Interacting Quantum Atoms—A Review



Abstract
1. Chemical Interactions and Energy Decompositions
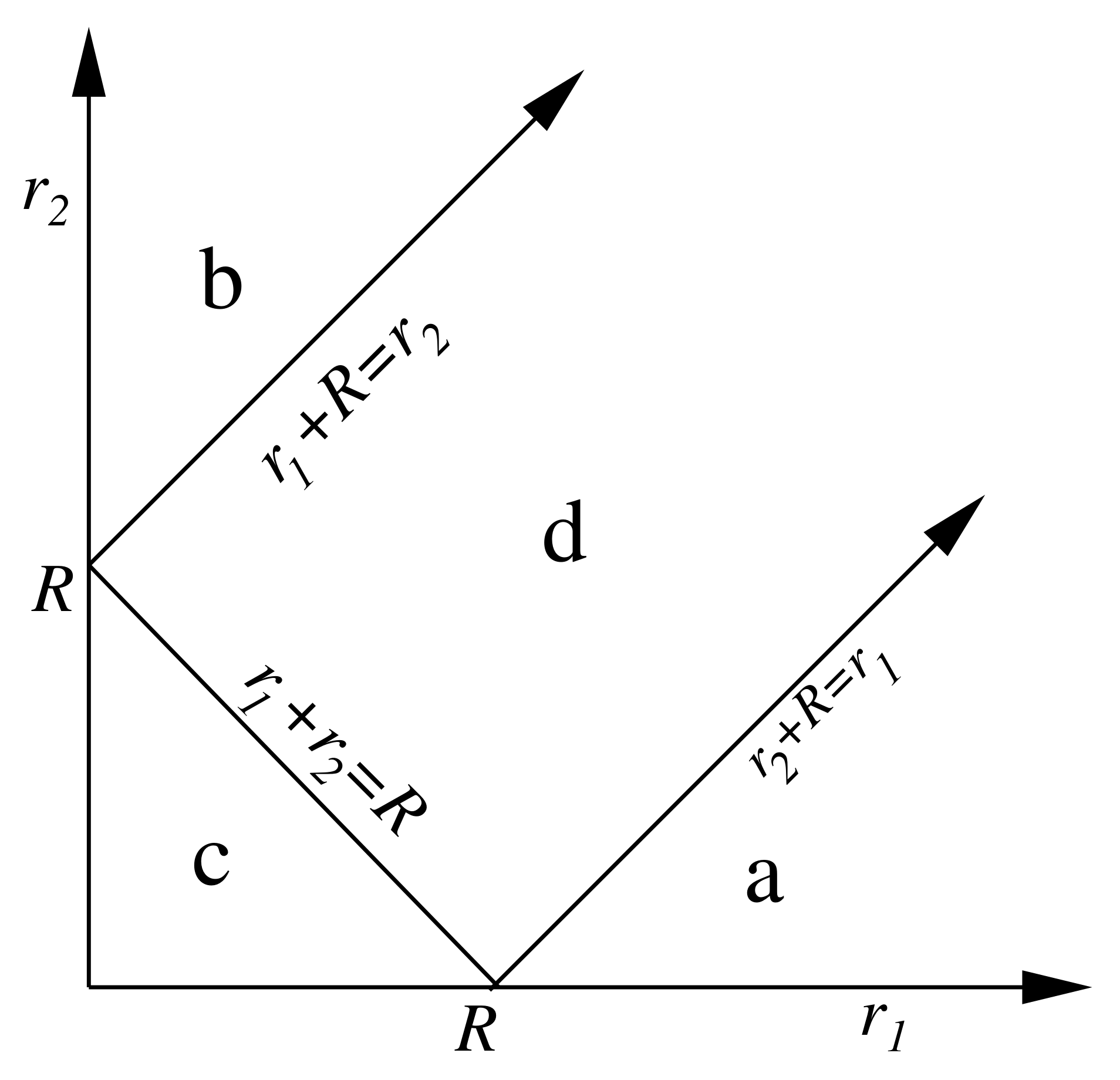
2. The Spatial or Real Space Point of View
3. The IQA Methodology
3.1. The Iqa Energy Partition
3.2. Iqa from Different Electronic Structure Approximations
3.2.1. Densities for Single- and Multideterminantal Wavefunctions
3.2.2. Coupled Cluster (Cc) Densities
3.2.3. Møller-Plesset Densities (Mpn)
3.2.4. Kohn-Sham Densities (Dft)
3.2.5. Other Approximate Densities
3.3. Practical Aspects of Iqa Implementation
3.3.1. Integration Schemes: Monoelectronic Terms
3.3.2. Integration Schemes: Bielectronic Terms
3.3.3. The Multipolar Approach for the Classical and Exchange-Correlation Energies
3.3.4. Increasing the Precision of Iqa Integrations: Atomic -Spheres
4. Selected Applications of the Iqa Methodology
4.1. Electronic Correlation
4.2. Relationships between V and Di
4.3. The Nature of Chemical Bonding
4.4. Non-Covalent Interactions
4.4.1. Hydrogen Bonding
4.4.2. Halogen Bonding
4.4.3. Other Bonding Situations
4.5. Bonding of Metallic Elements
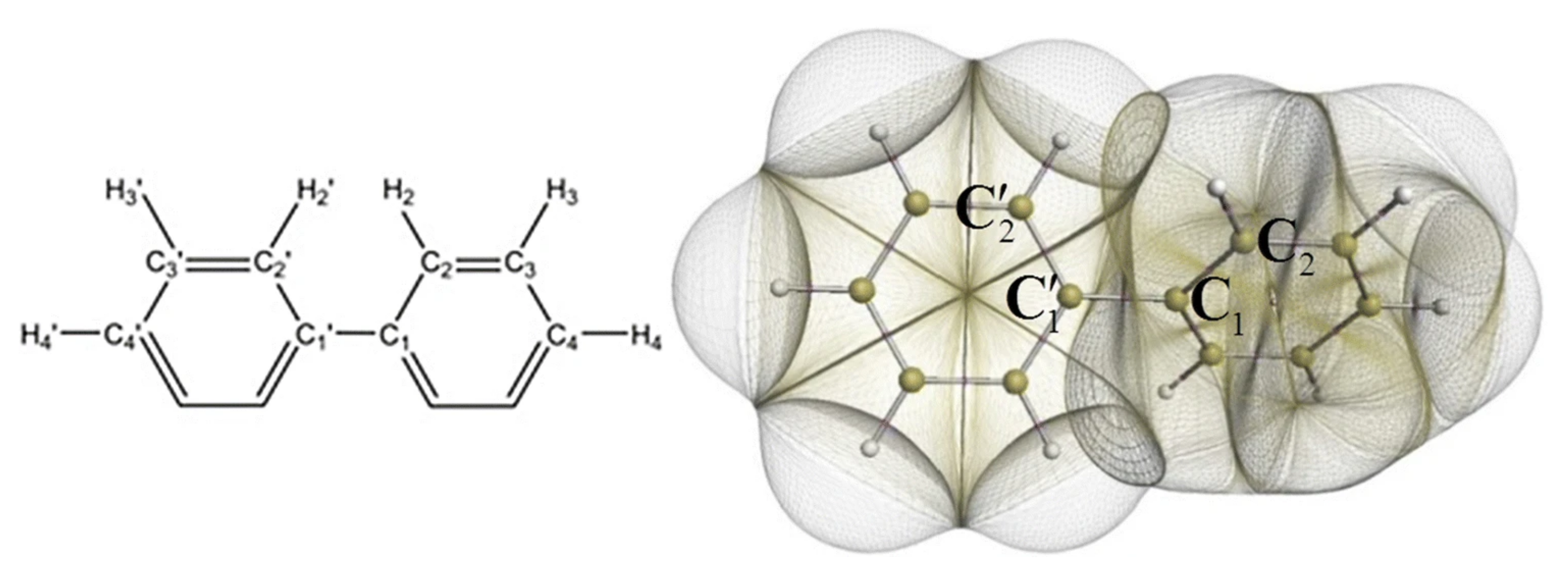
4.6. Organic Chemistry and Reactivity
4.7. Excited State
4.8. Steric Repulsion
4.9. Machine Learning
Author Contributions
Funding
Conflicts of Interest
References
- Hayes, I.; Stone, A. An intermolecular perturbation theory for the region of moderate overlap. Mol. Phys. 1984, 53, 83–105. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Stone, A.J. The Theory of Intermolecular Forces; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Hobza, P.; Muller-Dethlefs, K. Non-Covalent Interactions; Royal Society of Chemistry: London, UK, 2009. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chem. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Reviews in Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 1–86. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Streitwieser, A. Natural energy decomposition analysis: An energy partitioning procedure for molecular interactions with application to weak hydrogen bonding, strong ionic, and moderate donor–acceptor interactions. J. Chem. Phys. 1994, 100, 2900–2909. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Gatti, C.; Macchi, P. (Eds.) Modern Charge-Density Analysis; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- McWeeny, R. Methods of Molecular Quantum Mechanics; Academic Press: London, UK, 1992. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Heidar-Zadeh, F.; Ayers, P.W.; Verstraelen, T.; Vinogradov, I.; Vöhringer-Martinez, E.; Bultinck, P. Information-Theoretic Approaches to Atoms-in-Molecules: Hirshfeld Family of Partitioning Schemes. J. Phys. Chem. A 2017, 122, 4219–4245. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Francisco, E.; Pendás, A.M.; Blanco, M.A. A Molecular Energy Decomposition Scheme for Atoms in Molecules. J. Chem. Theory Comput. 2005, 2, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Parr, R.G. The atom in a molecule: A density matrix approach. J. Chem. Phys. 1986, 84, 1704–1711. [Google Scholar] [CrossRef]
- Rico, J.F.; López, R.; Ramírez, G. Analysis of the molecular density. J. Chem. Phys. 1999, 110, 4213–4220. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, Á.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, Á.; Blanco, M.A.; Francisco, E. Chemical fragments in real space: Definitions, properties, and energetic decompositions. J. Comput. Chem. 2006, 28, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Massa, L.; Matta, C.F. Exploiting the full quantum crystallography. Can. J. Chem. 2018, 96, 599–605. [Google Scholar] [CrossRef]
- Polkosnik, W.; Matta, C.F.; Huang, L.; Massa, L. Fast quantum crystallography. Int. J. Quantum Chem. 2019, 119. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef]
- Fernández-Alarcón, A.; Casals-Sainz, J.L.; Guevara-Vela, J.M.; Costales, A.; Francisco, E.; Pendás, Á.M.; Rocha-Rinza, T. Partition of electronic excitation energies: The IQA/EOM-CCSD method. Phys. Chem. Chem. Phys. 2019, 21, 13428–13439. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Kosov, D.S. Atom–atom partitioning of intramolecular and intermolecular Coulomb energy. J. Chem. Phys. 2001, 114, 6539–6547. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Joubert, L.; Kosov, D.S. Convergence of the Electrostatic Interaction Based on Topological Atoms. J. Phys. Chem. A 2001, 105, 8254–8261. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Francisco, E.; Blanco, M.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef] [PubMed]
- García-Revilla, M.; Francisco, E.; Popelier, P.L.A.; Martín Pendás, Á. Domain-Averaged Exchange-Correlation Energies as a Physical Underpinning for Chemical Graphs. ChemPhysChem 2013, 14, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- García-Revilla, M.; Popelier, P.L.A.; Francisco, E.; Martín Pendás, Á. Nature of Chemical Interactions from the Profiles of Electron Delocalization Indices. J. Chem. Theory Comput. 2011, 7, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Outeiral, C.; Vincent, M.A.; Martín Pendás, Á.; Popelier, P.L.A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Crespo, D.; Costales, A.; Francisco, E.; Martín Pendás, Á. Real-Space In Situ Bond Energies: Toward A Consistent Energetic Definition of Bond Strength. Chem. Eur. J. 2018, 24, 9101–9112. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Chávez-Calvillo, R.; García-Revilla, M.; Hernández-Trujillo, J.; Christiansen, O.; Francisco, E.; Martín Pendás, A. Hydrogen-Bond Cooperative Effects in Small Cyclic Water Clusters as Revealed by the Interacting Quantum Atoms Approach. Chem. Eur. J. 2013, 19, 14304–14315. [Google Scholar] [CrossRef]
- Löwdin, P.O. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys. Rev. 1955, 97, 1474–1489. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Francisco, E.; Blanco, M.A. Two-electron integrations in the Quantum Theory of Atoms in Molecules with correlated wave functions. J. Comput. Chem. 2005, 26, 344–351. [Google Scholar] [CrossRef]
- Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Dynamical correlation within the Interacting Quantum Atoms method through coupled cluster theory. Comput. Theor. Chem. 2015, 1053, 90–95. [Google Scholar] [CrossRef]
- Holguín-Gallego, F.J.; Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Electron correlation in the interacting quantum atoms partition via coupled-cluster lagrangian densities. J. Comput. Chem. 2016, 37, 1753–1765. [Google Scholar] [CrossRef]
- Helgaker, T. Molecular Electronic-Structure Theory; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- McDonagh, J.L.; Vincent, M.A.; Popelier, P.L. Partitioning dynamic electron correlation energy: Viewing Møller-Plesset correlation energies through Interacting Quantum Atom (IQA) energy partitioning. Chem. Phys. Lett. 2016, 662, 228–234. [Google Scholar] [CrossRef]
- Silva, A.F.; Popelier, P.L.A. MP2-IQA: Upscaling the analysis of topologically partitioned electron correlation. J. Mol. Model. 2018, 24. [Google Scholar] [CrossRef] [PubMed]
- Casals-Sainz, J.L.; Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza, T.; Martín Pendás, Á. Efficient implementation of the interacting quantum atoms energy partition of the second-order Møller–Plesset energy. J. Comput. Chem. 2020, 41, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.; Pendás, Á.M.; Popelier, P.L.A. Extension of the interacting quantum atoms (IQA) approach to B3LYP level density functional theory (DFT). Phys. Chem. Chem. Phys. 2016, 18, 20986–21000. [Google Scholar] [CrossRef]
- Francisco, E.; Casals-Sainz, J.L.; Rocha-Rinza, T.; Pendás, A.M. Partitioning the DFT exchange-correlation energy in line with the interacting quantum atoms approach. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- García-Revilla, M.; Francisco, E.; Costales, A.; Martín Pendás, Á. Performance of the Density Matrix Functional Theory in the Quantum Theory of Atoms in Molecules. J. Phys. Chem. A 2011, 116, 1237–1250. [Google Scholar] [CrossRef]
- Müller, A. Explicit approximate relation between reduced two- and one-particle density matrices. Phys. Lett. A 1984, 105, 446–452. [Google Scholar] [CrossRef]
- Buijse, M.A.; Baerends, E.J. An approximate exchange-correlation hole density as a functional of the natural orbitals. Mol. Phys. 2002, 100, 401–421. [Google Scholar] [CrossRef]
- Goedecker, S.; Umrigar, C.J. Natural Orbital Functional for the Many-Electron Problem. Phys. Rev. Lett. 1998, 81, 866–869. [Google Scholar] [CrossRef]
- Csányi, G.; Arias, T.A. Tensor product expansions for correlation in quantum many-body systems. Phys. Rev. B 2000, 61, 7348–7352. [Google Scholar] [CrossRef]
- Csányi, G.; Goedecker, S.; Arias, T.A. Improved tensor-product expansions for the two-particle density matrix. Phys. Rev. A 2002, 65. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E. Assessment of simple exchange-correlation energy functionals of the one-particle density matrix. J. Chem. Phys. 2002, 117, 2489–2495. [Google Scholar] [CrossRef]
- Gritsenko, O.; Pernal, K.; Baerends, E.J. An improved density matrix functional by physically motivated repulsive corrections. J. Chem. Phys. 2005, 122, 204102. [Google Scholar] [CrossRef] [PubMed]
- Piris, M. A new approach for the two-electron cumulant in natural orbital functional theory. Int. J. Quantum Chem. 2006, 106, 1093–1104. [Google Scholar] [CrossRef]
- Biegler-Konig, F.W.; Nguyen-Dang, T.T.; Tal, Y.; Bader, R.F.W.; Duke, A.J. Calculation of the average properties of atoms in molecules. J. Phys. B 1981, 14, 2739–2751. [Google Scholar] [CrossRef]
- Lebedev, V. Quadratures on a sphere. USSR Comp. Math. Math. Phys. 1976, 16, 10–24. [Google Scholar] [CrossRef]
- Favati, P.; Lotti, G.; Romani, F. Algorithm 691: Improving QUADPACK automatic integration routines. ACM Trans. Math. Softw. 1991, 17, 218–232. [Google Scholar] [CrossRef]
- Chisholm, C. Group Theoretical Techniques in Quantum Chemistry; Academic Press: Cambrige, MA, USA, 1976. [Google Scholar]
- Kay, K.G.; Todd, H.D.; Silverstone, H.J. Bipolar Expansion for r12nYlm(θ12, ϕ12). J. Chem. Phys. 1969, 51, 2363–2367. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Francisco, E. Overlap, effective-potential, and projection-operator bicentric integrals over complex Slater-type orbitals. Phys. Rev. A 1991, 43, 3384–3391. [Google Scholar] [CrossRef] [PubMed]
- Pendás, A.M.; Blanco, M.A.; Francisco, E. Two-electron integrations in the quantum theory of atoms in molecules. J. Chem. Phys. 2004, 120, 4581–4592. [Google Scholar] [CrossRef] [PubMed]
- Francisco, E.; Crespo, D.M.; Costales, A.; Martín Pendás, Á. A multipolar approach to the interatomic covalent interaction energy. J. Comput. Chem. 2017, 38, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, Á.; Francisco, E.; Blanco, M.A. Binding Energies of First Row Diatomics in the Light of the Interacting Quantum Atoms Approach. J. Phys. Chem. A 2006, 110, 12864–12869. [Google Scholar] [CrossRef] [PubMed]
- García-Revilla, M.A.; Francisco, E.; Martín Pendás, Á.; Recio, J.M.; Bartolomei, M.; Hernández, M.I.; Campos-Martínez, J.; Carmona-Novillo, E.; Hernández-Lamoneda, R. Chemical Interactions and Spin Structure in (O2)4: Implications for the ϵ-O2Phase. J. Chem. Theory Comput. 2013, 9, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Belyakov, A.V.; Gureev, M.A.; Garabadzhiu, A.V.; Losev, V.A.; Rykov, A.N. Determination of the molecular structure of gaseous proline by electron diffraction, supported by microwave and quantum chemical data. Struct. Chem. 2015, 26, 1489–1500. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Martín Pendás, Á.; Tsirelson, V.G. An anatomy of intramolecular atomic interactions in halogen-substituted trinitromethanes. Phys. Chem. Chem. Phys. 2014, 16, 16780–16789. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Krumm, B.; Moll, R.; Rest, S.F.; Vishnevskiy, Y.V.; Reuter, C.; Stammler, H.G.; Mitzel, N.W. Halogenotrinitromethanes: A Combined Study in the Crystalline and Gaseous Phase and Using Quantum Chemical Methods. Chem. Eur. J. 2014, 20, 12962–12973. [Google Scholar] [CrossRef]
- Cukrowski, I.; Mangondo, P. Interacting quantum fragments-rooted preorganized-interacting fragments attributed relative molecular stability of the BeIIcomplexes of nitrilotriacetic acid and nitrilotri-3-propionic acid. J. Comput. Chem. 2016, 37, 1373–1387. [Google Scholar] [CrossRef]
- Massa, L.; Keith, T.; Cheng, Y.; Matta, C.F. The kernel energy method applied to quantum theory of atoms in molecules–energies of interacting quantum atoms. Chem. Phys. Lett. 2019, 734, 136650. [Google Scholar] [CrossRef]
- Ruiz, I.; Matito, E.; Holguín-Gallego, F.J.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Fermi and Coulomb correlation effects upon the interacting quantum atoms energy partition. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- McDonagh, J.L.; Silva, A.F.; Vincent, M.A.; Popelier, P.L.A. Quantifying Electron Correlation of the Chemical Bond. J. Phys. Chem. Lett. 2017, 8, 1937–1942. [Google Scholar] [CrossRef]
- Vincent, M.A.; Silva, A.F.; McDonagh, J.L.; Popelier, P.L.A. The effects of higher orders of perturbation theory on the correlation energy of atoms and bonds in molecules. Int. J. Quantum Chem. 2017, 118, e25519. [Google Scholar] [CrossRef]
- Vincent, M.A.; Silva, A.F.; Popelier, P.L.A. A Comparison of the Interacting Quantum Atoms (IQA) Analysis of the Two-Particle Density-Matrices of MP4SDQ and CCSD. Z. Anorg. Allg. Chem. 2020. [Google Scholar] [CrossRef]
- Casalz-Sainz, J.L.; Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza, T.; Martín Pendás, Á. Where Does Electron Correlation Lie? Some Answers from a Real Space Partition. ChemPhysChem 2017, 18, 3553–3561. [Google Scholar] [CrossRef]
- Jara-Cortés, J.; Hernández-Trujillo, J. Energetic Analysis of Conjugated Hydrocarbons Using the Interacting Quantum Atoms Method. J. Comput. Chem. 2017, 39, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Badri, Z.; Foroutan-Nejad, C. Unification of ground-state aromaticity criteria–structure, electron delocalization, and energy–in light of the quantum chemical topology. Phys. Chem. Chem. Phys. 2016, 18, 11693–11699. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, Á.; Francisco, E. Real space bond orders are energetic descriptors. Phys. Chem. Chem. Phys. 2018, 20, 16231–16237. [Google Scholar] [CrossRef]
- Matta, C.F.; Hernández-Trujillo, J.; Tang, T.H.; Bader, R.F.W. Hydrogen–Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals. Chem. Eur. J. 2003, 9, 1940–1951. [Google Scholar] [CrossRef]
- Hernández-Trujillo, J.; Matta, C.F. Hydrogen–hydrogen bonding in biphenyl revisited. Struct. Chem. 2007, 18, 849–857. [Google Scholar] [CrossRef]
- Eskandari, K.; Alsenoy, C.V. Hydrogen-hydrogen interaction in planar biphenyl: A theoretical study based on the interacting quantum atoms and Hirshfeld atomic energy partitioning methods. J. Comput. Chem. 2014, 35, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Popelier, P.L.A.; Maxwell, P.I.; Thacker, J.C.R.; Alkorta, I. A relative energy gradient (REG) study of the planar and perpendicular torsional energy barriers in biphenyl. Theor. Chem. Acc. 2018, 138. [Google Scholar] [CrossRef] [PubMed]
- Mallia, A.R.; Ramakrishnan, R.; Niyas, M.A.; Hariharan, M. Crystalline triphenylamine substituted arenes: Solid state packing and luminescence properties. CrystEngComm 2017, 19, 817–825. [Google Scholar] [CrossRef]
- Matczak, P. Intramolecular C–H⋯H–C Contacts in Diheteroaryl Ketones and Thioketones: A Theoretical Analysis. Bull. Chem. Soc. Jpn. 2016, 89, 92–102. [Google Scholar] [CrossRef]
- Demyanov, P.I.; Polestshuk, P.M. Forced Bonding and QTAIM Deficiencies: A Case Study of the Nature of Interactions in He@Adamantane and the Origin of the High Metastability. Chem. Eur. J. 2013, 19, 10945–10957. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. On the physical role of exchange in the formation of an intramolecular bond path between two electronegative atoms. J. Chem. Phys. 2013, 138, 024102. [Google Scholar] [CrossRef]
- Dem’yanov, P.; Polestshuk, P. A Bond Path and an Attractive Ehrenfest Force Do Not Necessarily Indicate Bonding Interactions: Case Study on M2X2 (M = Li, Na, K: X = H, OH, F, Cl). Chem. Eur. J. 2012, 18, 4982–4993. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Badri, Z.; Marek, R. Multi-center covalency: Revisiting the nature of anion-π interactions. Phys. Chem. Chem. Phys. 2015, 17, 30670–30679. [Google Scholar] [CrossRef]
- Badri, Z.; Foroutan-Nejad, C.; Kozelka, J.; Marek, R. On the non-classical contribution in lone-pair–π interaction: IQA perspective. Phys. Chem. Chem. Phys. 2015, 17, 26183–26190. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N.; Francisco, E.; Martín Pendás, Á. Application of the Interacting Quantum Atoms Approach to the S66 and Ionic-Hydrogen-Bond Datasets for Noncovalent Interactions. ChemPhysChem 2018, 19, 973–987. [Google Scholar] [CrossRef]
- Sagan, F.; Filas, R.; Mitoraj, M. Non-Covalent Interactions in Hydrogen Storage Materials LiN(CH3)2BH3 and KN(CH3)2BH3. Crystals 2016, 6, 28. [Google Scholar] [CrossRef]
- Javadi, N.; Najafi, M.; Yourdkhani, S. On the role of substituent in noncovalent functionalization of graphene and organophosphor recognition: IQA and SAPT perspective. Int. J. Quantum Chem. 2017, 117, e25379. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Blanco, M.A.; Francisco, E. The nature of the hydrogen bond: A synthesis from the interacting quantum atoms picture. J. Phys. Chem. 2006, 125, 184112. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Gómez, V.A.M.; Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Hydrogen bond cooperativity and anticooperativity within the water hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566. [Google Scholar] [CrossRef]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; del Río Lima, A.; Martín Pendás, Á.; Hernández-Rodríguez, M.; Rocha Rinza, T. Hydrogen-Bond Weakening through π Systems: Resonance-Impaired Hydrogen Bonds (RIHB). Chem. Eur. J. 2017, 23, 16605–16611. [Google Scholar] [CrossRef]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. Cooperative and anticooperative effects in resonance assisted hydrogen bonds in merged structures of malondialdehyde. Phys. Chem. Chem. Phys. 2017, 19, 97–107. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Dabbagh, H.A.; Eskandari, K. Nature of intramolecular interactions of vitamin C in view of interacting quantum atoms: The role of hydrogen bond cooperativity on geometry. Phys. Chem. Chem. Phys. 2016, 18, 18278–18288. [Google Scholar] [CrossRef]
- Syzgantseva, O.A.; Tognetti, V.; Joubert, L. On the Physical Nature of Halogen Bonds: A QTAIM Study. J. Phys. Chem. A 2013, 117, 8969–8980. [Google Scholar] [CrossRef]
- Yahia-Ouahmed, M.; Tognetti, V.; Joubert, L. Intramolecular halogen bonding: An interacting quantum atoms study. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- Yahia-Ouahmed, M.; Tognetti, V.; Joubert, L. Halogen–halogen interactions in perhalogenated ethanes: An interacting quantum atoms study. Comput. Theor. Chem. 2015, 1053, 254–262. [Google Scholar] [CrossRef]
- Alkorta, I.; Silva, A.F.; Popelier, P.L.A. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation. Molecules 2020, 25, 2674. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, K.; Lesani, M. Does Fluorine Participate in Halogen Bonding? Chem. Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef]
- Madzhidov, T.I.; Chmutova, G.A.; Martín Pendás, Á. The Nature of the Interaction of Organoselenium Molecules with Diiodine. J. Phys. Chem. A 2011, 115, 10069–10077. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Ochoa-Resendiz, D.; Costales, A.; Hernández-Lamoneda, R.; Martín Pendás, Á. Halogen Bonds in Clathrate Cages: A Real Space Perspective. ChemPhysChem 2018, 19, 2512–2517. [Google Scholar] [CrossRef]
- Bartashevich, E.; Troitskaya, E.; Martín Pendás, Á.; Tsirelson, V. Understanding the bifurcated halogen bonding N…Hal…N in bidentate diazaheterocyclic compounds. Comput. Theor. Chem. 2015, 1053, 229–237. [Google Scholar] [CrossRef]
- Bora, P.L.; Novák, M.; Novotný, J.; Foroutan-Nejad, C.; Marek, R. Supramolecular Covalence in Bifurcated Chalcogen Bonding. Chem. Eur. J. 2017, 23, 7315–7323. [Google Scholar] [CrossRef]
- Niyas, M.A.; Ramakrishnan, R.; Vijay, V.; Sebastian, E.; Hariharan, M. Anomalous Halogen–Halogen Interaction Assists Radial Chromophoric Assembly. J. Am. Chem. Soc. 2019, 141, 4536–4540. [Google Scholar] [CrossRef]
- Buralli, G.; Petelski, A.; Peruchena, N.; Sosa, G.; Duarte, D. Multicenter (FX)n/NH3 Halogen Bonds (X = Cl, Br and n = 1–5). QTAIM Descriptors of the Strength of the X⋯N Interaction. Molecules 2017, 22, 2034. [Google Scholar] [CrossRef]
- Casals-Sainz, J.L.; Jiménez-Grávalos, F.; Costales, A.; Francisco, E.; Martín Pendás, Á. Beryllium Bonding in the Light of Modern Quantum Chemical Topology Tools. J. Phys. Chem. A 2018, 122, 849–858. [Google Scholar] [CrossRef]
- Casals-Sainz, J.L.; Castro, A.C.; Francisco, E.; Martín Pendás, Á. Tetrel Interactions from an Interacting Quantum Atoms Perspective. Molecules 2019, 24, 2204. [Google Scholar] [CrossRef] [PubMed]
- Madzhidov, T.I.; Chmutova, G.A. The Nature of the Interaction of Dimethylselenide with IIIA Group Element Compounds. J. Phys. Chem. A 2013, 117, 4011–4024. [Google Scholar] [CrossRef]
- Tiana, D.; Francisco, E.; Blanco, M.A.; Macchi, P.; Sironi, A.; Martín Pendás, Á. Bonding in Classical and Nonclassical Transition Metal Carbonyls: The Interacting Quantum Atoms Perspective. J. Chem. Theory Comput. 2010, 6, 1064–1074. [Google Scholar] [CrossRef]
- Tiana, D.; Francisco, E.; Blanco, M.A.; Macchi, P.; Sironi, A.; Martín Pendás, Á. Restoring orbital thinking from real space descriptions: Bonding in classical and non-classical transition metal carbonyls. Phys. Chem. Chem. Phys. 2011, 13, 5068. [Google Scholar] [CrossRef]
- Cukrowski, I.; de Lange, J.H.; Mitoraj, M. Physical Nature of Interactions in ZnII Complexes with 2, 2′-Bipyridyl: Quantum Theory of Atoms in Molecules (QTAIM), Interacting Quantum Atoms (IQA), Noncovalent Interactions (NCI), and Extended Transition State Coupled with Natural Orbitals for Chemical Valence (ETS-NOCV) Comparative Studies. J. Phys. Chem. A 2014, 118, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Chemey, A.T.; Celis-Barros, C.; Huang, K.; Sperling, J.M.; Windorff, C.J.; Baumbach, R.E.; Graf, D.E.; Páez-Hernández, D.; Ruf, M.; Hobart, D.E.; et al. Electronic, Magnetic, and Theoretical Characterization of (NH4)4UF8, a Simple Molecular Uranium(IV) Fluoride. Inorg. Chem. 2018, 58, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Tiana, D.; Francisco, E.; Macchi, P.; Sironi, A.; Martín Pendás, Á. An Interacting Quantum Atoms Analysis of the Metal–Metal Bond in [M2(CO)8]n Systems. J. Phys. Chem. A 2015, 119, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.R.; Cardoso-Gil, R.; Boucher, B.; Wagner-Reetz, M.; Sichelschmidt, J.; Gille, P.; Baenitz, M.; Grin, Y. On Fe–Fe Dumbbells in the Ideal and Real Structures of FeGa3. Inorg. Chem. 2018, 57, 12908–12919. [Google Scholar] [CrossRef]
- Jouanno, L.A.; Mascio, V.D.; Tognetti, V.; Joubert, L.; Sabot, C.; Renard, P.Y. Metal-Free Decarboxylative Hetero-Diels–Alder Synthesis of 3-Hydroxypyridines: A Rapid Access to N-Fused Bicyclic Hydroxypiperidine Scaffolds. J. Org. Chem. 2014, 79, 1303–1319. [Google Scholar] [CrossRef]
- Alkorta, I.; Montero-Campillo, M.M.; Elguero, J. Trapping CO2 by Adduct Formation with Nitrogen Heterocyclic Carbenes (NHCs): A Theoretical Study. Chem. Eur. J. 2017, 23, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Tognetti, V.; Bouzbouz, S.; Joubert, L. A theoretical study of the diastereoselective allylation of aldehydes with new chiral allylsilanes. J. Mol. Model. 2016, 23. [Google Scholar] [CrossRef] [PubMed]
- Barquera-Lozada, J.E. Torquoselectivity in Cyclobutene Ring Openings and the Interatomic Interactions That Control Them. J. Phys. Chem. A 2016, 120, 8450–8460. [Google Scholar] [CrossRef] [PubMed]
- Munarriz, J.; Velez, E.; Casado, M.A.; Polo, V. Understanding the reaction mechanism of the oxidative addition of ammonia by (PXP)Ir(i) complexes: The role of the X group. Phys. Chem. Chem. Phys. 2018, 20, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Thacker, J.C.R.; Popelier, P.L.A. An interacting quantum atom study of model SN2 reactions (X-⋯CH3X, X = F, Cl, Br, and I). J. Comput. Chem. 2017, 39, 546–556. [Google Scholar] [CrossRef]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Vallejo Narváez, W.E.; Costales, A.; Martín Pendás, Á.; Hernández-Rodríguez, M.; Rocha-Rinza, T. The bifunctional catalytic role of water clusters in the formation of acid rain. Chem. Commun. 2017, 53, 3516–3519. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Vincent, M.A.; Popelier, P.L.A. Using the Relative Energy Gradient Method with Interacting Quantum Atoms to Determine the Reaction Mechanism and Catalytic Effects in the Peptide Hydrolysis in HIV-1 Protease. Chem. Eur. J. 2018, 24, 11200–11210. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Popelier, P.L.A. Fluorine Gauche Effect Explained by Electrostatic Polarization Instead of Hyperconjugation: An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study. J. Phys. Chem. A 2018, 122, 1439–1450. [Google Scholar] [CrossRef]
- Cukrowski, I.; Sagan, F.; Mitoraj, M.P. On the Stability ofCis-andTrans-2-Butene Isomers. An Insight Based on the FAMSEC, IQA, and ETS-NOCV Schemes. J. Comput. Chem. 2016, 37, 2783–2798. [Google Scholar] [CrossRef]
- Matczak, P.; Domagała, M.; Domagała, S. Conformers of diheteroaryl ketones and thioketones: A quantum chemical study of their properties and fundamental intramolecular energetic effects. Struct. Chem. 2015, 27, 855–869. [Google Scholar] [CrossRef]
- Vishnevskiy, Y.V.; Schwabedissen, J.; Rykov, A.N.; Kuznetsov, V.V.; Makhova, N.N. Conformational and Bonding Properties of 3, 3-Dimethyl- and 6, 6-Dimethyl-1, 5-diazabicyclo[3.1.0]hexane: A Case Study Employing the Monte Carlo Method in Gas Electron Diffraction. J. Phys. Chem. A 2015, 119, 10871–10881. [Google Scholar] [CrossRef] [PubMed]
- Uhlemann, T.; Seidel, S.; Müller, C.W. Laser desorption single-conformation UV and IR spectroscopy of the sulfonamide drug sulfanilamide, the sulfanilamide–water complex, and the sulfanilamide dimer. Phys. Chem. Chem. Phys. 2017, 19, 14625–14640. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.I.; Popelier, P.L.A. Unfavorable regions in the ramachandran plot: Is it really steric hindrance? The interacting quantum atoms perspective. J. Comput. Chem. 2017, 38, 2459–2474. [Google Scholar] [CrossRef] [PubMed]
- Passmore, J.; Rautiainen, J.M. On The Lower Lewis Basicity of Siloxanes Compared to Ethers. Eur. J. Inorg. Chem. 2012, 2012, 6002–6010. [Google Scholar] [CrossRef]
- Waerder, B.; Steinhauer, S.; Bader, J.; Neumann, B.; Stammler, H.G.; Vishnevskiy, Y.V.; Hoge, B.; Mitzel, N.W. Pentafluoroethyl-substituted α-silanes: Model compounds for new insights. Dalton Trans. 2015, 44, 13347–13358. [Google Scholar] [CrossRef] [PubMed]
- Vallejo Narváez, W.E.; Jiménez, E.I.; Romero-Montalvo, E.; de la Vega, A.S.; Quiroz-García, B.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Acidity and basicity interplay in amide and imide self-association. Chem. Sci. 2018, 9, 4402–4413. [Google Scholar] [CrossRef]
- Narváez, W.E.V.; Jiménez, E.I.; Cantú-Reyes, M.; Yatsimirsky, A.K.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Stability of doubly and triply H-bonded complexes governed by acidity–basicity relationships. Chem. Commun. 2019, 55, 1556–1559. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Francisco, E.; Blanco, M. Spatial localization, correlation, and statistical dependence of electrons in atomic domains: The X1Eg+ and b3Eu+ states of H2. Chem. Phys. Lett. 2007, 437, 287–292. [Google Scholar] [CrossRef]
- Ferro-Costas, D.; Martín Pendás, Á.; González, L.; Mosquera, R.A. Beyond the molecular orbital conception of electronically excited states through the quantum theory of atoms in molecules. Phys. Chem. Chem. Phys. 2014, 16, 9249–9258. [Google Scholar] [CrossRef]
- Ferro-Costas, D.; Francisco, E.; Martín Pendás, Á.; Mosquera, R.A. How Electronic Excitation Can be Used to Inhibit Some Mechanisms Associated to Substituent Effects. ChemPhysChem 2016, 17, 2666–2671. [Google Scholar] [CrossRef]
- Jara-Cortés, J.; Guevara-Vela, J.M.; Martín Pendás, Á.; Hernández-Trujillo, J. Chemical bonding in excited states: Energy transfer and charge redistribution from a real space perspective. J. Comput. Chem. 2017, 38, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, Á.; Blanco, M.A.; Francisco, E. Steric repulsions, rotation barriers, and stereoelectronic effects: A real space perspective. J. Comput. Chem. 2009, 30, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Dillen, J. Congested molecules. Where is the steric repulsion? An analysis of the electron density by the method of interacting quantum atoms. Int. J. Quantum Chem. 2013, 113, 2143–2153. [Google Scholar] [CrossRef]
- Symons, B.C.B.; Williamson, D.J.; Brooks, C.M.; Wilson, A.L.; Popelier, P.L.A. Does the Intra-Atomic Deformation Energy of Interacting Quantum Atoms Represent Steric Energy? ChemistryOpen 2019, 8, 560–570. [Google Scholar] [CrossRef]
- Maxwell, P.; Popelier, P.L. Transferable atoms: An intra-atomic perspective through the study of homogeneous oligopeptides. Mol. Phys. 2015, 114, 1304–1316. [Google Scholar] [CrossRef]
- Maxwell, P.; di Pasquale, N.; Cardamone, S.; Popelier, P.L.A. The prediction of topologically partitioned intra-atomic and inter-atomic energies by the machine learning method kriging. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- Davie, S.J.; Pasquale, N.D.; Popelier, P.L.A. Incorporation of local structure into kriging models for the prediction of atomistic properties in the water decamer. J. Comput. Chem. 2016, 37, 2409–2422. [Google Scholar] [CrossRef]
- McDonagh, J.L.; Silva, A.F.; Vincent, M.A.; Popelier, P.L.A. Machine Learning of Dynamic Electron Correlation Energies from Topological Atoms. J. Chem. Theory Comput. 2017, 14, 216–224. [Google Scholar] [CrossRef]
- Zielinski, F.; Maxwell, P.I.; Fletcher, T.L.; Davie, S.J.; Pasquale, N.D.; Cardamone, S.; Mills, M.J.L.; Popelier, P.L.A. Geometry Optimization with Machine Trained Topological Atoms. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Wilson, A.L.; Hughes, Z.E.; Burn, M.J.; Maxwell, P.I.; Popelier, P.L.A. Towards the simulation of biomolecules: Optimisation of peptide-capped glycine using FFLUX. Mol. Simul. 2018, 44, 881–890. [Google Scholar] [CrossRef]
- Di Pasquale, N.; Davie, S.J.; Popelier, P.L.A. The accuracy of ab initio calculations without ab initio calculations for charged systems: Kriging predictions of atomistic properties for ions in aqueous solutions. J. Phys. Chem. 2018, 148, 241724. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.F.; Duarte, L.J.; Popelier, P.L.A. Contributions of IQA electron correlation in understanding the chemical bond and non-covalent interactions. Struct. Chem. 2020, 31, 507–519. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| oo Pairs | ov Pairs | vv Pairs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ● | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | |
| ○ | ● | ⋯ | ○ | ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | |
| ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | |
| ○ | ○ | ⋯ | ● | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | |
| ○ | ○ | ⋯ | ○ | ● | ● | ⋯ | ● | ○ | ○ | ⋯ | ○ | |
| ○ | ○ | ⋯ | ○ | ● | ● | ⋯ | ● | ○ | ○ | ⋯ | ○ | |
| ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | |
| ○ | ○ | ⋯ | ○ | ● | ● | ● | ● | ○ | ○ | ○ | ○ | |
| ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | |
| ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | ○ | ○ | ⋯ | ○ | |
| ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | ⋮ | ⋮ | ⋱ | ⋮ | |
| ○ | ○ | ⋯ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza , T.; Martín Pendás, Á. Interacting Quantum Atoms—A Review. Molecules 2020, 25, 4028. https://doi.org/10.3390/molecules25174028
Guevara-Vela JM, Francisco E, Rocha-Rinza T, Martín Pendás Á. Interacting Quantum Atoms—A Review. Molecules. 2020; 25(17):4028. https://doi.org/10.3390/molecules25174028
Chicago/Turabian StyleGuevara-Vela, José Manuel, Evelio Francisco, Tomás Rocha-Rinza , and Ángel Martín Pendás. 2020. "Interacting Quantum Atoms—A Review" Molecules 25, no. 17: 4028. https://doi.org/10.3390/molecules25174028
APA StyleGuevara-Vela, J. M., Francisco, E., Rocha-Rinza , T., & Martín Pendás, Á. (2020). Interacting Quantum Atoms—A Review. Molecules, 25(17), 4028. https://doi.org/10.3390/molecules25174028