Abstract
Based on the ligand H4Salen-8tBu (salen-4), a new dinuclear cobalt complex (salen-4)[Co(III)TFA]2 (salen-4 = 3,5-di-tert-butylsalicylaldehyde-3,3′-diaminobiphenylamine; TFA = trifluoroacetic acid) has been firstly synthesized and characterized. It shows high catalytic activity for the copolymerization of propylene oxide (PO) and carbon dioxide (CO2), yielding regioregular poly(propylene carbonate) (PPC) with little generation of propylene carbonate (PC) by-product. It has been found that (salen-4)[Co(III)TFA]2 shows higher activity at milder conditions, generating a polymer with maximum Mn of 293 kg/mol and a narrow molecular weight distribution PDI of 1.35. The influences of reaction time, CO2 pressure, reaction temperature, nature of the cocatalyst, catalyst dosage and substrate concentration on the molecular weight, yield and selectivity of the polymer were explored in detail. The results showed that the (salen-4)[Co(III)TFA]2/[PPN]TFA catalyst system demonstrated a remarkable TOF as high as 735 h–1. In addition, a hypothetical catalytic reaction mechanism was proposed based on density functional theory (DFT) calculations and the catalytic reaction results of the (salen-4)[Co(III)TFA]2.
1. Introduction
With the development of social industrialization, the concentration of CO2 in the atmosphere keeps rising, resulting in the greenhouse effect and a series of associated environmental disasters [1]. Nevertheless, CO2 is an inexpensive, a nontoxic, rich and special renewable C1 building block [2,3,4]. So far, wide attention has been paid to the chemical conversion of CO2 into high value-added energy, materials and chemical products. The beneficial use of CO2 including the alternating copolymerization of CO2 and epoxides to make degradable polycarbonates were popular (Scheme 1), which is considered one of the most potential green polymerization processes [5,6,7,8,9,10,11]. In addition, polycarbonates have become important topics because of their environment-friendly properties, such as atom-economy, energy-saving and degradation, as well as their several biomedical and pharmaceutical applications [12].
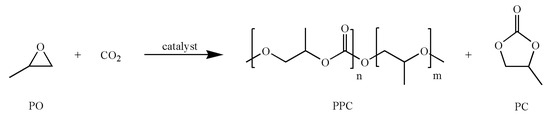
Scheme 1.
Reaction of propylene oxide (PO) and CO2 to yield poly(propylene carbonate) (PPC) and propylene carbonate (PC).
The problem is that CO2 is a highly stable and low-reactivity molecule, making it difficult to use CO2 as a reagent in a chemical reaction. Inspired by Inoue’s pioneering work, researchers have developed many catalysts [13,14,15,16,17,18,19,20,21,22,23]. Nevertheless, it is still a big challenge for scientists to develop low-cost, easily prepared and highly efficient catalysts for fixation and conversion of CO2 under mild conditions. Among them, salen metal complexes were widely used due to their easy modification, mild polymerization conditions, high activity and selectivity of poly(propylene carbonate) (PPC), excellent regioselectivity and stereoselectivity, as well as the high molecular weight with narrow molecular weight distribution PDI [24,25,26,27,28,29,30,31]. To improve the performance of salen-Co catalysts, structures with piperidinium end-capping arms, quaternary ammonium salts or other functional groups were explored [32,33,34,35,36].
Recently, dinuclear catalysts have aroused much attention. It is found that the two metal centers could work cooperatively in propylene oxide (PO) and CO2 copolymerization [37,38]. Nakano et al. synthesized a series of dinuclear salen-Co catalysts [9] and proposed a bimetallic mechanism for the dinuclear catalyst in the absence of cocatalyst. Klaus and coworkers developed a dinuclear salen-Cr complex. They found that with a suitable distance, the two metal centers showed a perfect synergy in high dilution, but a low selectivity of carbonate linkages [39]. Furthermore, in Li’s research, the optimal dinuclear catalyst showed a TOF of 254 h–1 at a [PO]/[Co] ratio of 2000, and the TOF increased to 406 h–1 at a [PO]/[Co] ratio of 20,000. In contrast, the TOF of the mononuclear catalyst system dropped from 674 h–1 to 3 h–1 upon dilution from 2500 to 20,000 [40]. Although these catalysts were impressive, they were less active and no high-molecular-weight PPC was obtained.
Previous studies have shown that reaction conditions play very important roles in binary systems [41]. Therefore, it is of great significance to explore the effect of reaction conditions on dinuclear catalytic activity. Inspired by Li’s efficient salph-co(III) catalysts [40], we firstly synthesized a new dinuclear complex (salen-4)[Co(III)TFA]2 (Figure 1). For comparison, we also synthesized a series of mononuclear complexes (salen 1-3)Co(III)X (X = Cl, DNP = 2,4-dinitrophenol, TFA) and two dinuclear complexes (salen-4)[Co(III)Cl]2, (salen-4)[Co(III)DNP]2 (Figure 1). And the influences of reaction conditions (time, pressure, temperature, cocatalyst, catalyst dosage and substrate concentration) were explored in detail. When (salen-4)[Co(III)TFA]2 was used as a catalyst for PO and CO2 copolymerization under mild conditions, it displayed excellent catalytic performance with high activity and selectivity. Furthermore, the relationship between the molecular structures and catalysis performance was analyzed based on experiments and DFT calculations, and a reaction mechanism was proposed.
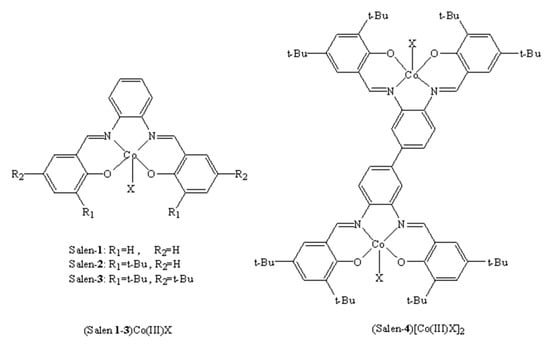
Figure 1.
(Salen)Co(III)X catalyst for PO/CO2 copolymerization (X = Cl, DNP, TFA).
2. Results and Discussion
2.1. Synthesis and Characterization of (Salen-4)[Co(III)TFA]2
Based on the ligand H4Salen-8tBu (salen-4), a new dinuclear cobalt complex (salen-4)[Co(III)TFA]2 has been firstly synthesized and characterized. The synthesis of (salen-4)[Co(III)TFA]2 was achieved as shown in Scheme 2. Reaction of 3, 5-di-tert-butylsalicylaldehyde with 3,3′-diaminobiphenylamine gave ligand salen-4. Treatment of ligand salen-4 with cobalt acetate gave bimetallic complexes (salen-4)[Co(II)]2, which when treated with trifluoroacetate under oxygen atmosphere gave the desired complex (salen-4)[Co(III)TFA]2.
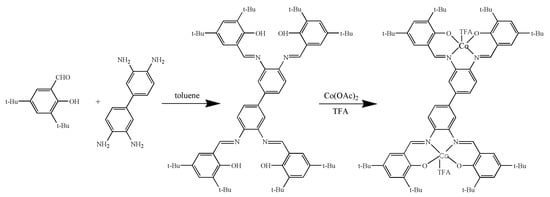
Scheme 2.
Synthesis of (salen-4)[Co(III)TFA]2.
Complex (salen-4)[Co(III)TFA]2 was characterized by FT-IR (Figure 2). As is readily observed, compared with the ligand salen-4, 2957 cm−1 and 2870 cm−1 can be considered as the stretching vibration of C-H in the methyl group on the tert-butyl; 1682 cm−1 can be considered as the characteristic absorption peak of axial ligand CF3COO−. Due to the coordination of the ligand with Co(III), the characteristic absorption peak of the 1634 cm−1 v(C=N) in salen-4 shifted to the lower wavenumber by 21 cm−1, and peaked at 1613 cm−1. Here, 1522 cm−1 and 1484 cm−1 can be considered as the skeleton vibration of the benzene, and 1387 cm−1 can be considered as the inplane bending vibration of the C-H on -CH3. Due to the drop of the hydrogen atom on Ph-OH during the coordination process, salen-4 v(C-O) stretching vibration of Ph-OH at 1272 cm−1 is shifted by 16 cm−1 to the lower wavenumber, and the peak appeared at 1253 cm−1. Here, 1175 cm−1 can be considered as the C-H inplane bending vibration of the benzene and 783 cm−1 can be considered as the out-of-plane bending vibration of C-H on the benzene. Therefore, Salen-4[Co(III)TFA]2 was successfully synthesized.
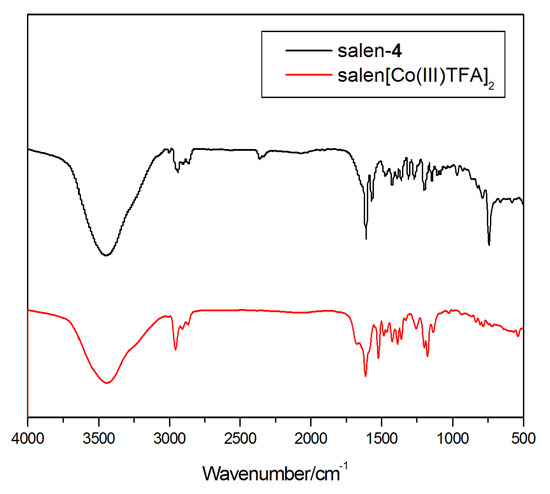
Figure 2.
IR of ligand salen-4 and complex (salen-4)[Co(III)TFA]2.
2.2. (Salen)Co(III)X Catalyzed PO/CO2 Copolymerization
The copolymerization of PO/CO2 was conducted in the presence of various catalysts, and the results are listed in Table 1. We designed a series of experiments to explore the potential catalytic performance of (salen)Co(III)X. As shown in Table 1, the catalyst and the cocatalyst had a significant effect on the copolymerization of PO/CO2. Dinuclear complex (salen-4)[Co(III)DNP]2 showed higher activity and selectivity than (salen-3)Co(III)DNP (which is one half of the dinuclear (salen-4)[Co(III)DNP]2 complexes structurally) (Entries 4 and 7, Table 1). The conversion of PO was 99%, the selectivity of PPC was 91% and TOF was 225 h–1. Importantly, with [PPN]TFA as the cocatalyst, the activity of dinuclear complex (salen-4)[Co(III)TFA]2 was much higher than that of mononuclear complex (salen-3)Co(III)TFA (Entries 5 and 8, Table 1) with TOF reaching 230 h–1, which was two times of that for (salen-3)Co(III)TFA.

Table 1.
(Salen)Co(III)X catalyzed PO/CO2 copolymerization a.
Interestingly, we found that as the number of tert-butyl groups with large steric hindrance on ligands of the complexes (Entries 1–3, Table 1) increased, the conversion of PO and selectivity of PPC both increased. With the rising of electron absorption capacity of axial groups of the complexes (Entries 3–5, Table 1), the conversion of PO increased continuously, so did the selectivity of PPC. Meanwhile, the dinuclear complexes (Entries 6–8, Table 1) showed the same results. By comparing mononuclear and dinuclear metal complexes of the same salen ligands and the same axial anions (Entries 3 and 6, 4 and 7, 5 and 8, Table 1), it was found that the conversion of PO and selectivity of PPC both increased slightly. The TOF also increased by two times, indicating that the dinuclear catalytic system was much more efficient than the mononuclear catalytic system for CO2 and PO copolymerization.
Furthermore, the optimal catalyst (salen-4)[Co(III)TFA]2 was selected. Next, the influences of reaction time, CO2 pressure, reaction temperature, cocatalyst, catalyst dosage and substrate concentration on PO/CO2 copolymerization were explored to improve the yield and selectivity of PPC.
2.2.1. Effect of Reaction Temperature
The conversion of PO increased along with the rising temperature and remained stable until it reached 25 °C. By contrast, the selectivity of PPC increased first and then decreased (Table 2). That is because the binding of PO and metal center was blocked when the temperature was too low, which lowered the activation rate of PO, further resulting in the lower conversion of PO, the selectivity of PPC and polymerization rate. By contrast, with the rising temperature, the selectivity of PPC increased first and then decreased. That is because when the temperature is too high, the product tends to generate propylene carbonate (PC) with higher thermodynamic stability. It is known that the thermodynamic stability of PPC is lower than PC, which reduces the selectivity of PPC. Therefore, the optimal polymerization temperature of (salen-4)[Co(III)TFA]2/[PPN]TFA is 25 °C.
Table 2.
(Salen-4)[Co(III)TFA]2/[PPN]TFA catalyzed PO/CO2 copolymerization at different temperatures a.
2.2.2. Effect of CO2 Pressure
The conversion of PO and selectivity of PPC increased (Table 3) as the CO2 pressure rose. When the pressure of CO2 was too low, the combination of PO and metal center was blocked, thereby lowering the activation rate of PO, the conversion of PO, the selectivity of PPC and the polymerization rate. The yield of PPC increased along with the CO2 pressure. When the CO2 pressure exceeded 3.0 MPa, the yield of PPC did not increase significantly. Given that there is a potential safety hazard in laboratory operations when the CO2 pressure is too high, thus, the optimal polymerization pressure of (salen-4)[Co(III)TFA]2/[PPN]TFA is 3.0 MPa.
Table 3.
(Salen-4)[Co(III)TFA]2/[PPN]TFA catalyzed PO/CO2 copolymerization under different pressure a.
2.2.3. Effect of Reaction Time
With the increase of reaction time, the conversion of PO increased gradually (Table 4). When the reaction time exceeded 4 h, the conversion of PO did not change much. The possible reason is that when the reaction time is too long, the polymer will adhere to the surface of the catalyst and reduces the contact area between the active center and PO, resulting in a slower catalytic rate. This indicates that the catalytic system basically reached equilibrium after 4 h. Therefore, the optimal reaction time of (salen-4)[Co(III)TFA]2/[PPN]TFA is 4 h.
Table 4.
(Salen-4)[Co(III)TFA]2/[PPN]TFA catalyzed PO/CO2 copolymerization at different reaction times a.
2.2.4. Effect of Cocatalyst
It was found that cocatalysts played very important roles in binary systems. During the CO2 copolymerization with PO, the CO2 pressure was 3.0 MPa, the reaction temperature was 25 °C, the reaction time lasted 4 h and n(PO)/n(Cat.)/n(Co-cat.) = 3000:1:1. The system with [PPN]TFA as the cocatalyst exhibited much higher catalytic activity than those with TBAB or [PPN]Cl as the cocatalyst (Table 5). Therefore, [PPN]TFA was selected as the best cocatalyst to form a two-component catalytic system.
Table 5.
(Salen-4)[Co(III)TFA]2 catalyzed PO/CO2 copolymerization under different cocatalyst a.
2.2.5. Effect of Substrate Concentration
When n(Cat.)/n(Co-cat.) = 1:1, as the molar amount of reaction substrate increased, the conversion of PO increased and then decreased. When n(PO)/n(Cat.)/n(Co-cat.) = 3000:1:1 (Entry 2, Table 6), the conversion of PO and selectivity of PPC reached the maximum. However, when n(PO)/n(Cat.)/n(Co-cat.) = 3000:0.5:1 (Entry 3, Table 6), the conversion of PO and selectivity of PPC further increased and the TOF reached 735 h–1. It is probably because the (salen-4)[Co(III)TFA]2 as a dinuclear catalyst, has two metal active centers. When n(Cat.)/n(Co-cat.) = 1:2, the TFA anion in [PPN]TFA formed a 1:1 molar ratio to the active center, which accelerated the activation of PO as well as the chain growth of polymerization reaction, thereby leading to a further increase in the conversion of PO and selectivity of PPC. Therefore, the optimal substrate concentration for (salen-4)[Co(III)TFA]2/[PPN]TFA is n(PO)/n(Cat.)/n(Co-cat.) = 3000:0.5:1. In addition, when n(PO)/n(Cat.)/n(Co-cat.) = 10,000:1:1, the (salen-4)[Co(III)TFA]2/[PPN]TFA catalytic system still performed well, whereas, the mononuclear complex (salen-3)Co(III)TFA activity would be greatly reduced or even inactivated [42].
Table 6.
(Salen-4)[Co(III)TFA]2/[PPN]TFA catalyzed PO/CO2 copolymerization at different substrate concentrations a.
2.3. Theoretical Calculations of Quantum Chemistry
In this part, (salen-4)[Co(III)TFA]2 was selected. In order to make the configuration simple and clear, the H atoms in the figure were omitted, but they were still included in the calculation process. Gaussian 16 [43] calculation program was used to calculate the (salen-4)[Co(III)TFA]2 model (Figure 3), and the DFT B3LYP method [44] was used to optimize the geometric configuration of the (salen-4)[Co(III)TFA]2 model at the level of pseudopotential group GENECP (the nonmetal atoms use the 6-31G** basis group [45] level, the Co atoms are heavy metal and the LanL2DZ basis group based on the effective core is adopted).
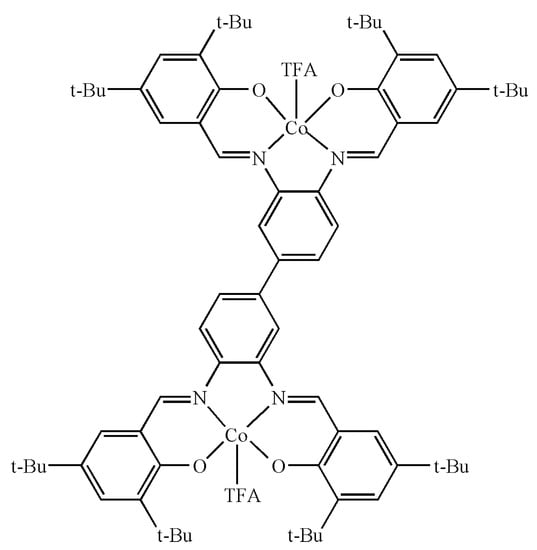
Figure 3.
Calculation model of (salen-4)[Co(III)TFA]2.
From the optimized geometry of (salen-4)[Co(III)TFA]2 (Figure 4), it can be seen that the two central metal Co atoms are in pentacoordinated structures [46] in which two N atoms and two O atoms of the ligand form the equatorial plane and the O atoms on the axial anion TFA occupy the axial position. Notably, the bond angles between the central metal Co atoms and the four coordination atoms of the ligand are about 90° or 180°, indicating that the Co atom is in a deformed planar quadrilateral field. The four bond angles formed by the metal Co atom and four coordination atoms on the ligand totals approximately about 360°, that is, the metal Co atom and the four coordination atoms are substantially in the same plane. In addition, the bond angles between the O atoms of TFA and metal Co atom, and the four coordination atoms on the ligand are about 90°. This indicates that the O atoms on the axial anion are basically perpendicular to the four coordination atoms on the ligand (Table S3).
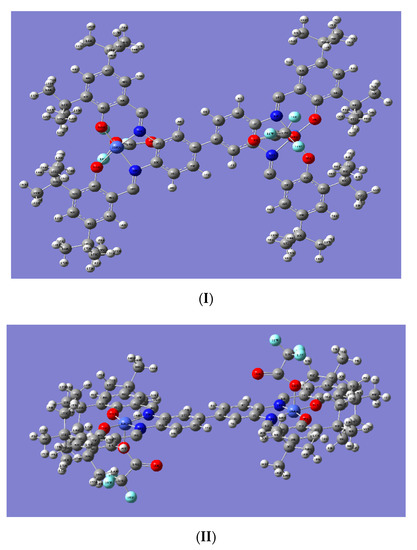
Figure 4.
Structure of optimized (salen-4)[Co(III)TFA]2; (I) is the main view, (II) is the top view.
From the energy of the frontal molecular orbitals (Table 7), it can be seen that the occupied orbitals are all negative, suggesting that the electronic state of the complexes is stable and its photoelectron spectrum is meaningful [47]. It can also be seen that the order of the LUMO orbital energy of the complexes is: (salen-4)[Co(III)TFA]2 > (salen-4)[Co(III)DNP]2 > (salen-4)[Co(III)Cl]2, but the stability of the system after receiving electrons is in the opposite order. By comparing the magnitude of the energy difference ΔE (ΔE = ELUMO − EHOMO) of the complexes, it is found that there is: (salen-4)[Co(III)Cl]2 > (salen-4)[Co(III)DNP]2 > (salen-4)[Co(III)TFA]2. The larger ΔE, the harder it is to excite the electrons of the molecule, the more stable the molecule and the weaker the activity. Therefore, the order of the ability of the complexes to accept the lone pair of electrons of O on PO is: (salen-4)[Co(III)TFA]2 > (salen-4)[Co(III)DNP]2 > (salen-4)[Co(III)Cl]2. In other words, (salen-4)[Co(III)TFA]2 has the strongest ability to activate PO, which affects the catalytic reaction activity and is consistent with the results of catalytic experiments.
Table 7.
Frontline molecular orbital energy.
2.4. Proposed Mechanism for the Catalyst
Based on the above research, we proposed mechanisms for the dinuclear catalyst systems. In previous reports, a great deal of supported that CO2 “insertion” actually occurs via a dissociative mechanism where a free nucleophilic chain end (alkoxide) attacks the CO2 reversibly to give a carbonate end that is stabilized by the charge of the metal complex [16,37,41,48,49,50,51]. As shown in Figure 5, first, the dinuclear complex interacted with the cocatalyst [PPN]Y to form two active centers (I), and the two active centers simultaneously activated the attacking PO to form a pair of hexacoordinate structures (intermediate II). Subsequently, the nucleophile Y− attacked the C atom with a smaller steric hindrance on PO. Then, CO2 was inserted into the metal alkoxide bond (intermediate III), forming the carbonate unit (intermediate IV). The growing copolymer chain could migrate to the other mental center and attack the activated PO (intermediate V), which was supposed to be the key process for the bimetallic synergism. In the chain growth stage: CO2 and PO were alternately copolymerized and M-O was broken to form poly(propylene carbonate) (VI) finally. The two active sites activated CO2 and PO at the same time and greatly improved the activation rate, which also explained the reason why the catalytic performance of the dinuclear system was significantly better than that of the mononuclear system.
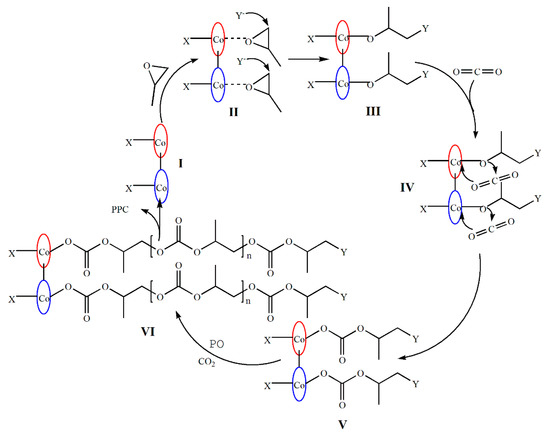
Figure 5.
Proposed mechanism of CO2/PO copolymerization with the dinuclear catalyst.
3. Materials and Methods
All reagents and solvents were analytical grade and were ready to use. The ligands H2Salen (salen-1), H2Salen-2tBu (salen-2), H2Salen-4tBu (salen-3), H4Salen-8tBu (salen-4) were synthesized following the reported procedure [41,52,53]. Propylene oxide was refluxed over a mixture of KOH/CaH2, and fractionally distilled under an argon atmosphere prior to use. Carbon dioxide (99.99%) and oxygen (99.99%) were purchased from Shaanxi Heping Glass Co., Ltd. (Shaanxi, China). and used as received. All other reagents were purchased from commercial sources and used as received. FT-IR spectra (Madison, WI, USA) were obtained on a VERTEX-70 Fourier transform infrared spectrometer with a band ranging from 4000 to 400 cm–1. The elemental analysis of complexes was performed by Elementar Germany’s Vario MICRO elemental analyzer (Hanau, Germany). The UV spectrum data of complexes were obtained by a Japan Shimadzu UV-2600 ultraviolet spectrophotometer (Kyoto, Japan). NMR spectra were recorded on a JEOL ECS400M spectrometer (Tokyo, Japan) with reference to the solvent signals.
3.1. Catalyst Preparation
3.1.1. Synthesis of Ligands
H2Salen (salen-1). Under nitrogen atmosphere, o-phenylenediamine (7.03 g, 65 mmol) was added into a three-port flask containing 100 mL anhydrous ethanol. The flask was heated to reflux. After o-phenylenediamine was completely dissolved, salicylaldehyde (1.22 g, 10 mmol) was dropped into the flask. After 12 h, the mixture was cooled to room temperature and then transferred into a clean beaker for standing. A large number of orange crystals precipitated and were collected after being filtered, washed with anhydrous ethanol and dried in vacuo. Yield (based on o-phenylenediamine): 82%. EA (%) C20H16N2O2: calc. C 75.92, H 5.11, N 8.85, found C 75.98, H 5.04, N 8.81. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 240 (0.23 × 105), 267 (0.29 × 105), 334 (0.23 × 105). 1H NMR (400 MHz, CDCl3, TMS) δ (ppm): 6.90 (t, 2H, Ar-H), 7.04 (d, 2H, Ar-H), 7.21 (q, 2H, Ar-H), 7.24 (s, 2H, Ar-H), 7.32 (q, 2H, Ar-H), 7.35 (s, 2H, Ar-H), 8.61 (s, 2H, N=C-H), 13.04 (s, 2H, -OH). IR (KBr, v, cm–1): 3448(s), 2930(w), 2780(w), 1630(s), 1566(m), 1478(s), 1388(s), 1278(s), 1180(s), 762(s).
H2Salen-2tBu (salen-2). Salen-2 was obtained with the similar procedure for salen-1 except that salicylaldehyde was replaced by 3-tert-butylsalicylaldehyde (1.78 g, 10 mmol). Yield (based on o-phenylenediamine): 79%. EA (%) C28H32N2O2: calc. C 78.46, H 7.54, N 6.53, found C 78.51, H 7.52, N 6.48. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 241 (0.24 × 105), 275 (0.31 × 105), 366 (0.24 × 105). 1H NMR (400 MHz, CDCl3, TMS) δ (ppm): 1.43 (t, 18H, t-Bu), 6.85 (t, 2H, -OH), 7.23 (t, 2H, Ar-H), 7.25 (s, 2H, Ar-H), 7.31 (d, 2H, Ar-H), 7.34 (t, 2H, Ar-H), 7.39 (d, 2H, Ar-H), 8.66 (s, 2H, N=C-H). IR (KBr, v, cm–1): 3450(s), 3010(w), 2948(m), 2867(w), 1628(s), 1566(s), 1482(m), 1432(m), 1368(m), 1268(s), 1191(s), 753(s).
H2Salen-4tBu (salen-3). Salen-3 was obtained with the similar procedure for salen-1 except that salicylaldehyde was replaced by 3, 5-di-tert-butylsalicylaldehyde (2.34 g, 10 mmol). Yield (based on o-phenylenediamine): 75%. EA (%) C36H48N2O2: calc. C 79.94, H 8.96, N 5.18, found C 80.01, H 9.01, N 5.16. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 242 (0.23 × 105), 279 (0.22 × 105), 338 (0.16 × 105). 1H NMR (400 MHz, CDCl3, TMS) δ (ppm): 1.33 (t, 18H, t-Bu), 1.44 (t, 18H, t-Bu), 7.21 (d, 2H, -OH), 7.22 (t, 2H, Ar-H), 7.25 (d, 2H, Ar-H), 7.31 (t, 2H, Ar-H), 7.44 (d, 2H, Ar-H), 8.67 (s, 2H, N=C-H). IR (KBr, v, cm–1): 3452(s), 2961(m), 2866(w), 1634(s), 1566(w), 1477(w), 1438(w), 1382(s), 1272(m), 1172(s), 753(m).
H4Salen-8tBu (salen-4). Under nitrogen atmosphere, 3, 3′-diaminobenzidine (2.5 g, 11.67 mmol), 3, 5-di-tert-butylsalicylaldehyde (15 g, 64.01 mmol) and p-toluenesulfonic acid (0.1 g, 0.58 mmol) were added into a 1000 mL flask. Freshly distilled toluene was added to set up an azeotropic dewatering device. After 72 h, the solvent was removed in vacuo and solids were washed with ethanol. The mixture was recrystallized by dissolving it in methylene chloride and tetrahydrofuran, respectively. A high-purity ligand was obtained. Yield (based on 3, 3′-diaminobenzidine): 60%. EA (%) C72H94N4O4: calc. C 80.09, H 8.79, N 5.19, found C 80.14, H 8.86, N 5.12. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 230 (0.95 × 105), 281 (0.58 × 105), 365 (0.54 × 105). 1H NMR (400 MHz, CDCl3, TMS) δ (ppm): 1.32 (s, 36H, t-Bu), 7.15~7.62 (m, 50H, Ar-H), 8.44 (s, 4H, N=C-H), 11.51 (s, 4H, -OH). IR (KBr, v, cm–1): 3448(s), 2953(s), 2864(w), 1635(s), 1580(w), 1471(m), 1432(m), 1384(m), 1269(w), 1168(s), 768(w).
3.1.2. Synthesis of Complexes
(Salen-1)Co(III)Cl. The ligand salen-1 (0.07 g, 0.22 mmol) and decrystallized water Co(OAc)2 (0.05 g, 0.27 mmol) were dissolved in 10 mL anhydrous methanol. The mixture was stirred at room temperature for 12 h, and then anhydrous LiCl (0.05 g, 1.10 mmol) was added and passed in oxygen to continue the reaction for 12 h. Solvent was removed under reduced pressure and the residue was dissolved in CH2Cl2. The organic layer was rinsed with saturated aqueous Na2SO4 (50 mL × 3) and NaCl (50 mL × 3), respectively. The organic layer was dried over Na2SO4, filtered and dried in vacuo. The crude product was recrystallized from CH2Cl2 and anhydrous CH3CH2OH to obtain a brown-red solid [54]. Yield: 82%. EA (%) C20H14CoN2O2Cl: calc. C 58.77, H 3.45, N 6.85, found C 58.83, H 3.48, N 6.81. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 254 (0.41 × 105), 369 (0.11 × 105), 387 (0.11 × 105), 473 (0.08 × 105). IR (KBr, v, cm–1): 3440(s), 2920(w), 2850(w), 1614(s), 1560(m), 1477(s), 1386(s), 1269(s), 1145(s), 762(s).
(Salen-2)Co(III)Cl. (Salen-2)Co(III)Cl was obtained with the similar procedure for (salen-1)Co(III)Cl except that ligand salen-1 was replaced by ligand salen-2 (0.09 g, 0.22 mmol). Yield: 75%. EA (%) C28H30CoN2O2Cl: calc. C 64.56, H 5.80, N 5.38, found C 64.49, H 5.73, N 5.43. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 260 (0.40 × 105), 369 (0.14 × 105), 391 (0.16 × 105), 508 (0.07 × 105). IR (KBr, v, cm–1): 3440(s), 2949(m), 2863(w), 1613(s), 1527(s), 1489(m), 1387(s), 1261(w), 1145(s), 741(s).
(Salen-3)Co(III)Cl. (Salen-3)Co(III)Cl was obtained with the similar procedure for (salen-1)Co(III)Cl except that ligand salen-1 was replaced by ligand salen-3 (0.12 g, 0.22 mmol). Yield: 72%. EA (%) C36H46CoN2O2Cl: calc. C 68.29, H 7.32, N 4.42, found C 68.23, H 7.40, N 4.39. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 264 (0.27 × 105), 369 (0.11 × 105), 411 (0.08 × 105), 513 (0.05 × 105). IR (KBr, v, cm–1): 3450(s), 2955(s), 2860(w), 1610(s), 1527(s), 1489(w), 1387(s), 1261(w), 1145(w), 743(s).
(Salen-3)Co(III)DNP. The ligand salen-3 (1.00 g, 1.85 mmol) and Co(OAc)2.4H2O (0.33 g, 1.3 mmol) were dissolved in 100 mL absolute ethanol. The mixture was heated to 80 °C for 30 min and then cooled to room temperature. The solvent was removed in vacuo and washed with cold methanol (15 mL × 3). The solid was dissolved in 10 mL CH2Cl2 and recrystallized by 200 mL n-hexane. After 24 h, a dark red crystal (salen-4)Co(II) was obtained. The (salen-4)Co(II) (0.07 g, 0.12 mmol) was dissolved in 50 mL CH2Cl2 and 2, 4-dinitrophenol sodium (0.02 g, 0.12 mmol) was added. The resulting solution was stirred for 2 h at room temperature in an oxygen atmosphere. Solvent was removed in vacuo to obtain the black crude product. The crude product was recrystallized from CH2Cl2 and n-hexane, respectively, and then dried in vacuo at 60 °C for 24 h [40]. Yield: 71%. EA (%) C42H49CoN4O7: calc. C 64.61, H 6.33, N 7.18, found C 64.53, H 6.29, N 7.14. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 263 (0.48 × 105), 321 (0.33 × 105), 430 (0.30 × 105), 533 (0.17 × 105). IR (KBr, v, cm–1): 3450(s), 2955(m), 2866(w), 1615(s), 1572(w), 1521(s), 1457(m), 1387(s), 1257(s), 1170(s), 753(w).
(Salen-3)Co(III)TFA. (Salen-3)Co(III)Cl (0.08 g, 0.12 mmol) and CF3COOAg (0.03 g, 0.12 mmol) were dissolved in 15 mL CH2Cl2. Under oxygen atmosphere, the mixture was stirred for 24 h in the dark. Solvent was removed by vacuum filtration. A red-black solid was obtained and recrystallized from CH2Cl2 and n-hexane, respectively [55]. Yield: 73%. EA (%) C38H46CoN2O4F3: calc. C 64.22, H 6.52, N 3.94, found C 64.29, H 6.61, N 4.01. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 263 (0.40 × 105), 308 (0.14 × 105), 401 (0.16 × 105), 506 (0.07 × 105). IR (KBr, v, cm–1): 3450(s), 2955(s), 2866(m), 1687(m), 1628(s), 1526(s), 1459(s), 1387(s), 1252(s), 1182(s), 753(m).
(Salen-4)[Co(III)Cl]2. The ligand salen-4 (0.24 g, 0.22 mmol) was dissolved in 100 mL anhydrous methanol. The mixture was heated to reflux at 65 °C. After the ligand was completely dissolved, decrystallized water Co(OAc)2 (0.09 g, 0.50 mmol) was dissolved in. Under oxygen atmosphere, anhydrous LiCl (0.02 g, 0.50 mmol) was added. After 12 h, the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 and recrystallized by anhydrous CH3CH2OH. The resulting solution was filtered and washed to obtain a brown-red solid [40]. Yield: 82%. EA (%) C72H90Co2N4O4Cl2: calc. C 68.40, H 7.18, N 4.43, found C 68.46, H 7.23, N 4.49. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 232 (1.02 × 105), 268 (0.72 × 105), 421 (0.17 × 105), 513 (0.12 × 105). IR (KBr, v, cm–1): 3440(s), 2955(s), 2859(w), 1610(s), 1515(s), 1489(m), 1381(s), 1259(s), 1171(s), 761(m).
(Salen-4)[Co(III)DNP]2. Under nitrogen atmosphere, the ligand salen-4 (2.16 g, 2.0 mmol) was dissolved in 20 mL CH2Cl2, and Co(OAc)2 (0.72 g, 4.04 mmol) was dissolved in 80 mL anhydrous methanol. The mixture was stirred for 3 h at room temperature to obtain the (salen-4)Co(II) complex. Under oxygen atmosphere, (salen-4)Co(II) (1.20 g, 1.0 mmol) was dissolved in 50 mL CH2Cl2, and 2, 4-dinitrophenol sodium (0.41 g, 2.0 mmol) was added. The mixture was stirred for 3 h. Solvent was removed in vacuo to obtain the crude product. The crude product was recrystallized from CH2Cl2 and n-hexane, respectively to obtain a dark green solid [40]. Yield: 76%. EA (%) C42H49Co2N4O7: calc. C 68.69, H 6.20, N 7.18, found C 68.60, H 6.13, N 7.09. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 235 (1.07 × 105), 270 (1.00 × 105), 418 (0.47 × 105), 533 (0.26 × 105). IR (KBr, v, cm–1): 3443(s), 2955(s), 2868(w), 1614(s), 1573(m), 1522(s), 1483(m), 1386(s), 1253(s), 1197(s), 783(m).
(Salen-4)[Co(III)TFA]2. (Salen-3)Co(III)TFA (0.09 g, 0.12 mmol) and CF3COOAg (0.06 g, 0.25 mmol) were dissolved in 15 mL CH2Cl2. Under oxygen atmosphere, the mixture was stirred for 24 h in dark and filtered to remove the insoluble substances. Solvent was removed in vacuo to obtain a red-black solid. The solid was recrystallized from CH2Cl2 and n-hexane, respectively. Yield: 68%. EA (%) C76H90Co2N4O8F6: calc. C 64.31, H 6.39, N 3.95, found C 64.41, H 6.32, N 3.88. UV-Vis (CH3Cl3) λmax/nm (ε/L.mol–1.cm–1): 231 (1.01 × 105), 269 (0.68 × 105), 425 (0.22 × 105), 519 (0.17 × 105). 1H NMR (400 MHz, CDCl3, TMS) δ (ppm): 1.33 (s, 36H, t-Bu), 1.43 (s, 36H, t-Bu), 6.99 (s, 6H, Ar-H), 7.26 (s, 4H, Ar-H), 7.34 (s, 4H, Ar-H), 7.79 ((s, 4H, C = N-H). IR (KBr, v, cm–1): 3453(s), 2957(s), 2870(w), 1682(w), 1613(s), 1522(s), 1484(m), 1387(s), 1253(s), 1175(s), 783(m).
3.2. Catalytic Procedure
The catalyst, cocatalyst and epoxide were sequentially added into a 100 mL high-pressure reactor, and then the reactor was pressurized to a required pressure with CO2. The mixture was heated and stirred for a while. After completion of the reaction, unreacted CO2 was evacuated to remove the pressure. A small aliquot of the resultant polymerization mixture was removed from the reactor for 1H NMR and GPC analysis. The remaining polymerization mixture was then dissolved in tetrahydrofuran, quenched with 5% HCl solution in absolute ethanol, and precipitated from absolute ethanol (30 mL). The polymer was collected and dried in vacuo to constant weight, and the polymer yield was determined.
4. Conclusions
In summary, we have compared the mononuclear and dinuclear catalysts systems in PO and CO2 copolymerization. The dinuclear catalyst system has higher activity, higher selectivity and PPC with higher molecular weight. A mechanism based on the conjugated dinuclear structure was proposed. At the same time, the effects of reaction temperature, CO2 pressure, reaction time, catalyst’s type and substrate concentration on the dinuclear catalytic system were studied. The DFT method was used to optimize the geometries of the complexes. Moreover, the stable structure of the complexes was further recognized in theory and its effect on the catalytic reaction was analyzed. Furthermore, an ideal catalyst model for PO/CO2 copolymerization was constructed. The greater the number of tert-butyl groups on the ligand and the greater the anion electronegativity of the axial ligand, the higher the catalytic activity of the complex. The activity of the catalysts is expected to be further improved by changing the steric hindrances and electronic structures of the ligands, which will be further studied in the future.
Supplementary Materials
The following are available online. Figures S1–S4: Infrared spectrum characterization of ligands and complexes, Figures S5–S8: Ultraviolet spectrum characterization of ligands and complexes, Figures S9–S12: 1H NMR spectrum characterization of ligands and complexes, Figures S13–S17: Characterization of the crude product PPC, Table S1: GPC of the crude product PPC, Table S2: Bond lengths [Å] and angle [°] after optimizing for (salen-4)[Co(III)TFA]2, Table S3: NBO charges distribution of some atoms in complex (salen-4)[Co(III)X]2.
Author Contributions
Data curation, K.-Y.Z.; formal analysis, K.-Y.Z., X.-G.J., and L.W.; funding acquisition, W.-Z.W. and L.-L.L.; investigation, W.F. and L.X.; methodology, K.-Y.Z.; project administration, W.-Z.W.; software, X.-G.J.; writing—original draft, K.-Y.Z.; writing—review and editing, W.-Z.W., L.W. and L.-L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Shaanxi Province, China (No: 2019JZ-44; 2017JQ2009; 18JK0607).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sanz-Perez, E.S.; Murdock, C.R.; Didas, S.A. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11877. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, S.H.; Ebrahimiasl, S.; Arshad, S.; Hosseinian, A. Synthesis of six-membered cyclic carbamates employing CO2 as building block: A review. J. CO2 Util. 2019, 33, 37–45. [Google Scholar] [CrossRef]
- Chen, Q.; Li, S.; Xu, H. Co-MOF as an electron donor for promoting visible-light photoactivities of g-C3N4 nanosheets for CO2 reduction. Chin. J. Catal. 2020, 41, 514–523. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Guo, L. Solar-heating boosted catalytic reduction of CO2 under full-solar spectrum. Chin. J. Catal. 2020, 41, 131–139. [Google Scholar] [CrossRef]
- Jessop, I.A.; Chong, A.; Graffo, L. Synthesis and Characterization of a 2,3-Dialkoxynaphthalene-Based Conjugated Copolymer via Direct Arylation Polymerization (DAP) for Organic Electronics. Polymers 2020, 12, 1377. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Y.Q.; Wang, H. Novel Conjugated Polymers Prepared by Direct (Hetero) arylation: An Eco-Friendly Tool for Organic Electronics. Molecules 2018, 23, 408. [Google Scholar] [CrossRef]
- Albéniz, A.C.; Carrera, N. Polymers for Green C–C Couplings. Eur. J. Inorg. Chem. 2011, 2011, 2347–2360. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. Acyl Sonogashira Cross-Coupling: State of the Art and Application to the Synthesis of Heterocyclic Compounds. Catalysts 2020, 10, 25. [Google Scholar] [CrossRef]
- Vogt, C.G.; Grätz, S.; Lukin, S. Direct Mechanocatalysis: Palladium as Milling Media and Catalyst in the Mechanochemical Suzuki Polymerization. Angew. Chem. Int. Ed. 2019, 58, 18942–18947. [Google Scholar] [CrossRef]
- Pucci, A.; Albano, G.; Pollastrini, M. Supported Tris-Triazole Ligands for Batch and Continuous-Flow Copper-Catalyzed Huisgen 1,3-Dipolar Cycloaddition Reactions. Catalysts 2020, 10, 434. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, B.; Zhu, C. Introducing mercaptoacetic acid locking imine reaction into polymer chemistry as a green click reaction. Polym. Chem. 2014, 5, 2695–2699. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Jin, L.; Jing, H.; Chang, T. Metal porphyrin/phenyltrimethyl ammonium tribromide: High efficient catalysts for coupling reaction of CO2 and epoxides. J. Mol. Catal. A Chem. 2007, 261, 262–266. [Google Scholar] [CrossRef]
- Fan, W.; Wang, W.Z.; Wang, L. Novel Cobalt Complex as an Efficient Catalyst for Converting CO2 into Cyclic Carbonates under Mild Conditions. Catalysts 2019, 9, 951. [Google Scholar] [CrossRef]
- Allen, S.D.; Moore, D.R.; Lobkovsky, E.B. High-Activity, Single-Site Catalysts for the Alternating Copolymerization of CO2 and Propylene Oxide. J. Am. Chem. Soc. 2002, 124, 14284–14285. [Google Scholar] [CrossRef]
- Nakano, K.; Hashimoto, S.; Nozaki, K. Bimetallic mechanism operating in the copolymerization of propylene oxide with carbon dioxide catalyzed by cobalt–salen complexes. Chem. Sci. 2010, 1, 369–373. [Google Scholar] [CrossRef]
- Sugimoto, H.; Ohtsuka, H.; Inoue, S. Alternating copolymerization of carbon dioxide and epoxide catalyzed by an aluminum Schiff base-ammonium salt system. J. Poly. Sci. Part A Polym. Chem. 2005, 43, 4172–4186. [Google Scholar] [CrossRef]
- Guo, J.T.; Wang, X.Y.; Xu, Y.S. Copolymerizations of carbon dioxide and epoxides in the presence of rare earth coordinate catalyst. J. Appl. Polym. Sci. 2003, 87, 2356–2359. [Google Scholar] [CrossRef]
- Lu, X.B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
- Zhong, X.; Dehghani, F. Solvent free synthesis of organometallic catalysts for the copolymerisation of carbon dioxide and propylene oxide. Appl. Catal. B Environ. 2010, 98, 101–111. [Google Scholar] [CrossRef]
- Wan, Y.; Qin, Y.; Wang, X. Trivalent Titanium Salen Complex: Thermally Robust and Highly Active Catalyst for Copolymerization of CO2 and Cyclohexene Oxide. ACS Catal. 2015, 5, 393–396. [Google Scholar]
- Childers, M.I.; Longo, J.M.; Van Zee, N.J. Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev. 2014, 114, 8129–8152. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J. Salen Metal Complexes as Catalysts for the Synthesis of Polycarbonates from Cyclic Ethers and Carbon Dioxide. Adv. Polym. Sci. 2012, 245, 1–27. [Google Scholar]
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Wu, G.P. A One-Pot Synthesis of a Triblock Copolymer from Propylene Oxide/Carbon Dioxide and Lactide: Intermediacy of Polyol Initiators. Angew. Chem. Int. Ed. 2013, 52, 10602–10606. [Google Scholar] [CrossRef]
- Decortes, A.; Castilla, A.M.; Kleij, A.W. Salen-Complex-Mediated Formation of Cyclic Carbonates by Cycloaddition of CO2 to Epoxides. Angew. Chem. Int. Ed. 2010, 49, 9822–9837. [Google Scholar] [CrossRef]
- Lu, X.B.; Ren, W.M.; Wu, G.P. CO2 Copolymers from Epoxides: Catalyst Activity, Product Selectivity, and Stereochemistry Control. Acc. Chem. Res. 2012, 45, 1721–1735. [Google Scholar] [CrossRef]
- Wu, G.P.; Ren, W.M.; Luo, Y. Enhanced Asymmetric Induction for the Copolymerization of CO2 and Cyclohexene Oxide with Unsymmetric Enantiopure SalenCo(III) Complexes: Synthesis of Crystalline CO2-Based Polycarbonate. J. Am. Chem. Soc. 2012, 134, 5682–5688. [Google Scholar] [CrossRef]
- Qin, Z.; Thomas, C.M.; Lee, S. Cobalt-Based Complexes for the Copolymerization of Propylene Oxide and CO2: Active and Selective Catalysts for Polycarbonate Synthesis. Angew. Chem. Int. Ed. 2003, 42, 5484–5487. [Google Scholar] [CrossRef]
- Cohen, C.T.; Chu, T.; Coates, G.W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127, 10869–10878. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Poater, A.; Childers, M.I. Enantioselective Polymerization of Epoxides Using Biaryl-Linked Bimetallic Cobalt Catalysts: A Mechanistic Study. J. Am. Chem. Soc. 2013, 135, 18901–18911. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Lehenmeier, M.W.; Anderson, C.E. Recent advances in CO2/epoxide copolymerization-New strategies and cooperative mechanisms. Coord. Chem. Rev. 2011, 255, 1460–1479. [Google Scholar] [CrossRef]
- Noh, E.K.; Na, S.J.; Kim, S.W. Two Components in a Molecule: Highly Efficient and Thermally Robust Catalytic System for CO2/Epoxide Copolymerization. J. Am. Chem. Soc. 2007, 129, 8082–8083. [Google Scholar] [CrossRef] [PubMed]
- Sujith, S.; Min, J.K.; Seong, J.E. A Highly Active and Recyclable Catalytic System for CO2/Propylene Oxide Copolymerization. Angew. Chem. Int. Ed. 2008, 47, 7306–7309. [Google Scholar]
- Ren, W.M.; Zhang, X.; Liu, Y. Highly Active, Bifunctional Co(III)-Salen Catalyst for Alternating Copolymerization of CO2 with Cyclohexene Oxide and Terpolymerization with Aliphatic Epoxides. Macromolecules 2010, 43, 1396–1402. [Google Scholar] [CrossRef]
- Wu, G.P.; Wei, S.H.; Ren, W.M. Perfectly Alternating Copolymerization of CO2 and Epichlorohydrin Using Cobalt(III)-Based Catalyst Systems. J. Am. Chem. Soc. 2011, 133, 15191–15199. [Google Scholar] [CrossRef]
- Kember, M.R.; Jutz, F.; Buchard, A. Di-cobalt(II) catalysts for the copolymerisation of CO2 and cyclohexene oxide: Support for a dinuclear mechanism? Chem. Sci. 2012, 3, 1245–1255. [Google Scholar] [CrossRef]
- Meléndez, J.; North, M.; Villuendas, P. One-component bimetallic aluminium(salen)-based catalysts for cyclic carbonate synthesis and their immobilization. Dalton Trans. 2011, 40, 3885–3902. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Vagin, S.I.; Lehenmeier, M.W.; Deglmann, P.; Brym, A.K.; Rieger, B. Kinetic and Mechanistic Investigation of Mononuclear and Flexibly Linked Dinuclear Complexes for Copolymerization of CO2 and Epoxides. Macromolecules 2011, 44, 9508–9516. [Google Scholar] [CrossRef]
- Li, X.; Duan, R.; Pang, X. Rigid linked dinuclear salph-co(III) catalyst for carbondioxide/epoxides copolymerization. Appl. Catal. B Environ. 2016, 182, 580–586. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Mackiewicz, R.M. Role of the Cocatalyst in the Copolymerization of CO2 and Cyclohexene Oxide Utilizing Chromium Salen Complexes. J. Am. Chem. Soc. 2005, 127, 14026–14038. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Chen, X.; Zheng, Y. One-pot atom-efficient synthesis of bio-renewable polyesters and cyclic carbonates through tandem catalysis. Chem. Commun. 2015, 51, 8504–8507. [Google Scholar] [CrossRef]
- Stepanian, S.G.; Adamowicz, L. Rich structural topology of the anion formed by the complex of acetonitrile with two water molecules. Chem. Phys. Lett. 2020, 740, 137082–137087. [Google Scholar] [CrossRef]
- De Souza, K.M.N.; de Carvalho, L.P.; da Silva, J.A.B. On the structures of dinuclear symmetric lanthanide complexes and the selectivity towards heterodinuclear complexes based on molecular modeling. Inorg. Chim. Acta 2019, 494, 65–73. [Google Scholar] [CrossRef]
- Xia, L.; Wang, W.Z.; Liu, S. New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides. Molecules 2018, 23, 2304. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Das, L.K.; Drew, M.G.B. Synthesis, Crystal Structures, Magnetic Properties and Catecholase Activity of Double Phenoxido-Bridged Penta-Coordinated Dinuclear Nickel(II) Complexes Derived from Reduced Schiff-Base Ligands: Mechanistic Inference of Catecholase Activity. Inorg. Chem. 2012, 51, 7993–8001. [Google Scholar] [CrossRef] [PubMed]
- Aihara, J. Reduced HOMO-LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Decortes, A.; Belmonte, M.M.; Benet-Buchholz, J.; Kleij, A.W. Efficient carbonate synthesis under mild conditions through cycloaddition of carbon dioxide to oxiranes using a Zn(salphen) catalyst. Chem. Commun. 2010, 46, 4580–4582. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, W.M.; Liu, C.; Fu, S.; Wang, M.; He, K.K.; Li, R.R.; Zhang, R.; Lu, X.B. Mechanistic Understanding of Dinuclear Cobalt(III) Complex Mediated Highly Enantioselective Copolymerization of meso-Epoxides with CO2. Macromolecules 2014, 47, 7775–7788. [Google Scholar] [CrossRef]
- Fu, X.; Jing, H. Quaternary onium modified SalenCoXY catalysts for alternating copolymerization of CO2 and propylene oxide: A kinetic study. J. Catal. 2015, 329, 317–324. [Google Scholar] [CrossRef]
- Lehenmeier, M.W.; Kissling, S.; Altenbuchner, P.T.; Bruckmeier, C.; Deglmann, P.; Brym, A.K.; Rieger, B. Flexibly Tethered Dinuclear Zinc Complexes: A Solution to the Entropy Problem in CO2/Epoxide Copolymerization Catalysis? Angew. Chem. Int. Ed. 2013, 52, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Peretti, K.L.; Ajiro, H.; Cohen, C.T. A Highly Active, Isospecific Cobalt Catalyst for Propylene Oxide Polymerization. J. Am. Chem. Soc. 2005, 127, 11566–11567. [Google Scholar] [CrossRef] [PubMed]
- Curreli, S.; Escudero-Adán, E.C.; Benet-Buchhlz, J.; Kleij, A.W. Facile Isolation of Bisimines Based on 3,3‘-Diaminobenzidine: Direct Access to Unsymmetrical Bimetallic Salphen Building Blocks. J. Org. Chem. 2007, 72, 7018–7021. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Moore, D.R. Discrete Metal-Based Catalysts for the Copolymerization of CO2 and Epoxides: Discovery, Reactivity, Optimization, and Mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Brandenburg, M. Catalytic Asymmetric Addition of Carbon Dioxide to Propylene Oxide with Unprecedented Enantioselectivity. Org. Lett. 2006, 8, 4401–4404. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).