholar] [CrossRef] [PubMed]
- Mancic, B.; Stevanovic, I.; Ilic, T.V.; Djuric, A.; Stojanovic, I.; Milanovic, S.; Ninkovic, M. Transcranial theta-burst stimulation alters GLT-1 and vGluT1 expression in rat cerebellar cortex. Neurochem. Int. 2016, 100, 120–127. [Google Scholar] [CrossRef]
- Djukic, M. Diagnostic characteristics and application of alcohol biomarkers. Clin. Lab. 2013, 59, 233–245. [Google Scholar] [CrossRef]
- Lavrnja, I.; Savic, D.; Bjelobaba, I.; Dacic, S.; Bozic, I.; Parabucki, A.; Nedeljkovic, N.; Pekovic, S.; Rakic, L.; Stojiljkovic, M. The effect of ribavirin on reactive astrogliosis in experimental autoimmune encephalomyelitis. J. Pharmacol. Sci. 2012, 119, 221–232. [Google Scholar] [CrossRef]
- Jakovljevic, M.B.; Jovanovic, M.; Nikic, K.; Dejanovic, S.D.; Radovanovic, A.; Pirkovic, I.; Yamada, T. Acute alcohol detoxification costs in upper-middle income: Western Balkans. J. Health Behav. Public Health 2011, 1, 1–7. [Google Scholar]
- Huang, Y.-Z.; Chen, R.-S.; Rothwell, J.C.; Wen, H.-Y. The after-effect of human theta burst stimulation is NMDA receptor dependent. Clin. Neurophysiol. 2007, 118, 1028–1032. [Google Scholar] [CrossRef]
- Hammer, L.A.; Zagon, I.S.; McLaughlin, P.J. Improved clinical behavior of established relapsing-remitting experimental autoimmune encephalomyelitis following treatment with endogenous opioids: Implications for the treatment of multiple sclerosis. Brain Res. Bull. 2015, 112, 42–51. [Google Scholar] [CrossRef]
- Gurd, J.; Jones, L.R.; Mahler, H.; Moore, W. Isolation and partial characterization of rat brain synaptic plasma membranes. J. Neurochem. 1974, 22, 281–290. [Google Scholar] [CrossRef]
- Navarro-Gonzálvez, J.A.; García-Benayas, C.; Arenas, J. Semiautomated measurement of nitrate in biological fluids. Clin. Chem. 1998, 44, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.; Khan, N.; McLellan, B. Early measurement of systemic lipid peroxidation products in the plasma of major blunt trauma patients. J. Trauma 1991, 31, 32–35. [Google Scholar] [CrossRef]
- Kono, Y.; Kobayashi, K.; Tagawa, S.; Adachi, K.; Ueda, A.; Sawa, Y.; Shibata, H. Antioxidant activity of polyphenolics in diets: Rate constants of reactions of chlorogenic acid and caffeic acid with reactive species of oxygen and nitrogen. Biochim. Biophys. Acta Gen. Subj. 1997, 1335, 335–342. [Google Scholar] [CrossRef]
- Sun, M.; Zigman, S. An improved spectrophotometric assay for superoxide dismutase based on epinephrine autoxidation. Anal. Biochem. 1978, 90, 81–89. [Google Scholar] [CrossRef]
- Elman, G. Tissue sulphydryl groups. Arch. Biochem. Biophys. 1959, 82, 70. [Google Scholar] [CrossRef]
- Stohs, S.J.; Lawson, T.A.; Anderson, L.; Bueding, E. Effects of oltipraz, BHA, ADT and cabbage on glutathione metabolism, DNA damage and lipid peroxidation in old mice. Mech. Ageing Dev. 1986, 37, 137–145. [Google Scholar] [CrossRef]
- Holmgren, A.; Bjornstedt, M. Thioredoxin and thioredoxin reductase. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1995; Volume 252, pp. 199–208. [Google Scholar]
- Das, M.; Rastogi, S.; Khanna, S.K. Mechanism to study 1:1 stoichiometry of NADPH and alkoxyphenoxazones metabolism spectrophotometrically in subcellular biological preparations. Biochim. Biophys. Acta Gen. Subj. 2004, 1675, 1–11. [Google Scholar] [CrossRef]
- Lowry, O.; Rosebrough, N.J.; Farr, A.l.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Xiao, S.; MacNair, L.; McGoldrick, P.; McKeever, P.M.; McLean, J.R.; Zhang, M.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Isoform-specific antibodies reveal distinct subcellular localizations of C 9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 78, 568–583. [Google Scholar] [CrossRef]
- Gentner, R.; Wankerl, K.; Reinsberger, C.; Zeller, D.; Classen, J. Depression of human corticospinal excitability induced by magnetic theta-burst stimulation: Evidence of rapid polarity-reversing metaplasticity. Cereb. Cortex 2008, 18, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Sahel, A.; Ortiz, F.C.; Kerninon, C.; Maldonado, P.P.; Angulo, M.C.; Nait-Oumesmar, B. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Nasios, G.; Messinis, L.; Dardiotis, E.; Papathanasopoulos, P. Repetitive transcranial magnetic stimulation, cognition, and multiple sclerosis: An overview. Behav. Neurol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Lehner, C.; Gehwolf, R.; Tempfer, H.; Krizbai, I.; Hennig, B.; Bauer, H.-C.; Bauer, H. Oxidative stress and blood–brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid. Redox Signal. 2011, 15, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.; Mahad, D.J. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett. 2018, 592, 1113–1121. [Google Scholar] [CrossRef]
- Stanojevic, P.B.; Stevanovic, I.; Mijuskovic, Z.; Djuric, A.; Gobeljic, B.; Banovic, T.; Vojvodic, D. Association between oxidative stress and melanoma progression. J. Med. Biochem. 2018, 37, 12–20. [Google Scholar]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgärtner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 2013, 136, 147–167. [Google Scholar] [CrossRef]
- Thimm, A.; Funke, K. Multiple blocks of intermittent and continuous theta-burst stimulation applied via transcranial magnetic stimulation differently affect sensory responses in rat barrel cortex. J. Physiol. 2015, 593, 967–985. [Google Scholar] [CrossRef]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.Á.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the blood–brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Metabolic signaling in the brain and the role of astrocytes in control of glutamate and GABA neurotransmission. Neurosci. Lett. 2019, 689, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Ruszkiewicz, J.; Fręśko, I.; Hilgier, W.; Albrecht, J. Decrease of glutathione content in the prefrontal cortical mitochondria of rats with acute hepatic encephalopathy: Prevention by histidine. Metab. Brain Dis. 2013, 28, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Cullen, C.L.; Young, K.M. How does transcranial magnetic stimulation influence glial cells in the central nervous system? Front. Neural Circuits 2016, 10, 26. [Google Scholar] [CrossRef]
- Lillig, C.; Holmgren, A. Thioredoxin and related molecules–from biology to health and disease. Antioxid Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef]
- Bendix, I.; Weichelt, U.; Strasser, K.; Serdar, M.; Endesfelder, S.; von Haefen, C.; Heumann, R.; Ehrkamp, A.; Felderhoff-Mueser, U.; Sifringer, M. Hyperoxia changes the balance of the thioredoxin/peroxiredoxin system in the neonatal rat brain. Brain Res. 2012, 1484, 68–75. [Google Scholar] [CrossRef]
- Kanamori, K.; Ross, B.D. Chronic electrographic seizure reduces glutamine and elevates glutamate in the extracellular fluid of rat brain. Brain Res. 2011, 1371, 180–191. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Kimura, J.; Hori, K.; Yodoi, J. Astroglial expression of ATL-derived factor, a human thioredoxin homologue, in the gerbil brain after transient global ischemia. Brain Res. 1993, 625, 1–8. [Google Scholar] [CrossRef]
- Hori, K.; Katayama, M.; Sato, N.; Ishii, K.; Waga, S.; Yodoi, J. Neuroprotection by glial cells through adult T cell leukemia-derived factor/human thioredoxin (ADF/TRX). Brain Res. 1994, 652, 304–310. [Google Scholar] [CrossRef]
- Ljubisavljevic, S.; Stojanovic, I.; Pavlovic, D.; Sokolovic, D.; Stevanovic, I. Aminoguanidine and N-acetyl-cysteine supress oxidative and nitrosative stress in EAE rat brains. Redox Rep. 2011, 16, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Shangari, N.; Mehta, R.; O’Brien, P.J. Hepatocyte susceptibility to glyoxal is dependent on cell thiamin content. Chem. Biol. Interact. 2007, 165, 146–154. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 822–829. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | Maximal Clinical Severity Score | Paralysis | Body Mass Loss and Gained | |||
|---|---|---|---|---|---|---|
| Started (dpi) | Lasted (Days) | Started (dpi) | Lasted (Days) | Min. Weight (%) | Gained Weight (%) | |
| EAE EAE+i/cTBSsh | 11.8 | 3.08 ± 0.97 | 12 | ~3.5 ~2.5 | ~70 (14th dpi) | ~96 (20–24th dpi) |
| EAE+i/cTBS | 11.5 | 2.79 ± 0.76 | 11.8 | ~1.7 a | ~81 (15th dpi) | ~90 (20–24th dpi) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevanovic, I.; Ninkovic, M.; Mancic, B.; Milivojevic, M.; Stojanovic, I.; Ilic, T.; Vujovic, M.; Djukic, M. Compensatory Neuroprotective Response of Thioredoxin Reductase against Oxidative-Nitrosative Stress Induced by Experimental Autoimmune Encephalomyelitis in Rats: Modulation by Theta Burst Stimulation. Molecules 2020, 25, 3922. https://doi.org/10.3390/molecules25173922
Stevanovic I, Ninkovic M, Mancic B, Milivojevic M, Stojanovic I, Ilic T, Vujovic M, Djukic M. Compensatory Neuroprotective Response of Thioredoxin Reductase against Oxidative-Nitrosative Stress Induced by Experimental Autoimmune Encephalomyelitis in Rats: Modulation by Theta Burst Stimulation. Molecules. 2020; 25(17):3922. https://doi.org/10.3390/molecules25173922
Chicago/Turabian StyleStevanovic, Ivana, Milica Ninkovic, Bojana Mancic, Marija Milivojevic, Ivana Stojanovic, Tihomir Ilic, Maja Vujovic, and Mirjana Djukic. 2020. "Compensatory Neuroprotective Response of Thioredoxin Reductase against Oxidative-Nitrosative Stress Induced by Experimental Autoimmune Encephalomyelitis in Rats: Modulation by Theta Burst Stimulation" Molecules 25, no. 17: 3922. https://doi.org/10.3390/molecules25173922
APA StyleStevanovic, I., Ninkovic, M., Mancic, B., Milivojevic, M., Stojanovic, I., Ilic, T., Vujovic, M., & Djukic, M. (2020). Compensatory Neuroprotective Response of Thioredoxin Reductase against Oxidative-Nitrosative Stress Induced by Experimental Autoimmune Encephalomyelitis in Rats: Modulation by Theta Burst Stimulation. Molecules, 25(17), 3922. https://doi.org/10.3390/molecules25173922